 Jiashen Tan
Jiashen Tan Weiqin Zhu1†
Weiqin Zhu1† Zhuofeng Ke
Zhuofeng Ke Yan Liu
Yan Liu- 1School of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou, China
- 2PCFM Lab, School of Materials Science and Engineering, Sun Yat-sen University, Guangzhou, China
- 3Graduate School of Pharmaceutical Sciences, Kyoto University, Kyoto, Japan
A binary hybrid system comprising a hypervalent iodine(III) reagent and BF3•OEt2 Lewis acid was found to be effective for the diastereoselective α-acetoxylation of cyclic ketones. In this hybrid system, BF3•OEt2 Lewis acid allowed the activation of the hypervalent iodine(III) reagent and cyclic ketones for smooth α-acetoxylation reaction, achieving high diastereoselectivity. This hypervalent iodine-mediated α-acetoxylation of the cyclic ketone reaction plausibly undergoes an SN2 substitution mechanism via an α-C-bound hypervalent iodine intermediate. The diastereoselectivity of the reaction mainly originates from thermodynamic control.
Introduction
The direct α-acetoxylation of ketones provides an efficient and synthetically practical method for α-oxygen functionalization of carbonyl compounds, which exhibits various applications as synthetic key precursors and versatile synthons in organic synthesis (Mizukami et al., 1978; Hecker and Werner, 1993; Varma et al., 1998; Kaila et al., 2007; Edwards et al., 2008; Richter et al., 2018; Huang et al., 2019). Therefore, considerable efforts have been made in the development of synthetic protocols for α-acetoxylation of ketones (Scheme 1). Traditional methods to prepare α-acetoxy ketones are the acetolysis of α-diazo ketone (Newman and Beal, 1950; Erickson et al., 1951; Corey and Knapp, 1976; Kitamura et al., 2012; Wang et al., 2014; Tan et al., 2016; Yuan et al., 2016; Zhang et al., 2016; Hu et al., 2018), α-bromo ketone (Tanner et al., 1991; Valgimigli et al., 2003; Ahmed and Langer, 2006; Chen et al., 2013, 2016; Nolla-Saltiel et al., 2014; Carneiro et al., 2015; Liu et al., 2016; Yuan et al., 2019), and in situ generated α-iodo derivatives (Du et al., 2015; Ren et al., 2016; Chen et al., 2017; Tan et al., 2017; Pogaku et al., 2019), which usually require the pre-functionalization of ketones (Scheme 1A). In contrast, umpolung reactions have taken a prominent place for the direct α-oxygenation of carbonyl compounds via oxidation of the enolates or related derivatives with transition metal complexes (Littler, 1962; Heiba and Dessau, 1971; Ng and Henry, 1976; Rubottom et al., 1983; El-Qisairi and Qaseer, 2002; Hamed et al., 2012), or hypervalent iodine reagents (Mizukami et al., 1978; Nicolaou et al., 2002; Bogevig et al., 2004; Sunden et al., 2004; Ochiai et al., 2005; Huang et al., 2007; Yu et al., 2010; Izquierdo et al., 2016; Arava et al., 2017) (Scheme 1B). Metal-free hypervalent iodine compounds as classical oxidative functionalization reagents have attracted considerable interest due to a variety of advantages relative to conventional oxidants such as heavy metals (Pb, Tl, or Hg), including low toxicity, ready availability, mild conditions, excellent selectivity, and a comparable reactivity (Du et al., 2015; Ren et al., 2016; Chen et al., 2017). However, efficient and practical methods for the diastereoselective α-acetoxylation of cyclic ketones have not yet been well-developed.
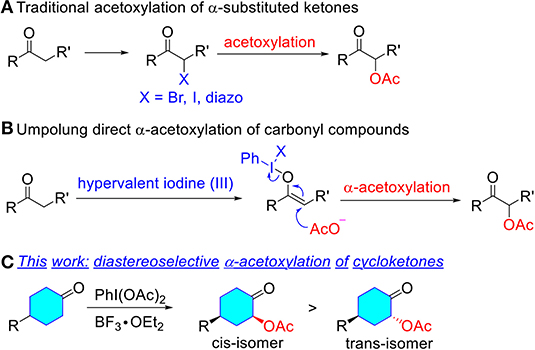
Scheme 1. Different methods for the α-acetoxylation reaction of ketones. (A) Traditional acetoxylation of α-substituted ketones. (B) Umpolung direct α-acetoxylation of carbonyl compounds. (C) This work: diastereoselective α-acetoxylation of cycloketones.
As one of our ongoing interests (Sakamoto et al., 2016, 2017, 2018; Liu et al., 2017; Selvakumar et al., 2017; Shu et al., 2019) to construct selective methods for α-functionalization of carbonyl compounds, we have become keenly interested in the possibility of the diastereoselective α-acetoxylation of cyclic ketones (Scheme 1C). In this context, we wish to report our initial study on creating a hybrid system comprised of hypervalent iodine(III) reagent and BF3•OEt2.
Results and Discussion
We began our study on the diastereoselective α-acetoxylation reaction of cyclic ketone with hypervalent iodine(III) reagent and certain acid as an activator. As shown in Table 1, the reaction of 4-phenylcyclohexanone with (diacetoxyiodo)benzene in acetic acid solvent at room temperature for 24 h resulted in almost recovery of the starting ketone (Table 1, entry 1). In the presence of additives such as CF3CO2H, α-acetoxylation product 1 was obtained in low yields (Table 1, entry 2), whereas the use of Lewis acids ZnCl2 and AlCl3 as additives resulted in no target product even though 4-phenylcyclohexanone was fully consumed under the applied reaction conditions (Table 1, entries 5 and 6). Fortunately, addition of Brønsted acid TfOH (Table 1, entry 3), and Lewis acids AgOTf, Sc(OTf)3, Mg(OTf)2, Cu(OTf)2, Zn(OTf)2, and BF3•OEt2 (Table 1, entries 4, 7–11) improved the yield of the product 1. BF3•OEt2 gave rise to desired cis-isomer of α-acetoxylation product, cis-1, in good yield (Table 1, entry 11). Increasing the loading of BF3•OEt2 from 1 equiv to 3 equiv resulted in an increased selectivity (Table 1, entry 13). However, reducing the reaction time from 24 h to 12 or 6 or 2 h resulted in a gradual decrease in selectivity toward cis-1 (Table 1, entries 15–17). Further increasing the BF3•OEt2 loading to 5 equiv, or extending the reaction time (48 h), or enhancing the reaction temperature to 50°C, however, did not increase the transformation or selectivity (Table 1, entries 14, 18, and 19). Lewis acid BF3•OEt2 plays a key role in the α-acetoxylation of cyclohexanone, which should be attributed to the enolization of cyclohexanone to enol that is essential for the reaction to smoothly take place (Ochiai et al., 2005). Changing acetic acid solvent to other organic solvents such as CH2Cl2, EtOAc, Et2O, toluene, CH3CN, CF3CH2OH, and (CF3)2CHOH even in the presence of 10 equiv acetic acid resulted in the decrease of both yield and selectivity (Table 1, entries 20–32). Therefore, the optimal condition for the synthesis of cis-1 was established as follows: Reaction of 4-phenylcyclohexanone (1 equiv) with PhI(OAc)2 (1.5 equiv) and BF3•OEt2 (3 equiv) in acetic acid solvent at room temperature for 24 h. The α-acetoxylation of 4-phenylcyclohexanone proceeded smoothly in ten-gram scale under this optimal reaction condition, without a lose of reactivity and selectivity. The configuration of the major α-acetoxylation product was determined as cis-isomer by a single-crystal X-ray diffraction analysis (Table 1).
