 Andreas Hennig1,2,3*
Andreas Hennig1,2,3* Werner M. Nau1
Werner M. Nau1- 1Department of Life Sciences and Chemistry, Jacobs University Bremen gGmbH, Bremen, Germany
- 2Institute of Chemistry of New Materials, School of Biology/Chemistry, Universität Osnabrück, Osnabrück, Germany
- 3Center of Cellular Nanoanalytics (CellNanOs), Universität Osnabrück, Osnabrück, Germany
We report the use of the macrocyclic host cucurbit[7]uril (CB7) as a supramolecular additive in nanosecond time-resolved fluorescence (Nano-TRF) assays for proteases to enhance the discrimination of substrates and products and, thereby, the sensitivity. A peptide substrate was labeled with 2,3-diazabicyclo[2.2.2]oct-2-ene (DBO) as a long-lived (>300 ns) fluorescent probe and 3-nitrotyrosine was established as a non-fluorescent fluorescence resonance energy transfer (FRET) acceptor that acts as a “dark quencher.” The substrate was cleaved by the model proteases trypsin and chymotrypsin and the effects of the addition of CB7 to the enzyme assay mixture were investigated in detail using UV/VIS absorption as well as steady-state and time-resolved fluorescence spectroscopy. This also allowed us to identify the DBO and nitrotyrosine residues as preferential binding sites for CB7 and suggested a hairpin conformation of the peptide, in which the guanidinium side chain of an arginine residue is additionally bound to a vacant ureido rim of one of the CB7 hosts.
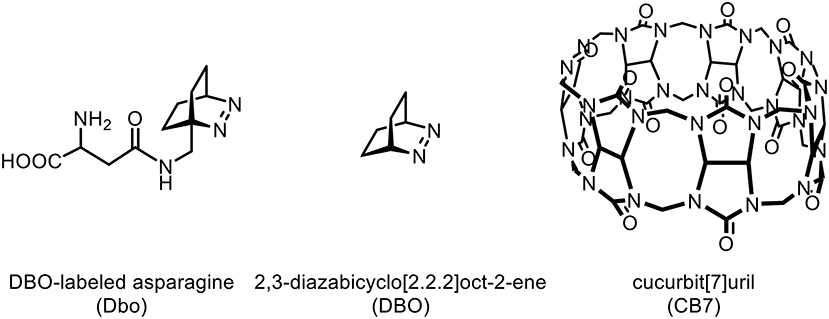
Graphical Abstract. Chemical structures of Dbo, DBO, and CB7.
Introduction
Time-resolved fluorescence (TRF) detection has become an indispensable tool in biochemical research (Liu and Hennig, in press; Hemmilä and Webb, 1997; Steinkamp and Karst, 2004), which has been applied to enzyme assays (Karvinen et al., 2004; Sadler et al., 2004; Terai et al., 2006; Vuojola et al., 2013; Hewitt and Butler, 2018), fluoroimmunoassays (Hildebrandt et al., 2005; Charbonnière et al., 2006; Geißler et al., 2010), and assays involving oligonucleotides (Johansson et al., 2004; Wu et al., 2013). Commonly, lanthanide chelates have been employed as luminescent probes with lifetimes in the millisecond time range (Bünzli, 2010). Since typical fluorescence lifetimes are <10 ns, such exceedingly long probe lifetimes allow to record the probe emission after a short delay time during which the short-lived background emission has already significantly decayed (Hemmilä and Laitala, 2005). Such a time-gated measurement affords an efficient reduction of short-lived background emission and an increased sensitivity (Hemmilä and Webb, 1997; Steinkamp and Karst, 2004) However, lanthanide chelates suffer from several drawbacks, i.e., a large size, a cationic center, and hydrophobic ligands, which may interfere with the biomolecular enzyme-substrate recognition. Thus, organic fluorescent probes with contrasting properties are often desirable for complementary applications (Turconi et al., 2001; Gribbon and Sewing, 2003; Kainmüller et al., 2005; Hennig et al., 2006, 2007b; Sahoo et al., 2007a).
With the advent of fast-switching electric circuits, shorter lifetimes in the range of 100 ns to several microseconds are nowadays also accessible for TRF detection, and ruthenium complexes have been suggested as suitable probes, which are, however, similarly problematic (Bannwarth et al., 1988; Hennig and Zeckert, 2000; Yun et al., 2003; Kainmüller et al., 2005; Kainmüller and Bannwarth, 2006; Clima et al., 2007; Martí et al., 2007a,b; Kramer et al., 2008; Kolpashchikov, 2010; Guo et al., 2011). Further, pyrene has been explored for its utility in TRF assays with nanosecond lifetimes (Yang et al., 2005; Martí et al., 2006, 2007a,b; Conlon et al., 2008; Guo et al., 2011), but large hydrophobic, aromatic surfaces, such as that of pyrene, are well-known to interfere with biomolecular recognition (Daugherty and Gellman, 1999; Sahoo et al., 2007a). As an appealing alternative, we have introduced 2,3-diazabicyclo[2.2.2]oct-2-ene labeled asparagine (Dbo), which exhibits an unquenched lifetime of ca. 330 ns in water (Hudgins et al., 2002; Nau and Wang, 2002). Application of Dbo to TRF assays for proteases, kinases, and phosphatases demonstrated for the first time the practical feasibility of this concept, for which we have coined the name nanosecond time-resolved fluorescence (Nano-TRF) assays (Hennig et al., 2006, 2007b; Sahoo et al., 2007a).
From previous work, it was known that the parent molecule 2,3-diazabicyclo[2.2.2]oct-2-ene (DBO) forms a strong supramolecular host-guest inclusion complex with the macrocyclic host cucurbit[7]uril (CB7) (Márquez and Nau, 2001; Marquez et al., 2004; Mohanty and Nau, 2004). The extremely low polarizability of the CB7 cavity, which lies between that of perfluorohexane and the gas phase, leads—according to the Strickler-Berg relation—to a prolonged radiative decay rate of fluorescent dyes resulting in an increased fluorescence lifetime of the CB7/DBO complex of up to 1,050 ns (Strickler and Berg, 1962; Márquez and Nau, 2001; Marquez et al., 2004; Mohanty and Nau, 2004; Nau and Mohanty, 2005). In addition, other desirable effects of CB7 complexation on fluorescent dyes have been observed, e.g., an enhanced stability, increased brightness, deaggregation, and a reduced intermolecular quenching (Márquez et al., 2003; Mohanty and Nau, 2005; Nau and Mohanty, 2005; Koner and Nau, 2007). It has consequently been proposed that the advantageous effects of CB7 on fluorescent dyes could be exploited for the construction of improved enzyme assays (Marquez et al., 2004). Herein, we explore the feasibility of this concept by applying CB7 to a Nano-TRF protease assay in which we introduce 3-nitrotyrosine as a “dark,” that is, non-fluorescent quencher of the DBO chromophore. Proteases were selected due to their importance as targets in drug discovery (Hennig and Zeckert, 2000; Hildebrandt et al., 2005; Clima et al., 2007; Hennig et al., 2007b; Sahoo et al., 2007a; Vuojola et al., 2013). We provide a detailed photophysical characterization of this novel assay and elucidate the interactions of CB7 with the substrate. Finally, we demonstrate that the lifetime-enhancing effect of CB7 presents a powerful enhancement strategy for Nano-TRF assays.
Materials and Methods
Peptide Substrates and Enzymes
DBO-labeled asparagine was synthesized according to a literature procedure (Hudgins et al., 2002). The peptide was prepared by Biosyntan GmbH (Berlin, Germany) and obtained in >95% purity. Trypsin (from bovine pancreas, 2,500 U/mg) and chymotrypsin (CT, from bovine pancreas, 1,500 U/mg) were from AppliChem (Darmstadt, Germany). We used a 67 mM phosphate buffer (pH 7) for CT and a 112 mM borate buffer (pH 8) for trypsin. CB7 was synthesized according to the literature procedure (Kim et al., 2000; Day et al., 2001; Marquez et al., 2004).
Fluorescence Spectroscopy
Absorption measurements of enzyme and peptide stock solutions were performed with a Varian Cary 4000 spectrophotometer. The following extinction coefficients were used to derive the concentrations of the enzymes: ε280 = 33,600 M−1cm−1 for trypsin (Labouesse and Gervais, 1967) and ε280 = 50,000 M−1cm−1 for CT (Martin and Marini, 1967). For 3-nitrotyrosine we determined an extinction coefficient of ε378 = 2,260 ± 50 M−1cm−1 (the isosbestic point in titrations with CB7), which we used for measuring the peptide concentrations. Fluorescence lifetimes were determined by time-correlated single photon counting (FLS920, Edinburgh Instruments) with a Picoquant picosecond pulsed diode laser (λexc = 373 nm, λem = 450 nm, ca. 50 ps pulse width) for excitation. The reported lifetimes were recovered by tail-fitting with the instrument-specific software. The goodness-of-fit was judged by a reduced χ2 of <1.1 and a random distribution of the weighted residuals around zero, if not otherwise specified. Kinetic traces were collected with a Varian Eclipse spectrofluorometer, equipped with a thermostatted cell holder (λexc = 365 nm, λem = 450 nm).
Kinetic Assays
1 millimolar stock solutions of the peptide were prepared in water and stored at +4°C. For the assays, an appropriate amount was diluted with the respective buffer solution and the sample was allowed to equilibrate for at least 10 min in a thermostatted cell holder at 25.0 ± 0.1°C. Enzyme stock solutions were freshly prepared on a daily basis in 1 mM HCl and small aliquots of the enzyme stock solution were added to the reaction mixture in 67 mM sodium phosphate buffer, pH 7.0 (for chymotrypsin) or 112 mM sodium borate buffer, pH 8.0 (for trypsin). It was ensured that the small aliquot of 1 mM HCl has no influence on the pH of the reaction mixture.
