 Jeong A Yoon1
Jeong A Yoon1 Changjin Lim
Changjin Lim Young Taek Han
Young Taek Han- 1College of Pharmacy, Dankook University, Cheonan-si, South Korea
- 2School of Pharmacy, Jeonbuk National University, Jeonju-si, South Korea
A preliminary study to develop a novel synthetic method for 3-aryl-5-azaisocoumarins was performed herein. The cycloisomerization of N-pyranonyl propargylamines in the AgOTf-catalyzed system efficiently afforded the desired 3-aryl-5-azaisocoumarins in a highly regioselective manner. This unprecedented method is expected as an expedient alternative synthetic route to 5-azaisocoumarins because the regioselectivity problem is circumvented, and it is easier to introduce substituents on the pyridine ring compared to previously reported intramolecular lactonization approaches.
Introduction
Isocoumarins, including 3,4-dihydroisocoumarin, are naturally abundant flavonoid scaffolds, that exhibit diverse pharmacological activities (Saeed, 2016; Saddiqa et al., 2017). In particular, 3-aryl isocoumarins possess unique and potent biological functions including antifungal (Saeed, 2003), anti-diabetic (Kim et al., 2017), immunomodulatory (Matsuda et al., 1998), antiallergic (Yoshikawa et al., 1992, 1994, 1996), and antimicrobial (Yoshikawa et al., 1992, 1994, 1996) activities, attracting the attention of synthetic and medicinal chemists. In medicinal chemistry, pyridine is considered to be the most efficient and popular bioisosteres with a phenyl moiety to improve biological activities and PK/PD profiles (Foye, 2008; Gaikwad et al., 2012). In this regard, azaisocoumarins, including 3-aryl-5-azaisocoumarin, are potential privileged scaffolds for developing drug candidates to treat a diverse range of diseases. Hence, there has been significant interest in the development of an efficient and novel synthetic route to 3-aryl-5-azaisocoumarin.
Despite the continual development of synthetic strategies for 3-aryl-isocoumarin analogs because of their interesting biological activities (Pal et al., 2011; Saddiqa et al., 2017), only a few methods and examples have been reported for the synthesis of 3-aryl-5-azaisocoumarin (Hellal et al., 2008; Li et al., 2010; Begouin and Maria-João, 2011; Panda et al., 2011; Park et al., 2011; Singh et al., 2018). To date, the synthesis of 3-aryl-5-azaisocoumarins has mostly been accomplished in the same manner as that of 3-aryl-isocoumarins, i.e., via construction of the 2-pyrone moiety in the final step via the intramolecular lactonization of 2-alkylnyl nicotinate derivatives. Because lactonization between the activated alkyne and ortho-carboxylate can occur by the 6-endo-dig or 5-exo-dig cyclization sequence (pathways a and b, respectively, in Figure 1), selectivity against methylenefuranone skeleton is a major challenge in these 2-pyrone formation routes. For instance, Li et al. reported the synthesis of 3-aryl-5-azaisocoumarin the Pinnick-type oxidative lactonization of 2-(phenylethynyl)-nicotinaldehyde in 2010 (Figure 1a) (Li et al., 2010). The transition metal-mediated intramolecular lactonizations of 2-alkynyl nicotinates or 2-alkynyl nicotinaldehyde are popular approaches to obtain 5-azaisocoumarins by construction of the 2-pyrone moiety. Queiroz et al. attempted to make the 2-pyrone moiety of 3-aryl-5-azaisocoumarin via the one-pot Sonogashira coupling/transition metal catalyzed alkyne activation cascade (Figure 1b) (Begouin and Maria-João, 2011). These two methods produced 5-exo-cyclized furanone as a major product. Youn et al. adopted NHC-catalyzed alkyne activation for the synthesis of 5-azaisocoumarins, which selectively afforded a 6-endo or 5-exo cyclized product with appropriate reaction conditions (Figure 1c) (Park et al., 2011). 3-Butyl-5-azaisocoumarin was selectively synthesized via 6-endo cyclization mode from the corresponding precursor. However, for the arylethynyl precursor, the major product was the 5-exo-cyclized lactone instead of 3-aryl-5-azaisocoumarin. Several successful examples of 3-aryl-5-azaisocoumarin synthesis via selective formation of the 2-pyrone moiety have been reported. The Bihel (Figure 1d) (Hellal et al., 2008) and Sarkar groups (Figure 1e) (Panda et al., 2011) reported 3-aryl-5-azaisocoumarin synthesis by transition metal-catalyzed 6-endo-lactonization from ethynylnicotinate or ethynylnicotinaldehyde precursors, respectively. In addition, selective 6-endo-lactonization toward pyrano[4,3-b]quinolin-1-one, a phenyl-fused azaisocoumarin, was accomplished under metal/additive-free domino reaction conditions (Figure 1f) (Singh et al., 2018). In addition to the selectivity problem, previous 2-pyrone formation approaches are inefficient for the installation of substituents on pyridine moiety of 5-azaisocoumarins.
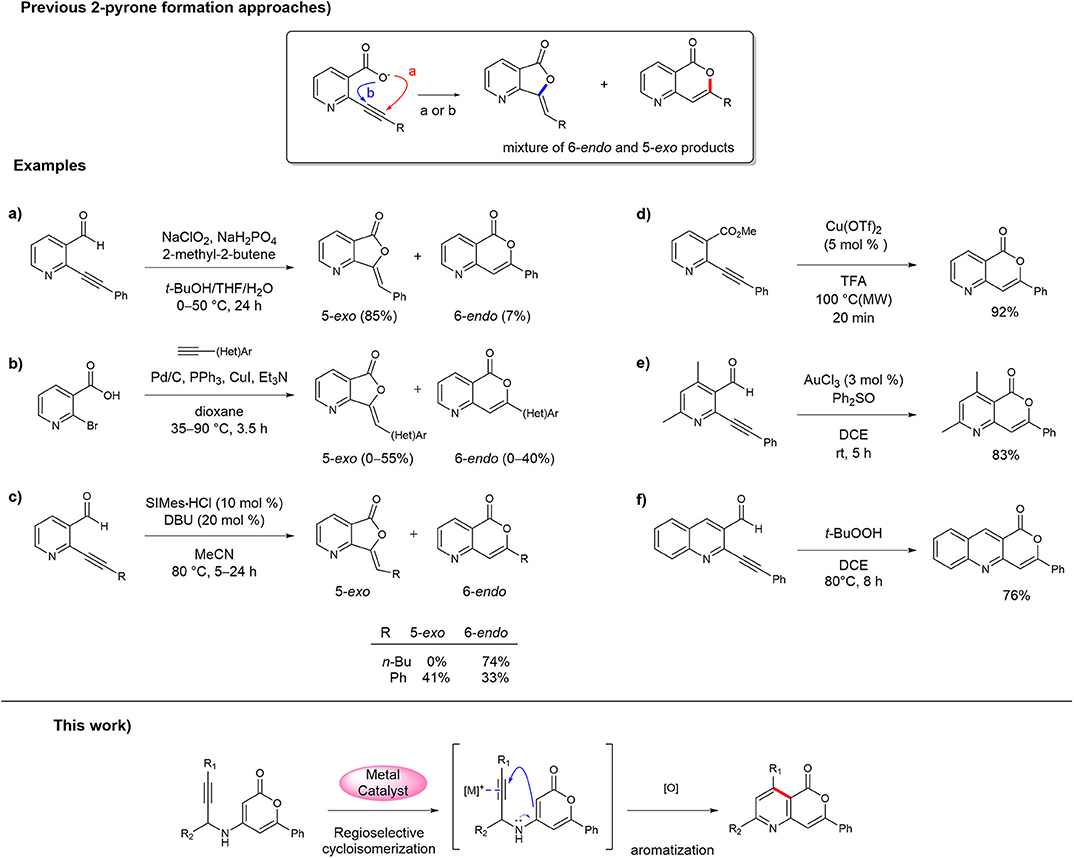
Figure 1. Approaches for the synthesis of 5-azaisocoumarins. (a) Li's synthesis of 5-azaisocoumarins; (b) Queiroz's synthesis of 5-azaisocoumarins; (c) Youn's synthesis of 5-azaisocoumarins; (d) Bihel's synthesis of 5-azaisocoumarins; (e) Sarkar's synthesis of 5-azaisocoumarins; (f) Singh's synthesis of 5-azaisocoumarins.