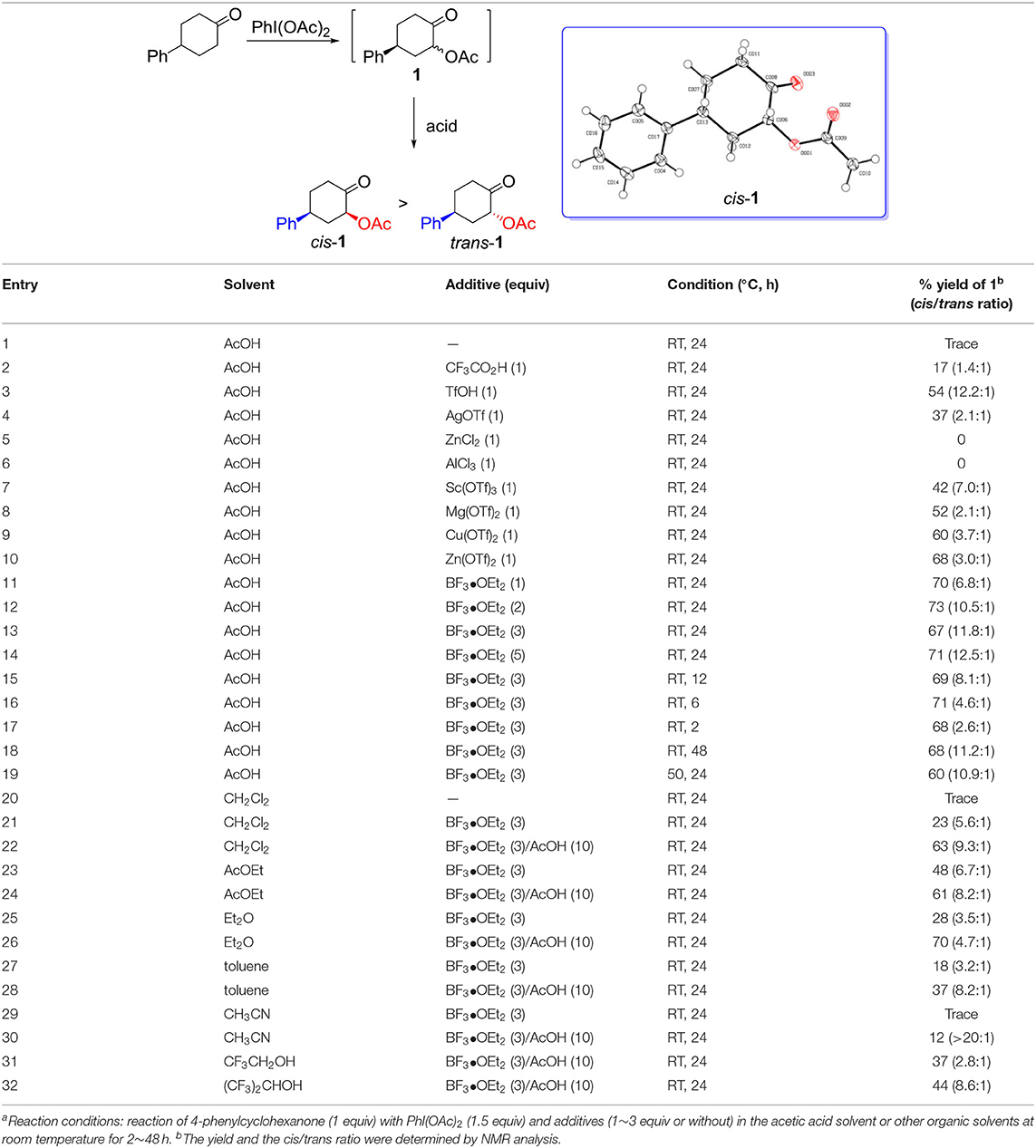
Table 1. Diastereoselective α-acetoxylation of ketone with hypervalent iodine(III) reagents in the presence of additivesa.
Having established a practical method for the selective α-acetoxylation of 4-phenylcyclohexanone by hybrid catalysis, we became interested in the substrate generality for diastereoselective α-acetoxylation of cyclic ketones with the aforementioned hybrid system: hypervalent iodine (III) reagents and BF3•OEt2. The results are summarized (Table 2). Firstly, we tested several substituents at C4 position of cyclohexanones to probe the versatility of this hybrid system. Switching phenylcyclohexanone to 4-tert-butylcyclohexanone resulted in a slight decrease in the yield to 57% with similar cis-selectivity (Table 2, entry 2). Further replacing the substituent to the dimethylphenylsilyl group resulted in a further decrease in both yield and selectivity (Table 2, entry 3). Extending the reaction time to 24 h, better diastereoselectivity (>9: 1) was observed by lowing the yield to 21%, presumably due to a decomposition of the starting material under the reaction conditions. In the case of 3-substituted cyclohexanones, good to high selectivities were observed (Table 2, entries 4 and 5).
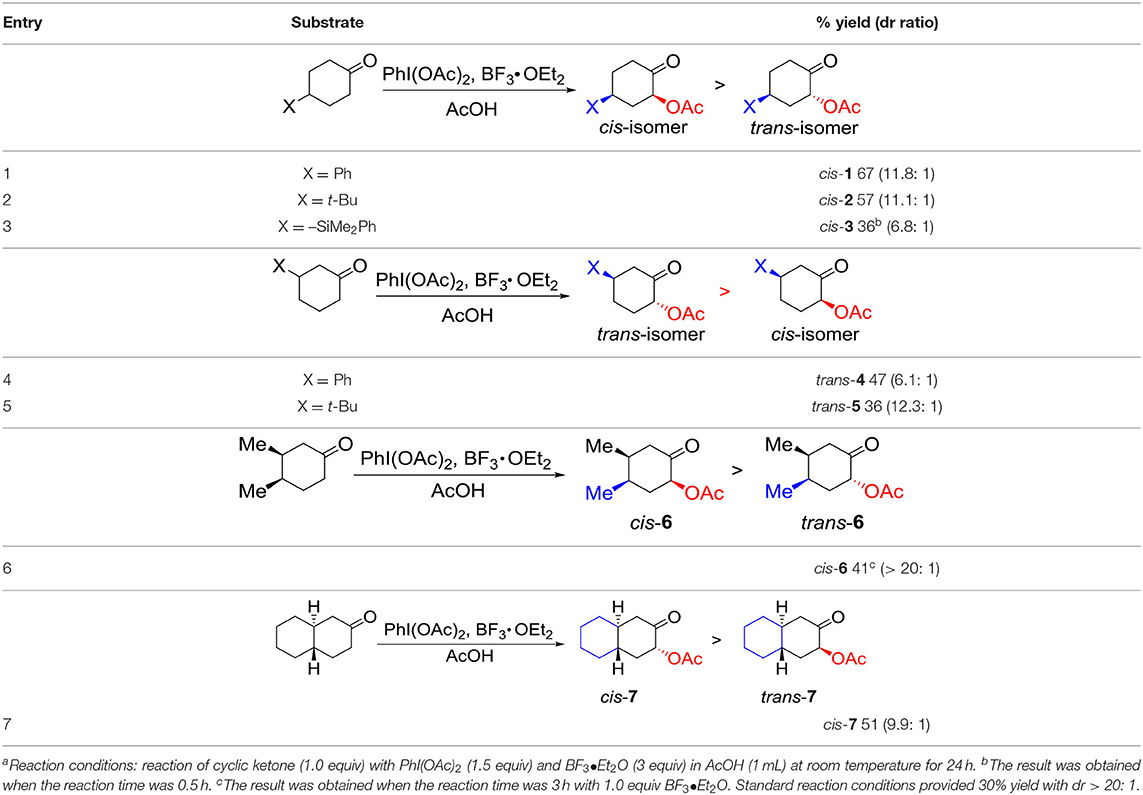
Table 2. Substrate scope for diastereoselective α-acetoxylation of cyclic ketones with hypervalent iodine(III) reagents and BF3•O.