Nano-TRF assays were carried out in black 384-well microplates (Corning NBS) using 50 μl final sample volume with an LF 402 NanoScan FI microplate reader (IOM, Berlin, Germany). An external nitrogen laser (MNL 200, Laser Technik Berlin, Germany) was coupled by glass fibers to a dye laser module, which had a maximum dye emission at 365 nm. The emission was detected at 450 nm with a gate time of 2 μs.
Results and Discussion
Choice and Characterization of the Probe-Quencher Pair
The natural amino acids tryptophan and tyrosine have been exploited as intrinsic contact-based quenchers of the DBO chromophore in the previously reported Nano-TRF assays (Liu and Hennig, in press; Hennig et al., 2006, 2007b; Sahoo et al., 2007a). The use of these natural quenching sites is, however, not feasible for the current approach, because CB7 acts not only as a lifetime-enhancing agent but also as an efficient “protection shield” against contact-based quenching (Márquez et al., 2003; Marquez et al., 2004; Nau et al., 2003). Both effects are inherently related to the formation of supramolecular host-guest inclusion complexes, where the DBO chomophore, as a guest, is deeply immersed inside the cavity of CB7, the host. We therefore needed to resort to conventional fluorescence resonance energy transfer (FRET) to construct substrates for proteases (Matayoshi et al., 1990; Gershkovich and Kholodovych, 1996). FRET does not require an intimate probe-quencher contact but acts through space (Förster, 1948; Stryer and Haugland, 1967; Selvin and Hearst, 1994) and also through the walls of the macrocyclic host (Zhang et al., 2019).
While FRET with Dbo as an acceptor (note that Dbo designates the entire amino acid, while DBO designates the neat chromophore, see structure chart) is being used to assess peptide folding rates and distance distributions (Sahoo et al., 2006, 2007b; Jacob et al., 2013, 2018; Norouzy et al., 2015), the use of Dbo as a donor has not been reported so far, and we thus sought for an efficient energy acceptor. We selected 3-nitrotyrosine (NO2)Tyr, for several reasons: First, the absorption of (NO2)Tyr excellently overlaps with the emission of Dbo (Figure 1) in the biologically relevant pH range, ensuring efficient quenching. Second, (NO2)Tyr is a non-fluorescent dark quencher, which by-passes complications owing to residual acceptor fluorescence (Grüninger-Leitch et al., 2002; Johansson et al., 2004). Third, (NO2)Tyr has a very low affinity toward CB7, which allows the selective complexation of Dbo in the presence of the acceptor (vide infra). And fourth, (NO2)Tyr is a relatively small and not too hydrophobic molecule compared to many fluorescent dyes. The latter has the added advantage that a small residue is less prone to interfere with the enzyme-substrate recognition than a larger molecule (Gershkovich and Kholodovych, 1996; Hennig et al., 2006; Christopher et al., 2011). In other words, while we needed to sacrifice the possibility of employing an intrinsic quencher like tryptophan or tyrosine, we selected a quencher that has a similarly low propensity to interfere with the enzyme activity as the previously employed quenchers (Hennig et al., 2006, 2007b; Sahoo et al., 2007a) but that exhibits a different quenching mechanism; the latter allowed, as an added advantage, that other beneficial effects of CB7 encapsulation, namely its lifetime-enhancing effect on Dbo, could be fully exploited, while maintaining effective quenching.
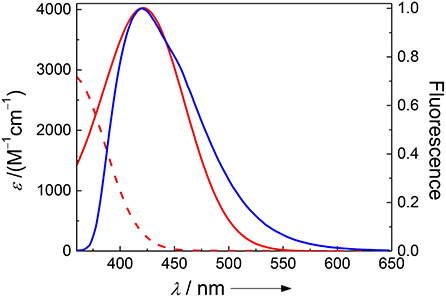
Figure 1. Spectral overlap between the pH-independent emission of Dbo (blue) and the absorption of 3-nitrotyrosine (red) at pH 2 (dashed line) and pH 8 (solid line).
The absorption spectra of (NO2)Tyr were measured in the pH range from 2.0 to 9.0 (Table 1) to assess the expected assay performance of the Dbo/(NO2)Tyr FRET pair. The absorption spectral properties of (NO2)Tyr compare well with literature data (ε420 = 4189 M−1cm−1 for pH > 8 and ε365 = 2761 M−1cm−1 for pH < 6.8) (Fisher and Naughton, 2003), and the reported pKa of (NO2)Tyr of 6.8 was precisely reproduced (6.79 ± 0.02). The Förster distance, R0, was calculated from the donor emission spectrum and the acceptor absorption spectra according to the established methods, using an orientation factor of 2/3 and a refractive index of 1.333 (Lakowicz, 2006; Sahoo et al., 2006; Sloniec et al., 2013). This provided an estimate of the energy transfer efficiency E for two typical lengths (10 and 15 Å) of a terminally probe-quencher-labeled protease substrate (Table 1). Details on the determination of R0 and the energy transfer efficiency have been previously reported and are included as Supporting Information (Birks and Georghiou, 1968). Quenching efficiencies >95% in the relevant pH range (pH 7–8) predict a minimum 20-fold increase in fluorescence intensity for the Dbo/(NO2)Tyr probe/quencher pair in protease assays, which represents a considerable improvement to our previously reported assays with tryptophan and tyrosine as quenchers (Hennig et al., 2007b), and which is comparable to the best available protease assays based on FRET (Matayoshi et al., 1990; Gershkovich and Kholodovych, 1996; Christopher et al., 2011).
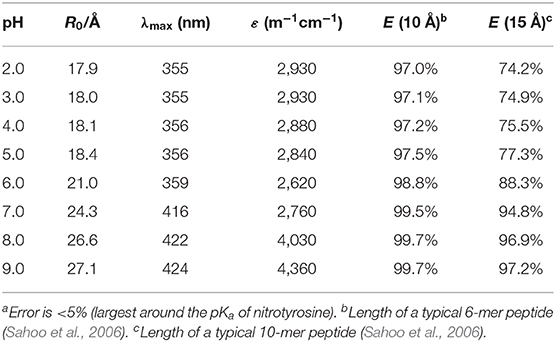
Table 1. Dependence of the Förster critical radius R0, the absorption maximum λmax, and the extinction coefficient ε of the Dbo/3-nitrotyrosine FRET pair on pH.a
Effects of Donor/Acceptor Diffusion on FRET
For a long-lived fluorescent probe like Dbo in connection with FRET, it is necessary to take the effects of mutual probe-acceptor diffusion into consideration (Yokota and Tanimoto, 1967; Birks and Georghiou, 1968; Steinberg and Katchalski, 1968; Thomas et al., 1978). Therefore, we determined the fluorescence lifetimes of the parent chromophore DBO and of the CB7/DBO complex in the presence of 0–100 μM (NO2)Tyr at pH 8. The decay traces were in all cases monoexponential, as judged by a reduced χ2 close to 1.0 and a random distribution of the weighted residuals around zero. A tentative Stern-Volmer analysis revealed apparent intermolecular quenching rate constants of 8.0 × 109 M−1s−1 for DBO and 4.7 × 109 M−1s−1 for the CB7/DBO complex.
Such monoexponential decay traces can, however, not be expected a priori for FRET-based quenching. The theory on the effects of diffusion on FRET is complex, but several limiting cases have been described (Yokota and Tanimoto, 1967; Birks and Georghiou, 1968; Steinberg and Katchalski, 1968; Thomas et al., 1978). Stryer and co-workers introduced for two freely diffusing molecules in solution the parameter Dτ0/s2, in which D is the sum of the diffusion coefficients of donor Dd and acceptor Da, τ0 is the fluorescence lifetime of the donor in absence of FRET, and s is the mean distance between donor and acceptor [s = 0.5(cANA)1/3, with cA being the acceptor concentration and NA the Avogadro number] (Thomas et al., 1978). A long fluorescence lifetime and a rapid diffusion lead to Dτ0/s2 >> 1. This allows the acceptor molecules to reach an equilibrium distribution during the lifetime and monoexponential fluorescence lifetimes are expected for such a case. If Dτ0/s2 << 1, the donor molecules can be regarded as static and a multiexponential distribution of fluorescence lifetimes results, which reflects the uneven distribution of acceptor molecules during the fluorescence lifetime. The former case applies to extremely long excited-state lifetimes, e.g., those of lanthanide chelates, and the latter case best describes common fluorescent dyes with lifetimes <25 ns.
Medium-range lifetimes such as that of Dbo, for which Dτ0/s2 is in the range of 1.1–6.2 require, however, a more elaborate treatment. Birks and Georghiou (1968) suggested the use of the model of Yokota and Tanimoto (1967) for the limiting case that (2Dτ0)1/2 > 3R0. This criterion was met for all our donor-acceptor combinations, and we accordingly performed simulations with this model. The simulated decay traces could be fitted with a monoexponential decay function as well and followed the Stern-Volmer equation, as did our experimental traces. The simulated quenching rate constants of 4.9 × 109 M−1s−1 for DBO as donor and 3.7 × 109 M−1s−1 for the CB7/DBO complex as a donor are in satisfactory agreement with the experimental values (vide supra).
Most important to note, the experimental as well as the simulated values lie distinctly above the theoretical rate constants for intermolecular diffusion-limited reactions, kdiff, in water (4.7 × 109 M−1s−1 for the DBO/(NO2)Tyr encounter and 2.9 × 109 M−1s−1 for the encounter of (NO2)Tyr with the larger CB7/DBO complex at 20°C) (Lapidus et al., 2000; Lee et al., 2003; Huang et al., 2004; Sahoo et al., 20061. This conclusively demonstrates that the quenching proceeds primarily by FRET and that there is at best a small contribution of contact-based quenching. Interestingly, ruthenium-based complexes have been used as FRET donors as well as acceptors (Bannwarth et al., 1988; Hennig and Zeckert, 2000; Yun et al., 2003; Kainmüller et al., 2005; Kainmüller and Bannwarth, 2006; Clima et al., 2007; Martí et al., 2007a,b; Kramer et al., 2008; Kolpashchikov, 2010; Guo et al., 2011), but the effects of diffusion in this time range has yet not been characterized in the context of bioassays. The present study might therefore provide a basis for the further application of biolabels in the microsecond time range.
