 Kristel M. Rabanzo-Castillo
Kristel M. Rabanzo-Castillo Vipin B. Kumar
Vipin B. Kumar Tilo Söhnel
Tilo Söhnel Erin M. Leitao
Erin M. Leitao- 1School of Chemical Sciences, University of Auckland, Auckland, New Zealand
- 2The MacDiarmid Institute for Advanced Materials and Nanotechnology, Auckland, New Zealand
The utility of (C6F5)3B(OH2) as catalyst for the simple and environmentally benign synthesis of oligosiloxanes directly from hydrosilanes, is reported. This protocol offers several advantages compared to other methods of synthesizing siloxanes, such as mild reaction conditions, low catalyst loading, and a short reaction time with high yields and purity. The considerable H2O-tolerance of (C6F5)3B(OH2) promoted a catalytic route to disiloxanes which showed >99% conversion of three tertiary silanes, Et3SiH, PhMe2SiH, and Ph3SiH. Preliminary data on the synthesis of unsymmetrical disiloxanes (Si-O-Si') suggests that by modifying the reaction conditions and/or using a 1:1 combination of silane to silanol the cross-product can be favored. Intramolecular reactions of disilyl compounds with catalytic (C6F5)3B(OH2) led to the formation of novel bridged siloxanes, containing a Si-O-Si linkage within a cyclic structure, as the major product. Moreover, the reaction conditions enabled recovery and recycling of the catalyst. The catalyst was re-used 5 times and demonstrated excellent conversion for each substrate at 1.0 mol% catalyst loading. This seemingly simple reaction has a rather complicated mechanism. With the hydrosilane (R3SiH) as the sole starting material, the fate of the reaction largely depends on the creation of silanol (R3SiOH) from R3SiH as these two undergo dehydrocoupling to yield a disiloxane product. Generation of the silanol is based on a modified Piers-Rubinsztajn reaction. Once the silanol has been produced, the mechanism involves a series of competitive reactions with multiple catalytically relevant species involving water, silane, and silanol interacting with the Lewis acid and the favored reaction cycle depends on the concentration of various species in solution.
Introduction
Organo(poly)siloxanes (silicones), bearing the repeating Si-O bond motif, are considered one of the most important classes of functional materials that have influenced many technological industries (Sawama et al., 2016; Wang et al., 2017). Industrially, polysiloxanes are generated by acid- or base-catalyzed ring opening polymerization of cyclic siloxanes, or by hydrolysis of chlorosilanes (Grubb, 1954; Brinker and Scherer, 1990). However, these methods have limited control over the oligomeric or polymeric siloxane sequence being formed. The substitution process is catalyzed either by an acid or a base under equilibrium in which the polysiloxanes can also be degraded (Brinker and Scherer, 1990). Although a step-wise synthesis of oligosiloxane has been reported, it is still based on conventional condensation of silanols with chlorosilanes (Uchida et al., 1990; Matsumoto et al., 2018, 2019). Other methods available in literature for the preparation of oligosiloxanes involve catalytic cross-coupling reactions of oxygen nucleophiles (e.g., alkoxysilanes, silanols, phenols, and ethers) with hydrosilanes (Brook, 2018; Zhang et al., 2020). The synthesis of symmetrical disiloxanes directly from hydrosilanes have been reported using InBr3, a reaction which involves Lewis acid-catalyzed air oxidation of hydrosilanes (Scheme 1, A, conditions I) (Sridhar et al., 2009). Reduction of CO2 with hydrosilanes and a zirconium complex/B(C6F5)3 as the catalyst have also been reported to produce methane and oligosiloxanes as products (Matsuo and Kawaguchi, 2006). Other direct syntheses reported the use of H2O as the solvent and oxidant, however, generally these involved expensive transition metal catalysts and are conducted at elevated temperature (Scheme 1, A, conditions II–IV) (Lee et al., 2000, 2004; Ison et al., 2005; Mitsudome et al., 2008, 2009; Chauhan et al., 2009; Asao et al., 2010; John et al., 2011; Tan et al., 2011; Jeon et al., 2012; Shimizu et al., 2012a,b; Liu et al., 2014; Sawama et al., 2016; Tsuchido et al., 2020). The development of synthetic methods catalyzed by abundant and cheap base-metal complexes then emerged to form symmetrical and unsymmetrical disiloxanes and other oligosiloxanes (Pattanaik and Gunanathan, 2019) (Scheme 1, B, conditions V). Catalytic routes toward unsymmetrical siloxanes (Si-O-Si') have also been demonstrated (Scheme 1, B, conditions VI–VIII) using Sc(OTf)3 (Hreczycho et al., 2013; Hreczycho, 2015), Nafion (Kaźmierczak and Hreczycho, 2018), or Amberlyst-15 (Kuciński and Hreczycho, 2019a) with silanols and alkylsilanes at room temperature. Transition metal catalyzed routes to unsymmetrical disiloxanes have been achieved through Pd-catalyzed arylation of hydroxysiloxanes (Kurihara et al., 2013), nonhydrolytic Pd/C-catalyzed cross-coupling reactions (Igarashi et al., 2014), Ba-mediated dehydrocoupling of hydrosilanes and silanols (Le Coz et al., 2019) and silylation of silanols catalyzed by a ruthenium complex (Marciniec et al., 2008). Other routes to form Si-O-Si' bonds include the use of fluoride and azidosilanes (Abele et al., 2003) as well as a catalyst-free, chlorine-free option using disilazanes (Kuciński and Hreczycho, 2019b) (Scheme 1, B, conditions IX–X).
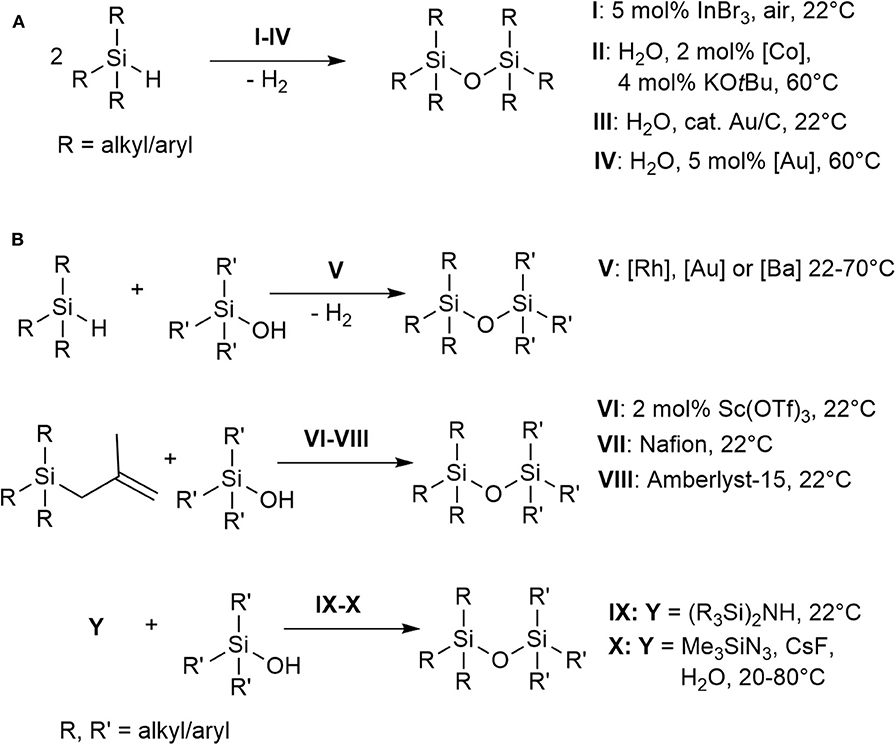
Scheme 1. Selected examples of the catalytic synthesis of disiloxanes.
Main group (and metal-free) catalysts such as B(C6F5)3 have gained great interest as a catalyst activator and as strong Lewis acid for many purposes (Piers, 2004; Lawson and Melen, 2017; Brook, 2018). Several studies have been published for the preparation of siloxanes and hyperbranched siloxanes involving catalytic cross-coupling reactions of oxygen nucleophiles (e.g., alkoxysilanes, silanols, phenols, and ethers) with hydrosilanes (Kawakami et al., 2004; Chojnowski et al., 2005, 2006, 2008; Zhou and Kawakami, 2005; Shinke et al., 2007; Thompson and Davies, 2007; Cella and Rubinsztajn, 2008; Kurjata et al., 2009; Feghali and Cantat, 2014; Madsen et al., 2014; Zhang et al., 2014, 2020; Feghali et al., 2015; Laengert et al., 2017; Szawiola et al., 2017; Wu et al., 2017; Brook, 2018; Ai et al., 2019). Recently, Matsumoto and co-workers reported a sequence-controlled synthesis of oligosiloxanes via dehydrocarbonative coupling of alkoxysilanes and hydrosilanes, named the Piers-Rubinsztajn reaction, and also via hydrosilylation of carbonyl compounds using B(C6F5)3 (Matsumoto et al., 2018). Several mechanistic studies have been demonstrated for the formation of siloxanes depending on the type of substrates and catalysts used (Brook, 2018; Pattanaik and Gunanathan, 2019). The standard Piers-Rubinsztajn reaction involves the condensation of a hydrosilane (R3SiH) and alkoxysilane (R3SiOR) to form a siloxane, with subsequent removal of an alkane, RH (R = alkyl/aryl). Knowledge of this mechanism has allowed for the design of controlled synthetic routes to polysiloxanes using B(C6F5)3 as the catalyst (Chojnowski et al., 2005; Rubinsztajn and Cella, 2005; Cella and Rubinsztajn, 2008; Yi et al., 2018; Schneider et al., 2019). The strong affinity of B(C6F5)3 with H2O can lead to the formation of Brønsted acids which seems to not be a problem in terms of siloxane or polymer synthesis (Neumann et al., 2004; Chojnowski et al., 2005; Longuet et al., 2007). However, B(C6F5)3 can form several complexes with H2O which serve to remove the active catalyst from the reaction (Brook, 2018). In the presence of excess H2O therefore, the Piers-Rubinsztajn reaction is not observed or leads to lower rate of reaction.
In this article, we report the synthesis of oligomeric siloxanes starting from hydrosilanes and tethered hydrosilanes using controlled amounts of H2O. Specifically, we explored the possibility of replacing the B(C6F5)3 catalyst with (C6F5)3B(OH2) aimed at understanding the effect of the moisture stable (C6F5)3B(OH2) catalyst on the selectivity, the recyclability and the mechanism of siloxane formation.