Thirdly, we studied the substrates with substituents at both C3 and C4 positions of cyclohexanones. cis-3,4-Dimethylcyclohexene as substrate gave 30% yield of the product cis-6 with high diastereoselectivity (>20:1) under standard reaction conditions. Lowering the loading of BF3•OEt2 to 1.0 equiv resulted in a full consumption of starting material in 3 h with 41% yield and high diastereoselectivity (>20:1) (Table 2, entry 6). To our delight, trans-octahydro-naphthalen-2(1H)-one as substrate resulted in 51% yield of the product cis-7 with 9.9: 1 diastereoselectivity under the reaction conditions (Table 2, entry 7). We tried the application of this hybrid system of PIDA and BF3•OEt2 to both cyclopentanone and cycloheptanone. Unfortunately, the yield of the target molecules were quiet low, indicating that this hybrid system was not effective for the α-acetoxylation of cyclopentanones or cycloheptanones.
Having investigated the substrate scope for diastereoselective α-acetoxylation of cyclic ketones, we are interested in the application of the hypervalent iodine(III) reagent–BF3•OEt2 hybrid system. Changing AcOH solvent to i-PrCO2H, 2-acyloxy-4-phenylcyclohexanone 8 was obtained smoothly in 50% yield with high diastereoselectivity (11.1:1) (Scheme 2).
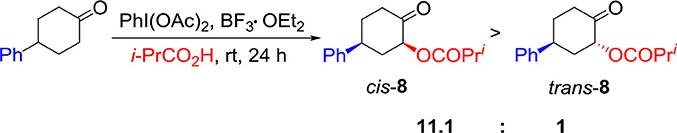
Scheme 2. Synthesis of 2-acyloxy-4-phenylcyclohexanone 8 by using the hypervalent iodine (III) reagent–BF3•OEt2 hybrid system.
It should be fundamentally interesting to understand the origin of the diastereoselectivity of the hypervalent iodine-mediated α-acetoxylation of cyclic ketones. With the assistance of Lewis acid BF3•OEt, the keto-enol equilibrium would lead to the activated enol to react with hypervalent iodine PhI(OAc)2. Generally, it is believed that an iodonium enolate (O-bound intermediate) or an α-iodanyl ketone (α-C-bound intermediate) could be formed for the subsequent α-acetoxylation. The O-bound hypervalent iodine intermediate can be traced back to Mizukami's proposal in their work in tosylation reactions in 1978. (Mizukami et al., 1978) The C-bound hypervalent iodine intermediate was first proposed by Moriarty et al. (1981). Accordingly, the possible mechanism for hypervalent iodine-mediated α-acetoxylation of cyclic ketones is proposed (Scheme 3). The O-bound intermediate would lead to the α-acetoxylation via the sigmatropic rearrangement pathway, while the C-bound intermediate will undergo an SN2 substitution by the acetate in the solution to form α-acetoxylation product.
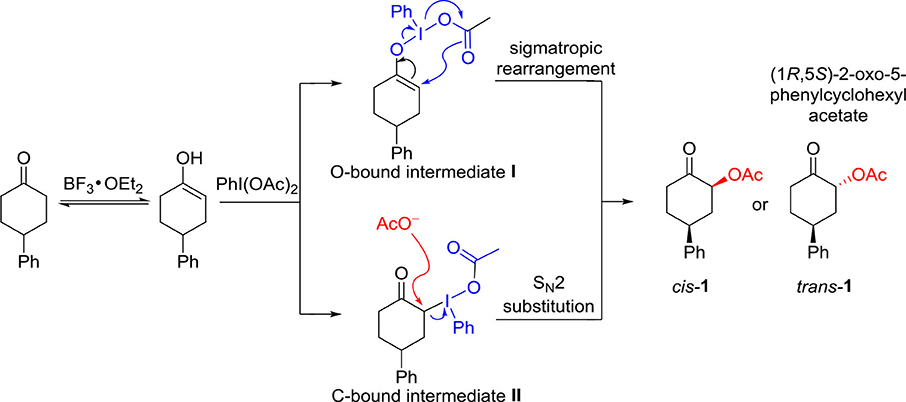
Scheme 3. The possible mechanism for the hypervalent iodine-mediated α-acetoxylation of cyclic ketones.
To further understand the diastereoselectivity α-acetoxylation of cyclic ketones, density functional theory (DFT) calculations were carried out with Gaussian 16 program. Frisch et al. (2016) Geometry optimizations and frequency calculations were carried out using the M06-2X functional (Zhao and Truhlar, 2007; Walker et al., 2013) in solution by the SMD continuum solvent model (solvent = acetic acid) (Marenich et al., 2009), with the basis sets of SDD (Andrae et al., 1991) for iodine atom and 6–311++G** for other atoms.
The calculated free energies of the hypervalent iodine intermediates I (O-bound) and II (C-bound) are depicted (Scheme 4). In the stable chair-conformer of 4-phenylcyclohexanone, different isomeric intermediates I (Itransoidal, Icisoidal, and Ivert) are calculated to be close in free energy, among which Icisoidal is predicted to be the most stable one. In contrast, the C-bound intermediates II (IIcisoidal and IIvert) are much more thermodynamically stable than the O-bound ones (by 22.6 (7) kcal/mol) in acetic acid. DFT results interestingly revealed a case of preferred C-bound species for the interaction between hypervalent iodine with cyclo-enol, which is complementary to previous suggestions that the enolate-like O-bound intermediates were more likely to be formed in the reaction of non-cyclic ketones. Norrby et al. (2010); Beaulieu and Legault (2015); Shneider et al. (2015); Arava et al. (2017) This predicted preference of α-C-bound hypervalent iodine species is also interestingly supported by the experimentally observed structures of the isolable iodonium ylides (Ivanov et al., 2014).
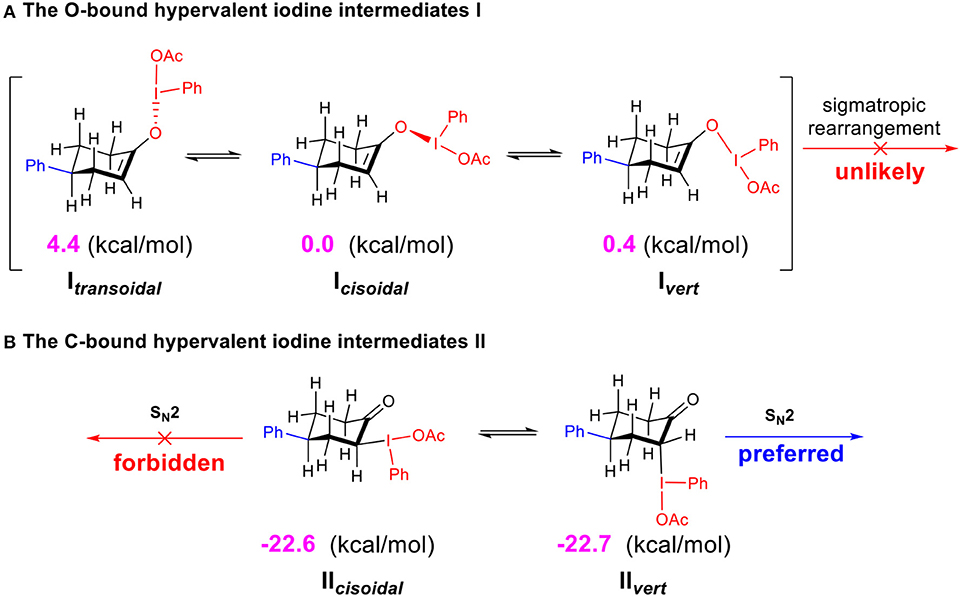
Scheme 4. Relative free energies (in kcal/mol) of the O-bound intermediates I and C-bound intermediates II calculated at the M06-2X/6311++G**&SDD(I)/SMD(solvent = acetic acid) level of theory. (A) The O-bound hypervalent iodine intermediates I. (B) The C-bound hypervalent iodine intermediates II.