Design of Protease Substrate and Expected Assay Performance
We designed a substrate for trypsin and chymotrypsin, two well-accepted model proteases (Fischer et al., 2002; Hedstrom, 2002; Park et al., 2002). The peptide sequence for the protease substrate was derived from known (Smyth, 1967) recognition motifs; appending a (NO2)Tyr N-terminal and a Dbo C-terminal to the recognition sequence afforded the desired FRET substrate: H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2. As expected, the substrate is cleaved by chymotrypsin and trypsin, and the fluorescence intensity increased 15 and 35 times, respectively, as expected from the quenching behavior of the neat fluorophores.
The increase in fluorescence intensity is accompanied by an increase in fluorescence lifetime to 325 ns after proteolytic cleavage, which resembles the lifetime of previously reported values for an unquenched Dbo and which is sufficiently long to allow Nano-TRF detection (Hudgins et al., 2002; Hennig et al., 2006, 2007b; Sahoo et al., 2007a). The fluorescence lifetime after proteolytic cleavage can be practically regarded constant (within 10% error) up to a substrate concentration of 50 μM. Knowledge of the upper concentration range is, however, essential for a reliable determination of enzyme kinetic parameters or inhibition constants (Grüninger-Leitch et al., 2002). Alternatively, the lifetime dependence on the (NO2)Tyr concentration can be corrected for, if necessary, according to the intermolecular diffusion model (vide supra) (Yokota and Tanimoto, 1967).
For the uncleaved peptide, however, an unexpected result was obtained (Birks and Georghiou, 1968; Thomas et al., 1978). We expected a monoexponential fluorescence lifetime, because Dτ0/s2 is ca. 10–100 for the uncleaved peptide (Wang et al., 20032) which should allow for a better description by the rapid diffusion model (Thomas et al., 1978). In contrast, we observed a pronounced multiexponential decay, which could not be satisfactorily fitted (reduced χ2 >> 1 and a non-random distribution of the weighted residuals around zero in case of all attempted fittings). We therefore presume the formation of several relatively long-lived low-energy conformations of the peptide, each contributing with a different fluorescence lifetime. This is not unlikely in view of the possibility of ground-state complexes between the electron-rich phenylalanine group and the electron-poor nitrotyrosine (Yaron et al., 1979; Packard et al., 1996; Johansson and Cook, 2003); interestingly, dissociation rate constants in the relevant range for tryptophan and oxazine dyes in peptides have, in fact, been detected by single-molecule spectroscopy (Neuweiler et al., 2003). Despite the difficulties to reliably fit and interpret the fluorescence lifetime of the uncleaved peptide, it is important to note that the lifetime is nonetheless sufficiently short (ca. 5 ns, reduced χ2 = 10) to be efficiently suppressed by Nano-TRF detection.
Substrate Binding Studies With CB7
Cucurbit[n]urils constitute a class of macrocyclic oligomers composed of n glycoluril units, which are supramolecular host molecules with outstanding molecular recognition properties that have been applied as stimuli-responsive systems, e.g., in catalysis, bioanalysis, drug delivery, or in polymeric and nano-sized materials (Bhasikuttan et al., 2011; Masson et al., 2012; Isaacs, 2014; Kaifer, 2014; Assaf and Nau, 2015; Barrow et al., 2015; Cong et al., 2016; van Dun et al., 2017; Kim, 2020). The most studied homologs with n = 6, 7, and 8 are known to bind to various amino acids, peptides, and proteins with affinities in the range of 103-109 M−1, and the molecular recognition properties indicated a preference for hydrophobic and cationic amino acid residues (Urbach and Ramalingam, 2011; Gamal-Eldin and Macartney, 2013; Logsdon and Urbach, 2013; Lee et al., 2015a,b; Smith et al., 2015; Li et al., 2016; Webber et al., 2016; Hirani et al., 2018). In our own work, we have exploited the interactions of cucurbiturils with amino acids, peptides, and proteins for the sensing of enzyme activity, membrane transport, and chirality (Hennig et al., 2007a; Bailey et al., 2008; Nau et al., 2009; Dsouza et al., 2012; Biedermann and Nau, 2014; Ghale et al., 2014; Schnurr et al., 2015; Nilam et al., 2017; Barba-Bon et al., 2019; Liu et al., 2019; Biedermann et al., 2020). It was therefore considered to be very likely that CB7 will bind to several sites of the polyfunctional substrate H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2 with different affinity, in particular because DBO is also known to strongly bind with CB7 (Marquez et al., 2004).
It often presents a major challenge to dissect the contribution of each amino acid side chain to the overall binding, but we will show that the properties of the present substrate allow the combined use of several independent analytical techniques. Straightforward to interpret are the changes on the absorption spectrum in the range of the (NO2)Tyr absorption (Figure 2A). The 430 nm absorption band of (NO2)Tyr decreases upon addition of increasing concentrations of CB7, while the absorption below 385 nm increases with an apparent isosbestic point at 385 nm3. The initial lag phase, in which the absorption does not change upon CB7 addition immediately suggests a 2:1 host-guest binding (Bakirci and Nau, 2005). The weaker binding constant of (2,200 ± 500) M −1 is associated with encapsulation of the (NO2)Tyr side chain into the CB7 cavity, because the change in the (NO2)Tyr absorption is most pronounced at high CB7 concentrations. At low CB7 concentrations, no change in the (NO2)Tyr absorption is observed suggesting that the stronger binding constant of (1.2 ± 0.7) × 104 M−1 does not involve the (NO2)Tyr side chain but must take place at a different position of the substrate (vide infra). Unfortunately, there is yet not much available data for a comprehensive interpretation, but a binding constant of 2,200 M−1 with CB7 is reasonable in view of a binding constant of 2,400 M−1 for Trp-OMe with CB7 (Hennig et al., 2007c). The formation of a 2:1 complex is further supported by time-correlated single photon counting (TCSPC) experiments. At low CB7 concentrations (up to ca. 100 μM), there is virtually no change in the fluorescence decay traces (see inset of Figure 2B), but at higher CB7 concentrations a long-lived component evolves (see Figure 2B). A quantitative analysis was not attempted at this point, because of the complex multiexponential decay (vide supra), but it is obvious that the decrease in the (NO2)Tyr absorption band centered at 430 nm (compare Figure 2A), which is the maximum of the Dbo emission, will lead to a less efficient FRET and, thus, to a prolonged fluorescence lifetime.
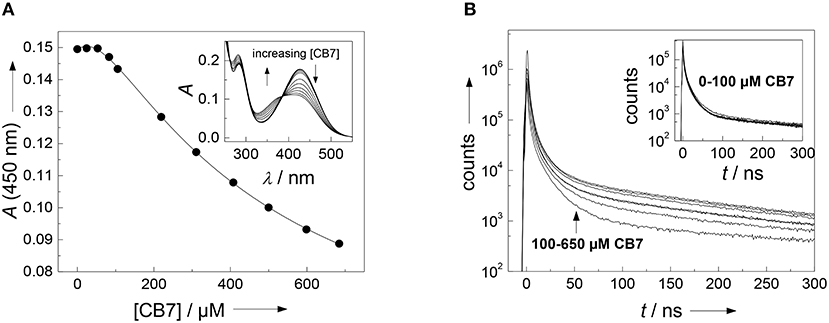
Figure 2. (A) Determination of the binding constant between the FRET acceptor 3-nitrotyrosine and CB7. Shown is the titration curve of a 40 μM peptide solution with varying amounts of CB7 in borate buffer, pH 8. The solid line indicates the fitting according to a 2:1 host-guest binding model. The inset shows the respective UV absorption spectra. (B) Dependence of fluorescence decay traces of the peptide H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2 (30 μM) on CB7 concentration. The experiments were performed in 67 mM phosphate buffer, pH 7.
Since it was not clear, which part of the intact peptide is responsible for the higher binding constant, titrations with the cleaved peptide fragments were performed. Chymotrypsin preferentially hydrolyzes peptide bonds N-terminally to hydrophobic residues, which results in the peptide fragments H-(NO2)Tyr-Gly-Ser-Gly-Phe-OH and H-Arg-Gly-Dbo-NH2. Trypsin preferentially hydrolyzes peptide bonds N-terminally to cationic residues, which results in the peptide fragments H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-OH and H-Gly-Dbo-NH2. Addition of small amounts of CB7 after cleavage to either mixture led to a biexponential decay with one component of 325 ns and a second component with 950 ns. The contribution as expressed by the preexponential factor of the longer-lived component increased with increasing concentrations of CB7, while the shorter-lived decreased, which clearly suggests the formation of a Dbo/CB7 inclusion complex.
The titrations (inset of Figure 3) could be excellently described according to a 1:1 host-guest binding model and revealed a binding constant of (1.9 ± 0.5) × 104 M−1. This binding constant of the cleaved peptide fragments is in excellent agreement with the independently determined first binding constant from the 2:1 fitting of the UV absorption titration for the uncleaved peptide. Based on the evident encapsulation of Dbo after cleavage and because the cleavage site is remote from the complexation site, it can, thus, be inferred that Dbo is also preferentially complexed in the uncleaved peptide. Although the complexation of Dbo in the uncleaved peptide had no effect on the fluorescence decay traces, this was not deteriorating for the desired application, for which solely the effect on the fluorescence lifetime after cleavage is essential. In the Discussion section, we will provide a comprehensive model to explain the peculiar effects supported by enzyme kinetic measurements (vide infra).