Materials and Methods
All reactions and manipulations were performed under a nitrogen atmosphere in an MBraun Unilab 1200/780 glovebox or using conventional Schlenk techniques, unless otherwise specified. Dry solvents were obtained using a solvent purification system. Reagents were purchased from Sigma-Aldrich, AK Scientific, Arcos and TCI and used as received. (C6F5)3B(OH2) (Beringhelli et al., 2001) was made by adding a stoichiometric amount of H2O to B(C6F5)3 followed by purification by sublimation. 1H, 13C, and 29Si NMR spectra were recorded on a Bruker DPX-400 (400MHz) spectrometer. Chemical shifts for protons are reported in parts per million (ppm) downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (e.g., CHCl3 = 7.26 ppm). Chemical shifts for carbon are reported in ppm downfield from CDCl3 (77.3 ppm). Chemical shifts for silicon are reported in ppm downfield to the silicon resonance of tetramethylsilane (TMS δ 0.0). The silicon NMR resonances were determined with a DEPT pulse sequence. Data are represented as follows: chemical shift, multiplicity (app = apparent, br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constants in Hertz (Hz), and integration. High resolution mass spectrometry measurements were made on a Bruker microTOF-QII mass spectrometer, equipped with a KD Scientific syringe pump, in positive ion ESI mode. Hard ionization mass spectrometry analysis was done on Agilent 7890A GC + 5975C EI-MS with Agilent auto-sampler. X-ray diffraction analysis of single crystals of 7a and 8a were performed on a Rigaku Oxford Diffraction XtaLAB-Synergy-S single crystal diffractometer with a PILATUS 200 K hybrid pixel array detector using Cu Kα radiation (Supplementary Table 1). The data was processed with the SHELX2016 (Sheldrick, 2015) and Olex2 (Dolomanov et al., 2009) software packages. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were inserted at calculated positions and refined with a riding model or without restrictions. 8a was refined on a HKL5 dataset extracted from PLATON (Spek, 2003). Mercury 4.2.0 (Macrae et al., 2006) was used to visualize the molecular structures.
Intermolecular Reactions
Synthesis of Tertiary Disiloxanes, 3a-3c
Et3SiOEt3 (3a) and PhMe2SiOSiMe2Ph (3b)
To a mixture of hydrosilane (Et3SiH, 1a, 5.0 mmol, 0.80 mL or PhMe2SiH, 1b, 5.0 mmol, 0.77 mL) and 0.1–5.0 mol% (C6F5)3B(OH2), was added H2O (2.5 mmol, 0.050 mL) while stirring at room temperature (22°C). The reaction was monitored using 1H and 29Si{1H} NMR spectroscopy at specific time interval using an insert containing deuterated solvent. Yield: 3a: 70.6%, 3b: 94.0%.
3a (Sridhar et al., 2009; Jorapur and Shimada, 2012)
1H NMR (400 MHz CDCl3): δ 0.95–0.91 (t, CH3, 6H), δ 0.55–0.49 (q, CH2, 4H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 6.7 (s, CH2), δ 6.4 (s, CH3). 29Si{1H} NMR (79.5 MHz, CDCl3): δ 8.9 ppm (s). GC-MS: cal'd: 246.1835 m/z; observed 246.1900 m/z.
3b (Jorapur and Shimada, 2012; Sawama et al., 2016)
1H NMR (400 MHz CDCl3): δ 7.57–7.36 (m, Ph, 10H), δ 0.35 (s, CH3, 12H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 139.8, δ 133.0, δ 129.4, δ 127.7, δ 0.85 (s, CH3). 29Si{1H} NMR (79.5 MHz, CDCl3): δ 0.01 ppm (s). HRMS-ESI: [C16H22OSi2Na]+ = cal'd: 309.1101 m/z; observed 309.1091 m/z.
Ph3SiOSiPh3, 3c
To a mixture of Ph3SiH (1c; 2.0 mmol, 0.26 g) and 0.1–5.0 mol% (C6F5)3B(OH2) dissolved in 0.50 mL toluene-d8, was added H2O (1.0 mmol, 0.02 mL) while stirring at room temperature (22°C). The reaction was monitored using 1H and 29Si{1H} NMR spectroscopy at specific time interval. Yield: 98.0%.
3c (Jorapur and Shimada, 2012)
1H NMR (400 MHz CDCl3): δ 7.49–7.24 (m, Ph, 30H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 135.5, δ 135.2, δ 129.8, δ 127.7. 29Si{1H} NMR (79.5 MHz, CDCl3): δ −18.6 ppm (s). HRMS-ESI: [C36H30OSi2Na]+ cal'd: 557.1733 m/z; observed 557.1701 m/z.
Synthesis of Secondary Oligosiloxanes, (Cyclo) 3d-3f, (Cyclo) 4e-4f, 5e
To a mixture of 4.0 mmol hydrosilane (0.52 mL Et2SiH2, 1d; 0.55 mL PhMeSiH2, 1e; 0.76 mL Ph2SiH2, 1f) and 0.1–5.0 mol% (C6F5)3B(OH2), was added 2.0 mmol, 0.04 mL H2O while stirring at room temperature (22°C). The reaction was monitored using ESI-MS at specific time interval. All products were filtered through Florisil to remove the catalyst using n-pentane or hexanes (10 ml) as eluent. ESI-MS: 3d (trimer): [C14H36O4Si3Na]+ cal'd: 375.1819 m/z; observed 375.1812 m/z, cyclo-3d (cyclic trimer): [C13H33O3Si3Na]+ cal'd: 321.1737 m/z; observed 321.1731 m/z, 3e (trimer): [C23H30O4Si3Na]+ cal'd: 477.1350 m/z; observed 477.1337 m/z, 4e (tetramer): [C28H34O5Si4Na]+ cal'd: 585.1381 m/z; observed 585.1357 m/z, 5e (pentamer): [C35H42O6Si5Na]+ cal'd: 721.1725 m/z; observed 721.1657 m/z, cyclo-3f (cyclic trimer): [C36H30O3Si3Na]+ cal'd: 617.1400 m/z; observed 617.1356 m/z, cyclo-4f (cyclic tetramer): [C48H40O4Si4Na]+ cal'd: 815.1901 m/z; observed 815.1837 m/z.
Intramolecular Reactions
Synthesis of Disilyl Precursors, 7a-7c
To an oven dried two neck round bottom flask purged with nitrogen was added magnesium turnings (12.0 mmol, 0.29 g), dry THF (5 mL) and diphenylchlorosilane (7a; 12.0 mmol, 2.3 mL) or dimethylchlorosilane (7b, 7c; 12.0 mmol, 1.3 mL). To this suspension was added 2-bromobenzylbromide (6a; 3.0 mmol, 0.75 g) or α,α'-dibromo-o-xylene (6b; 3.0 mmol, 0.79 g) dissolved in dry THF (10 mL) dropwise over a period of 15 min. The mixture was refluxed for 1 h and then stirred at 22°C overnight. The reaction mixture was quenched with saturated solution of NaHCO3 (5 mL) and the aqueous layer was extracted with diethyl ether (3 ×10 mL). All the organic layers were combined and dried over Na2SO4. The crude product was obtained upon removal of solvents under vacuum.
(2-(diphenylsilyl)benzyl)diphenylsilane (7a)
The crude product was dissolved in n-pentane (20 mL) and the mixture was stored at −20°C overnight. Colorless crystals were filtered off and were washed with cold pentane (2 ×5 mL) to obtain the pure product in 72.3% yield.
1H NMR (400 MHz CDCl3): δ 7.45–6.97 (m, Ph, 24H), δ 5.53 (s, SiH, 1H), δ 4.8 (t, SiH, 1H), 2.8 (d, CH2, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 145.67, δ 137.16, δ 135.95, δ 135.38, δ 134.32, δ 133.58, δ 133.39, δ 131.36, δ 129.98, δ 129.66, δ 128.01, δ 127.89, δ 124.28, δ 23.06. 29Si{1H} NMR (79.5 MHz, CDCl3): δ −13.70, δ −22.48 ppm. HRMS-ESI: [C31H28Si2Na]+ cal'd: 479.1626 m/z; observed: 479.1615 m/z.
(2-(dimethylsilyl)benzyl)diphenylsilane (7b)
The crude product was purified over silica gel column chromatography using hexanes as the eluent and was obtained as a colorless oil in 80.8% yield.
1H NMR (400 MHz CDCl3): δ 7.45–7.05 (m, Ph, 4H), δ 4.5 (sep, SiH, 1H), δ 3.9 (sep, SiH, 1H), 2.3 (d, CH2, 2H), δ 0.3 (d, CH3, 6H), δ 0.1 (d, CH3, 6H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 146.04, δ 134.77, δ 129.34, δ 128.32, δ 123.85, δ 24.39, δ −3.06, δ −4.26. 29Si{1H} NMR (79.5 MHz, CDCl3): δ −11.04, δ −21.63 ppm. HRMS-ESI: [C11H20Si2H]+ cal'd: 209.1181 m/z; observed: 209.1171 m/z.
1,2-bis((dimethylsilyl)methyl)benzene (7c)
The crude product was purified over silica gel column chromatography using hexanes as the eluent and was obtained as colorless oil in 52.5% yield.
1H NMR (400 MHz CDCl3): δ 6.99 (s, Ph, 4H), δ 4.0 (sep, SiH, 1H), δ 2.1 (d, CH2, 2H), δ 0.1 (d, CH3, 12H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 136.71, δ 129.19, δ 124.47, δ 21.61, δ −4.21. 29Si{1H} NMR (79.5 MHz, CDCl3): δ−13.28 ppm. GC-MS: cal'd: 222.1260 m/z; observed: 222.1000 m/z.
Synthesis of Bridged Siloxanes, 8a-8c
1,1,3,3-tetraphenyl-3,4-dihydro-1H-2,1,3-benzoxadisiline (8a)
To a round bottom flask was added 7a (1.0 mmol, 0.45 g) and 5.0 mol% (C6F5)3B(OH2). The solids were dissolved in toluene (10 mL) and the reaction mixture was stirred at 90°C for 24 h. Toluene was removed under vacuum and the crude product was dissolved in hexanes (10 mL) which was then filtered through a Florisil pad with hexanes as eluent. Upon removal of volatiles the product was obtained as white solids. After recrystallization in ethyl acetate a pure crystalline product with 45.2% yield was obtained.
1H NMR (400 MHz CDCl3): δ 7.57–7.06 (m, Ph, 24H), δ 2.64 (s, CH2, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 145.04, δ 135.42, δ 135.14, δ 134.97, δ 134.31, δ 130.56, δ 130.48, δ 130.13, δ 130.02, δ 127.86, δ 124.63, δ 23.66. 29Si{1H} NMR (79.5 MHz, CDCl3): δ −9.91, −14.11 ppm. HRMS-ESI: [C31H26OSi2Na]+ cal'd: 493.1419 m/z; observed: 493.1421 m/z.
1,1,3,3-tetramethyl-3,4-dihydro-1H-2,1,3-benzoxadisiline (8b)
To a vial was added 1.0 mol% (C6F5)3B(OH2), 0.5 mL toluene and 7b (1.0 mmol, 0.21 g). The reaction mixture was stirred for 3 h at 22°C. Toluene was then removed under reduced pressure and hexanes (3 mL) was added to the mixture. Upon Florisil filtration with hexanes as eluent and removal of volatiles the product was obtained as clear oil with 61.8% yield.