Recently, the metal-catalyzed 6-endo-dig cycloisomerization of N-propargyl enaminone derivatives has been widely used for the synthesis of pyridine-fused complex heterocycles, including azaanthraquinone, and steroidal pyridine (Abbiati et al., 2003; Yan et al., 2007; Jiang et al., 2010; Fei et al., 2011). In addition, intensive efforts to establish efficient synthetic methods for pyridine-fused coumarins and natural products using the metal-catalyzed cycloisomerization of N-propargyl enaminone intermediates have been reported (Ahn et al., 2019; Yoon and Han, 2019). In these studies, the pyridine moiety was constructed in a highly 6-endo-selective manner, which can be explained by σ-bond formation between the nucleophilic enolizable α-position and exo-position of the electrophilic alkyne-metal complex and subsequent spontaneous oxidation to form a stable aromatic pyridine ring (Abbiati et al., 2003; Yan et al., 2007; Cacchi et al., 2008; Jiang et al., 2010; Fei et al., 2011; Mikušek et al., 2016; Lyubov'N et al., 2018; Ahn et al., 2019). It was anticipated that construction of the pyridine moiety via cycloisomerization of N-propargylamine intermediates in the last-stage would be an efficient strategy for the synthesis of 3-aryl-5-azaisocoumarins. As part of our effort to develop a novel synthetic approach toward privileged 3-aryl-5-azaisocoumarin scaffolds, we investigated the metal-catalyzed cycloisomerization of N-pyranonyl propargylamines.
Herein, we describe a concise synthesis of 3-aryl-5-azaisocoumarins via AgOTf-catalyzed cycloisomerization, which to the best of our knowledge, has not been reported to date.
Materials and Methods
General Information
Unless noted otherwise, all starting materials and reagents were obtained commercially and were used without further purification. All solvents utilized for routine product isolation and chromatography were of reagent grade and glass-distilled, and reaction flasks were dried at 100°C before use. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck, Kenilworth, NJ, USA) with the indicated solvents. Thin-layer chromatography (TLC) was performed using 0.25-mm silica gel plates (Merck, Kenilworth, NJ, USA). Mass spectra were obtained using an Agilent 6530 Q-TOF (Santa Clara, CA, USA) instrument. Infrared spectra were recorded on a JASCO FT-IR-4200 spectrometer (Tokyo, Japan). 1H and 13C spectra were recorded on a Brucker Analytik ADVANCE digital 500 (500 MHz) (Billerica, MA, USA) and BRUKER AVANCE-800 (Billerica, MA, USA). Chemical shifts are expressed in parts per million (ppm, δ) downfield from tetramethylsilane and are referenced to the deuterated solvent. 1HNMR data are reported in the order of chemical shift, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; bs, broad singlet; and/or multiple resonances), number of protons, and coupling constant in hertz (Hz).
2-Oxo-6-phenyl-2H-pyran-4-yl 4-methylbenzenesulfonate (2)
To a stirred solution of 1 (500 mg, 2.66 mmol) in CH2Cl2 (20 mL) were added tosyl chloride (507 mg, 2.66 mmol) and Et3N (1.11 mL, 7.98 mmol) at ambient temperature. After stirring for 3 h, the reaction mixture was quenched with water and extracted with CH2Cl2. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/n-Hexane = 1: 5) to give 838 mg (92%) of 2 as a yellow solid.
m.p.: 128–130°C; Rf = 0.67 (EtOAc/n-Hexane = 1: 1); 1H-NMR (500 MHz, CDCl3) δ 7.86 (d, 2H, J = 8.3 Hz, Ar-H), 7.78 (d, 2H, J = 6.9 Hz, Ar-H), 7.51–7.45 (m, 3H, Ar-H), 7.40 (d, 2H, J = 8.0 Hz, Ar-H), 6.61 (d, 1H, J = 1.8 Hz, vinyl H), 5.91 (d, 1H, J = 1.8 Hz, cyclic-CH), 2.48 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3) δ 162.4, 162.3, 162.2, 146.9, 131.9, 131.8, 130.5, 130.5, 129.2, 128.6, 126.0, 101.7, 98.3, 22.0; IR (thin film, neat) νmax 1,736, 1,631, 1,556, 1,496, 1,386, 1,198, 1,139, 1,091, 916, 748 cm−1; LR-MS (ESI+) m/z 343 (M + H+); HR-MS (ESI+) calcd for C18H15O5S (M + H+) 343.0635; found 343.0631.
General Procedure I for Nucleophilic Aromatic Substitution
To a stirred solution of 2 (1 equiv.) and N,N-diisopropylethylamine (3 equiv.) in MeCN (0.1 M solution) was added propargylamine hydrochloride (1.5 equiv.) at ambient temperature. After stirring for 14 h at 80°C, the reaction mixture was quenched with 1N HCl and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/n-Hexane = 1: 1) to give title compounds (3a–3i).
6-Phenyl-4-(prop-2-yn-1-ylamino)-3,4-dihydro-2H-pyran-2-one (3a)
Yellow solid of 3a (1.13 g, 69%) was obtained via General Procedure I from 2.50 g of 2.
m.p.: 169–171°C; Rf = 0.29 (EtOAc/n-Hexane = 1: 1); 1H-NMR (500 MHz, DMSO-d6) δ 7.72 (s, 3H, Ar-H), 7.54–7.51 (m, 2H, Ar-H), 6.55 (s, 1H, vinyl H), 5.03 (d, 1H, J = 1.3 Hz, vinyl H), 4.01 (s, 2H, CH2), 3.30 (t, 1H, J = 2.3 Hz, CH); 13C-NMR (125 MHz, DMSO-d6) δ 162.5, 157.8, 157.1, 131.6, 130.6, 129.1, 125.1, 97.0, 80.7, 79.5, 74.6, 31.3; IR (thin film, neat) νmax 3,289, 3,089, 1,666, 1,549, 1,298, 1,198, 1,063, 847, 767, 691 cm−1; LR-MS (ESI+) m/z 226 (M + H+); HR-MS (ESI+) calcd for C14H12NO2 (M + H+) 226.0863; found 226.0856.