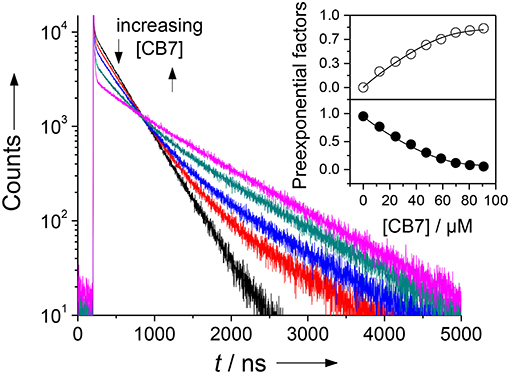
Figure 3. Fluorescence lifetime enhancing effect upon addition of increasing amounts of CB7 to an assay mixture after complete cleavage of 30 μM H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2 by chymotrypsin in phosphate buffer, pH 7. The different traces represent CB7 concentrations of 0, 12, 24, 36, and 70 μM. The sharp peak immediately after the laser pulse is attributed to a minor fluorescent impurity. The inset shows the dependence of the normalized preexponential factors on the CB7 concentration (upper panel, open circles: long-live component with 950 ns; lower panel, filled circles: short-lived component with 325 ns fluorescence lifetime), which provided a suitable titration curve. The solid lines were obtained by fitting according to a 1:1 host-guest binding model.
Enzyme Kinetics With Trypsin
We and others have previously described that CB7 can inhibit the activity of certain proteases by complexation of the substrate, which results in a “masking” of the enzyme recognition site in the substrate (Hennig et al., 2007c; Ghale et al., 2012; Logsdon and Urbach, 2013; Lee et al., 2015a). Conversely, the inhibition can be exploited to determine the binding constant with the enzyme recognition site of the protease substrate (McGarraghy and Darcy, 2000; Hennig et al., 2007c). The uncomplexed peptide H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2 is a good substrate for trypsin having a catalytic turnover number of kcat = 188 min−1 and a Michaelis-Menten constant of KM = 229 μM (determined according to a Lineweaver-Burk plot, n = 7, r > 0.989). However, in the presence of varying amounts of CB7, the initial cleavage rate, v0, decreases below the detection limit (Figure 4) (Hennig et al., 2007c). A fitting according to a 1:1 binding model provided a binding constant of (3.2 ± 1.7) × 104 M−1, which falls nicely in the range determined for the cleaved peptide and the strong binding constant from the UV absorption titration (vide supra). However, note that the recognition site for trypsin is the arginine side chain, while the complexation site of CB7 is the Dbo side chain. This seeming incongruity will be explained by the comprehensive binding model in the Discussion section.
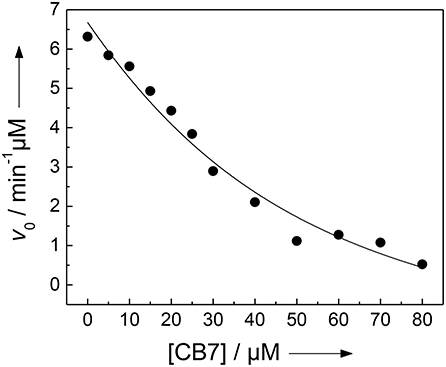
Figure 4. Inhibition of the activity of 300 nM trypsin against 30 μM H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2 by CB7 (varying concentrations) in 112 mM borate buffer, pH 8.0. The solid line represents the fitted curve according to a 1:1 host-guest binding model.
Enzyme Kinetics With Chymotrypsin
The results with chymotrypsin are clear-cut and show potential for application to Nano-TRF assays (vide infra). The pseudo first-order rate constant (kcat/KM), which is relevant at such low concentrations also used in many screening applications, is not much influenced by CB7 (1.90 min−1μM−1 in the absence and 2.36 min−1μM−1 in presence of 1 mM CB7). A more detailed investigation by Lineweaver-Burk plots (see Figure 5), however, revealed, that the CB7 complexation influences the kinetic properties in such a way that the influence on kcat and KM nearly compensate each other. In the absence of CB7, the peptide is cleaved with kcat = 479 min−1 and KM = 252 μM (n = 9, r > 0.999). Under conditions of complete complexation, i.e., in the presence of 1 mM CB7, the kinetic parameters were kcat = 1,780 min−1 and KM = 755 μM (n = 7, r > 0.998). This provides another independent evidence for the complexation of the peptide substrate and indicates that the transition state of the complex is more efficiently bound at the expense of the complexed substrate; in particular it suggests that the complexation takes place at other positions than at the phenylalanine residue, which is the recognition site for chymotrypsin. This finding is in accordance with a report pointing to a much lower binding affinity of phenylalanine residues within a peptide chain than at the N-terminal position (Urbach and Ramalingam, 2011).
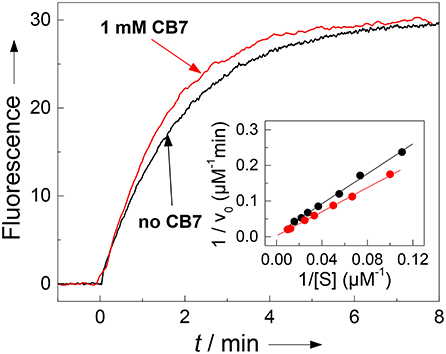
Figure 5. Influence of complexation of 30 μM H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2 by CB7 on enzyme kinetics. The pseudo first-order rate constant kcat/KM for cleavage by chymotrypsin (3 μM) is not significantly affected by complexation as can be seen from the fluorescence traces without CB7 (slower black trace, kcat/KM = 1.90 μM−1min−1) and in the presence of 1 mM CB7 (faster red trace, kcat/KM = 2.36 μM−1min−1). The Lineweaver-Burk plot without CB7 (black circles) and in presence of 1 mM CB7 (red circles) reveals the influence on the catalytic turnover number kcat and on the Michaelis-Menten constant KM. The experiments were performed in 67 mM phosphate buffer, pH 7.0.
Model for the Substrate-CB7 Binding
According to the combined results from steady-state and time-resolved fluorescence spectroscopy as well as absorption spectroscopy and from the enzyme kinetic measurements it became evident that CB7 forms a 2:1 host-guest complex with the peptide substrate. The prolonged fluorescence lifetime confirms that binding of the Dbo side chain is strongest and that Dbo occupies the inner cavity of CB7. The second binding takes place at the (NO2)Tyr side chain, which can be most conveniently seen from the effects on the absorption spectrum.
However, one problem still persists: How can it be that complexation of the Dbo residue leads to an inhibition of the arginine-recognizing enzyme trypsin, but not of the phenylalanine-recognizing chymotrypsin? We presume that the complexation properties of CB7 induce the formation of a “bidentate” inclusion/exclusion host-guest complex, in which DBO resides in the inner cavity of CB7 and the guanidinium group of the arginine side chain is bound to the remaining free portal (see Figure 6). NMR observations on other substrates have already suggested portal binding for the guanidinium side chain of an arginine residue and cavity binding of a hydrophobic residue (Hennig et al., 2007c). This model now also provides an excellent explanation for the seemingly different inhibition behavior of the substrate reported in this paper [inhibition as expressed by the binding constant is (3.2 ± 1.7) × 104 M−1] and the previously communicated substrate Nα-benzoyl-L-arginine p-nitroanilide [inhibition as expressed by the binding constant was (0.38 ± 0.06) × 104 M−1]. The different inhibition of the activity of trypsin against those substrates does therefore not reflect the binding toward the arginine side chain, but reflects the binding of the hydrophobic residues and the subsequent intramolecular cyclization as proposed in Figure 6.
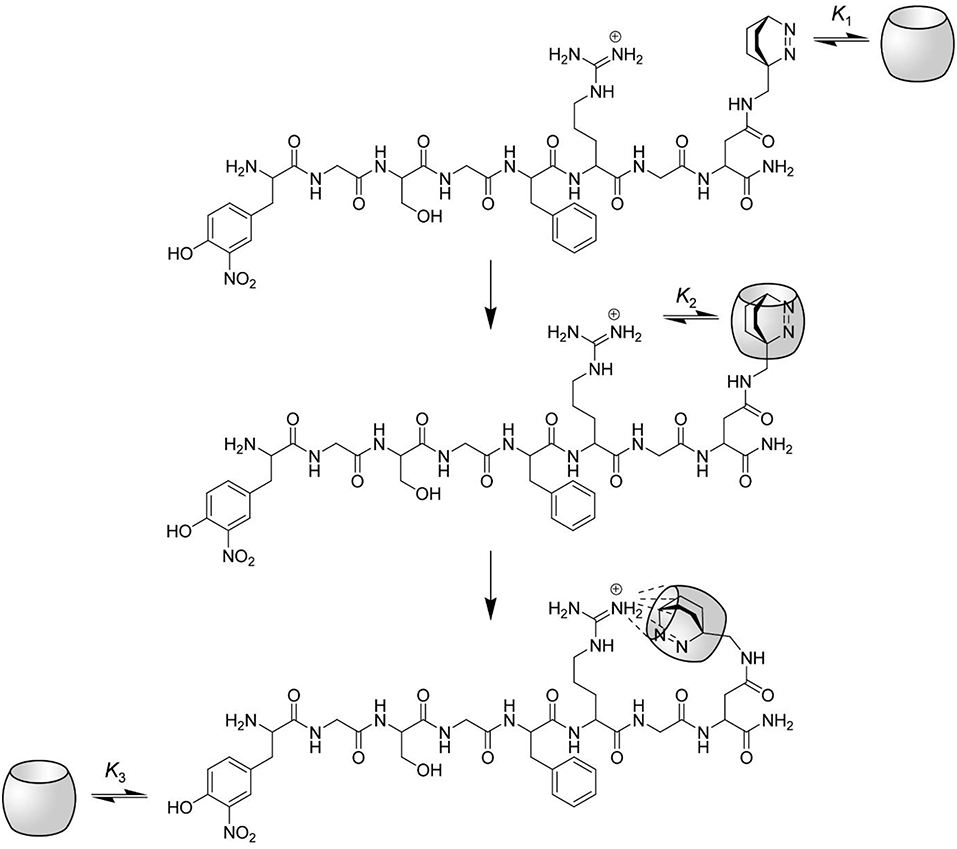
Figure 6. Proposed interactions between CB7 and the protease substrate H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2. The complexation of the substrate by CB7 proceeds in three steps. At low CB7 concentration, the substrate is selectively complexed at the C-terminal Dbo residue with a binding constant K1. The upper rim of CB7 enables the complexation of small cationic residues (compare structure), although the inner cavity is already occupied. Intramolecular host-guest complexation with the arginine side chain takes thus place in the second step with a binding constant K2, which also provides an efficient masking of the arginine side chain against enzymatic cleavage by trypsin. Note that this mode of intramolecular binding is highly preferred (see text). Finally, at high CB7 concentrations the (NO2)Tyr residue is also complexed with a binding constant K3.