1H NMR (400 MHz CDCl3): δ 7.40–7.12 (m, Ph, 4H), δ 2.19 (s, CH2, 2H), δ 0.38 (s, CH3, 6H), δ 0.16 (s, CH3, 6H). 29Si{1H} NMR (79.5 MHz, CDCl3): δ 10.85, δ 3.66 ppm. HRMS-ESI: [C11H18OSi2H]+ cal'd: 223.0974 m/z; observed: 223.0925 m/z.
2,2,4,4-tetramethyl-1,2,4,5-tetrahydrobenzoxadisilepine (8c)
To a vial was added 1.0 mol% (C6F5)3B(OH2), 0.5 mL toluene and 7c (1.0 mmol, 0.22 g). The reaction mixture was stirred for 3 h at 22°C. Toluene was then removed under reduced pressure and hexanes (3 mL) was added to the mixture. Upon Florisil filtration with hexanes as eluent and removal of volatiles the product was obtained as clear oil with 30.6% yield.
1H NMR (400 MHz CDCl3): δ 7.03–6.95 (m, Ph, 4H), δ 2.16 (s, CH2, 2H), δ 0.07 (s, CH3, 12H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 137.38, δ 129.35, δ 125.04, δ 27.48, δ 0.0. 29Si{1H} NMR (79.5 MHz, CDCl3): δ 7.23 ppm. HRMS-ESI: [C12H20OSi2H]+ cal'd: 237.1130 m/z; observed: 237.1123 m/z.
Cross-Condensation Reactions
1:1 reaction of 1a and 1c
To a mixture of 1 equiv. 1a, 1 equiv. 1c and 5.0 mol% (C6F5)3B(OH2) dissolved in 0.5 mL C6D6, was added 1.0 mmol, 0.02 mL H2O. The reaction was stirred for 5 min at 22°C and was analyzed by 1H and 29Si{1H} NMR spectroscopy.
1:1 reaction of 2a and 1b
A mixture of 1 equiv. 2a, 1 equiv. 1b and 5.0 mol% (C6F5)3B(OH2) was stirred for 1 h at 22°C and was analyzed by 1H and 29Si{1H} NMR spectroscopy.
1:1 reaction of 1b and 2b
A mixture of 1 equiv. 1b, 1 equiv. 2b and 5.0 mol% (C6F5)3B(OH2) was stirred for 5 min at 22°C and was analyzed by 1H and 29Si{1H} NMR spectroscopy.
Control Reactions
A mixture of silanol (Et3SiOH, 2a, 2.6 mmol, 0.40 mL or PhMe2SiOH, 2b, 2.6 mmol, 0.40 mL) and 0.1–0.5 mol% (C6F5)3B(OH2) was stirred and allowed to react at room temperature (22°C). The reaction was monitored using 1H and 29Si{1H} NMR spectroscopy at a specific time interval.
Catalyst Recycling Studies
3a
To 0.1–5.0 mol% (C6F5)3B(OH2) was added 5.0 mmol, 0.80 mL Et3SiH while stirring at room temperature (22°C). The reaction was monitored using 1H NMR after 1 and 3 h. At the end of the 3 h period, each product from different catalyst loading was isolated by extraction with pentane while recovering back the (C6F5)3B(OH2) catalyst used. The recovered catalyst at 1.0 mol% loading was recycled and re-used 4 times to give a total of 5 cycles and 5 isolated yields for Et3SiOSiEt3.
3b
To 0.1–5.0 mol% (C6F5)3B(OH2) was added 2.0 mmol, 0.31 mL PhMe2SiH while stirring at room temperature (22°C). The reaction was monitored using 1H NMR after 1 h. At the end of the 1 h period, each product from different catalyst loading was isolated by extraction with pentane. At 1.0 mol% catalyst loading, another 0.31 mL (2.0 mmol) of PhMe2SiH was added to the same vial. This procedure was done 4 times at 1 h intervals. The reported % yield at 1.0 mol% catalyst loading was the average of 5 cycles.
3c
To 0.1–5.0 mol% (C6F5)3B(OH2) was added 1.0 mmol, 0.26 g Ph3SiH, dissolved in 0.50 mL toluene, while stirring at room temperature (22°C). The reaction was monitored using 1H NMR after 1 and 2 h. At the end of the 2 h period, each product from different catalyst loading was isolated by extraction with dichloromethane (5 mL). At 1.0 mol% catalyst loading, another 0.26 g (1.0 mmol) of Ph3SiH dissolved in 0.50 mL toluene was added to the same vial. This procedure was done 4 times at a 2 h intervals. The reported % yield at 1.0 mol% catalyst loading was the average of 5 cycles.
Results and Discussion
Intermolecular Synthesis of Oligosiloxanes Using a (C6F5)3B(OH2) Catalyst
Three different types of tertiary hydrosilanes (Et3SiH, 1a; PhMe2SiH, 1b; and Ph3SiH, 1c) were reacted with varying equivalents of H2O (0.0–1.0) and varying concentrations of catalyst (0.1–10.0 mol %), at 22°C to yield the corresponding disiloxane (3a-c; Scheme 2). For each completed reaction, the catalyst was removed by Florisil filtration. For 1a and 1b, the siloxane product was isolated by dissolving the reaction mixture in n-pentane to selectively precipitate out the (C6F5)3B(OH2) catalyst. 1c is a solid so the catalytic reactions were enabled by the addition of minimum amount of dry toluene (~1 mL). The disiloxane products formed (3a-3c) from this synthetic route were isolated and confirmed by several characterization techniques in conjunction (Supplementary Figures 1–28) (Jorapur and Shimada, 2012). Generally, the complete conversion to disiloxane is much slower in the absence of H2O and no reaction can be observed in the absence of the catalyst (Table 1). The transformation of neat 1b to the corresponding disiloxane appeared to be the fastest and most facile at 0.1 mol% catalyst loading. This may be attributed to the fact that (C6F5)3B(OH2) is quite soluble in 1b.
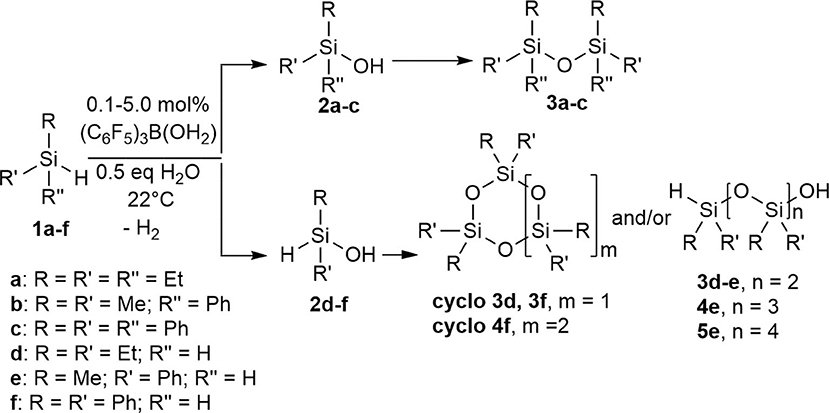
Scheme 2. Intermolecular synthesis of siloxanes using a (C6F5)3B(OH2) catalyst.
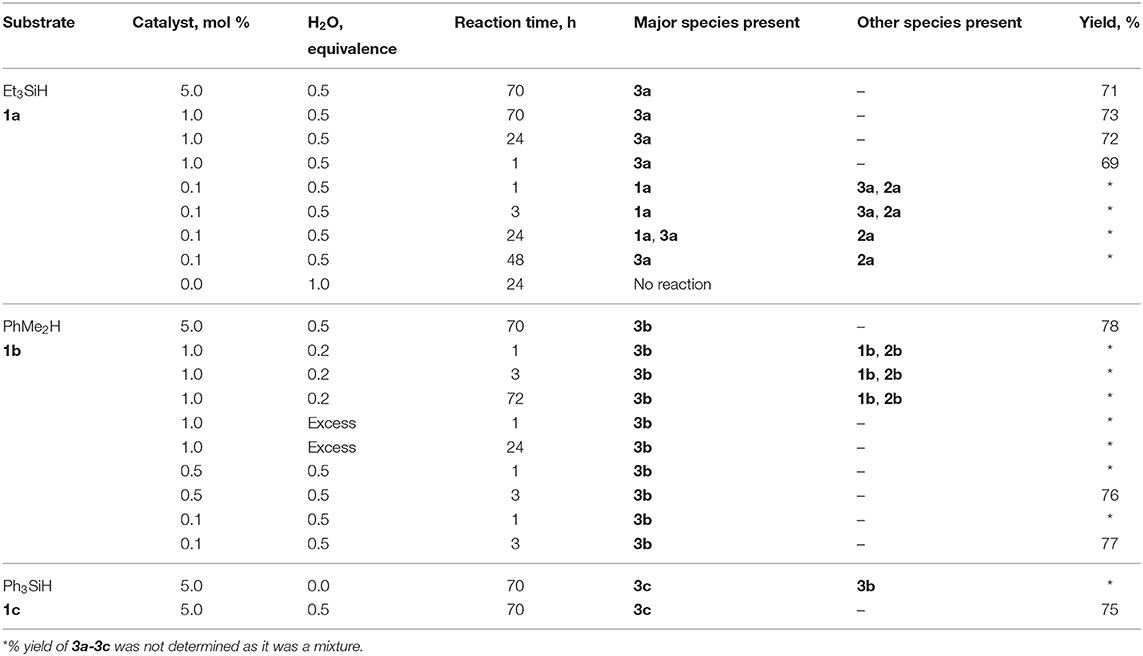
Table 1. Scope of substrates and products in the synthesis of disiloxanes, 3a-3c.
The reaction of 1a was slow at a low catalyst loading, 0.1 mol% (C6F5)3B(OH2), in the presence of 0.5 eq H2O. However, this allowed the for the detection of the silanol intermediate, Et3SiOH, 2a, along with the formation of the disiloxane, Et3SiOSiEt3, 3a (Supplementary Figures 6, 7). After 3 h, the formation of both 2a (δSi = 19.8 ppm) and 3a (δSi = 8.9 ppm) was more evident with a significant amount of unreacted 1a (δSi = 0.01 ppm). By contrast, the reaction was almost complete after 1 h when the catalyst loading was increased to 1.0 mol% (C6F5)3B(OH2), with >99% conversion and the 3a as the major product. There was no significant change observed when the same reaction was left to react further for 24 h (Supplementary Figures 8, 9). When the catalyst loading was further increased to 5.0 mol% (C6F5)3B(OH2) the results were the same as those found at the 1.0 mol% catalyst loading. As suggested by the Piers-Rubinsztajn mechanism, the condensation of silanols to form oligo- or polysiloxanes occurs at relatively low catalyst concentration (Brook, 2018). A control reaction wherein 1a was reacted with 1.0 eq H2O in the absence of the catalyst gave no reaction, showing only the presence of unreacted 1a by NMR spectroscopy. Similarly, no reaction was observed when 2a was reacted with an equivalent of H2O without a catalyst. These studies prove that both the conversion of 1a to 2a and the condensation step to 3a require a catalyst (Supplementary Figures 10, 11).