4-(But-2-yn-1-ylamino)-6-phenyl-3,4-dihydro-2H-pyran-2-one (3b)
Yellow solid of 3b (25 mg, 72%) was obtained via General Procedure I from 50 mg of 2.
m.p.: 181–183°C; Rf = 0.38 (EtOAc/n-Hexane = 1: 1); 1H-NMR (500 MHz, DMSO-d6) δ 7.72 (s, 2H, Ar-H), 7.68 (t, 1H, J = 5.3 Hz, NH), 7.54–7.49 (m, 3H, Ar-H), 6.54 (s, 1H, vinyl H), 4.99 (s, 1H, vinyl H), 3.94 (s, 2H, CH2), 1.81 (t, 3H, J = 2.1 Hz, CH3); 13C-NMR (125 MHz, DMSO-d6) δ 162.5, 157.7, 157.2, 131.6, 130.5, 129.1, 125.1, 97.0, 80.4, 79.6, 74.6, 31.6, 3.0; IR (thin film, neat) νmax 3,269, 3,090, 1,658, 1,547, 1,298, 1,194, 1,063, 992, 850, 768, 692 cm−1; LR-MS (ESI+) m/z 240 (M + H+); HR-MS (ESI+) calcd for C15H14NO2 (M + H+) 240.1019; found 240.1018.
4-(But-3-yn-2-ylamino)-6-phenyl-3,4-dihydro-2H-pyran-2-one (3c)
Yellow solid of 3c (17 mg, 48%) was obtained via General Procedure I from 50 mg of 2.
m.p.: 190–191°C; Rf = 0.30 (EtOAc/n-Hexane = 1: 1); 1H-NMR (500 MHz, DMSO-d6) δ 7.72 (s, 2H, Ar-H), 7.66 (d, 1H, J = 6.9 Hz, NH), 7.54–7.50 (m, 3H, Ar-H), 6.53 (s, 1H, vinyl H), 5.06 (d, 1H, J = 1.5 Hz, vinyl H), 4.37 (s, 1H, CH), 3.32 (d, 1H, J = 2.1 Hz, CH), 1.43 (d, 3H, J = 6.8 Hz, CH3); 13C-NMR (125 MHz, DMSO-d6) δ 162.5, 156.9, 156.4, 131.6, 130.5, 129.1, 126.9, 125.1, 97.0, 83.8, 81.0, 73.5, 20.9; IR (thin film, neat) νmax 3,283, 3,066, 1,667, 1,550, 1,298, 1,198, 1,176, 1,063, 847, 758, 691 cm−1; LR-MS (ESI+) m/z 240 (M + H+); HR-MS (ESI+) calcd for C15H14NO2 (M + H+) 240.1019; found 240.1029.
6-Phenyl-4-((3-phenylprop-2-yn-1-yl)amino)-3,4-dihydro-2H-pyran-2-one (3d)
Yellow solid of 3d (24 mg, 52%) was obtained via General Procedure I from 50 mg of 2.
m.p.: 194–196°C; Rf = 0.24 (EtOAc/n-Hexane = 1: 1); 1H-NMR (500 MHz, DMSO-d6), δ 7.83 (t, 1H, J = 5.3 Hz, Ar-H), 7.74 (s, 1H, Ar-H), 7.54–7.51 (m, 3H, Ar-H), 7.45–7.38 (m, 5H, Ar-H), 6.59 (d, 1H, J = 6.6 Hz, vinyl H), 5.12 (d, 1H, J = 1.3 Hz, vinyl H), 4.28 (s, 2H, CH2); 13C-NMR (125 MHz, DMSO-d6) δ 162.5, 157.9, 157.2, 131.6, 131.4, 130.6, 129.1, 128.8, 128.7, 125.1, 121.9, 97.1, 85.4, 83.1, 80.7, 32.1; IR (thin film, neat) νmax 3,271, 3,089, 1,663, 1,544, 1,451, 1,293, 1,199, 1,072, 849, 768, 691 cm−1; LR-MS (ESI+) m/z 324 (M + Na+); HR-MS (ESI+) calcd for C20H15NNaO2 (M + Na+) 324.0995; found 324.0990.
6-phenyl-4-((3-(o-tolyl)prop-2-yn-1-yl)amino)-2H-pyran-2-one (3e)
Yellow solid of 3e (69 mg, 75%) was obtained via General Procedure I from 100 mg of 2.
m.p.: 163–164°C; Rf = 0.40 (EtOAc/n-Hexane = 1: 1); 1H-NMR (800 MHz, DMSO-d6) δ 7.84 (s, 1H, NH), 7.74 (s, 2H, Ar-H), 7.55–7.48 (m, 3H, Ar-H), 7.38 (d, J = 7.6 Hz, 1H, Ar-H), 7.30–7.23 (m, 2H, Ar-H), 7.22 – 7.12 (m, 1H, Ar-H), 6.59 (s, 1H, vinyl H), 5.15 (d, J = 1.2 Hz, 1H, vinyl H), 4.31 (s, 2H, CH2), 2.36 (s, 3H, CH3); 13C-NMR (200 MHz, DMSO-d6) δ 162.5, 157.8, 157.1, 139.7, 131.6, 131.6, 130.6, 129.6, 129.1, 128.7, 125.9, 125.1, 121.8, 97.1, 89.2, 82.0, 80.9, 32.2, 20.2; IR (thin film, neat) νmax 3,263, 3,072, 1,664, 1,544, 1,289, 1,182, 1,027, 759, 689 cm−1; LR-MS (ESI+) m/z 316 (M + H+); HR-MS (ESI+) calcd for C21H18NO2 (M + H+) 316.1332; found 316.1333.
4-(hex-5-en-2-yn-1-ylamino)-6-phenyl-2H-pyran-2-one (3g)
Yellow solid of 3g (53 mg, 68%) was obtained via General Procedure I from 100 mg of 2.
m.p.: 119–120°C; Rf = 0.39 (EtOAc/n-Hexane = 1: 1); 1H-NMR (800 MHz, DMSO-d6) δ 7.82–7.65 (m, 3H, Ar-H & NH), 7.54–7.48 (m, 3H, Ar-H), 6.55 (s, 1H, vinyl H), 5.87–5.69 (ddt, J = 17.0, 10.1, 5.2 Hz, 1H, vinyl H), 5.30 (dd, J = 17.0, 1.7 Hz, 1H, vinyl H), 5.10 (d, J = 9.7 Hz, 1H, vinyl H), 5.03 (d, J = 1.5 Hz, 1H, vinyl H), 4.02 (s, 2H, CH2), 3.05–3.01 (m, 2H, CH2); 13C-NMR (200 MHz, DMSO-d6) δ 162.6, 157.7, 157.1, 132.8, 131.6, 130.6, 129.1, 125.1, 116.0, 97.0, 80.5, 80.3, 78.1, 31.7, 22.3; IR (thin film, neat) νmax 3,272, 3,092, 1,664, 1,548, 1,297, 1,182, 1,038, 918, 767, 691 cm−1; LR-MS (ESI+) m/z 266 (M + H+); HR-MS (ESI+) calcd for C17H16NO2 (M + H+) 266.1176; found 266.1169.
4-((3-(cyclohex-2-en-1-yl)prop-2-yn-1-yl)amino)-6-phenyl-2H-pyran-2-one (3h)
Yellow oil of 3h (39 mg, 44%) was obtained via General Procedure I from 100 mg of 2.