According to the Gaussian chain model, which relates the intermolecular binding constant Kinter to the intramolecular binding constant Kintra (Nau et al., 2003), the binding constants can be reduced to the ratio of their collision rate constants ( and ) by assuming that the dissociation rate constant is not significantly affected by the intramolecular chain connecting the two chain ends. It follows that
with N representing the number and b representing the length of each chain segment. Assuming N = 15 (the number of connecting bonds) and an average bond length of b = 1.5 Å (Lide, 2003) one obtains for the ratio of Kintra/Kinter = 3 M−1, where Kintra represents the ratio of the concentrations of the intramolecularly complexed vs. the uncomplexed form. This rough estimation demonstrates that there is no significant hindrance toward intramolecular complexation. Additional NMR studies with methylguanidinium (MeGua) chloride suggest that the formation of a ternary complex with CB7, DBO, and MeGua is indeed possible.
Nano-TRF Assays
With the knowledge of the binding preferences of CB7 toward the protease substrate, it was possible to select a CB7 concentration, at which the fluorescent probe Dbo is selectively complexed. Trypsin is efficiently inhibited under this condition such that the feasibility for a continuous measurement of the enzymatic activity is sacrificed. However, the possibility for a stopped assay is not affected, i.e., the assay can still be carried out in the absence of CB7, CB7 is then added at known intervals after enzyme addition and the progress curves is derived from these measurements. Interestingly, CB7 also acts as the stopping reagent in this case, which remedies the necessity to perform, e.g., pH-jumps to stop the assay. In contrast, the minor influence on the activity of chymotrypsin is not deteriorating for a continuous measurement for this enzyme.
The steady-state intensity (expressed as the fluorescence quantum yield) of the Dbo fluorescence is slightly decreased inside CB7 (Mohanty and Nau, 2004), while the fluorescence lifetime is increased by ca. 3 times. The enhancement of adding CB7 in a Nano-TRF measurement is thus dependent on the applied delay time; at short delay times, the Nano-TRF intensity is smaller than the steady-state intensity, while at long delay times the opposite applies. In general, the Nano-TRF intensity can be theoretically predicted according to Equation (2), which relates two Nano-TRF intensities (I1 and I2) to their respective fluorescence lifetimes (τ1 and τ2) at different instrumental settings of the delay time, tdelay, and gate time, tgate.
Considering the fluorescence quantum yields of Dbo (0.26) and the Dbo/CB7 complex (0.19) (Mohanty and Nau, 2004) and their respective fluorescence lifetimes (325 and 950 ns) one obtains a ratio of preexponential factors of ADbo/CB7/ADbo of 0.25. A calculation with these values affords that the Nano-TRF intensity of Dbo and the Dbo/CB7 complex are equal for tdelay ≈ 200 ns and tgate = 2 μs. With tdelay > 200 ns a higher Nano-TRF intensity is expected for the Dbo/CB7 complex, which was experimentally confirmed in a convincing manner (Figure 7). In this context, it is important to note that usually delay times of 150 ns are sufficient to suppress the short-lived background fluorescence, but longer delay times are beneficial to enhance the differentiation of substrate and product (Johansson et al., 2004; Hennig et al., 2007c).
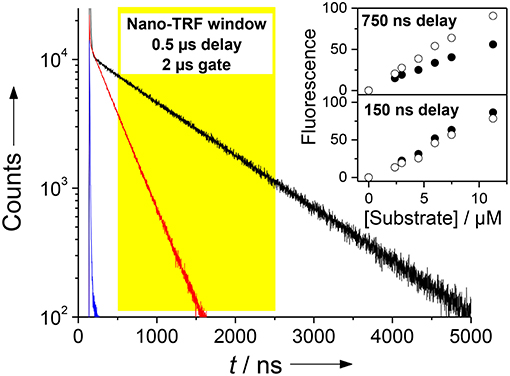
Figure 7. Demonstration of the Nano-TRF principle for a CB7-enhanced assay. Shown are the fluorescence decay traces for the uncleaved peptide (τ ≈ 5 ns, blue), the peptide after proteolytic cleavage in the absence of CB7 (τ = 325 ns, red), and after proteolytic cleavage in the presence of 100 μM CB7 (τ = 950 ns, black). The assay was performed with 30 μM H-(NO2)Tyr-Gly-Ser-Gly-Phe-Arg-Gly-Dbo-NH2 in 67 mM phosphate buffer, pH 7. Detection in the Nano-TRF mode is indicated by the yellow box (set to a delay time of 500 ns). The prolonged fluorescence lifetime translates directly into an enhanced fluorescence intensity in Nano-TRF measurements at delay times >150 ns, which is shown in the inset for delay times of 150 and 750 ns (solid circles without CB7, open circles with 100 μM CB7). Note that our instrumentation unfortunately only offered a gate time of 2 μs.
In conclusion, we have established herein CB7 as a supramolecular additive to enhance the sensitivity Nano-TRF assays. A peptide substrate for trypsin and chymotrypsin was doubly labeled with Dbo as a fluorescent probe and 3-nitrotyrosine as a FRET-based quencher. CB7 binds to the Dbo residue in the enzyme substrate, which has only a small effect on the fluorescence lifetime, whereas the fluorescence lifetime of the cleaved product increased from 325 to 950 ns. This facilitates the discrimination of substrates and products. While the presence of CB7 had only a small effect on the enzyme kinetics with chymotrypsin, the activity of trypsin was nearly completely inhibited, which suggested a hairpin conformation of the peptide, in which the guanidinium side chain of the arginine residue is additionally bound to a vacant ureido rim of one of the CB7 hosts.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
AH and WN jointly conceived the study, interpreted the results, and wrote the manuscript. The experiments were performed by AH. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a research collaboration of the F. Hoffmann-La Roche Ltd. and Jacobs University Bremen.
Conflict of Interest
The authors declare that this study received funding from F. Hoffmann-La Roche Ltd. The funder was not involved in the study design, collection, analysis, data interpretation, and writing of this article or the decision to submit it for publication.
Acknowledgments
We thank Dr. Garima Ghale for carrying out preliminary enzyme-kinetic measurements.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00806/full#supplementary-material
Footnotes
1. ^kdiff is determined from the Smoluchowski equation: kdiff = 4πNA(rA + rB)(DA + DB). The diffusion coefficient for 3-nitrotyrosine (6.30 × 10−6 cm2 s−1) was determined according an empirical formula for amino acids, peptides and proteins (D = 10−7) exp(α – β ln M, where α = 6.42, β = 0.42, and M is the molecular weight). The diffusion coefficients of DBO and the CB7/DBO complex were determined according to the Stokes-Einstein relation D = (kT)/(6πrη). The molecular shape of DBO is approximated by a sphere with a radius r = 2.3 Å. CB7 can be best described as an oblate spheroid with ra = 8.0 Å and rb = 4.55 Å, which gives an effective radius of r = (rb)1/3 = 6.6 Å. The diffusion coefficients are thus 9.32 × 10−6 cm2 s−1 for DBO and 3.25 × 10−6 cm2 s−1 for the CB7/DBO complex. The reaction radius (rA + rB) was taken as 4 Å = 4 × 10−8 cm [see (Stryer and Haugland, 1967; Yokota and Tanimoto, 1967; Steinberg and Katchalski, 1968; Thomas et al., 1978) for details].
2. ^Taking the reduced mutual diffusion coefficient of biopolymer chain ends into account (see Wang et al., 2003).
3. ^Note that the extinction coefficient of Dbo is ca. 50times smaller than the extinction coefficient of (NO2)Tyr; the absorption of Dbo is thus virtually invisible and can be neglected.