Staring from a low catalyst loading of 0.1 mol% (C6F5)3B(OH2) in the presence of 0.5 eq H2O, the conversion of PhMe2SiH, 1b, to the corresponding disiloxane, 3b (δSi = 0.01 ppm) was already evident after only 1 h reaction time. Even when the catalyst concentration was further increased to 0.5 and 5.0 mol%, the reaction proceeded in the same manner as when using a lower amount of catalyst (Supplementary Figures 17, 18). Decreasing the amount of H2O to 0.2 eq at 1.0 mol% (C6F5)3B(OH2) left some of the starting material, 1b, and presence of the silanol, PhMe2SiOH (2b), was also observed (Supplementary Figures 20, 21). It can be inferred that the addition of H2O plays an important role in the reaction, especially, in terms of the length of time needed to complete the reaction. Conversely, using an excess amount of H2O hastened the reaction while using the same catalyst concentration of 1.0 mol% (C6F5)3B(OH2). The reaction was observed to have been completed after 1 h (Supplementary Figure 22).
To further investigate the effect of reaction time and the necessity of H2O, the direct synthesis of Ph3SiOSiPh3, 3c, was conducted at 5.0 mol% (C6F5)3B(OH2), for 70 h with and without the addition of H2O. It was observed that for reactions left for a longer period of time, the addition of H2O seemed to be not necessary, and the presence of the catalyst (C6F5)3B(OH2), alone is sufficient enough to yield the desired product. As previously observed with the other silanes, a mixture of Ph3SiH (1c) with H2O in the absence of the catalyst did not show any sign of a reaction (Supplementary Figures 27, 28).
Using the same protocol as for the tertiary silanes (1a-c), the reaction of secondary hydrosilanes (1d-f) was performed and generally resulted to the direct synthesis of oligomeric siloxanes with 3-5 repeat units identified based on ESI-MS. Three different secondary silanes were used: Et2SiH2, 1d, PhMeSiH2, 1e, and Ph2SiH2, 1f. Depending on the substrate used, the products were observed to be linear and/or cyclic siloxane chains (Scheme 2; Supplementary Figures 29–31). Furthermore, in the case of 1e there was evidence of siloxanediol formation (Diemoz et al., 2016) indicating its role as an intermediate in the synthesis of the higher siloxanes under these conditions.
Similar to the synthesis of symmetrical disiloxanes, the reaction proceeded at a faster rate in the presence of H2O. The reaction with 1e gave mostly linear oligomers containing 3-5 repeat units, with a minor amount of oligosiloxanes containing 6-7 repeat units. The oligomerization of dialkylsilane, 1d, resulted to both linear and cyclic products with 3 Si-O units, while the diarylsilane, 1f, resulted to cyclic oligosiloxanes with n = 3 and 4. For 1d, it is interesting to note that after 48 h it forms a cyclic species but if left for longer reverts back to the linear species (Table 2).
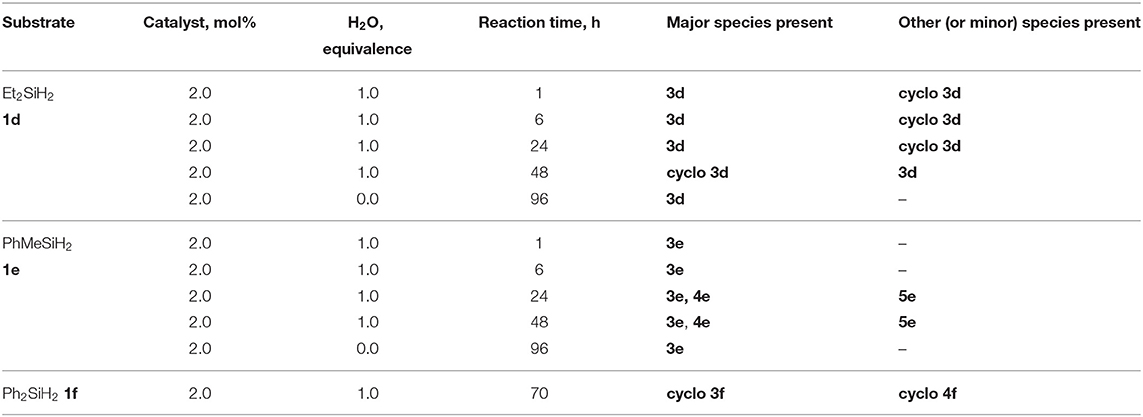
Table 2. Scope of substrates and products in the synthesis of oligosiloxanes, cyclo 3d, 3f, 4f, and linear 3d-3e, 4e, 5e.
Intramolecular Synthesis of Tethered Siloxanes Using a (C6F5)3B(OH2) Catalyst
We previously reported a preliminary study on the preparation of symmetrical naphthalene bridged disilanes with a Si-O-Si motif (Rabanzo-Castillo et al., 2019). Having success in forming linear siloxanes from tertiary and secondary silanes, it intrigued us to perform intramolecular reactions with catalytic amounts of (C6F5)3B(OH2) to produce tethered and unsymmetrical siloxanes. In order to perform such reactions, disilyl precursors (7a-c) that resemble tertiary silanes having Si-H bonds were prepared using Grignard reactions from dibrominated bridge precursors (6a-b; Scheme 3). The synthesized disilyl tertiary silane precursors were obtained in high yields (Supplementary Figures 32–44) and were later subjected to intramolecular reactions with the catalyst under aerobic conditions to obtain the desired tethered siloxanes (Supplementary Figures 45–57), cyclic structures with 6-7 membered rings (8a-c; Scheme 3).
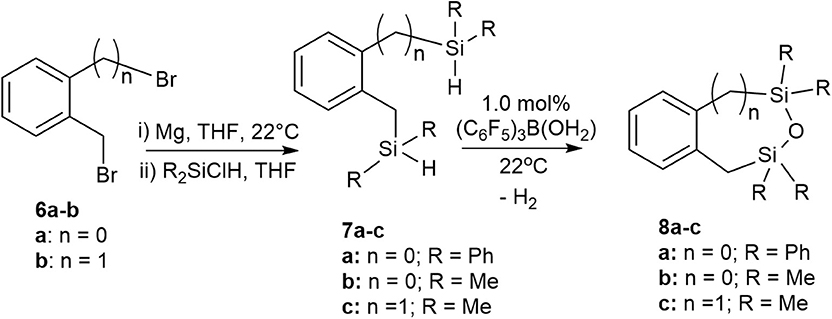
Scheme 3. Intramolecular synthesis of symmetric and asymmetric tethered siloxanes using a (C6F5)3B(OH2) catalyst.
The intramolecular dehydrocoupling step was optimized for each of the disilyl tertiary silane precursors. 7a (δSi = −13.7, −22.5 ppm) is a solid therefore was dissolved in toluene prior to the intramolecular dehydrocoupling using 5.0 mol% (C6F5)3B(OH2) at 90°C for 24 h. Under these conditions, the only product observed was 8a (δSi = −9.9, −14.1 ppm). The disilyl tertiary silane precursors 7b (δSi = −11.0, −21.6 ppm) and 7c (δSi = −13.3) are liquids and therefore were able to undergo intramolecular dehydrocoupling reactions without the addition of solvents, but the catalysis was more efficient with the addition of toluene along with 1.0 mol% (C6F5)3B(OH2) at 22°C for 3 h, yielding 8b (δSi = 10.9, 3.7 ppm) and 8c (δSi = 7.2), respectively. The crude siloxanes (8a-c) were filtered through a Florisil pad to remove the catalyst. Although cyclic siloxanes are known to degrade when subjected to purification by silica gel column chromatography, compounds 8a-8c could be purified by column chromatography, using hexanes as an eluent (Blackwell et al., 1999). The 29Si{1H} NMR chemical shifts of the purified product matched that of the filtered product suggesting that, in this case, the products do not degrade on silica gel (Supplementary Figures 45–57).
Single crystals were grown for compounds 7a and 8a by slow evaporation of pentane and ethyl acetate, respectively; the crystal structures were solved by single crystal X-ray diffraction (Figure 1; Supplementary Table 1). From the molecular structure of product 7a it is evident that due to the steric bulk from the phenyl groups on the silyl substituents and the flexibility at C19, the Si-H bonds are quite far from each other. The two silicon atoms are 4.292 Å apart. The molecular structure of 8a shows that the newly formed 6-membered cyclic structure, containing the Si1-O1-Si2 bond, results in a slightly distorted boat shaped geometry with Si1 and C19 as the central two atoms. The distortion can be attributed to the presence of the Si-O-Si linkage and phenyl substituents lengthening the bonds through these atoms in addition to the wider Si-O-Si bond angle of 124.41°.
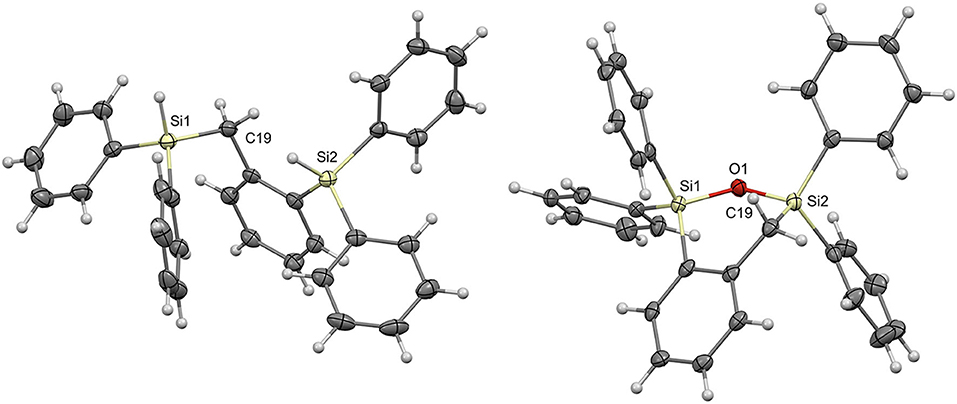
Figure 1. Molecular structures of 7a (Left) and 8a (Right) with thermal ellipsoids drawn at the 50% probability level.
Mechanistic Considerations
Looking at the simplest system, using the tertiary silanes (1a-c), the mechanism of the catalysis was investigated. The reaction process involves the formation of the silanol intermediate and generation of either hydrogen gas or H2O to give the disiloxanes, 3a-c, depending on which pathway is operating (route I or route II; Scheme 4). It is important to note that the two pathways are experimentally distinct. Creation of 2 from 1 involves the loss of H2. Similarly, with a 50% conversion to 2, the silanol (2) can react with the hydrosilane (1) to form the disiloxane (3) with concomitant loss of H2 in a dehydrocoupling reaction (route I; Scheme 4). However, if all of 1 becomes 2 (more likely under higher concentrations of H2O), then the subsequent condensation of the silanols eliminates H2O in a condensation reaction to form a disiloxane (route II; Scheme 4).