Rf = 0.35 (EtOAc/n-Hexane = 1: 1); 1H-NMR (800 MHz, DMSO-d6) δ 7.72 (s, 2H, Ar-H), 7.66 (s, 1H, NH), 7.54–7.48 (m, 3H, Ar-H), 6.55 (s, 1H, vinyl H), 5.72–5.68 (m, 1H, vinyl H), 5.58–5.55 (m, 1H, vinyl H), 5.00 (s, 1H, vinyl H), 3.98 (s, 2H, CH2), 3.18–3.11 (m, 1H, CH), 1.98–1.91 (m, 2H, 2 × CH2), 1.85–1.79 (m, 1H, 1/2 × CH2), 1.73–1.67 (m, 1H, 1/2 × CH2), 1.60–1.55 (m, 1H, 1/2 × CH2), 1.54–1.48 (m, 1H, 1/2 × CH2); 13C-NMR (200 MHz, DMSO-d6) δ 162.5, 157.7, 157.1, 131.6, 130.5, 129.1, 127.8, 126.8, 125.1, 97.1, 86.4, 80.4, 75.3, 31.7, 28.9, 26.6, 24.1, 20.0; IR (thin film, neat) νmax 3,117, 2,887, 1,670, 1,564, 1,337, 1,245, 767, 688 cm−1; LR-MS (ESI+) m/z 306 (M + H+); HR-MS (ESI+) calcd for C20H20NO2 (M + H+) 306.1489; found 306.1486.
6-phenyl-4-((3-(triisopropylsilyl)prop-2-yn-1-yl)amino)-2H-pyran-2-one (3i)
Yellow solid of 3i (80 mg, 72%) was obtained via General Procedure I from 100 mg of 2.
m.p.: 175–176°C; Rf = 0.60 (EtOAc/n-Hexane = 1: 1); 1H-NMR (800 MHz, DMSO-d6) δ 7.74 (s, 2H, Ar-H), 7.68 (s, 1H, NH), 7.54–7.47 (m, 3H, Ar-H), 6.57 (s, 1H, vinyl H), 5.07 (d, J = 1.9 Hz, 1H, vinyl H), 4.09 (s, 2H, CH2), 1.05–0.98 (m, 21H, TIPS-H); 13C-NMR (200 MHz, DMSO-d6) δ 162.4, 157.7, 157.0, 131.6, 130.5, 129.1, 125.1, 103.6, 97.0, 83.6, 81.1, 32.3, 18.4, 10.6; IR (thin film, neat) νmax 3,272, 3,075, 1,670, 1,549, 1,245, 1,182, 1,038, 883, 767, 677 cm−1; LR-MS (ESI+) m/z 382 (M + H+); HR-MS (ESI+) calcd for C23H32NO2Si (M + H+) 382.2197; found 382.2193.
General Procedure II for AgOTf-Catalyzed Cycloisomerization
To a stirred solution of 3a–3f (1 equiv.) in DMSO (0.06 M) was added AgOTf (0.2 equiv.) at ambient temperature. After stirring for 2–4 h at 120°C, the reaction mixture was cooled to ambient temperature, quenched with water and extracted with EtOAc. The combined organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc/n-Hexane = 1: 3) to give title compounds (4a–4f).
7-Phenyl-5H-pyrano[4,3-b]pyridin-5-one (4a)
Yellow solid of 4a (13 mg, 66%) was obtained via General Procedure II from 20 mg of 3a.
m.p.: 135–136°C; Rf = 0.47 (EtOAc/n-Hexane = 1: 1); 1H-NMR (800 MHz, CDCl3) δ 8.94 (dd, 1H, J = 1.8, 4.6 Hz, Ar-H), 8.55 (dd, 1H, J = 1.4, 7.8 Hz, Ar-H), 7.93–7.91 (m, 2H, Ar-H), 7.51–7.47 (m, 3H, Ar-H), 7.43 (dd, 1H, J = 4.6, 7.9 Hz, Ar-H), 7.23 (s, 1H, vinyl H); 13C-NMR (125 MHz, CDCl3) δ 162.1, 157.6, 156.3, 155.1, 137.9, 131.5, 131.0, 129.2, 125.9, 123.0, 117.2, 103.6; IR (thin film, neat) νmax 3,067, 1,735, 1,633, 1,561, 1,450, 1,216, 1,044, 1,035, 753, 688 cm−1; LR-MS (ESI+) m/z 224 (M + H+); HR-MS (ESI+) calcd for C14H10NO2 (M + H+) 224.0706; found 224.0707.
4-Methyl-7-phenyl-5H-pyrano[4,3-b]pyridin-5-one (4b)
Yellow solid of 4b (16 mg, 65%) was obtained via General Procedure II from 25 mg of 3b.
m.p.: 113–114°C; Rf = 0.58, (EtOAc/n-Hexane = 1: 1); 1H-NMR (500 MHz, CDCl3) δ 8.68 (d, 1H, J = 4.9 Hz, Ar-H), 7.93–7.91 (m, 2H, Ar-H), 7.51–7.47 (m, 3H, Ar-H), 7.21–7.20 (m, 2H, Ar-H & vinyl H), 2.86 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3) δ 161.3, 157.1, 156.2, 154.6, 153.6, 131.4, 130.9, 129.1, 125.8, 125.7, 116.2, 103.8, 22.7; IR (thin film, neat) νmax 3,060, 1,734, 1,637, 1,574, 1,465, 1,228, 1,058, 1,034, 847, 762, 681 cm−1; LR-MS (ESI+) m/z 238 (M + H+); HR-MS (ESI+) calcd for C15H12NO2 (M + H+) 238.0863; found 238.0860.
2-Methyl-7-phenyl-5H-pyrano[4,3-b]pyridin-5-one (4c)
Yellow solid of 4c (11 mg, 54%) was obtained via General Procedure II from 20 mg of 3c.
m.p.: 129–130°C; Rf = 0.53 (EtOAc/n-Hexane = 1: 1); 1H-NMR (500 MHz, CDCl3) δ 8.43 (d, 1H, J = 8.1 Hz, Ar-H), 7.93–7.90 (m, 2H, Ar-H), 7.51–7.47 (m, 3H, Ar-H), 7.30 (d, 1H, J = 8.1 Hz, Ar-H), 7.22 (s, 1H, vinyl H), 2.72 (s, 3H, CH3); 13C-NMR (125 MHz, CDCl3) δ 166.6, 162.2, 157.6, 154.8, 137.8, 131.6, 130.9, 129.1, 125.8, 123.3, 114.7, 103.6, 25.5; IR (thin film, neat) νmax 3,061, 1,752, 1,630, 1,570, 1,460, 1,241, 1,119, 1,064, 1,030, 762, 687 cm−1; LR-MS (ESI+) m/z 238 (M + H+); HR-MS (ESI+) calcd for C15H12NO2 (M + H+) 238.0863; found 238.0857.