References
Assaf, K. I., and Nau, W. M. (2015). Cucurbiturils: from synthesis to high-affinity binding and catalysis. Chem. Soc. Rev. 44, 394–418. doi: 10.1039/C4CS00273C
Bailey, D. M., Hennig, A., Uzunova, V. D., and Nau, W. M. (2008). Supramolecular tandem enzyme assays for multiparameter sensor arrays and enantiomeric excess determination of amino acids. Chem. Eur. J. 14, 6069–6077. doi: 10.1002/chem.200800463
Bakirci, H., and Nau, W. M. (2005). Chiral resolution through precipitation of diastereomeric capsules in the form of 2:1 b-cyclodextrin-guest complexes. J. Org. Chem. 70, 4506–4509. doi: 10.1021/jo050447w
Bannwarth, W., Schmidt, D., Stallard, R. L., Hornung, C., Knorr, R., and Müller, F. (1988). Bathophenanthroline-ruthenium(II) complexes as non-radioactive labels for oligonucleotides which can be measured by time-resolved fluorescence techniques. Helv. Chim. Acta 71, 2085–2099. doi: 10.1002/hlca.19880710826
Barba-Bon, A., Pan, Y.-C., Biedermann, F., Guo, D.-S., Nau, W. M., and Hennig, A. (2019). Fluorescence monitoring of peptide transport pathways into large and giant vesicles by supramolecular host–dye reporter pairs. J. Am. Chem. Soc. 141, 20137–20145. doi: 10.1021/jacs.9b09563
Barrow, S. J., Kasera, S., Rowland, M. J., del Barrio, J., and Scherman, O. A. (2015). Cucurbituril-based molecular recognition. Chem. Rev. 115, 12320–12406. doi: 10.1021/acs.chemrev.5b00341
Bhasikuttan, A. C., Pal, H., and Mohanty, J. (2011). Cucurbit[n]uril based supramolecular assemblies: tunable physico-chemical properties and their prospects. Chem. Commun. 47, 9959–9971. doi: 10.1039/c1cc12091c
Biedermann, F., Ghale, G., Hennig, A., and Nau, W. M. (2020). Fluorescent artificial receptor-based membrane assay (FARMA) for spatiotemporally resolved monitoring of biomembrane permeability. Comms. Biol. 3:383. doi: 10.1038/s42003-020-1108-9
Biedermann, F., and Nau, W. M. (2014). Noncovalent chirality sensing ensembles for the detection and reaction monitoring of amino acids, peptides, proteins, and aromatic drugs. Angew. Chem. Int. Ed. 53, 5694–5699. doi: 10.1002/anie.201400718
Birks, J. B., and Georghiou, S. (1968). Energy transfer in organic systems VII. Effects of diffusion on fluorescence decay. J. Phys. B 1, 958–965. doi: 10.1088/0022-3700/1/5/324
Bünzli, J.-C. G. (2010). Lanthanide luminescence for biomedical analyses and imaging. Chem. Rev. 110, 2729–2755. doi: 10.1021/cr900362e
Charbonnière, L. J., Hildebrandt, N., Ziessel, R. F., and Löhmannsröben, H.-G. (2006). Lanthanides to quantum dots resonance energy transfer in time-resolved fluoro-immunoassays and luminescence microscopy. J. Am. Chem. Soc. 128, 12800–12809. doi: 10.1021/ja062693a
Christopher, R. D., David, C. M., and Ella, F. J. (2011). Activatable optical probes for the detection of enzymes. Curr. Org. Synth. 8, 498–520. doi: 10.2174/157017911796117232
Clima, L., Hirtz-Haag, C., Kienzler, A., and Bannwarth, W. (2007). Application of a highly robust and efficient fluorescence-resonance-energy-transfer (FRET) system in DNA. Helv. Chim. Acta 90, 1082–1098. doi: 10.1002/hlca.200790107
Cong, H., Ni, X. L., Xiao, X., Huang, Y., Zhu, Q.-J., Xue, S.-F., et al. (2016). Synthesis and separation of cucurbit[n]urils and their derivatives. Org. Biomol. Chem. 14, 4335–4364. doi: 10.1039/C6OB00268D
Conlon, P., Yang, C. J., Wu, Y., Chen, Y., Martinez, K., Kim, Y., et al. (2008). Pyrene excimer signaling molecular beacons for probing nucleic acids. J. Am. Chem. Soc. 130, 336–342. doi: 10.1021/ja076411y
Daugherty, D. L., and Gellman, S. H. (1999). A fluorescence assay for leucine zipper dimerization: avoiding unintended consequences of fluorophore attachment. J. Am. Chem. Soc. 121, 4325–4333. doi: 10.1021/ja990178p
Day, A., Arnold, A. P., Blanch, R. J., and Snushall, B. (2001). Controlling factors in the synthesis of cucurbituril and its homologues. J. Org. Chem. 66, 8094–8100. doi: 10.1021/jo015897c
Dsouza, R. N., Hennig, A., and Nau, W. M. (2012). Supramolecular tandem enzyme assays. Chem. Eur. J. 18, 3444–3459. doi: 10.1002/chem.201103364
Fischer, N. O., McIntosh, C. M., Simard, J. M., and Rotello, V. M. (2002). Inhibition of chymotrypsin through surface binding using nanoparticle-based receptors. Proc. Natl. Acad. Sci. U.S.A. 99, 5018–5023. doi: 10.1073/pnas.082644099
Fisher, A. E. O., and Naughton, D. P. (2003). EDTA bis-(methyl tyrosinate): a chelating peptoid peroxynitrate scavenger. Bioorg. Med. Chem. Lett. 13, 1733–1735. doi: 10.1016/S0960-894X(03)00250-6
Förster, T. (1948). Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. 437, 55–75. doi: 10.1002/andp.19484370105
Gamal-Eldin, M. A., and Macartney, D. H. (2013). Selective molecular recognition of methylated lysines and arginines by cucurbit[6]uril and cucurbit[7]uril in aqueous solution. Org. Biomol. Chem. 11, 488–495. doi: 10.1039/C2OB27007B
Geißler, D., Charbonnière, L. J., Ziessel, R. F., Butlin, N. G., Löhmannsröben, H.-G., and Hildebrandt, N. (2010). Quantum dot biosensors for ultrasensitive multiplexed diagnostics. Angew. Chem. Int. Ed. 49, 1396–1401. doi: 10.1002/anie.200906399
Gershkovich, A. A., and Kholodovych, V. V. (1996). Fluorogenic substrates for proteases based on intramolecular fluorescence energy transfer (IFETS). J. Biochem. Biophys. Methods 33, 135–162. doi: 10.1016/S0165-022X(96)00023-1
Ghale, G., Kuhnert, N., and Nau, W. M. (2012). Monitoring stepwise proteolytic degradation of peptides by supramolecular domino tandem assays and mass spectrometry for trypsin and leucine aminopeptidase. Nat. Prod. Commun. 7, 343–348. doi: 10.1177/1934578X1200700315
Ghale, G., Lanctôt, A. G., Kreissl, H. T., Jacob, M. H., Weingart, H., Winterhalter, M., et al. (2014). Chemosensing ensembles for monitoring biomembrane transport in real time. Angew. Chem. Int. Ed. 53, 2762–2765. doi: 10.1002/anie.201309583
Gribbon, P., and Sewing, A. (2003). Fluorescence readouts in HTS: no gain without pain? Drug Discov. Today 8, 1035–1043. doi: 10.1016/S1359-6446(03)02895-2
Grüninger-Leitch, F., Schlatter, D., Küng, E., Nelböck, P., and Döbeli, H. (2002). Substrate and inhibitor profile of BACE (β-secretase) and comparison with other mammalian aspartic proteases. J. Biol. Chem. 277, 4687–4693. doi: 10.1074/jbc.M109266200
Guo, J., Ju, J., and Turro, N. J. (2011). Fluorescent hybridization probes for nucleic acid detection. Anal. Bioanal. Chem. 402, 3115–3125. doi: 10.1007/s00216-011-5526-x
Hedstrom, L. (2002). Serine protease mechanism and specificity. Chem. Rev. 102, 4501–4524. doi: 10.1021/cr000033x
Hemmilä, I., and Laitala, V. (2005). Progress in lanthanides as luminescent probes. J. Fluoresc. 15, 529–542. doi: 10.1007/s10895-005-2826-6
Hemmilä, I., and Webb, S. (1997). Time-resolved fluorometry: an overview of the labels and core technologies for drug screening applications. Drug Discov. Today 2, 373–381. doi: 10.1016/S1359-6446(97)01080-5
Hennig, A., Bakirci, H., and Nau, W. M. (2007a). Label-free continuous enzyme assays with macrocycle-fluorescent dye complexes. Nat. Methods 4, 629–632. doi: 10.1038/nmeth1064
Hennig, A., Florea, M., Roth, D., Enderle, T., and Nau, W. M. (2007b). Design of peptide substrates for nanosecond time-resolved fluorescence assays of proteases: 2,3-diazabicyclo[2.2.2]oct-2-ene as a noninvasive fluorophore. Anal. Biochem. 360, 255–265. doi: 10.1016/j.ab.2006.09.015
Hennig, A., Ghale, G., and Nau, W. M. (2007c). Effects of cucurbit[7]uril on enzymatic activity. Chem. Commun. 1614–1616. doi: 10.1039/b618703j
Hennig, A., Roth, D., Enderle, T., and Nau, W. M. (2006). Nanosecond time-resolved fluorescence protease assays. ChemBioChem 7, 733–737. doi: 10.1002/cbic.200500561
Hennig, H., and Zeckert, K. (2000). Luminescence quenching of ruthenium complexes through fluorescence resonance energy transfer (FRET). J. Inf. Rec. 25, 391–395.