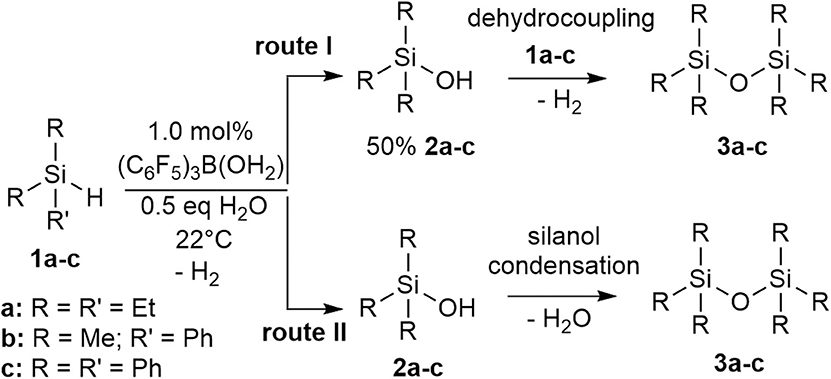
Scheme 4. Two potential routes to disiloxane from silane via silanol.
Kinetic studies were attempted to better probe the postulated mechanism, however, were not possible due to the difficulty in matching the experimental conditions by NMR spectroscopy. Moreover, each individual reactant (1a-c) gave reactions that were too fast with (C6F5)3B(OH2). To elaborate the routes occurring, neat reactions of individual silanols (2a, 2b), under the same reaction conditions of the hydrosilanes, were also performed. Hydrosilanes (1, R3SiH) are said to be more susceptible to Brønsted acid-catalyzed reactions (Muzafarov, 2011) which is generally what was observed (Table 3). Interestingly, the addition of 0.5 eq. of H2O increased the rate of reaction of 1a to 3a, from 50% conversion to >99% after 1 h, while the same addition of H2O did not have an impact on the already rapid conversion of 1b to 3b. For the case of a catalytic reaction starting with Et3SiOH, 2a, the reaction took longer than the reaction starting with 1a to reach completion (24 and 3 h, respectively). However, the same reaction starting with PhMe2SiOH, 2b, proceeded to completion within an hour. Limiting air in the reactions starting with the silanol (e.g., 2a and 2b) only made a significant difference in the reaction rate for 2a; 2b was unaffected. When a 1:1 mixture of PhMe2SiH, 1b, and PhMe2SiOH, 2b, were reacted, a rapid exotherm and build-up of pressure was observed resulting to almost full conversion after only a few seconds (Table 3). These results suggest, not only that the reaction with 1b is the most rapid in the series, but also, that both routes to form the disiloxane are possible under the catalytic conditions.
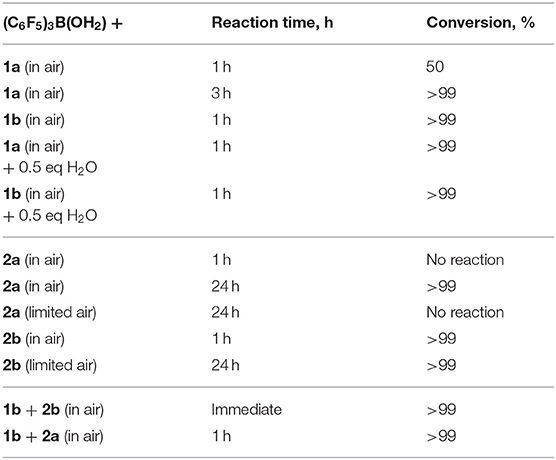
Table 3. Comparative reactivities of SiH and SiOH groups in the presence of catalytic (C6F5)3B(OH2).
To elaborate on the relative rates of reaction for substrates 1a-c, and to have a better indication of chemoselectivity, a cross-condensation reaction of a 1:1:1 mixture of Ph3SiH (1c), Et3SiH (1a), and H2O was performed with 1 mol% catalyst loading. The reaction resulted in the rapid evolution of H2 and was observed to reach completion soon after the addition of H2O into the reaction mixture (Supplementary Figures 58–61). As expected, the reaction resulted to three different products—two homo-coupling products, Ph3SiOSiPh3 (3c) and Et3SiOSiEt3 (3a) and a cross-coupling product, Ph3SiOSiEt3. A crude estimate using the 1H NMR spectrum of the product mixture revealed a ratio of 2.5 3c: 1 Ph3SiOSiEt3: 2 3a, instead of a 1:1:1 ratio which would be expected if the 1a and 1c reacted at the same rate. This deviation can be explained by the difference in kinetics and solubilities of the two reactants. 1c alone and 1a alone require 2 and 3 h reaction time, respectively, to reach full conversion, while 1b is even more rapid at 1 h. To further confirm this claim, cross-condensation reaction between 1b and 1a (Supplementary Figure 62) resulted to 3b and PhMe2SiOSiEt3 as the major products in a 1:6 ratio and, no 3a was observed. Therefore, the reactivity to form disiloxanes (3a-c) from the three tertiary silane substrates under the catalytic conditions follows the trend 1b > 1c > 1a.
Cross-coupling between PhMe2SiH, 1b, and Et3SiOH, 2a, on the other hand, resulted to a 1:1:1 ratio of 3b to the cross product, PhMe2SiOSiEt3 to 3a. In this case, both homo-coupling products were obtained along with the expected unsymmetrical cross-coupling product (Supplementary Figures 63, 64) and the ratio suggests that the difference in the aforementioned reactivity trend is mostly likely due to the rate determining formation of the silanol (2) from the silane (1), which is most sluggish for the conversion of 1a to 2a.
Monitoring the catalytic intermediates and the formation of silanol experimentally in situ in order to capture the impact of ppm-level changes of the substrates was challenging. Nevertheless, NMR tube reactions wherein 2.6 mmol each of 1b and 2b were reacted, separately, with 0.1 mol% of (C6F5)3B(OH2). Experimentally, partial conversion to 2b and 3b after 4 h was observed with 1b, which is significantly slower compared to the reaction performed with constant stirring. It is interesting to note that when using a hydrosilane (e.g., 1b) as the starting material, the products that have been formed (i.e., 2b and 3b) after 4 h tend to revert back to PhMe2SiH after letting the reaction stand for a total of 96 h (Supplementary Figure 65). This indicates that the reaction is not quite as simple as implied, with reversibility at play along with the direct siloxane formation. In contrast, using the silanol (2b) as the starting material, full conversion was observed after 24 h and the product (3b) did not convert back to the silanol substrate after 96 h (Supplementary Figure 66).
In summary, the results obtained from cross-coupling and control reactions indicate that the prevalent mechanism is where the hydrosilane (R3SiH) and silanol (R3SiOH) undergo dehydrocoupling to yield the disiloxane product (3), but under high concentration regimes of some silanol substrates (e.g., as confirmed for 2b) the silanol condensation route is possible. The postulated reaction mechanism, therefore, for the transformation is a modified Pier-Rubinsztajn reaction (Scheme 5). The process involves: i) the hydrolysis of Si-H to Si-OH using (C6F5)3B(OH2) with subsequent release of H2 gas, followed by ii) nucleophilic attack of silanol (2, R3SiOH) to the hydrosilane (1, R3SiH) to form a disiloxane and another equivalent of H2 gas (Scheme 5).
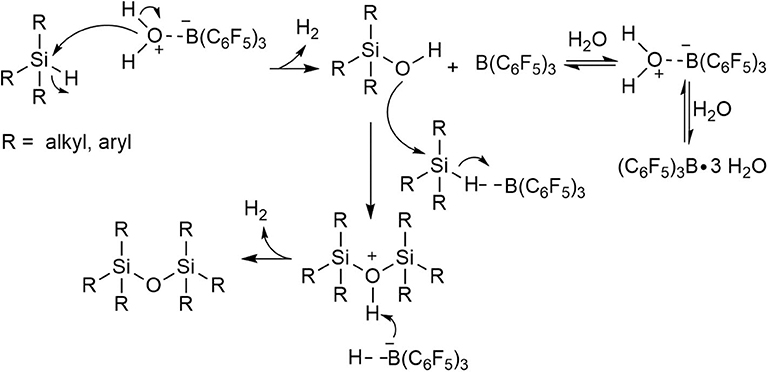
Scheme 5. Proposed mechanism for the catalytic formation of disiloxanes using (C6F5)3B(OH2).
In addition to the conversion of the three components (1, 2, 3) over time, the catalyst can have different interactions with the substrates (1, 2) and H2O that are present during the reaction. Decomposition of (C6F5)3B(OH2) catalyst via B-C bond protonolysis is highly probable in strongly basic conditions and high temperatures (Bradley et al., 1996; Ashley et al., 2009; Scott et al., 2015). Generally, Frustrated Lewis Pairs (FLP's) demonstrate high sensitivity to moisture. The high Lewis acidity of B(C6F5)3 leads to strong complexation with H2O and deprotonation even with moderately strong bases can occur irreversibly (Bergquist et al., 2000). However, the method described in this article demonstrated moisture tolerance of the (C6F5)3B(OH2) catalyst as it was conducted at room temperature, and due to the lack of any strong base in a reaction with a hydrosilane as the sole substrate.
Another way to view the direct synthesis of disiloxanes is through a non-conventional mechanism which features multiple catalytically relevant species and series of competitive reactions (Scheme 6). The formation of disiloxanes can be regarded as a Lewis acid-catalyzed reaction, a water-mediated or a silanol-mediated type of catalysis (Yu et al., 2018). In the Lewis acid catalysis (Scheme 6), the Si-H bond of the silane is activated by B(C6F5)3 and then will further react with a silanol to produce the disiloxane with concomitant release of H2. By contrast, hydrosilylation reactions catalyzed by B(C6F5)3 are generally characterized by the formation of borane-silane complex (Parks and Piers, 1996; Parks et al., 2000; Rendler and Oestreich, 2008; Sakata and Fujimoto, 2013; Zhang et al., 2016; Cheng et al., 2018). Furthermore, several experimental and theoretical mechanistic studies have suggested that borane-catalyzed reactions of siloxanes are initiated by activation of the silane Si-H bond (Parks et al., 2000; Hog and Oestreich, 2009; Mewald and Oestreich, 2012; Sakata and Fujimoto, 2013; Mathew et al., 2017).
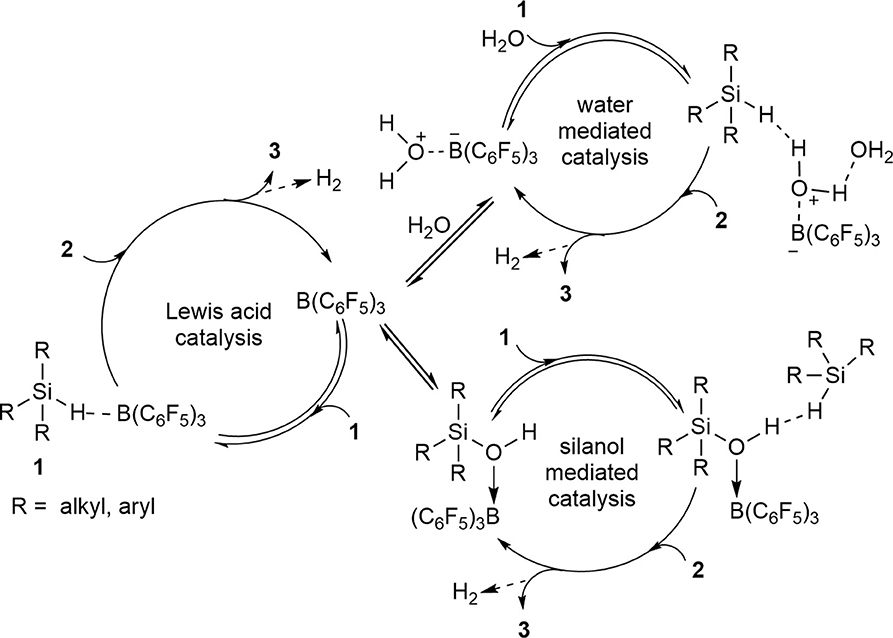
Scheme 6. A catalytic mechanism with multiple competing catalytic pathways to form disiloxanes.