4,7-Diphenyl-5H-pyrano[4,3-b]pyridin-5-one (4d)
Yellow solid of 4d (12 mg, 60%) was obtained via General Procedure II from 20 mg of 3d.
m.p.: 142–144°C; Rf = 0.61 (EtOAc/n-Hexane = 1: 1); 1H-NMR (500 MHz, CDCl3) δ 8.86 (d, 1H, J = 4.9 Hz, Ar-H), 7.94–7.92 (m, 2H, Ar-H), 7.50–7.48 (m, 6H, Ar-H), 7.40–7.37 (m, 3H, Ar-H), 7.28 (d, 1H, J = 4.2 Hz, vinyl H); 13C-NMR (125 MHz, CDCl3) δ 160.1, 157.5, 156.4, 154.7, 154.6, 138.5, 131.3, 131.0, 129.1, 128.8, 128.3, 128.2, 125.8, 125.5, 114.3, 103.7; IR (thin film, neat) νmax 3,058, 1,745, 1,637, 1,568, 1,463, 1,222, 1,060, 1,029, 856, 764, 696 cm−1; LR-MS (ESI+) m/z 300 (M + H+); HR-MS (ESI+) calcd for C20H14NO2 (M + H+) 300.1019; found 300.1017.
7-phenyl-4-(o-tolyl)-5H-pyrano[4,3-b]pyridin-5-one (4e)
Yellow solid of 4e (34 mg, 50%) was obtained via General Procedure II from 68 mg of 3e.
m.p.: 137–139°C; Rf = 0.62 (EtOAc/n-Hexane = 1: 1); 1H-NMR (800 MHz, CDCl3) δ 8.90 (s, 1H, Ar-H), 7.91–7.88 (m, 2H, Ar-H), 7.49–7.45 (m, 3H, Ar-H), 7.36–7.32 (m, 2H, Ar-H), 7.30–7.25 (m, 2H, Ar-H), 7.22–7.18 (m, 1H, Ar-H), 7.06 (d, J = 6.9 Hz, 1H, vinyl H), 2.09 (s, 3H, CH3); 13C-NMR (200 MHz, CDCl3) δ 159.7, 157.9, 156.0, 154.9, 154.4, 138.5, 134.7, 131.2, 131.1, 123.0, 129.2, 128.6, 127.3, 125.9, 125.8, 125.3, 115.5, 103.3, 20.0; IR (thin film, neat) νmax 3,066, 1,746, 1,636, 1,568, 1,495, 1,223, 1,027, 761, 688 cm−1; LR-MS (ESI+) m/z 314 (M + H+); HR-MS (ESI+) calcd for C21H16NO2 (M + H+) 314.1176; found 314.1174.
4-(3-nitrophenyl)-7-phenyl-5H-pyrano[4,3-b]pyridin-5-one (4f)
N-pyranonyl propargylamine 3f was obtained via General Procedure I from 100 mg of 2. The crude 3f was used for the next step without further purification. Yellow solid of 4f (32 mg, 32% for 2 steps) was obtained via General Procedure II from the crude 3f.
m.p.: 261–262°C; Rf = 0.48 (EtOAc/n-Hexane = 1: 1); 1H-NMR (800 MHz, DMSO-d6) δ 9.02 (d, J = 4.8 Hz, 1H, Ar-H), 8.38 (t, J = 1.9 Hz, 1H, Ar-H), 8.32 (ddd, J = 8.3, 2.3, 0.9 Hz, 1H, Ar-H), 8.01 (dd, J = 8.1, 1.5 Hz, 2H, Ar-H), 7.95 (ddd, J = 7.6, 1.4, 1.1 Hz, 1H, Ar-H), 7.77 (t, J = 7.9 Hz, 1H, Ar-H), 7.60 (s, 1H, Ar-H), 7.58–7.53 (m, 3H, Ar-H), 7.47 (d, J = 4.8 Hz, 1H, vinyl H); 13C-NMR (200 MHz, DMSO-d6) δ 159.9, 156.1, 155.8, 155.6, 149.9, 147.3, 140.2, 135.1, 130.9, 130.8, 129.4, 129.2, 125.4, 125.3, 123.5, 123.1, 114.0, 103.9; IR (thin film, neat) νmax 3,115, 1,796, 1,637, 1,542, 1,446, 1,219, 1,038, 856, 759, 676 cm−1; LR-MS (ESI+) m/z 345 (M + H+); HR-MS (ESI+) calcd for C20H13N2O4 (M + H+) 345.0870; found 345.0867.
Results and Discussions
Our research on the development of a novel synthetic route toward 3-aryl-5-azaisocoumarin commenced with the preparation of cycloisomerization precursors. As shown in Scheme 1, N-pyranonyl propargylamine precursors (3a–3k) were prepared according to literature procedures with slight modifications (Dong et al., 2011). The known hydroxy-2-pyrone 1 (Prasad et al., 1995) was treated with tosyl chloride and triethylamine to afford tosylate 2 in 92% yield. Nucleophilic aromatic substitution of pyrone 2 with commercially available hydrochloride salts of propargylamine or diverse known substituted propargylamine (Experimental details: see Supplementary Materials) readily afforded the corresponding N-pyranonyl propargylamines 3a–3h in moderate yields (48–74%). However, 3-bromopropargyl- and 1-phenylpropargyl analogs (3j–3k) were not obtained. The nitrophenyl substituted analog 3i was used for the next reaction without further purification due to inseparable impurities.
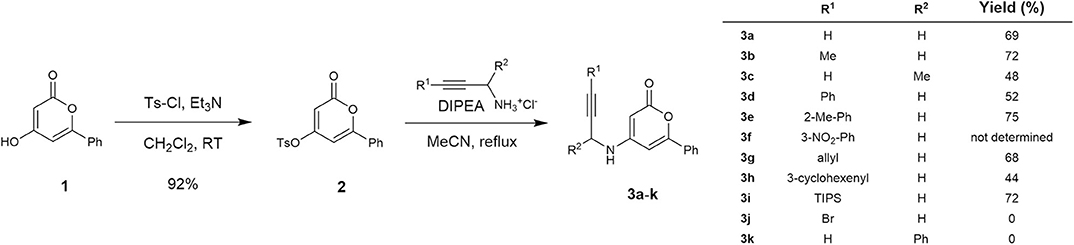
Scheme 1. Synthesis of N-pyranonyl propargylamines 3a−3k.