Hewitt, S. H., and Butler, S. J. (2018). Application of lanthanide luminescence in probing enzyme activity. Chem. Commun. 54, 6635–6647. doi: 10.1039/C8CC02824A
Hildebrandt, N., Charbonnière, L. J., Beck, M., Ziessel, R. F., and Löhmannsröben, H.-G. (2005). Quantum dots as efficient energy acceptors in a time-resolved fluoroimmunoassay. Angew. Chem. Int. Ed. 44, 7612–7615. doi: 10.1002/anie.200501552
Hirani, Z., Taylor, H. F., Babcock, E. F., Bockus, A. T., Varnado, C. D., Bielawski, C. W., et al. (2018). Molecular recognition of methionine-terminated peptides by cucurbit[8]uril. J. Am. Chem. Soc. 140, 12263–12269. doi: 10.1021/jacs.8b07865
Huang, F., Hudgins, R. R., and Nau, W. M. (2004). Primary and secondary structure dependence of peptide flexibility assessed by fluorescence-based measurement of end-to-end collision rates. J. Am. Chem. Soc. 126, 16665–16675. doi: 10.1021/ja0466053
Hudgins, R. R., Huang, F., Gramlich, G., and Nau, W. M. (2002). A fluorescence-based method for direct measurement of submicrosecond intramolecular contact formation in biopolymers: an exploratory study with polypeptides. J. Am. Chem. Soc. 124, 556–564. doi: 10.1021/ja010493n
Isaacs, L. (2014). Stimuli responsive systems constructed using cucurbit[n]uril-type molecular containers. Acc. Chem. Res. 47, 2052–2062. doi: 10.1021/ar500075g
Jacob, M. H., Dsouza, R. N., Ghosh, I., Norouzy, A., Schwarzlose, T., and Nau, W. M. (2013). Diffusion-enhanced förster resonance energy transfer and the effects of external quenchers and the donor quantum yield. J. Phys. Chem. B 117, 185–198. doi: 10.1021/jp310381f
Jacob, M. H., D'Souza, R. N., Schwarzlose, T., Wang, X., Huang, F., Haas, E., et al. (2018). Method-unifying view of loop-formation kinetics in peptide and protein folding. J. Phys. Chem. B 122, 4445–4456. doi: 10.1021/acs.jpcb.8b00879
Johansson, M. K., and Cook, R. M. (2003). Intramolecular dimers: a new design strategy for fluorescence-quenched probes. Chem. Eur. J. 9, 3466–3471. doi: 10.1002/chem.200304941
Johansson, M. K., Cook, R. M., Xu, J., and Raymond, K. N. (2004). Time gating improves sensitivity in energy transfer assays with terbium chelate/dark quencher oligonucleotide probes. J. Am. Chem. Soc. 126, 16451–16455. doi: 10.1021/ja0452368
Kaifer, A. E. (2014). Toward reversible control of cucurbit[n]uril complexes. Acc. Chem. Res. 47, 2160–2167. doi: 10.1021/ar5001204
Kainmüller, E. K., Ollé, E. P., and Bannwarth, W. (2005). Synthesis of a new pair of fluorescence resonance energy transfer donor and acceptor dyes and its use in a protease assay. Chem. Commun. 5459–5461. doi: 10.1039/b509234e
Kainmüller, Eva, K., and Bannwarth, W. (2006). A new robust and highly sensitive FRET donor-acceptor pair: synthesis, characterization, and application in a thrombin assay. Helv. Chim. Acta 89, 3056–3070. doi: 10.1002/hlca.200690275
Karvinen, J., Laitala, V., Mäkinen, M.-L., Mulari, O., Tamminen, J., Hermonen, J., et al. (2004). Fluorescence quenching-based assays for hydrolyzing enzymes. Application of time-resolved fluorometry in assays for caspase, helicase, and phosphatase. Anal. Chem. 76, 1429–1436. doi: 10.1021/ac030234b
Kim, J., Jung, I.-S., Kim, S.-Y., Lee, E., Kang, J.-K., Sakamoto, S., et al. (2000). New cucurbituril homologues: syntheses, isolation, characterization, and X-ray crystal structures of cucurbit[n]uril (n) 5, 7, and 8. J. Am. Chem. Soc. 122, 540–541. doi: 10.1021/ja993376p
Kolpashchikov, D. M. (2010). Binary probes for nucleic acid analysis. Chem. Rev. 110, 4709–4723. doi: 10.1021/cr900323b
Koner, A. L., and Nau, W. M. (2007). Cucurbituril encapsulation of fluorescent dyes. Supramol. Chem. 19, 53–65. doi: 10.1080/10610270600910749
Kramer, R. A., Kainmüller, E. K., Flehr, R., Kumke, M. U., and Bannwarth, W. (2008). Quenching of the long-lived Ru(ii)bathophenanthroline luminescence for the detection of supramolecular interactions. Org. Biomol. Chem. 6, 2355–2360. doi: 10.1039/b800357b
Labouesse, J., and Gervais, M. (1967). Preparation of chemically defined ε N-acetylated trypsin. Eur. J. Biochem. 2, 215–223. doi: 10.1111/j.1432-1033.1967.tb00127.x
Lapidus, L. J., Eaton, W. A., and Hofrichter, J. (2000). Measuring the rate of intramolecular contact formation in polypeptides. Proc. Natl. Acad. Sci. U.S.A. 97, 7220–7225. doi: 10.1073/pnas.97.13.7220
Lee, J. W., Lee, H. H. L., Ko, Y. H., Kim, K., and Kim, H. I. (2015b). Deciphering the specific high-affinity binding of cucurbit[7]uril to amino acids in water. J. Phys. Chem. B 119, 4628–4636. doi: 10.1021/acs.jpcb.5b00743
Lee, J. W., Samal, S., Selvapalam, N., Kim, H.-J., and Kim, K. (2003). Cucurbituril homologues and derivatives: new opportunities in supramolecular chemistry. Acc. Chem. Res. 36, 621–630. doi: 10.1021/ar020254k
Lee, J. W., Shin, M. H., Mobley, W., Urbach, A. R., and Kim, H. I. (2015a). Supramolecular enhancement of protein analysis via the recognition of phenylalanine with cucurbit[7]uril. J. Am. Chem. Soc. 137, 15322–15329. doi: 10.1021/jacs.5b10648
Li, W., Bockus, A. T., Vinciguerra, B., Isaacs, L., and Urbach, A. R. (2016). Predictive recognition of native proteins by cucurbit[7]uril in a complex mixture. Chem. Commun. 52, 8537–8540. doi: 10.1039/C6CC03193E
Liu, Y.-C., and Hennig, A. (in press). Optical Spectroscopic Microscopic Techniques - Analysis of Biological Molecules. Springer Series on Fluorescence, ed H. Sahoo.
Liu, Y.-C., Peng, S., Angelova, L., Nau, W. M., and Hennig, A. (2019). Label-free fluorescent kinase and phosphatase enzyme assays with supramolecular host-dye pairs. ChemistryOpen 8, 1350–1354. doi: 10.1002/open.201900299
Logsdon, L. A., and Urbach, A. R. (2013). Sequence-specific inhibition of a nonspecific protease. J. Am. Chem. Soc. 135, 11414–11416. doi: 10.1021/ja406032x
Marquez, C., Huang, F., and Nau, W. M. (2004). Cucurbiturils: molecular nanocapsules for time-resolved fluorescence-based assays. IEEE Trans. Nanobiosci. 3, 39–45. doi: 10.1109/TNB.2004.824269
Márquez, C., and Nau, W. M. (2001). Polarizabilities inside molecular containers. Angew. Chem. Int. Ed. 40, 4387–4390. doi: 10.1002/1521-3773(20011203)40:23<4309::AID-ANIE4309>3.0.CO;2-L
Márquez, C., Pischel, U., and Nau, W. M. (2003). Selective fluorescence quenching of 2,3-diazabicyclo[2.2.2]oct-2-ene by nucleotides. Org. Lett. 5, 3911–3914. doi: 10.1021/ol035454q
Martí, A. A., Li, X., Jockusch, S., Li, Z., Raveendra, B., Kalachikov, S., et al. (2006). Pyrene binary probes for unambiguous detection of mRNA using time-resolved fluorescence spectroscopy. Nucl. Acids Res. 34, 3161–3168. doi: 10.1093/nar/gkl406
Martí, A. A., Li, X., Jockusch, S., Stevens, N., Li, Z., Raveendra, B., et al. (2007b). Design and characterization of two-dye and three-dye binary fluorescent probes for mRNA detection. Tetrahedron 63, 3591–3600. doi: 10.1016/j.tet.2006.08.109
Martí, A. A., Puckett, C. A., Dyer, J., Stevens, N., Jockusch, S., Ju, J., et al. (2007a). Inorganic–organic hybrid luminescent binary probe for DNA detection based on spin-forbidden resonance energy transfer. J. Am. Chem. Soc. 129, 8680–8681. doi: 10.1021/ja0717257
Martin, C. J., and Marini, M. A. (1967). Spectral detection of the reaction of formaldehyde with the histidine residues of α-chymotrypsin. J. Biol. Chem. 242, 5736–5743.