Since the B(C6F5)3 catalyst has strong affinity with H2O, the Lewis-acid mediated pathway (Scheme 6) is not enough on its own to describe the formation of the disiloxane. The reaction could also possibly proceed via a water-mediated catalytic cycle (Scheme 6). For this reaction pathway, R3SiH is initially activated by the (C6F5)3B(OH2) catalyst, which is in equilibrium with B(C6F5)3 in the presence of H2O. While B(C6F5)3 is a potent Lewis acid (C6F5)3B(OH2), can be regarded as a strong Brønsted acid (Bergquist et al., 2000). The Si-H activation step can lead to Lewis acidic silicon atom, which for this case then interacts with oxygen lone pair of the pre-formed silanol to generate 3 and eliminate H2. One final catalytic possibility, under higher concentrations of silanol (2) is that it can mediate the catalysis. Here the silanol (2) interacts with (C6F5)3B(OH2) catalyst via the oxygen atom which activates it toward a reaction with silane (1) to produce 3 and H2 while regenerating the catalyst/silanol adduct (Scheme 6).
The observations from the control and competition reactions suggest that the water-mediated catalysis (Scheme 6) is the most probable catalytic pathway which will lead to the formation of disiloxane. Nonetheless, there also might be a competition between more than one catalytic cycle happening simultaneously or under different concentration regimes of substrates 1 (e.g., toward the start of the reaction) and 2 (e.g., toward full conversion to 3). Therefore, this seemingly simple reaction may have an incredibly complex mechanism.
Catalyst Recycling Studies
Catalyst recycling, that is the reuse of the (C6F5)3B(OH2) catalyst, was considered to demonstrate the efficiency and sustainability of the direct synthetic route to oligosiloxanes. For this purpose, all experiments were conducted with constant stirring to allow efficient removal of hydrogen gas and favor the formation of disiloxane product. The reactions were monitored by 1H NMR spectroscopy to estimate the reaction time needed for each substrate to reach completion. From these studies, 3, 1 and 2 h reaction time was applied for Et3SiH (1a), PhMe2SiH (1b) and Ph3SiH (1c), respectively.
For Et3SiOSiEt3, 3a, the catalyst was recovered conveniently as it settled at the bottom of the flask after the stirring was stopped. The product was isolated by addition of n-pentane followed by decantation using a cannula. Looking at catalyst loading (0.1–5.0 mol%) there was very little discrepancy in isolated yield over this range and the best TOF achieved was 167 h−1 with 0.1 mol% catalyst loading (Figure 2; Supplementary Figure 67; Supplementary Table 2). The dependence on catalyst loading for the reaction of 1b to form PhMe2SiOSiMe2Ph, 3b, is far less pronounced with the given experimental conditions allowing for an increase in TOF with a decrease in catalyst loading; 900 h−1 TOF was achieved at 0.1 mol% loading (3b, Figure 2; Supplementary Figure 68; Supplementary Table 3). The reaction was completed after 1 h as observed by 1H NMR spectroscopy. In this case, the (C6F5)3B(OH2) remained soluble even after the formation of 3b. Therefore, due to the challenges of directly testing the catalyst recyclability on such a small scale (e.g., 0.1 mol% loading), the activity of the catalyst (at 1.0 mol% loading) was demonstrated by adding a constant amount of 1b to the same flask every hour after testing the degree of completion by 1H and 29Si NMR spectroscopy. After 5 cycles, 3b was isolated and the % yield for each trial was reported as the average of 5 cycles (94%). Similarly, using 1c as the substrate, the reaction rate exhibited independence of catalyst loading with a TOF of 485 h−1 at 0.1 mol% loading (3c, Figure 2; Supplementary Figure 69; Supplementary Table 4). However, the reaction required the addition of toluene to dissolve the two solids (1c and catalyst). Similar to the reaction with 1b, given that the reaction was performed in a mmol scale, a constant volume of 3.0 M solution of 1c in toluene was added sequentially to the same flask at 1.0 mol% catalyst loading. The product Ph3SiOSiPh3, 3c, was again isolated collectively at the end of the fifth cycle (98%).
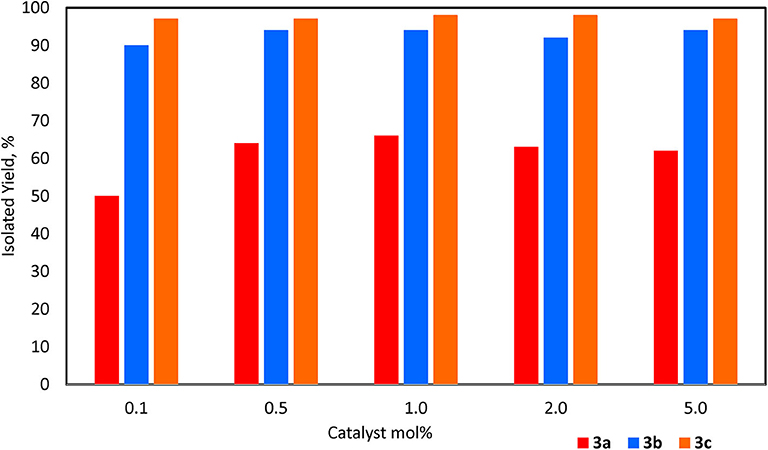
Figure 2. Yields for 3a-c with catalyst loadings 0.1–5.0 mol%.
To demonstrate the recycling by separating the catalyst and product after each cycle, both 0.5 mol% and 1.0 mol% catalyst loadings of (C6F5)3B(OH2) were chosen. The results showed that the activity of the catalyst for each cycle is almost the same (within experimental error) for up to five repeats (Figure 3). The volatility and solubility of the substrate plays an important role in facilitating the formation of the disiloxane product in excellent yields. Since the reactions were performed with constant stirring, it can be assumed that some of the Et3SiH may have been lost prior to its conversion to product (see lower yield for 3a, Figure 2). In addition, the solubility of the catalyst was enhanced in the presence of a solvent as compared to a neat reaction. Overall, these experiments successfully demonstrated the longevity of the (C6F5)3B(OH2) catalyst and that it is possible to recycle it at least five times in reactions with the given substrates.
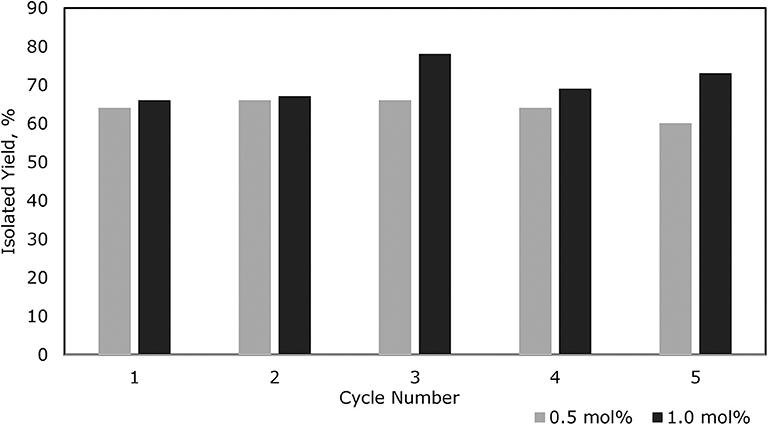
Figure 3. Yields for 3a over 5 cycles of recycling using 0.5 and 1.0 mol% catalyst loading.
Conclusions
The study presented in this article has demonstrated an efficient route from secondary and tertiary hydrosilanes into the corresponding oligosiloxanes using a moisture-stable and recyclable (C6F5)3B(OH2) catalyst through either an intermolecular or intramolecular reaction depending on the substrate. Multiple catalytically relevant species and a series of competitive reactions are probable when considering the mechanism involved for the direct synthesis of siloxanes from only hydrosilanes and water. A single catalytic pathway is simply not enough to describe the formation of the disiloxane. The experimental results gathered from control studies suggest that the most feasible route to disiloxanes is where the Si-H is partially converted to Si-OH followed by a heterodehydrocoupling reaction of Si-H/Si-OH. Moreover, the cross-coupling results show promise for the selective synthesis of unsymmetrical siloxanes under controlled conditions directly from silanes or using a mixture of silanes and silanols or alcohols and this will be the subject of future studies in our group. Nonetheless, given the wide range of hydrosilanes that are commercially available, this protocol should provide an easy route to a large variety of oligosiloxanes.
Data Availability Statement
The datasets generated for this study can be found in the CCDC repository (1987430, 1987431) or are included in article/Supplementary Material.
Author Contributions
KR-C and VK designed and performed the experiments under the supervision of EL. TS refined the crystal structures. KR-C and VK wrote the paper with editorial support from EL.
Funding
This work was supported by Royal Society of New Zealand, Marsden Fast Start Fund (16-UOA-042).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the School of Chemical Sciences at the University of Auckland for a doctoral scholarship (KR-C) as well as the Royal Society of New Zealand Marsden Fast-Start grant for providing financial support as well as a doctoral scholarship (VK). We thank Tatiana Groutso for collecting the single crystal X-ray diffraction data. We would also like to acknowledge the MacDiarmid Institute for financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00477/full#supplementary-material
References
Abele, R., Abele, E., Fleisher, M., Geinberga, S., and Lukevics, E. (2003). Novel fluoride ion mediated synthesis of unsymmetrical siloxanes under phase transfer catalysis conditions. J. Organometal. Chem. 686, 52–57. doi: 10.1016/S0022-328X(03)00286-9
Ai, L., Chen, Y., He, L., Luo, Y., Li, S, and Xu, C. (2019). Synthesis of structured polysiloxazanes via a Piers–Rubinsztajn reaction. Chem. Comm. 55, 14019–14022. doi: 10.1039/C9CC07312D
Asao, N., Ishikawa, Y., Hatakeyama, N., Menggenbateer, Yamamoto, Y., Chen, M., et al. (2010). Nanostructured materials as catalysts: nanoporous-gold-catalyzed oxidation of organosilanes with water. Angew. Chem. Int. Ed. 49, 10093–10095. doi: 10.1002/anie.201005138
Ashley, A. E., Thompson, A. L., and O'Hare, D. (2009). Non-metal-mediated homogeneous hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 48, 9839–9843. doi: 10.1002/anie.200905466
Bergquist, C., Bridgewater, B. M., Harlan, C. J., Norton, J. R., Friesner, R. A, and Parkin, G. (2000). Aqua, alcohol, and acetonitrile adducts of Tris(perfluorophenyl)borane: evaluation of brønsted acidity and ligand lability with experimental and computational methods. J. Am. Chem. Soc. 122, 10581–10590. doi: 10.1021/ja001915g
Beringhelli, T., Maggioni, D., and D'alfonso, G. (2001). 1H and 19F NMR investigation of the reaction of B(C6F5)3 with water in toluene solution. Organometallics 20, 4927–4938. doi: 10.1021/om010610n
Blackwell, J. M., Foster, K. L., Beck, V. H., and Piers, W. E. (1999). B(C6F5)3-catalyzed silation of alcohols: a mild, general method for synthesis of silyl ethers. J. Org. Chem. 64, 4887–4892. doi: 10.1021/jo9903003
Bradley, D. C., Harding, I. S., Keefe, A. D., Motevalli, M., and Zheng, D. H. (1996). Reversible adduct formation between phosphines and triarylboron compounds. J. Chem. Soc. Dalton Trans. 3931–3936. doi: 10.1039/dt9960003931
Brinker, C. J., and Scherer, G. W. (1990). Sol–Gel Science: The Physics and Chemistry of Sol–Gel Processing. New York, NY: Academic Press.