Since the O-bound intermediate I is more than 22 kcal/mol higher than the C-bound intermediate II in free energy, the sigmatropic rearrangement reaction pathway should be unlikely to operate in this system. The α-acetoxylation of cyclic ketones should prefer the SN2 substitution mechanism via the α-C-bound intermediate. This explains well the importance of acetic acid as solvent to facilitate the SN2 substitution in our studied system (Table 1, entries 20–27).
Although the C-bound intermediates IIcisoidal and IIvert are close in free energy, the equatorial iodanyl group (IIcisoidal) is forbidden for SN2 substitution. The axial iodanyl intermediate IIvert should play a key role in the diastereoselective α-acetoxylation of cyclic ketones. Relative free energies (in kcal/mol) of the isomers of α-C-bound intermediates IIvert are depicted (Scheme 5). The equatorial phenyl intermediate IIvert corresponds to the cis product for the SN2 substitution, while the axial phenyl intermediate axial-IIvert leads to the trans product. IIvert is thermodynamically more stable than axial-IIvert by 2.2 kcal/mol, due to the steric effect of the axial phenyl on the chair conformation of the cyclic ketone. According to the Hammond–Leffler postulate, this implies the kinetic preference of the formation of the cis product from intermediate IIvert. Indeed, the calculated free energy of the transition state (cis-TS) for the cis pathway is lower than that (trans-TS) for the trans Pathway by 1.0 kcal/mol. The free energy of cis-TS is −12.0 kcal/mol and the activation free energy of the SN2 step is only 10.7 kcal/mol, which further supports that the SN2 substitution mechanism should be more plausible than the sigmatropic rearrangement mechanism via the O-bound intermediate. More importantly, our experimentally observed diastereoselective results strongly supported the influence of the thermodynamic control in selectivity. Extending the reaction time from 2 to 48 h, the cis/trans ratio increased from 2.6:1 to 11.2:1, with unchanged yield (Table 1, entries 15–18), when we performed a control experiment by subjecting the product 1 with low cis/trans ratio of 3.1:1 to the standard reaction condition for 24 h, a cis/trans ratio of 11.8:1 was observed. The equilibrium between the cis/trans products in acetic acid was observed, leading to the most table isomer as the major product. To further verify the thermodynamic control selectivity, we compared the relative free energy difference between cis product and trans product for representative substrates, i.e., 4-phenylcyclohexanone, 3-phenylcyclohexanone, and cis-3,4-dimethylcyclohexene, as shown in Scheme 6. The DFT-predicted free energy difference clearly demonstrated the thermodynamic control in selectivity, which is in good agreement with the experimentally observed diastereoselective ratio. The product cis-1 is lower in free energy than trans-1 by 1.9 kcal/mol, consistent with the experimentally observed cis/trans ratio of 11.2:1. When the phenyl substituent moves from the β position of the acetoxyl to the further γ position, the free energy difference decreases to 1.6 kcal/mol, in accordance with a slightly lower diastereoselective ratio of 6.1:1, and the inverse ratio of trans/cis is well-reproduced. With respect to cis-3,4-dimethylcyclohexene, DFT results suggest a free energy difference of 1.0 kcal/mol for the cis/trans products, in good agreement with the experimentally observed cis/trans ratio of 6.8:1 as well-indicating the β-substituent is predominant compared to the γ-position, probably due to the distance between the substituent and the acetoxyl group.
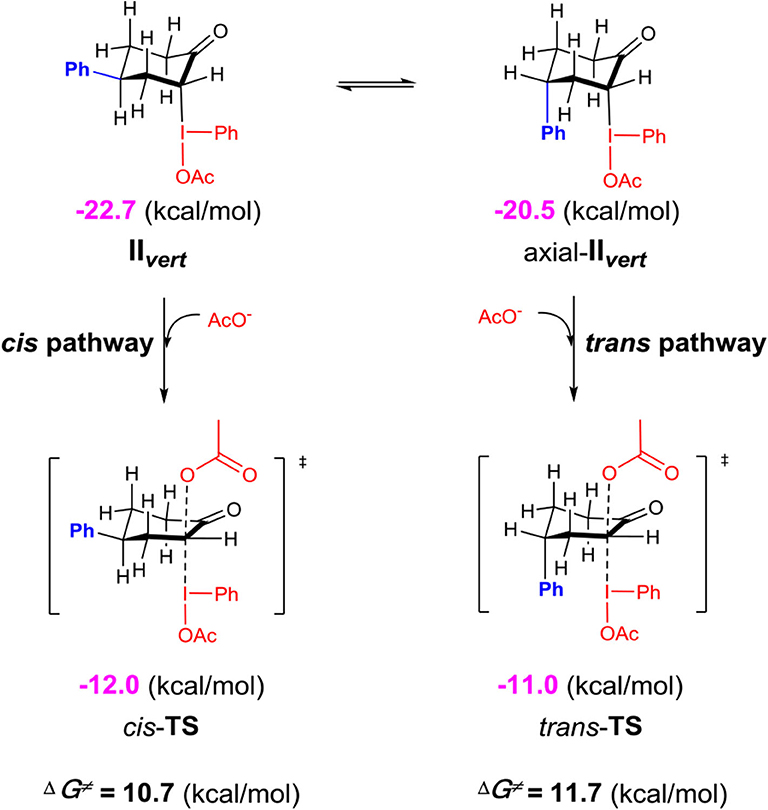
Scheme 5. Relative free energies (in kcal/mol) of the isomers of α-C-bound intermediates IIvert calculated at the M06-2X/6311++G**&SDD(I)/SMD(solvent = acetic acid) level of theory.
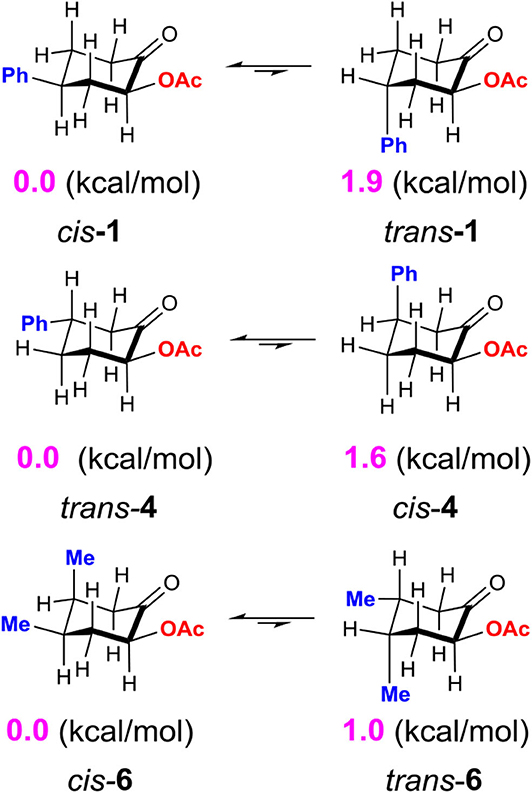
Scheme 6. Relative free energy difference (in kcal/mol) between cis product and trans product for representative cyclic ketones, calculated at the M06-2X/6311++G**/SMD (solvent = acetic acid) level of theory.