Masson, E., Ling, X., Joseph, R., Kyeremeh-Mensah, L., and Lu, X. (2012). Cucurbituril chemistry: a tale of supramolecular success. RSC Adv. 2, 1213–1247. doi: 10.1039/C1RA00768H
Matayoshi, E. D., Wang, G. T., Krafft, G. A., and Erickson, J. (1990). Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science 247, 954–958. doi: 10.1126/science.2106161
McGarraghy, M., and Darcy, R. (2000). Effects of cyclodextrins on chymotrypsin action. J. Inclusion Phenom. Macrocyclic Chem. 37, 259–264. doi: 10.1023/A:1008122203599
Mohanty, J., and Nau, W. M. (2004). Refractive index effects on the oscillator strength and radiative decay rate of 2,3-diazabicyclo[2.2.2]oct-2-ene. Photochem. Photobiol. Sci. 3, 1026–1031. doi: 10.1039/b412936a
Mohanty, J., and Nau, W. M. (2005). Ultrastable rhodamine with cucurbituril. Angew. Chem. Int. Ed. 44, 3750–3754. doi: 10.1002/anie.200500502
Nau, W. M., Ghale, G., Hennig, A., Bakirci, H., and Bailey, D. M. (2009). Substrate-selective supramolecular tandem assays: monitoring enzyme inhibition of arginase and diamine oxidase by fluorescent dye displacement from calixarene and cucurbituril macrocycles. J. Am. Chem. Soc. 131, 11558–11570. doi: 10.1021/ja904165c
Nau, W. M., Huang, F., Wang, X., Bakirci, H., Gramlich, G., and Márquez, C. (2003). Exploiting long-lived molecular fluorescence. Chimia 57, 161–167. doi: 10.2533/000942903777679424
Nau, W. M., and Mohanty, J. (2005). Taming fluorescent dyes with cucurbituril. Int. J. Photoenergy 7, 133–141. doi: 10.1155/S1110662X05000206
Nau, W. M., and Wang, X. (2002). Biomolecular and supramolecular kinetics in the submicrosecond time range: the fluorazophore approach. ChemPhysChem 3, 393–398. doi: 10.1002/1439-7641(20020517)3:5<393::AID-CPHC393>3.0.CO;2-4
Neuweiler, H., Schulz, A., Böhmer, M., Enderlein, J., and Sauer, M. (2003). Measurement of submicrosecond intramolecular contact formation of peptide at the single-molecule level. J. Am. Chem. Soc. 125, 5324–5330. doi: 10.1021/ja034040p
Nilam, M., Gribbon, P., Reinshagen, J., Cordts, K., Schwedhelm, E., Nau, W. M., et al. (2017). A label-free continuous fluorescence-based assay for monitoring ornithine decarboxylase activity with a synthetic putrescine receptor. SLAS Discov. 22, 906–914. doi: 10.1177/2472555216689288
Norouzy, A., Assaf, K. I., Zhang, S., Jacob, M. H., and Nau, W. M. (2015). Coulomb repulsion in short polypeptides. J. Phys. Chem. B 119, 33–43. doi: 10.1021/jp508263a
Packard, B. Z., Toptygin, D. D., Komoriya, A., and Brand, L. (1996). Profluorescent protease substrates: intramolecular dimers described by the exciton model. Proc. Natl. Acad. Sci. U.S.A. 93, 11640–11645. doi: 10.1073/pnas.93.21.11640
Park, H. S., Lin, Q., and Hamilton, A. D. (2002). Modulation of protein-protein interactions by synthetic receptors: design of molecules that disrupt serine-proteinaceous inhibitor interaction. Proc. Natl. Acad. Sci. U.S.A. 99, 5105–5109. doi: 10.1073/pnas.082675899
Sadler, T. M., Achilleos, M., Ragunathan, S., Pitkin, A., LaRocque, J., Morin, J., et al. (2004). Development and comparison of two nonradioactive kinase assays for I kappa B kinase. Anal. Biochem. 326, 106–113. doi: 10.1016/j.ab.2003.11.021
Sahoo, H., Hennig, A., Florea, M., Roth, D., Enderle, T., and Nau, W. M. (2007a). Single-label kinase and phosphatase assays for tyrosine phosphorylation using nanosecond time-resolved fluorescence detection. J. Am. Chem. Soc. 129, 15927–15934. doi: 10.1021/ja074975w
Sahoo, H., Roccatano, D., Hennig, A., and Nau, W. M. (2007b). A 10-Å spectroscopic ruler applied to short polyprolines. J. Am. Chem. Soc. 129, 9762–9772. doi: 10.1021/ja072178s
Sahoo, H., Roccatano, D., Zacharias, M., and Nau, W. M. (2006). Distance distributions of short polypeptides recovered by fluorescence resonance energy transfer in the 10.ANG. Domain J. Am. Chem. Soc. 128, 8118–8119. doi: 10.1021/ja062293n
Schnurr, M., Sloniec-Myszk, J., Döpfert, J., Schröder, L., and Hennig, A. (2015). Supramolecular assays for mapping enzyme activity by displacement-triggered change in hyperpolarized 129Xe magnetization transfer NMR spectroscopy. Angew. Chem. Int. Ed. 54, 13444–13447. doi: 10.1002/anie.201507002
Selvin, P. R., and Hearst, J. E. (1994). Luminescence energy transfer using a terbium chelate: improvements on fluorescence energy transfer. Proc. Natl. Acad. Sci. U.S.A. 91, 10024–10028. doi: 10.1073/pnas.91.21.10024
Sloniec, J., Schnurr, M., Witte, C., Resch-Genger, U., Schröder, L., and Hennig, A. (2013). Biomembrane interactions of functionalized cryptophane-A: combined fluorescence and 129Xe NMR studies of a bimodal contrast agent. Chem. Eur. J. 19, 3110–3118. doi: 10.1002/chem.201203773
Smith, L. C., Leach, D. G., Blaylock, B. E., Ali, O. A., and Urbach, A. R. (2015). Sequence-specific, nanomolar peptide binding via cucurbit[8]uril-induced folding and inclusion of neighboring side chains. J. Am. Chem. Soc. 137, 3663–3669. doi: 10.1021/jacs.5b00718
Smyth, D. G. (1967). Techniques in enzymic hydrolysis. Methods Enzymol. 11, 214–231. doi: 10.1016/S0076-6879(67)11026-4
Steinberg, I. Z., and Katchalski, E. (1968). Theoretical analysis of the role of diffusion in chemical reactions, fluorescence quenching, and nonradiative energy transfer. J. Chem. Phys. 48, 2404–2410. doi: 10.1063/1.1669460
Steinkamp, T., and Karst, U. (2004). Detection strategies for bioassays based on luminescent lanthanide complexes and signal amplification. Anal. Bioanal. Chem. 380, 24–30. doi: 10.1007/s00216-004-2682-2
Strickler, S. J., and Berg, R. A. (1962). Relationship between absorption intensity and fluorescence lifetime of molecules. J. Chem. Phys. 37, 814–822. doi: 10.1063/1.1733166
Stryer, L., and Haugland, R. P. (1967). Energy transfer - a spectroscopic ruler. Proc. Natl. Acad. Sci. U.S.A. 58, 719–726. doi: 10.1073/pnas.58.2.719
Terai, T., Kikuchi, K., Iwasawa, S.-Y., Kawabe, T., Hirata, Y., Urano, Y., et al. (2006). Modulation of luminescence intensity of lanthanide complexes by photoinduced electron transfer and its application to a long-lived protease probe. J. Am. Chem. Soc. 128, 6938–6946. doi: 10.1021/ja060729t
Thomas, D. D., Carlsen, W. F., and Stryer, L. (1978). Fluorescence energy transfer in the rapid-diffusion limit. Proc. Natl. Acad. Sci. U.S.A. 75, 5746–5750. doi: 10.1073/pnas.75.12.5746
Turconi, S., Bingham, R. P., Haupts, U., and Pope, A. J. (2001). Developments in fluorescence lifetime-based analysis for ultra-HTS. Drug Discov. Today 6, S27–S39. doi: 10.1016/S1359-6446(01)00159-3
Urbach, A. R., and Ramalingam, V. (2011). Molecular recognition of amino acids, peptides, and proteins by cucurbit[n]uril receptors. Isr. J. Chem. 51, 664–678. doi: 10.1002/ijch.201100035
van Dun, S., Ottmann, C., Milroy, L.-G., and Brunsveld, L. (2017). Supramolecular chemistry targeting proteins. J. Am. Chem. Soc. 139, 13960–13968. doi: 10.1021/jacs.7b01979
Vuojola, J., Syrjänpää, M., Lamminmäki, U., and Soukka, T. (2013). Genetically encoded protease substrate based on lanthanide-binding peptide for time-gated fluorescence detection. Anal. Chem. 85, 1367–1373. doi: 10.1021/ac302030q
Wang, X., Bodunov, E. N., and Nau, W. M. (2003). Fluorescence quenching kinetics in short polymer chains: dependence on chain length. Opt. Spectrosc. 95, 603–613. doi: 10.1134/1.1621440
Webber, M. J., Appel, E. A., Vinciguerra, B., Cortinas, A. B., Thapa, L. S., Jhunjhunwala, S., et al. (2016). Supramolecular PEGylation of biopharmaceuticals. Proc. Natl. Acad. Sci. U.S.A. 113, 14189–14194. doi: 10.1073/pnas.1616639113
Wu, M., Dul, B. E., Trevisan, A. J., and Fiedler, D. (2013). Synthesis and characterization of non-hydrolysable diphosphoinositol polyphosphate messengers. Chem. Sci. 4, 405–410. doi: 10.1039/C2SC21553E
Yang, C. J., Jockusch, S., Vicens, M., Turro, N. J., and Tan, W. (2005). Light-switching excimer probes for rapid protein monitoring in complex biological fluids. Proc. Nat. Acad. Sci. U.S.A. 102, 17278–17283. doi: 10.1073/pnas.0508821102
Yaron, A., Carmel, A., and Katchalski-Katzir, E. (1979). Intramolecularly quenched fluorogenic substrates for hydrolytic enzymes. Anal. Biochem. 95, 228–235. doi: 10.1016/0003-2697(79)90210-0
Yokota, M., and Tanimoto, O. (1967). Effects of diffusion on energy transfer by resonance. J. Phys. Soc. Jpn. 22, 779–784. doi: 10.1143/JPSJ.22.779
Yun, B. H., Kim, J. O., Lee, B. W., Lincoln, P., Norden, B., Kim, J. M., et al. (2003). Simultaneous binding of ruthenium(II) [(1,10-phenanthroline)2dipyridophenazine]2+ and minor groove binder 4',6-diamidino-2-phenylindole to poly[d(A-T)2] at high binding densities: observation of fluorescence resonance energy trasfer across the DNA stem. J. Phys. Chem. B 107, 9858–9864. doi: 10.1021/jp027828n
Keywords: cucurbiturils, enzyme assays, proteases, time-resolved fluorescence (TRF), peptides
Citation: Hennig A and Nau WM (2020) Interaction of Cucurbit[7]uril With Protease Substrates: Application to Nanosecond Time-Resolved Fluorescence Assays. Front. Chem. 8:806. doi: 10.3389/fchem.2020.00806
Received: 14 July 2020; Accepted: 31 July 2020;
Published: 10 September 2020.
Edited by:
Chusen Huang, Shanghai Normal University, ChinaReviewed by:
Apurba Lal Koner, Indian Institute of Science Education and Research, Bhopal, IndiaDong-Sheng Guo, Nankai University, China
Copyright © 2020 Hennig and Nau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Hennig, YW5kcmVhcy5oZW5uaWdAdW5pLW9zbmFicnVlY2suZGU=