With the cycloisomerization precursors 3a–3i in hand, the cycloisomerization conditions were examined using the simple 3a as a model substrate (Table 1). Initially, cycloisomerization of 3a was conducted under thermal rearrangement conditions. When 3a was heated at 120°C in DMSO for 10 h, the desired 5-azaisocoumarin 4a was obtained in poor yield (15%, Table 1, entry 1). The reaction was performed at higher temperature (220°C) in triethylene glycol (TEG) (Han, 2017), affording 5-azaisocoumarin 4a in a slightly higher yield (28%, entry 2). The structure of 4a was confirmed by comparison of its NMR spectrum with previously reported data (Table S1) (Hellal et al., 2008). Recently, many research groups have continuously reported the efficiency of various transition-metal Lewis acid catalysts for alkyne activation in cycloisomerization reaction (Abbiati et al., 2003; Yan et al., 2007; Cacchi et al., 2008; Saito et al., 2009; Jiang et al., 2010; Fei et al., 2011; Mikušek et al., 2016; Vessally et al., 2016; Nizami and Hua, 2017; Sakthivel et al., 2017; Gianni et al., 2018; Lyubov'N et al., 2018; Mancuso et al., 2018; Ahn et al., 2019). To increase the product yield, cycloisomerization was performed in the presence of various metal catalysts, including Ag(I), Au(I), Cu(I), Cu(II), and In(III). Based on our previous work (Ahn et al., 2019), the cycloisomerization of 3a was performed with 0.2 equiv. of AgSbF6 (entry 3), affording 4a in a higher yield (38%) than that without the catalyst after heating in DMSO at 120°C for 7 h. Encouraged by this yield improvement with the Ag(I) catalyst, various counter-anions was screened (entries 4–7) given that the alkyne-activating Lewis acidity of metal ions such as Ag(I) depends on the nature of the counter-anion (Yamamoto, 2000; Lu et al., 2017). When Ag2O was employed, the yield of 4a did not notably improve over that obtained with AgSbF6 (38%, entry 4). However, the yields increased slightly when AgCl (50%, entry 5) or AgNO3 (53%, entry 6) were used. The best result was obtained when the cycloisomerization was performed with AgOTf, furnishing 4a in 66% yield (entry 7). Moreover, the reaction proceeded to completion much faster (2 h) than with AgSbF6. Although regioisomeric byproducts were not isolated as expected, the optimal yield for the cycloisomerization of N-pyranonyl propargylamine precursors 3a was slightly lower than that reported in a previous study on pyridocoumarins (Ahn et al., 2019), likely because of the greater degree of decomposition of the acid-labile pyrone moiety. When the reaction was conducted with AuCl, the yield was reduced compared to that observed in the thermal aza-Claisen rearrangement in TEG (24%, entry 8). Furthermore, the desired cycloisomerization product 4a was not obtained when using InCl3 (entry 9). Cu(I) and Cu(II) catalysts afforded 5-azaisocoumarin 4a in better yields than with Au(I) or In(III) catalysts (33%−62%; entries 10–14). Although CuI afforded 4a in the best yield among the copper catalysts tested herein (62%, entry 11), the yield of 4a and reaction rate did not show any notable improvements over that obtained with AgOTf. Other polar solvents including DMF (entry 15), MeCN (entry 16), and ionic liquid systems (entries 17–18), resulted in lower reaction rates and yields of 4a. In particular, the use of an ionic liquid hampered the cycloisomerization, contrary to previous reports (Corma et al., 2005; Neatu et al., 2009; Mancuso et al., 2019). The lower yields obtained in ionic liquids was attributed to the inhibition of the interaction between the alkyne and metal catalyst by the highly polar solvent.
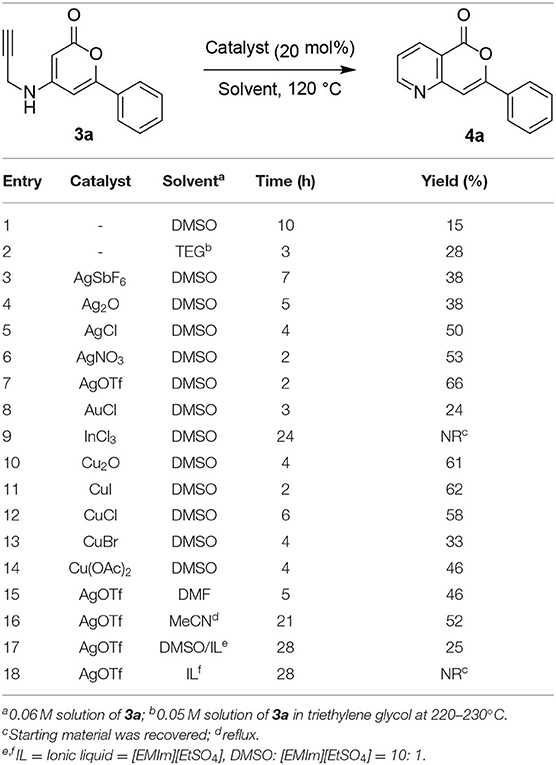
Table 1. Investigation of reaction conditions for the cycloisomerization of 3a.
Next, the scope and limitations of the newly developed Ag(I)-catalyzed 3-aryl-5-azaisocoumarin synthesis were examined (Table 2). We attempted to synthesize the 3-aryl-5-azaisocoumarins from corresponding substituted alkynes (3b–3i). Methyl (3b–3c; entry 1–2) and phenyl substituted (3d–3f; entry 3–5) propargylamines generated the corresponding 3-aryl-5-azaisocoumarins (4b–4f) without any notable yield decreases. However, cycloisomerization under standard condition of enynylamine precursors (3g–3h; entry 6–7) resulted in an unidentifiable mixture of byproducts, whereas only substrates were recovered when the reactions were conducted at ambient temperature. These results were attributed to preference of the metal-alkyne complex for reaction with the electron-rich alkene rather than enolizable α-position. Although the cycloisomerization reaction of triisopropylsilyl (TIPS) propargylamine (3i) was allowed to proceed for 24 h, only substrate 3i was recovered in 88% yield (entry 8). This implied that the bulky silyl substituents of alkyne can interfere with formation of the metal-alkyne complex.
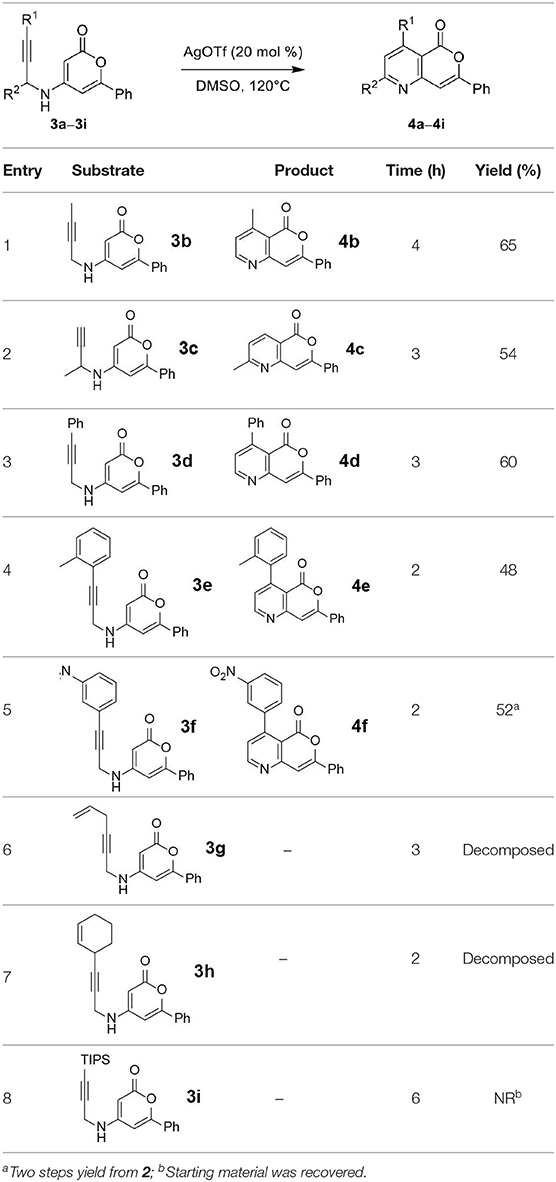
Table 2. Substrate scope of the AgOTf-catalyzed cycloisomerization toward 3-aryl-5-azaisocoumarin.