Conclusion
In conclusion, we have developed a practical approach for diastereoselective α-acetoxylation of cyclic ketones by a binary hybrid system comprising a hypervalent iodine(III) reagent and BF3•OEt2 Lewis acid. In this hybrid system, BF3•OEt2 Lewis acid allowed the activation of the hypervalent iodine(III) reagent for the smooth α-acetoxylation reaction of cyclic ketones and also for achieving high diastereoselectivity. The substrate scoping investigation showed that sterically hindered substituent groups on cyclic ketones are favorable for the diastereoselectivity. Cyclic ketone substrates bearing the mono-substituent group at C3 or C4 position, and substrates bearing di-substituent groups at C3 and C4 positions demonstrated good to high diastereoselectivity. This approach was successfully applied to the synthesis of other cis-substituted 2-acyloxycyclohexanones in moderate yield with high diastereoselectivity. Computational studies showed that this α-acetoxylation of cyclic ketone reaction plausibly undergoes an SN2 substitution mechanism, in which the α-C-bound hypervalent iodine species is the important intermediate. The diastereoselectivity of the reaction mainly originated from the thermodynamic control. This hypervalent iodine(III) reagent–BF3•OEt2 hybrid system can thus be applied to certain selective organic reactions and the efficient diastereoselective synthesis of cyclic ketone derivatives or other related biologically active compounds.
Materials and Methods
General
1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE III 400 MHz spectrometer [400 MHz for 1H NMR, 100 MHz for 13C NMR]. Tetramethylsilane (TMS) was used as an internal standard (0 ppm) for the 1H NMR spectra, and CDCl3 was used as the internal standard (77.0 ppm) for the 13C NMR spectra. High-resolution mass spectra (HRMS) were recorded on a Thermo MAT95XP or on an Agilent 6540 UHD Accurate-Mass Q-TOF LC-MS spectrometer. Infrared (IR) spectra were obtained on a Thermo Scientific Nicolet FT/IR-6700 spectrometer. Reactions were monitored by thin-layer chromatography (TLC). Reaction products were purified by column chromatography on silica gel. AcOH was dried before use. Other chemical reagents were purchased from common commercial suppliers and used as received.
General procedures for the α-acetoxylation of cyclic ketones: To a solution of cyclic ketones (0.5 mmol) and PhI(OAc)2 (241.5 mg, 0.75 mmol) in acetic acid (1 mL) was added BF3•OEt2 (212.9 mg, 1.5 mmol) dropwise, and the reaction mixture was stirred at room temperature for 24 h. The reaction progress was monitored by TLC. Upon completion, the reaction mixture was quenched with saturated aqueous Na2S2O3 and then saturated aqueous NaHCO3, washed with brine, extracted with dichloromethane, and dried over anhydrous Na2SO4. After filtration, the solvent was removed under reduced pressure to afford the crude product, which was purified by silica gel column chromatography using hexane/acetone and analyzed by 1H and 13C NMR spectroscopy.
2-Oxo-5-Phenylcyclohexyl Acetate (1)
The crude product was purified via flash chromatography, eluting with hexane/acetone = 30/1 to give a white solid (77.8 mg, 67%). Characterization of the major product (cis-isomer): Mp: 90–92°C; 1H NMR (400 MHz, CDCl3): δ = 7.35–7.23 (m, 5H), 5.38 (dd, J = 12.8, 6.4 Hz, 1H), 3.23 (t, J = 12.4 Hz, 1H), 2.62–2.59 (m, 2H), 2.49–2.44 (m, 1H), 2.27–2.24 (m, 1H), 2.17 (s, 3H), 2.08 (q, J = 12.4 Hz, 1H), 1.96–1.85 (m, 1H); 13C NMR (100 MHz, CDCl3): δ = 204.0, 170.1, 143.2, 128.9, 127.1, 126.8, 75.8, 42.1, 40.0, 39. 9, 34.5, 20.8; IR (KBr): 3,081, 3,021, 2,938, 2,875, 2,854, 1,757, 1,718, 1,641, 1,492, 1,445, 1,423, 1,385, 1,370, 1,324, 1,287, 1,263, 1,245, 1,150, 1,120, 1,072, 1,061, 982, 935, 911, 852, 756, 743, 695, 610, 517, 466 cm−1; HRMS (ESI) calcd for C14H17O3 [M+H]+: 233.1178; found: 233.1168; calcd for C14H16O3Na [M+Na]+: 255.0997; found: 255.0987.
5-(tert-Butyl)-2-Oxocyclohexyl Acetate (2)
The crude product was purified via flash chromatography, eluting with hexane/acetone = 30/1 to give a colorless oil (60.5 mg, 57%). Characterization of the major product (cis-isomer): 1H NMR (400 MHz, CDCl3): δ = 5.23–5.18 (m, 1H), 2.52–2.47 (m, 1H), 2.43–2.34 (m, 1H), 2.33–2.27 (m, 1H), 2.15 (s, 3H), 2.13–2.07 (m, 1H), 1.74–1.66 (m, 1H), 1.57 (q, J = 12.4 Hz, 1H), 1.48–1.37 (m, 1H), 0.93 (s, 9H); 13C NMR (100 MHz, CDCl3): δ = 205.0, 170.2, 76.3, 46.0, 39.7, 34.4, 32.6, 28.2, 27.7, 20.9.
5-(Dimethyl(Phenyl)Silyl)-2-Oxocyclohexyl Acetate (3)
The crude product was purified via flash chromatography, eluting with hexane/acetone = 30/1 to give a light yellow solid (52.3 mg, 36%). Characterization of the major product (cis-isomer): Mp: 121–123°C; 1H NMR (400 MHz, CDCl3): δ = 7.49–7.37 (m, 5H), 5.15 (dd, J = 12.4, 6.4 Hz, 1 H), 2.53–2.50 (m, 1H), 2.43–2.35 (m, 1H), 2.27–2.23 (m, 1H), 2.13 (s, 3H), 2.09–2.04 (m, 1H), 1.61 (q, J = 12.8 Hz, 1H), 1.53–1.42 (m, 1H), 1.38–1.32 (m, 1H), 0.33 (s, 6H); 13C NMR (100 MHz, CDCl3): δ = 204.9, 170.2, 136.5, 133.9, 129.9, 129.6, 128.1, 77.7, 42.5, 34.7, 28.9, 24.1, 20.9, −4.9, −5.0; IR (KBr): 3,071, 3,012, 2,949, 2,935, 2,865, 2,841, 1,748, 1,721, 1,427, 1,407, 1,376, 1,342, 1,321, 1,257, 1,233, 1,173, 1,143, 1,112, 1,102, 1,082, 1,050, 968, 912, 885, 850, 834, 821, 776, 763, 742, 728, 704, 661, 643, 605, 570, 482, 451, 436 cm−1; HRMS (ESI) calcd for C16H23O3Si [M+H]+: 291.1416; found: 291.1420; calcd for C16H22O3Na [M+Na]+: 313.1236; found: 313.1227.
2-Oxo-4-Phenylcyclohexyl Acetate (4)
The crude product was purified via flash chromatography, eluting with hexane/acetone = 30/1 to give a white solid (54.6 mg, 47%). Characterization of the major product (trans-isomer): Mp: 72–74°C; 1H NMR (400 MHz, CDCl3): δ = 7.35–7.31 (m, 2H), 7.25–7.20 (m, 3H), 5.29 (dd, J = 12.8, 6.4 Hz, 1H), 3.02–2.94 (m, 1H), 2.70–2.62 (m, 2H), 2.40–2.35 (m, 1H), 2.18 (s, 3H), 2.14 (m, 1H), 2.09–1.98 (m, 1H), 1.95–1.84 (m, 1H); 13C NMR (100 MHz, CDCl3): δ = 203.4, 170.3, 143.2, 129.0, 127.2, 126.6, 76.3, 47.9, 45.4, 31.8, 31.7, 20.9; IR (KBr): 3,033, 2,959, 2,941, 2,908, 1,745, 1,721, 1,602, 1,501, 1,458, 1,432, 1,376, 1,319, 1,281, 1,233, 1,174, 1,081, 1,046, 897, 763, 703, 665, 599, 531, 501 cm−1; HRMS (ESI) calcd for C14H17O3 [M+H]+: 233.1178; found: 233.1145; calcd for C14H16O3Na [M+Na]+: 255.0997; found: 255.0986.