Brook, M. A. (2018). New control over silicone synthesis using sih chemistry: the piers–rubinsztajn reaction. Chem. Eur. J. 24, 8458–8469. doi: 10.1002/chem.201800123
Cella, J., and Rubinsztajn, S. (2008). Preparation of polyaryloxysilanes and polyaryloxysiloxanes by B(C6F5)3 catalyzed polyetherification of dihydrosilanes and bis-phenols. Macromolecules 41, 6965–6971. doi: 10.1021/ma800833c
Chauhan, B. P. S., Sarkar, A., Chauhan, M., and Roka, A. (2009). Water as green oxidant: a highly selective conversion of organosilanes to silanols with water. Appl. Organomet. Chem. 23, 385–390. doi: 10.1002/aoc.1528
Cheng, G. J., Drosos, N., Morandi, B., and Thiel, W. (2018). Computational study of B(C6F5)3-catalyzed selective deoxygenation of 1,2-diols: cyclic and noncyclic pathways. ACS Catal. 8, 1697–1702. doi: 10.1021/acscatal.7b04209
Chojnowski, J., Fortuniak, W., Kurjata, J., Rubinsztajn, S., and Cella, J. A. (2006). Oligomerization of hydrosiloxanes in the presence of tris(pentafluorophenyl)borane. Macromolecules, 39, 3802–3807. doi: 10.1021/ma060080c
Chojnowski, J., Rubinsztajn, S., Cella, J. A., Fortuniak, W., Cypryk, M., Kurjata, J., et al. (2005). Mechanism of the B(C6F5)3-catalyzed reaction of silyl hydrides with alkoxysilanes. Kinetic and spectroscopic studies. Organometallics, 24, 6077–6084. doi: 10.1021/om050563p
Chojnowski, J., Rubinsztajn, S., Fortuniak, W., and Kurjata, J. (2008). Synthesis of highly branched alkoxysiloxane–dimethylsiloxane copolymers by nonhydrolytic dehydrocarbon polycondensation catalyzed by tris(pentafluorophenyl)borane. Macromolecules, 41, 7352–7358. doi: 10.1021/ma801130y
Diemoz, K. M., Wilson, S. O., and Franz, A. K. (2016). Synthesis of structurally varied 1,3-disiloxanediols and their activity as anion-binding catalysts. Chem Eur. J. 22, 18349–18353. doi: 10.1002/chem.201604103
Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K., and Puschmann, H. (2009). OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 42, 339–341. doi: 10.1107/S0021889808042726
Feghali, E., and Cantat, T. (2014). Unprecedented organocatalytic reduction of lignin model compounds to phenols and primary alcohols using hydrosilanes. Chem. Commun. 50, 862–865. doi: 10.1039/C3CC47655C
Feghali, E., Carrot, G., Thuéry, P., Genre, C., and Cantat, T. (2015). Convergent reductive depolymerization of wood lignin to isolated phenol derivatives by metal-free catalytic hydrosilylation. Energy Environ. Sci. 8, 2734–2743. doi: 10.1039/C5EE01304F
Grubb, W. T. (1954). A rate study of the silanol condensation reaction at 25° in alcoholic solvents1. J. Am. Chem. Soc. 76, 3408–3414. doi: 10.1021/ja01642a014
Hog, D. T., and Oestreich, M. (2009). B(C6F5)3-catalyzed reduction of ketones and imines using silicon-stereogenic silanes: stereoinduction by single-point binding. Eur. J. Org. Chem. 2009, 5047–5056. doi: 10.1002/ejoc.200900796
Hreczycho, G. (2015). An efficient catalytic approach for the synthesis of unsymmetrical siloxanes. Eur. J. Inorg. Chem. 2015, 67–72. doi: 10.1002/ejic.201402904
Hreczycho, G., Kucinski, K., Pawluć, P., and Marciniec, B. (2013). Catalytic synthesis of linear oligosiloxanes and germasiloxanes mediated by scandium trifluoromethanesulfonate. Organometallics 32, 5001–5004. doi: 10.1021/om400581g
Igarashi, M., Kubo, K., Matsumoto, T., Sato, K., Ando, W, and Shimada, S. (2014). Pd/C-catalyzed cross-coupling reaction of benzyloxysilanes with halosilanes for selective synthesis of unsymmetrical siloxanes. RSC Adv. 4, 19099–19102. doi: 10.1039/c4ra02126f
Ison, E. A., Corbin, R. A., and Abu-Omar, M. M. (2005). Hydrogen production from hydrolytic oxidation of organosilanes using a cationic oxorhenium catalyst. J. Am. Chem. Soc. 127, 11938–11939. doi: 10.1021/ja053860u
Jeon, M., Han, J., and Park, J. (2012). Transformation of silanes into silanols using water and recyclable metal nanoparticle catalysts. ChemCatChem 4, 521–524. doi: 10.1002/cctc.201100456
John, J., Gravel, E., Hagège, A., Li, H., Gacoin, T., and Doris, E. (2011). Catalytic oxidation of silanes by carbon nanotube–gold nanohybrids. Angew. Chem. Int. Ed. 50, 7533–7536. doi: 10.1002/anie.201101993
Jorapur, Y. R., and Shimada, T. (2012). An efficient method for the synthesis of symmetrical disiloxanes from alkoxysilanes using meerwein's reagent. Synlett 23, 1633–1638. doi: 10.1055/s-0031-1290668
Kawakami, Y., Li, Y., Liu, Y., Seino, M., Pakjamsai, C., Oishi, M., et al. (2004). Control of molecular weight, stereochemistry and higher order structure of siloxane-containing polymers and their functional design. Macromol. Res. 12, 156–171. doi: 10.1007/BF03218384
Kaźmierczak, J., and Hreczycho, G. (2018). Nafion as effective and selective heterogeneous catalytic system in O-metalation of silanols and POSS silanols. J. Cat. 367, 95–103. doi: 10.1016/j.jcat.2018.08.024
Kuciński, K., and Hreczycho, G. (2019a). A highly effective route to si-o-si moieties through o-silylation of silanols and polyhedral oligomeric silsesquioxane silanols with disilazanes. ChemSusChem 12, 1043–1048. doi: 10.1002/cssc.201802757
Kuciński, K., and Hreczycho, G. (2019b). O-metalation of silanols and POSS silanols over amberlyst-15 catalyst: a facile route to unsymmetrical siloxanes, borasiloxanes and germasiloxanes. Inorg. Chim. Acta 490, 261–266. doi: 10.1016/j.ica.2019.03.025
Kurihara, Y., Yamanoi, Y., and Nishihara, H. (2013). Pd-catalyzed synthesis of symmetrical and unsymmetrical siloxanes. Chem. Comm., 49, 11275–11277. doi: 10.1039/c3cc46294c
Kurjata, J., Fortuniak, W., Rubinsztajn, S., and Chojnowski, J. (2009). B(C6F5)3 catalyzed dehydrocarbon polycondensation of PhSiH3 with (MeO)4Si as model polyfunctional comonomers in new route to hydrophobic silicone TQ resins. Eur. Polym. J. 45, 3372–3379. doi: 10.1016/j.eurpolymj.2009.10.004
Laengert, S. E., Schneider, A. F., Lovinger, E., Chen, Y., and Brook, M. A. (2017). Sequential functionalization of a natural crosslinker leads to designer silicone networks. Chem. Asian J. 12, 1208–1212. doi: 10.1002/asia.201700160
Lawson, J. R., and Melen, R. L. (2017). Tris(pentafluorophenyl)borane and beyond: modern advances in borylation chemistry. Inorg. Chem. 56, 8627–8643. doi: 10.1021/acs.inorgchem.6b02911
Le Coz, E., Kahlal, S., Saillard, J. Y., Roisnel, T., Dorcet, V., Carpentier, J. F., et al. (2019). Barium siloxides and catalysed formation of Si-O-Si' motifs. Chem. Eur. J. 25, 13509–13513. doi: 10.1002/chem.201903676
Lee, M., Ko, S., and Chang, S. (2000). Highly selective and practical hydrolytic oxidation of organosilanes to silanols catalyzed by a ruthenium complex. J. Am. Chem. Soc. 122, 12011–12012. doi: 10.1021/ja003079g
Lee, Y., Seomoon, D., Kim, S., Han, H., Chang, S., and Lee, P. H. (2004). Highly efficient iridium-catalyzed oxidation of organosilanes to silanols. J. Org. Chem. 69, 1741–1743. doi: 10.1021/jo035647r
Liu, T., Yang, F., Li, Y., Ren, L., Zhang, L., Xu, K., et al. (2014). Plasma synthesis of carbon nanotube-gold nanohybrids: efficient catalysts for green oxidation of silanes in water. J. Mater. Chem. A 2, 245–250. doi: 10.1039/C3TA13693K
Longuet, C., Joly-Duhamel, C., and Ganachaud, F. (2007). Copolycondensation of regular functional silane and siloxane in aqueous emulsion using B(C6F5)3 as a catalyst. Macromol. Chem. Phys. 208, 1883–1892. doi: 10.1002/macp.200700202
Macrae, C. F., Edgington, P. R., Mccabe, P., Pidcock, E., Shields, G. P., Taylor, R., et al. (2006). Mercury: visualization and analysis of crystal structures. J. Appl. Crystallogr. 39, 453–457. doi: 10.1107/S002188980600731X
Madsen, F. B., Javakhishvili, I., Jensen, R. E., Daugaard, A. E., Hvilsted, S., and Skov, A. L. (2014). Synthesis of telechelic vinyl/allyl functional siloxane copolymers with structural control. Polym. Chem. 5, 7054–7061. doi: 10.1039/C4PY00919C
Marciniec, B., Pawluć, P., Hreczycho, G., Macina, A., and Adalska, M. (2008). Silylation of silanols with vinylsilanes catalyzed by a ruthenium complex. Tetrahedron Lett. 49, 1310–1313. doi: 10.1016/j.tetlet.2007.12.091
Mathew, J., Eguchi, K., Nakajima, Y., Sato, K., Shimada, S., and Choe, Y.-K. (2017). Tris(pentafluorophenyl)borane-catalyzed reactions of siloxanes: a combined experimental and computational study. Eur. J. Org. Chem. 2017, 4922–4927. doi: 10.1002/ejoc.201700760