Conclusion
In conclusion, an alternative synthetic route for 3-aryl-5-azaisocoumarins was developed which includes novel approach to install pyridine moieties on a readily available phenyl-2-pyrone precursor via regioselective metal-catalyzed cycloisomerization. Through an intensive preliminary investigation, AgOTf was determined to be an adequate alkyne-activation catalyst and was applicable in rearrangement of N-pyranonyl propargylamines bearing methyl or phenyl substituents to C-6 or C-8 substituted 3-aryl-5-azaisocoumarin. Derivatization of the pyridine moiety was accomplished efficiently via three-step reaction using the common starting material hydroxy-2-pyrone 1 and known propargylamines. Considering that 3-phenyl-5-azaisocoumarin 4a, which was synthesized previously as a regioisomeric mixture using conventional lactonization strategies, was obtained regioselectively in moderate yield, this novel step-economical procedure can be widely utilized in derivatization of C-3, C-6, and C-8 of 5-azaisocoumarins. Further studies on the synthetic applications of the newly developed method are currently underway.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
YH conceptualized the work. JY and CL synthesized all compound and performed m.p. Rf, NMR, IR, LRMS, and HRMS analysis. The manuscript was written by CL and YH. All authors approved the manuscript in its final form for publication.
Funding
This research was funded by the National Research Foundation of Korea, NRF-2020R1F1A1058295.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00772/full#supplementary-material
References
Abbiati, G., Arcadi, A., Bianchi, G., Di Giuseppe, S., Marinelli, F., and Rossi, E. (2003). Sequential amination/annulation/aromatization reaction of carbonyl compounds and propargylamine: A new one-pot approach to functionalized pyridines. J. Org. Chem. 68, 6959–6966. doi: 10.1021/jo0347260
Ahn, S., Yoon, J. A., and Han, Y. T. (2019). Total synthesis of the natural pyridocoumarins goniothaline A and B. Synthesis 51, 552–556. doi: 10.1055/s-0037-1610909
Begouin, A., Maria-João, and Queiroz, R. P. (2011). Tandem palladium/charcoal-copper(I) iodide (Pd/C-CuI) catalyzed Sonogashira coupling and intramolecular cyclization from 2-bromonicotinic acid (=2-bromopyridine-3-carboxylic acid) and ethynylarenes to 4-zzaphthalides (=furo[3,4-b]pyridin-5(7H)-ones) and 5-azaisocoumarins (=5H-pyrano[4, 3-b]pyridin-5-ones). Helv. Chim. Acta 94, 1792–1801. doi: 10.1002/hlca.201100060
Cacchi, S., Fabrizi, G., and Filisti, E. (2008). N-propargylic β-enaminones: Common intermediates for the synthesis of polysubstituted pyrroles and pyridines. Org. Lett. 10, 2629–2632. doi: 10.1021/ol800518j
Corma, A., García, H., and Leyva, A. (2005). Palladium catalyzed cycloisomerization of 2,2-diallylmalonates in imidazolium ionic liquids. J. Organomet. Chem. 690, 3529–3534. doi: 10.1016/j.jorganchem.2005.02.044
Dong, Y., Nakagawa-Goto, K., Lai, C.-Y., Morris-Natschke, S. L., Bastow, K. F., and Lee, K.-H. (2011). Antitumor agents 287. Substituted 4-amino-2H-pyran-2-one (APO) analogs reveal a new scaffold from neo-tanshinlactone with in vitro anticancer activity. Bioorg. Med. Chem. Lett. 21, 2341–2344. doi: 10.1016/j.bmcl.2011.02.084
Fei, N., Yin, H., Wang, S., Wang, H., and Yao, Z.-J. (2011). CuCl2-promoted 6-endo-dig chlorocyclization and oxidative aromatization cascade: Efficient construction of 1-azaanthraquinones from N-propargylaminoquinones. Org. Lett. 13, 4208–4211. doi: 10.1021/ol201542h
Foye, W. O. (2008). Foye's Principles of Medicinal Chemistry. Philadelphia, PA: Lippincott Williams & Wilkins.
Gaikwad, P. L., Gandhi, P. S., Jagdale, D. M., and Kadam, V. J. (2012). The use of bioisosterism in drug design and molecular modification. Am. J. Pharmtech Res. 2, 1–23. Available online at: http://ajptr.com/assets/upload/publish_article/AJPTR%20article%20Priyanka%20Gaikwad_9133.pdf
Gianni, J., Pirovano, V., and Abbiati, G. (2018). Silver triflate/p-TSA co-catalysed synthesis of 3-substituted isocoumarins from 2-alkynylbenzoates. Org. Biomol. Chem. 16, 3213–3219. doi: 10.1039/c8ob00436f
Han, Y. T. (2017). Synthesis of proposed structure of rennellianone B: A study on rearrangement of anthraquinonyl propargyl ether toward 2H-pyranoanthraquinone. Tetrahedron Lett. 58, 556–558. doi: 10.1016/j.tetlet.2016.12.080
Hellal, M., Bourguignon, J.-J., and Bihel, F. J.-J. (2008). 6-endo-dig cyclization of heteroarylesters to alkynes promoted by Lewis acid catalyst in the presence of Brønsted acid. Tetrahedron Lett. 49, 62–65. doi: 10.1016/j.tetlet.2007.11.020
Jiang, C., Xu, M., Wang, S., Wang, H., and Yao, Z.-J. (2010). Azaanthraquinone assembly from N-propargylamino quinone via a Au(I)-catalyzed 6-endo-dig cycloisomerization. J. Org. Chem. 75, 4323–4325. doi: 10.1021/jo1006637
Kim, E., Lim, S.-M., Kim, M.-S., Yoo, S.-H., and Kim, Y. (2017). Phyllodulcin, a natural sweetener, regulates obesity-related metabolic changes and fat browning-related genes of subcutaneous white adipose tissue in high-fat diet-induced obese mice. Nutrients 9:1049. doi: 10.3390/nu9101049
Li, J., Chin, E., Lui, A. S., and Chen, L. (2010). One-pot synthesis of phthalides via regioselective intramolecular cyclization from ortho-alkynylbenzaldehydes. Tetrahedron Lett. 51, 5937–5939. doi: 10.1016/j.tetlet.2010.09.023
Lu, Z., Han, J., Okoromoba, O. E., Shimizu, N., Amii, H., Tormena, C. F., et al. (2017). Predicting couterion effects using a gold affinity index and a hydrogen bonding basicity index. Org. Lett. 19, 5848–5851. doi: 10.1021/acs.orglett.7b02829
Lyubov'N, S, Sagitova, E. F., Markova, M. V., Igor,'A, U., et al. (2018). Acylethynylpyrroles as a platform for the one-pot access to 2-(pyrrol-2-yl)-3-acylpyridines via the dihydrogenative annelation with propargylamine. Tetrahedron Lett. 59, 4047–4049. doi: 10.1016/j.tetlet.2018.09.066