4-(Tert-Butyl)-2-Oxocyclohexyl Acetate (5)
The crude product was purified via flash chromatography, eluting with hexane/acetone = 30/1 to give a light yellow oil (38.2 mg, 36%). Characterization of the major product (trans-isomer): 1H NMR (400 MHz, CDCl3): δ = 5.15 (dd, J = 12.8, 6.8 Hz, 1H), 2.56–2.53 (m, 1H), 2.33–2.27 (m, 1H), 2.22–2.13 (m, 1H), 2.16 (s, 3H), 2.03–2.00 (m, 1H), 1.73–1.63 (m, 1H), 1.56–1.51 (m, 2H), 0.91 (s, 9H); 13C NMR (100 MHz, CDCl3): δ = 205.3, 170.3, 76.6, 50.0, 42.4, 32.9, 31.7, 27.4, 25.0, 20.9.
4,5-Dimethyl-2-Oxocyclohexyl Acetate (6)
The crude product was purified via flash chromatography, eluting with hexane/acetone = 30/1 to give a light yellow oil (37.8 mg, 41%). Characterization of the major isomer: 1H NMR (400 MHz, CDCl3): δ = 5.19 (dd, J = 12.4, 6.8 Hz, 1H), 2.67–2.62 (m, 1H), 2.35–2.21 (m, 3H), 2.14 (s, 3H), 2.07–2.03 (m, 1H), 1.74 (q, J = 12.8 Hz, 1H), 1.01 (d, J = 6.8 Hz, 3H), 0.83 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 204.6, 170.1, 75.6, 47.3, 36.5, 34.9, 33.2, 20.8, 18.5, 12.1; IR (KBr): 2,959, 2,928, 2,891, 2,871, 1,749, 1,721, 1,470, 1,455, 1,431, 1,380, 1,370, 1,243, 1,175, 1,102, 1,087, 1,075, 1,036, 975, 941, 885, 790, 715, 651, 609, 549, 510, 482, 436 cm−1; HRMS (ESI) calcd for C10H16O3Na [M+Na]+: 207.0997; found: 207.0988.
3-Oxodecahydronaphthalen-2-yl Acetate (7)
The crude product was purified via flash chromatography, eluting with hexane/acetone = 30/1 to give a light yellow solid (53.6 mg, 51%). Characterization of the major isomer: 1H NMR (400 MHz, CDCl3): δ = 5.19 (dd, J = 12.0, 6.8 Hz, 1H), 2.41–2.37 (m, 1H), 2.20–2.10 (m, 2H), 2.13 (s, 3H), 1.78–1.68 (m, 4H), 1.56–1.46 (m, 2H), 1.36–0.99 (m, 5H); 13C NMR (100 MHz, CDCl3): δ = 204.0, 170.2, 76.1, 47.2, 43.8, 40.5, 39.4, 33.7, 32.5, 25.8, 25.5, 20.8.
2-Oxo-5-Phenylcyclohexyl Isobutyrate (8)
The crude product was purified via flash chromatography, eluting with hexane/acetone = 30/1 to give a white solid (65.1 mg, 50%). Characterization of the major isomer: Mp: 63–65°C; 1H NMR (400 MHz, CDCl3): δ = 7.38–7.34 (m, 2H), 7.29–7.26 (m, 3H), 5.41(dd, J = 12.8, 6.0 Hz, 1H), 3.26 (t, J = 12.8 Hz, 1H), 2.71–2.62 (m, 3H), 2.51–2.46 (m, 1H), 2.30–2.26 (m, 1H), 2.11 (q, J = 12.8 Hz, 1H), 2.00–1.89 (m, 1H); 1.28 (d, J = 6.8 Hz, 3H), 1.23 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 204.1, 176.4, 143.3, 128.9, 127.1, 126.8, 75.4, 42.1, 40.0, 39.9, 34.5, 34.0, 19.2, 19.1; IR (KBr): 3,030, 2,977, 2,929, 2,866, 1,751, 1,727, 1,632, 1,605, 1,498, 1,462, 1,429, 1,385, 1,349, 1,293, 1,260, 1,200, 1,165, 1,147, 1,117, 1,069, 977, 918, 843, 763, 739, 701, 596, 540, 507 cm−1; HRMS (ESI) calcd for C16H20O3Na [M+Na]+: 283.1310; found: 283.1299.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Cambridge Crystallographic Data Centre (CCDC-1997827).
Author Contributions
JT, WZ, and WX were responsible for designing and performing the experiments. YJ and ZK were responsible for DPT calculation. YL and KM directed the project and wrote the manuscript.
Funding
This research was supported by the NSFC (21977019, 21502023), the Guangdong Provincial Key Research and Development Program (2019B020201005), and the Open Project of Guangdong Provincial Key Laboratory of New Drug Screening (GDKLNDS-2018OF004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00467/full#supplementary-material
References
Ahmed, Z., and Langer, P. (2006). Synthesis of functionalized 4-chlorophenols and 1,4-dihydroquinones by 3+3 cyclization of 1,3-bis-silyl enol ethers with 2-chloro- and 2-acyloxy-3-(silyloxy)alk-2-en-1-ones. Tetrahedron Lett. 47, 417–419. doi: 10.1016/j.tetlet.2005.11.077
Andrae, D., Häuβermann, U., Dolg, M., Stoll, H., and Preu,β, H. (1991). Energy-adjusted ab initio pseudopotentials for the second- and third-row transition elements. Theor. Chim. Acta 1991, 78, 247–266. doi: 10.1007/BF01112848
Arava, S., Kumar, J. N., Maksymenko, S., Iron, M. A., Parida, K. N., Fristrup, P., et al. (2017). Enolonium species-umpoled enolates. Angew. Chem. Int. Ed. 56, 2599–2603. doi: 10.1002/anie.201610274
Beaulieu, S., and Legault, C. Y. (2015). Mechanistic insights on the iodine(III)-mediated α-Oxidation of Ketones. Chem. Eur. J. 21, 11206–11211. doi: 10.1002/chem.201501177
Bogevig, A., Sunden, H., and Cordova, A. (2004). Direct catalytic enantioselective alpha-aminoxylation of ketones: a stereoselective synthesis of alpha-hydroxy and alpha,alpha'-dihydroxy ketones. Angew. Chem. Int. Ed. 43, 1109–1112. doi: 10.1002/anie.200353018
Carneiro, P. F., Gutmann, B., de Souza, R. O. M. A., and Kappe, C. O. (2015). Process intensified flow synthesis of 1H-4-substituted imidazoles: toward the continuous production of daclatasvir. Acs Sust. Chem. Eng. 3, 3445–3453. doi: 10.1021/acssuschemeng.5b01191
Chen, C., Liu, W. B., Zhou, P., and Liu, H. L. (2017). I-2/TBHP-mediated oxidative coupling of ketones and toluene derivatives: a facile method for the preparation of alpha-benzoyloxy ketones. RSC Adv. 7, 20394–20397. doi: 10.1039/C7RA02298K
Chen, J. Z., Liu, D. L., Butt, N., Li, C., Fan, D. Y., Liu, Y. G., et al. (2013). Palladium-catalyzed asymmetric hydrogenation of alpha-acyloxy-1-arylethanones. Angew. Chem. Int. Ed. 52, 11632–11636. doi: 10.1002/anie.201306231
Chen, J. Z., Zhang, Z. F., Liu, D. L., and Zhang, W. B. (2016). Palladium-catalyzed chemo- and enantioselective C-O bond cleavage of alpha-acyloxy ketones by hydrogenolysis. Angew. Chem. Int. Ed. 55, 8444–8447. doi: 10.1002/anie.201603590
Corey, E. J., and Knapp, S. (1976). α-functionalization of ketones via N, N-dimethylhydrazones. Tetrahedron Lett. 17, 4687–4690. doi: 10.1016/S0040-4039(00)92996-4
Du, J., Zhang, X., Sun, X., and Wang, L. (2015). Copper-catalyzed direct alpha-ketoesterification of propiophenones with acetophenones via C(sp(3))-H oxidative cross-coupling. Chem Commun. 51, 4372–4375. doi: 10.1039/C4CC09524C
Edwards, M. G., Kenworthy, M. N., Kitson, R. R. A., Scott, M. S., and Taylor, R. J. K. (2008). The telescoped intramolecular Michael/olefination (TIMO) approach to alpha-alkylidene-gamma-butyrolactones: synthesis of (+)-paeonilactone B. Angew. Chem. Int. Ed. 47, 1935–1937. doi: 10.1002/anie.200705329
El-Qisairi, A. K., and Qaseer, H. A. (2002). Oxidation of ketone by palladium(II). α-Hydroxyketone synthesis catalyzed by a bimetallic palladium(II) complex. J. Organomet. Chem. 659, 50–55. doi: 10.1016/S0022-328X(02)01691-1
Erickson, J. L. E., Dechary, J. M., and Kesling, M. R. (1951). Diazoketones as reagents for the identification of organic acids. J. Am. Chem. Soc. 73, 5301–5302. doi: 10.1021/ja01155a086
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 Revision A.03. Wallingford, CT: Gaussian, Inc.