Matsumoto, K., Oba, Y., Nakajima, Y., Shimada, S., and Sato, K. (2018). One-pot sequence-controlled synthesis of oligosiloxanes. Angew. Chem. Int. Ed. 57, 4637–4641. doi: 10.1002/anie.201801031
Matsumoto, K., Shimada, S., and Sato, K. (2019). Sequence-controlled catalytic one-pot synthesis of siloxane oligomers. Chem. Eur. J. 25, 920–928. doi: 10.1002/chem.201803565
Matsuo, T., and Kawaguchi, H. (2006). From carbon dioxide to methane: homogeneous reduction of carbon dioxide with hydrosilanes catalyzed by zirconium–borane complexes. J. Am. Chem. Soc. 128, 12362–12363. doi: 10.1021/ja0647250
Mewald, M., and Oestreich, M. (2012). Illuminating the mechanism of the borane-catalyzed hydrosilylation of imines with both an axially chiral borane and silane. Chem. Eur. J. 18, 14079–14084. doi: 10.1002/chem.201202693
Mitsudome, T., Arita, S., Mori, H., Mizugaki, T., Jitsukawa, K., and Kaneda, K. (2008). Supported silver-nanoparticle-catalyzed highly efficient aqueous oxidation of phenylsilanes to silanols. Angew. Chem. Int. Ed. 47, 7938–7940. doi: 10.1002/anie.200802761
Mitsudome, T., Noujima, A., Mizugaki, T., Jitsukawa, K., and Kaneda, K. (2009). Supported gold nanoparticlecatalyst for the selective oxidation of silanes to silanols in water. Chem. Commun. 5302–5304. doi: 10.1039/b910208f
Neumann, B., Vincent, B., Krustev, R., and Müller, H. J. (2004). Stability of various silicone oil/water emulsion films as a function of surfactant and salt concentration. Langmuir 20, 4336–4344. doi: 10.1021/la035517d
Parks, D. J., Blackwell, J. M., and Piers, W. E. (2000). Studies on the mechanism of B(C6F5)3-catalyzed hydrosilation of carbonyl functions. J. Org. Chem. 65, 3090–3098. doi: 10.1021/jo991828a
Parks, D. J., and Piers, W. E. (1996). Tris(pentafluorophenyl)boron-catalyzed hydrosilation of aromatic aldehydes, ketones, and esters. J. Am. Chem. Soc. 118, 9440–9441. doi: 10.1021/ja961536g
Pattanaik, S., and Gunanathan, C. (2019). Cobalt-catalyzed selective synthesis of disiloxanes and hydrodisiloxanes. ACS Cataly. 9, 5552–5561. doi: 10.1021/acscatal.9b00305
Piers, W. E. (2004). The chemistry of perfluoroaryl boranes. Adv. Organomet. Chem. 52, 1–76. doi: 10.1016/s0065-3055(04)52001-4
Rabanzo-Castillo, K. M., Hanif, M., Söhnel, T., and Leitao, E. M. (2019). Synthesis, characterisation and electronic properties of naphthalene bridged disilanes. Dalton Trans. 48, 13971–13980. doi: 10.1039/C9DT03058A
Rendler, S., and Oestreich, M. (2008). Conclusive evidence for an SN2-Si mechanism in the B(C6F5)3-catalyzed hydrosilylation of carbonyl compounds: implications for the related hydrogenation. Angew. Chem. Int. Ed. 47, 5997–6000. doi: 10.1002/anie.200801675
Rubinsztajn, S., and Cella, J. A. (2005). A new polycondensation process for the preparation of polysiloxane copolymers. Macromolecules 38, 1061–1063. doi: 10.1021/ma047984n
Sakata, K., and Fujimoto, H. (2013). Quantum chemical study of B(C6F5)3-catalyzed hydrosilylation of carbonyl group. J. Org. Chem. 78, 12505–12512. doi: 10.1021/jo402195x
Sawama, Y., Masuda, M., Yasukawa, N., Nakatani, R., Nishimura, S., Shibata, K., et al. (2016). Disiloxane synthesis based on silicon–hydrogen bond activation using gold and platinum on carbon in water or heavy water. J. Org. Chem. 81, 4190–4195. doi: 10.1021/acs.joc.6b00556
Schneider, A. F., Laidley, E., and Brook, M. A. (2019). Facile synthesis of Cx(AB)yCx triblock silicone copolymers utilizing moisture mediated living-end chain extension. Macromol. Chem. Phys. 220:1800575. doi: 10.1002/macp.201800575
Scott, D. J., Simmons, T. R., Lawrence, E. J., Wildgoose, G. G., Fuchter, M. J., and Ashley, A. E. (2015). Facile protocol for water-tolerant “frustrated lewis pair”-catalyzed hydrogenation. ACS Catal. 5, 5540–5544. doi: 10.1021/acscatal.5b01417
Sheldrick, G. (2015). SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 71, 3–8. doi: 10.1107/S2053273314026370
Shimizu, K., Kubo, T., and Satsuma, A. (2012a). Surface oxygen-assisted pd nanoparticle catalysis for selective oxidation of silanes to silanols. Chem. Eur. J. 18, 2226–2229. doi: 10.1002/chem.201103088
Shimizu, K.-I., Shimura, K., Imaiida, N., and Satsuma, A. (2012b). Heterogeneous nickel catalyst for selective hydration of silanes to silanols. J. Mol. Catal. A Chem. 365, 50–54. doi: 10.1016/j.molcata.2012.08.007
Shinke, S., Tsuchimoto, T., and Kawakami, Y. (2007). Stereochemistry in Lewis acid-catalyzed silylation of alcohols, silanols, and methoxysilanes with optically active methyl(1-naphthyl)phenylsilane. Silicon Chem. 3, 243–249. doi: 10.1007/s11201-007-9026-y
Spek, A. (2003). Single-crystal structure validation with the program PLATON. J. App. Cryst. 36, 7–13. doi: 10.1107/S0021889802022112
Sridhar, M., Ramanaiah, B. C., Narsaiah, C., Kumara Swamy, M., Mahesh, B., and Kumar Reddy, M. K. (2009). An efficient and simple method for the preparation of symmetrical disiloxanes from hydrosilanes by Lewis acid-catalyzed air oxidation. Tetrahedron Lett., 50, 7166–7168. doi: 10.1016/j.tetlet.2009.10.020
Szawiola, A. M., De Melo Souza, N., Lessard, B. H., and Bender, T. P. (2017). Phenoxylated siloxane-based polymers via the Piers–Rubinsztajn process. Polym. Int. 66, 1324–1328. doi: 10.1002/pi.5396
Tan, S. T., Kee, J. W., and Fan, W. Y. (2011). Catalytic hydrogen generation from the hydrolysis of silanes by ruthenium complexes. Organometallics, 30, 4008–4013. doi: 10.1021/om200256h
Thompson, J. L., and Davies, H. M. (2007). Enhancement of cyclopropanation chemistry in the silver-catalyzed reactions of aryldiazoacetates. J. Am. Chem. Soc. 129, 6090–6091. doi: 10.1021/ja069314y
Tsuchido, Y., Kanda, A., and Osakada, K. (2020). Gold (I) complexes with chloro(diaryl)silyl ligand. Stoichiometric reactions and catalysis for O-functionalization of organosilane. Tetrahedron 76:131076. doi: 10.1016/j.tet.2020.131076
Uchida, H., Kabe, Y., Yoshino, K., Kawamata, A., Tsumuraya, T., and Masamune, S. (1990). General strategy for the systematic synthesis of oligosiloxanes. Silicone dendrimers. J. Am. Chem. Soc. 112, 7077–7079. doi: 10.1021/ja00175a062
Wang, D., Klein, J., and Mejía, E. (2017). Catalytic systems for the cross-linking of organosilicon polymers. Chem. Asian J. 12, 1180–1197. doi: 10.1002/asia.201700304
Wu, C., Yu, J., Li, Q., and Liu, Y. (2017). High molecular weight cyclic polysiloxanes from organocatalytic zwitterionic polymerization of constrained spirocyclosiloxanes. Polym. Chem. 8, 7301–7306. doi: 10.1039/C7PY01499F
Yi, M., Chen, X., Wu, S., Ge, J., Zhou, X., and Yin, G. (2018). Fabrication of reactive poly(Phenyl-Substituted Siloxanes/Silsesquioxanes) with Si–H and alkoxy functional groups via the piers–rubinsztajn reaction. Polymers 10:1006. doi: 10.3390/polym10091006
Yu, Y., Zhu, Y., Bhagat, M. N., Raghuraman, A., Hirsekorn, K. F., Notestein, J. M., et al. (2018). Mechanism of regioselective ring-opening reactions of 1,2-epoxyoctane catalyzed by tris(pentafluorophenyl)borane: a combined experimental, density functional theory, and microkinetic study. ACS Catal. 8, 11119–11133. doi: 10.1021/acscatal.8b02632
Zhang, H., Xue, L., Li, J., and Ma, Q. (2020). Hyperbranched polycarbosiloxanes: synthesis by piers-rubinsztajn reaction and application as precursors to magnetoceramics. Polymers 12:672. doi: 10.3390/polym12030672
Zhang, J., Chen, Y., and Brook, M. A. (2014). Reductive degradation of lignin and model compounds by hydrosilanes. ACS Sust. Chem. Eng. 2, 1983–1991. doi: 10.1021/sc500302j
Zhang, Q., Fu, M. C., Yu, H. Z., and Fu, Y. (2016). Mechanism of boron-catalyzed N-alkylation of amines with carboxylic acids. J. Org. Chem. 81, 6235–6243. doi: 10.1021/acs.joc.6b00778
Keywords: siloxane, lewis acid catalysis, dehydrocoupling, catalyst recycling, silane, competing mechanisms
Citation: Rabanzo-Castillo KM, Kumar VB, Söhnel T and Leitao EM (2020) Catalytic Synthesis of Oligosiloxanes Mediated by an Air Stable Catalyst, (C6F5)3B(OH2). Front. Chem. 8:477. doi: 10.3389/fchem.2020.00477
Received: 14 March 2020; Accepted: 08 May 2020;
Published: 23 June 2020.
Edited by:
Eugene A. Goodilin, Lomonosov Moscow State University, RussiaReviewed by:
Grzegorz Hreczycho, Adam Mickiewicz University, PolandAziz M. Muzafarov, A. N. Nesmeyanov Institute of Organoelement Compounds (RAS), Russia
Copyright © 2020 Rabanzo-Castillo, Kumar, Söhnel and Leitao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erin M. Leitao, ZXJpbi5sZWl0YW9AYXVja2xhbmQuYWMubno=