Mancuso, R., Mili,è, R., Palumbo Piccionello, A., Olivieri, D., Della Ca,', N., Carfagna, C., et al. (2019). Catalytic carbonylative double cyclization of 2-(3-hydroxy-1-yn-1-yl) phenols in ionic liquid leading to furobenzofuranone derivatives. J. Org. Chem. 84, 7303–7311. doi: 10.1021/acs.joc.9b00952
Mancuso, R., Pomelli, C. S., Chiappetta, P., Gioia, K. F., Maner, A., Marino, N., et al. (2018). Divergent syntheses of (Z)-3-alkylideneisobenzofuran-1 (3H)-ones and 1H-isochromen-1-ones by copper-catalyzed cycloisomerization of 2-alkynylbenzoic acids in ionic liquids. J. Org. Chem. 83, 6673–6680. doi: 10.1021/acs.joc.8b00923
Matsuda, H., Shimoda, H., Yamahara, J., and Yoshikawa, M. (1998). Immunomodulatory activity of thunberginol a and related compounds isolated from Hydrangeae Dulcis Folium on splenocyte proliferation activated by mitogens. Bioorg. Med. Chem. Lett. 8, 215–220. doi: 10.1016/s0960-894x(97)10221-9
Mikušek, J., Matouš, P., Matoušová, E., Janoušek, M., Kuneš, J., and Pour, M. (2016). Substrate control in the gold (I)-catalyzed cyclization of β-propargylamino acrylic esters and further transformations of the resultant dihydropyridines. Adv. Synth. Catal. 358, 2912–2922. doi: 10.1002/adsc.201600412
Neatu, F., Pârvulescu, V. I., Michelet, V., Gênet, J.-P., Goguet, A., and Hardacre, C. (2009). Gold imidazolium-based ionic liquids, efficient catalysts for cycloisomerization of γ-acetylenic carboxylic acids. New J. Chem. 33, 102–106. doi: 10.1039/B812580E
Nizami, T. A., and Hua, R. (2017). Silver-catalyzed chemoselective annulation of propargyl amines with alkynes for access to pyridines and pyrroles. Tetrahedron 73, 6080–6084. doi: 10.1016/j.tet.2017.09.002
Pal, S., Chatare, V., and Pal, M. (2011). Isocoumarin and its derivatives: An overview on their synthesis and applications. Curr. Org. Chem. 15, 782–800. doi: 10.2174/138527211794518970
Panda, B., Bhadra, J., and Sarkar, T. K. (2011). An approach to highly functionalized quinolines and isoquinolines via a gold-catalyzed benzannulation. Synlett 5, 689–693. doi: 10.1055/s-0030-1259555
Park, J. H., Bhilare, S. V., and Youn, S. W. (2011). NHC-catalyzed oxidative cyclization reactions of 2-alkynylbenzaldehydes under aerobic conditions: Synthesis of O-heterocycles. Org. Lett. 13, 2228–2231. doi: 10.1021/ol200481u
Prasad, J. V., Para, K. S., Tummino, P. J., Ferguson, D., McQuade, T. J., Lumney, E. A., et al. (1995). Nonpeptidic potent HIV-1 protease inhibitors:(4-hydroxy-6-phenyl-2-oxo-2H-pyran-3-yl) thiomethanes that span P1-P2' subsites in a unique mode of binding. J. Med. Chem. 38, 898–905. doi: 10.1021/jm00006a007
Saddiqa, A., Çakmak, O., and Usman, M. (2017). Isocoumarins and 3, 4-dihydroisocoumarins, amazing natural products: A review. Turk. J. Chem. 41, 153–178. doi: 10.3906/kim-1604-66
Saeed, A. (2003). Synthesis of montroumarin. Z. Naturforsch. 58, 691–696. doi: 10.1515/znc-2003-9-1017
Saeed, A. (2016). Isocoumarins, miraculous natural products blessed with diverse pharmacological activities. Eur. J. Med. Chem. 116, 290–317. doi: 10.1016/j.ejmech.2016.03.025
Saito, A., Konishi, T., and Hanzawa, Y. (2009). Synthesis of pyrroles by gold(I)-catalyzed amino-Claisen rearrangement of N-propargyl enaminone derivatives. Org. Lett. 11, 372–374. doi: 10.1021/ol902716n
Sakthivel, S., Sharma, A., and Balamurugan, R. (2017). Silver-catalyzed synthesis of substituted pyridine derivatives from N-propargylic α-enamino esters. Eur. J. Org. Chem. 3941–3946. doi: 10.1002/ejoc.201700559
Singh, J. B., Mishra, K., Gupta, T., and Singh, R. M. (2018). TBHP-promoted oxidative cyclization of o-alkynylquinoline aldehydes: Metal/additive-free domino synthesis of pyrano [4,3-b] quinolin-1-ones. Tetrahedron Lett. 59, 1019–1022. doi: 10.1016/j.tetlet.2018.01.083
Vessally, E., Hosseinian, A., Edjlali, L., Bekhradnia, A., and Esrafili, M. D. (2016). New page to access pyridine derivatives: Synthesis from N-propargylamines. RSC Adv. 6, 71662–71675. doi: 10.1039/C6RA08720E
Yan, J.-Z., Li, J., and Rao, G.-W. (2007). One-pot synthesis of new A-ring fused steroidal pyridines. Steroids 72, 736–739. doi: 10.1016/j.steroids.2007.06.002
Yoon, J. A., and Han, Y. T. (2019). Efficient synthesis of pyrido[3,2-c]coumarins via silver nitrate catalyzed cycloisomerization and application to the first synthesis of polyneomarline C. Synthesis 51, 4611–4618. doi: 10.1055/s-0037-1610730
Yoshikawa, M., Harada, E., Naitoh, Y., Inoue, K., Matsuda, H., Shimoda, H., et al. (1994). Development of bioactive functions in Hydrangeae Dulcis Folium. III. On the antiallergic and antimicrobial principles of Hydrangeae Dulcis Folium. (1). Thunberginols A, B, and F. Chem. Pharm. Bull. 42, 2225–2230. doi: 10.1248/cpb.42.2225
Yoshikawa, M., Matsuda, H., Shimoda, H., Shimada, H., Harada, E., Naitoh, Y., et al. (1996). Development of bioactive functions in Hydrangeae Dulcis Folium. V. On the antiallergic and antimicrobial principles of hydrangeae dulcis folium. (2). Thunberginols C, D, and E, thunberginol G 3'-O-glucoside, (-)-hydrangenol 4'-O-glucoside, and (+)-hydrangenol 4'-O-glucoside. Chem. Pharm. Bull. 44, 1440–1447. doi: 10.1248/cpb.44.1440
Keywords: 5-azaisocoumarin, cycloisomerization, synthetic method, AgOTf, N-pyranonyl propargylamine, Ag(I)-catalysis
Citation: Yoon JA, Lim C and Han YT (2020) Preliminary Study on Novel Expedient Synthesis of 5-Azaisocoumarins by Transition Metal-Catalyzed Cycloisomerization. Front. Chem. 8:772. doi: 10.3389/fchem.2020.00772
Received: 11 June 2020; Accepted: 24 July 2020;
Published: 04 September 2020.
Edited by:
Jiyong Hong, Duke University, United StatesReviewed by:
Xiao-Wei Liang, Duke University, United StatesJongkook Lee, Kangwon National University, South Korea
Copyright © 2020 Yoon, Lim and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Changjin Lim, bGltY2pAamJudS5hYy5rcg==; Young Taek Han, aGFueXRAZGFua29vay5hYy5rcg==