Hamed, O. A., El-Qisairi, A., Qaseer, H., Hamed, E. M., Henry, P. M., and Becker, D. P. (2012). Asymmetric α-hydroxy ketone synthesis by direct ketone oxidation using a bimetallic palladium(II) complex. Tetrahedron Lett. 53, 2699–2701. doi: 10.1016/j.tetlet.2012.03.066
Hecker, S. J., and Werner, K. M. (1993). Total synthesis of (+/-)-leuhistin. J. Org. Chem. 58, 1762–1765. doi: 10.1021/jo00059a028
Heiba, E.-A. I., and Dessau, R. M. (1971). Oxidation by metal salts. VII. Syntheses based on the selective oxidation of organic free radicals. J. Am. Chem. Soc. 93, 524–527. doi: 10.1021/ja00731a042
Hu, X. W., Chen, X., Shao, Y. X., Xie, H. S., Deng, Y. F., Ke, Z. F., et al. (2018). Co(III)-catalyzed coupling-cyclization of aryl C-H bonds with alpha-diazoketones involving wolff rearrangement. ACS Catal. 8, 1308–1312. doi: 10.1021/acscatal.7b03668
Huang, G., Sheng, J., Li, X., Tang, M., and Gao, B. (2007). An efficient method for the α-acetoxylation of ketones. Synthesis 2007, 1165–1168. doi: 10.1055/s-2007-965984
Huang, X., Zhang, Y. G., Zhang, C. S., Zhang, L., Xu, Y., Kong, L. C., et al. (2019). The ortho-difluoroalkylation of aryliodanes with enol silyl ethers: rearrangement enabled by a fluorine effect. Angew. Chem. Int. Ed. 58, 5956–5961. doi: 10.1002/anie.201900745
Ivanov, A. S., Popov, I. A., Boldyrev, A. I., and Zhdankin, V. V. (2014). The I=X (X=O, N, C) double bond in hypervalent iodine compounds: is it real?. Angew. Chem. Int. Ed. 53, 9617–9621. doi: 10.1002/anie.201405142
Izquierdo, S., Essafi, S., Del Rosal, I., Vidossich, P., Pleixats, R., Vallribera, A., et al. (2016). Acid activation in phenyliodine dicarboxylates: direct observation, structures, and implications. J. Am. Chem. Soc. 138, 12747–12750. doi: 10.1021/jacs.6b07999
Kaila, N., Janz, K., DeBernardo, S., Bedard, P. W., Camphausen, R. T., Tam, S., et al. (2007). Synthesis and biological evaluation of quinoline salicylic acids as P-selectin antagonists. J. Med. Chem. 50, 21–39. doi: 10.1021/jm0602256
Kitamura, M., Kisanuki, M., and Okauchi, T. (2012). Synthesis of 1,2-naphthalenediol diacetates by rhodium(II)-catalyzed reaction of 1,2-diazonaphthoquinones with acetic anhydride. Eur. J. Org. Chem. 2012, 905–907. doi: 10.1002/ejoc.201101698
Littler, J. S. (1962). 156. The mechanisms of oxidation of cyclohexanone under acid conditions. Part I. Two-electron oxidants. J. Chem. Soc. 827–832. doi: 10.1039/jr9620000827
Liu, L., Feng, S. L., and Li, C. B. (2016). Practical approach for quantitative green esterifications. ACS Sust. Chem. Eng. 4, 6754–6762. doi: 10.1021/acssuschemeng.6b01718
Liu, Y., Huang, D., Huang, J., and Maruoka, K. (2017). Hypervalent iodine mediated chemoselective iodination of alkynes. J. Org. Chem. 82, 11865–11871. doi: 10.1021/acs.joc.7b01555
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 113, 6378–6396. doi: 10.1021/jp810292n
Mizukami, F., Ando, M., Tanaka, T., and Imamura, J. (1978). The acetoxylation ofp-substituted acetophenones and β-diketones with (diacetoxyiodo)benzene. Bull. Chem. Soc. Jpn. 51, 335–336. doi: 10.1246/bcsj.51.335
Moriarty, R. M., Hu, H., and Gupta, S. C. (1981). Direct α-hydroxylation of ketones using iodosobenzene. Tetrahedron Lett. 22, 1283–1286. doi: 10.1016/S0040-4039(01)90297-7
Newman, M. S., and Beal, P. F. (1950). A new synthesis of α-alkoxy ketones1. J. Am. Chem. Soc. 72, 5161–5163. doi: 10.1021/ja01167a100
Ng, F. T. T., and Henry, P. M. (1976). Kinetics and mechanism of the outer-sphere oxidation of cyclohexanone by tris(polypyridyl) complexes of iron(III) and ruthenium(III). J. Am. Chem. Soc. 98, 3606–3611. doi: 10.1021/ja00428a036
Nicolaou, K. C., Montagnon, T., Ulven, T., Baran, P. S., Zhong, Y. L., and Sarabia, F. (2002). Novel chemistry of alpha-tosyloxy ketones: applications to the solution- and solid-phase synthesis of privileged heterocycle and enediyne libraries. J. Am. Chem. Soc. 124, 5718–5728. doi: 10.1021/ja012146j
Nolla-Saltiel, R., Alonso Carrillo-Arcos, U., and Porcel, S. (2014). Silver acetate mediated acetoxylations of alkyl halides. Synth. Stuttgart 46, 165–169. doi: 10.1055/s-0033-1338551
Norrby, P. O., Petersen, T. B., Bielawski, M., and Olofsson, B. (2010). α-arylation by rearrangement: on the reaction of enolates with diaryliodonium salts. Chem. Eur. J. 16, 8251–8254. doi: 10.1002/chem.201001110
Ochiai, M., Takeuchi, Y., Katayama, T., Sueda, T., and Miyamoto, K. (2005). Iodobenzene-catalyzed alpha-acetoxylation of ketones. in situ generation of hypervalent (diacyloxyiodo)benzenes using m-chloroperbenzoic acid. J. Am. Chem. Soc. 127, 12244–12245. doi: 10.1021/ja0542800
Pogaku, N., Krishna, P. R., and Prapurna, Y. L. (2019). Iodine-mediated nucleophilic direct oxidative α-acetoxylation and α-alkoxylation of ketones. ChemistrySelect 4, 12333–12336. doi: 10.1002/slct.201903028
Ren, S. C., Song, S. J., Ye, L., Feng, C., and Loh, T. P. (2016). Copper-catalyzed oxyamination of electron-deficient alkenes with N-acyloxyamines. Chem. Commun. 52, 10373–10376. doi: 10.1039/C6CC04638J
Richter, M. J. R., Schneider, M., Brandstatter, M., Krautwald, S., and Carreira, E. M. (2018). Total synthesis of (-)-mitrephorone A. J. Am. Chem. Soc. 140, 16704–16710. doi: 10.1021/jacs.8b09685
Rubottom, G. M., Gruber, J. M., Marrero, R., Juve, H. D., and Kim, C. W. (1983). Oxidation of alkyl trimethylsilyl ketene acetals with lead(IV) carboxylates. J. Org. Chem. 48, 4940–4944. doi: 10.1021/jo00173a031
Sakamoto, R., Hirama, N., and Maruoka, K. (2018). The radical acylarylation of N-arylacrylamides with aliphatic aldehydes using the photolysis of hypervalent iodine(iii) reagents. Org. Biomol. Chem. 16, 5412–5415. doi: 10.1039/C8OB01420E
Sakamoto, R., Inada, T., Selvakumar, S., Moteki, S. A., and Maruoka, K. (2016). Efficient photolytic C-H bond functionalization of alkylbenzene with hypervalent iodine(III) reagent. Chem. Commun. 52, 3758–3761. doi: 10.1039/C5CC07647A
Sakamoto, R., Kashiwagi, H., and Maruoka, K. (2017). The direct C-H difluoromethylation of heteroarenes based on the photolysis of hypervalent iodine(III) reagents that contain difluoroacetoxy ligands. Org. Lett. 19, 5126–5129. doi: 10.1021/acs.orglett.7b02416
Selvakumar, S., Kang, Q. K., Arumugam, N., Almansour, A. I., Kumar, R. S., and Maruoka, K. (2017). Hypervalent iodine(III) catalyzed radical hydroacylation of chiral alkylidenemalonates with aliphatic aldehydes under photolysis. Tetrahedron Lett. 73, 5841–5846. doi: 10.1016/j.tet.2017.08.018
Shneider, O. S., Pisarevsky, E., Fristrup, P., and Szpilman, A. M. (2015). Oxidative Umpolung α-Alkylation of Ketones. Org. Lett. 17, 282–285. doi: 10.1021/ol503384c
Shu, S., Li, Y., Jiang, J., Ke, Z., and Liu, Y. (2019). Mechanism of hypervalent iodine promoted fluorocyclization of unsaturated alcohols: metathesis via double acids activation. J. Org. Chem. 84, 458–462. doi: 10.1021/acs.joc.8b02741
Sunden, H., Engqvist, M., Casas, J., Ibrahem, I., and Cordova, A. (2004). Direct amino acid catalyzed asymmetric alpha oxidation of ketones with molecular oxygen. Angew. Chem. Int. Ed. 43, 6532–6535. doi: 10.1002/anie.200460295
Tan, F., Liu, X. H., Hao, X. Y., Tang, Y., Lin, L. L., and Feng, X. M. (2016). Asymmetric catalytic insertion of alpha-diazo carbonyl compounds into O-H bonds of carboxylic acids. ACS Catal. 6, 6930–6934. doi: 10.1021/acscatal.6b02184
Tan, L., Chen, C., and Liu, W. (2017). alpha-acetoxyarone synthesis via iodine-catalyzed and tert-butyl hydroperoxide-mediateded self-intermolecular oxidative coupling of aryl ketones. Beilstein J. Org. Chem. 13, 1079–1084. doi: 10.3762/bjoc.13.107
Tanner, D. D., Chen, J. J., Chen, L., and Luelo, C. (1991). Fragmentation of substituted acetophenones and halobenzophenone ketyls. Calibration of a mechanistic probe. J. Am. Chem. Soc. 113, 8074–8081. doi: 10.1021/ja00021a038
Valgimigli, L., Brigati, G., Pedulli, G. F., DiLabio, G. A., Mastragostino, M., Arbizzani, C., et al. (2003). The effect of ring nitrogen atoms on the homolytic reactivity of phenolic compounds: understanding the radical-scavenging ability of 5-pyrimidinols. Chem.Eur. J. 9, 4997–5010. doi: 10.1002/chem.200304960
Varma, R. S., Kumar, D., and Liesen, P. J. (1998). Solid state synthesis of 2-aroylbenzo b furans, 1,3-thiazoles and 3-aryl-5,6-dihydroimidazo 2,1-b 1,3 thiazoles from alpha-tosyloxyketones using microwave irradiation. J. Chem. Soc. Perkin Trans. 1, 4093–4096. doi: 10.1039/a807563h
Walker, M., Harvey, A. J., Sen, A., and Dessent, C. E. (2013). Performance of M06, M06-2X, and M06-HF density functionals for conformationally flexible anionic clusters: M06 functionals perform better than B3LYP for a model system with dispersion and ionic hydrogen-bonding interactions. J. Phys. Chem. A. 117, 12590–12600. doi: 10.1021/jp408166m
Wang, Z. K., Bi, X. H., Liang, Y. J., Liao, P. Q., and Dong, D. W. (2014). A copper-catalyzed formal O-H insertion reaction of alpha-diazo-1,3-dicarbonyl compounds to carboxylic acids with the assistance of isocyanide. Chem. Commun. 50, 3976–3978. doi: 10.1039/C4CC00402G
Yu, J., Tian, J., and Zhang, C. (2010). Various α-oxygen functionalizations of β-dicarbonyl compounds mediated by the hypervalent iodine(III) reagentp-iodotoluene difluoride with different oxygen-containing nucleophiles. Adv. Synth. Catal. 352, 531–546. doi: 10.1002/adsc.200900737
Yuan, W. K., Zhu, M. H., Geng, R. S., Ren, G. Y., Zhang, L. B., Wen, L. R., et al. (2019). Construction of benzofuran-3(2H)-one scaffolds with a quaternary center via Rh/Co relay catalyzed C-H functionalization/annulation of N-aryloxyacetamides and propiolic acids. Org. Lett. 21, 1654–1658. doi: 10.1021/acs.orglett.9b00181
Yuan, W. M., Eriksson, L., and Szabo, K. J. (2016). Rhodium-catalyzed geminal oxyfluorination and oxytrifluoro-methylation of diazocarbonyl compounds. Angew. Chem. Int. Ed. 55, 8410–8415. doi: 10.1002/anie.201602137
Zhang, T. S., Hao, W. J., Wang, N. N., Li, G. G., Jiang, D. F., Tu, S. J., et al. (2016). Catalytic oxidative carbene coupling of alpha-diazo carbonyls for the synthesis of beta-amino ketones via C(sp(3))-H functionalization. Org. Lett. 18, 3078–3081. doi: 10.1021/acs.orglett.6b01189
Zhao, Y., and Truhlar, D. G. (2007). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi: 10.1007/s00214-007-0310-x
Keywords: α-acetoxylation, hypervalent iodine (III) reagent, cyclic ketones, diastereoselectivity, density functional theory
Citation: Tan J, Zhu W, Xu W, Jing Y, Ke Z, Liu Y and Maruoka K (2020) Hypervalent Iodine-Mediated Diastereoselective α-Acetoxylation of Cyclic Ketones. Front. Chem. 8:467. doi: 10.3389/fchem.2020.00467
Received: 05 March 2020; Accepted: 04 May 2020;
Published: 10 July 2020.
Edited by:
Jian-Wei Han, East China University of Science and Technology, ChinaReviewed by:
Chang-Hua Ding, Shanghai University, ChinaYin Wei, Shanghai Institute of Organic Chemistry (CAS), China
Chi Zhang, Nankai University, China
Copyright © 2020 Tan, Zhu, Xu, Jing, Ke, Liu and Maruoka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Liu, eWFubGl1QGdkdXQuZWR1LmNu; Keiji Maruoka, bWFydW9rYUBrdWNoZW0ua3lvdG8tdS5hYy5qcA==
†These authors share first authorship