 Huili Li1†Ling Fu2,3†
Huili Li1†Ling Fu2,3† Chaozheng He4,5*Jinrong Huo4Houyong Yang4,5Tingyue Xie6
Chaozheng He4,5*Jinrong Huo4Houyong Yang4,5Tingyue Xie6 Guozheng Zhao1*
Guozheng Zhao1* Guohui Dong7*
Guohui Dong7*- 1Key Laboratory of Magnetic Molecules, Magnetic Information Materials Ministry of Education, The School of Chemistry and Material Science, Shanxi Normal University, Linfen, China
- 2College of Agricultural Engineering, Nanyang Normal University, Nanyang, China
- 3College of Resources and Environmental Engineering, Tianshui Normal University, Tianshui, China
- 4Institute of Environmental and Energy Catalysis, School of Materials Science and Chemical Engineering, Xi’an Technological University, Xi’an, China
- 5Shaanxi Key Laboratory of Optoelectronic Functional Materials and Devices, School of Materials Science and Chemical Engineering, Xi’an Technological University, Xi’an, China
- 6School of Physics and Electronic Science, Shanxi Datong University, Shanxi, China
- 7School of Environmental Science and Engineering, Shaanxi University of Science and Technology, Xi’an, China
Based on the first principles of density functional theory, the adsorption behavior of H2CO on original monolayer MoS2 and Zn doped monolayer MoS2 was studied. The results show that the adsorption of H2CO on the original monolayer MoS2 is very weak, and the electronic structure of the substrate changes little after adsorption. A new kind of surface single cluster catalyst was formed after Zn doped monolayer MoS2, where the ZnMo3 small clusters made the surface have high selectivity. The adsorption behavior of H2CO on Zn doped monolayer MoS2 can be divided into two situations. When the H-end of H2CO molecule in the adsorption structure is downward, the adsorption energy is only 0.11 and 0.15 eV and the electronic structure of adsorbed substrate changes smaller. When the O-end of H2CO molecule is downward, the interaction between H2CO and the doped MoS2 is strong leading to the chemical adsorption with the adsorption energy of 0.80 and 0.98 eV. For the O-end-down structure, the adsorption obviously introduces new impurity states into the band gap or results in the redistribution of the original impurity states. All of these may lead to the change of the chemical properties of the doped MoS2 monolayer, which can be used to detect the adsorbed H2CO molecules. The results show that the introduction of appropriate dopant may be a feasible method to improve the performance of MoS2 gas sensor.
Introduction
In recent years, two-dimensional (2D) materials have attracted much attention due to their unique physical, chemical and electrical properties (Abbasi and Sardroodi, 2018a; Abbasi and Sardroodi, 2018b; Wu et al., 2018; Abbasi, 2019; Abbasi and Sardroodi, 2019). Among them, monolayer MoS2 belongs to hexagonal system, which is a typical 2D layered transition metal sulfide with sheet structure similar to graphene (Pan et al., 2020). It has been studied due to the excellent electronic structure, chemical and thermal stability, high surface activity and high strength (Xu et al., 2013). Compared with the zero gap of graphene, single-layer MoS2 has considerable direct band gap, which is suitable for light emitter, energy conversion and solar cell (Yu et al., 2016). Meanwhile, MoS2 has large specific surface area, surface activity and excellent adsorption capacity, so it is a special gas storage material or gas sensing material source (Zhang et al., 2017; Zhang et al., 2018; Zhou et al., 2018; Chen et al., 2019; Kathiravan et al., 2019; Tan et al., 2020). These excellent properties make the monolayer MoS2 have potential applications in the field of gas sensing.
However, due to the lack of free bonds on the surface of intact MoS2, which is chemically inert (Wang et al., 2013), the interaction with most gas molecules is limited to physical adsorption (Wanno and Tabtimsai, 2014), resulting in weak interaction between the adsorbent and monolayer MoS2, and the change of electronic properties is not obvious, and the original MoS2 cannot detect H2CO gas molecule (Ma et al., 2016a). Therefore, it is an effective and feasible method to adjust the electronic structure, chemical activity and sensitivity of MoS2 by introducing appropriate dopants into defect sites (Ma et al., 2016c; Sharma et al., 2018; Cui et al., 2019; Guo et al., 2019; Ren et al., 2019; Zhang et al., 2019; Guo et al., 2020; Zheng et al., 2020). For example, Lolla et al. have shown that Fe and Co. doped monolayer MoS2 can enhance the adsorption of O due to the partial occupation of d-orbitals on Fermi level, and the introduction of doping significantly improves the catalytic activity of MoS2 monolayer (Lolla and Luo, 2020). (Abbas et al., 2018) proposed that compared with the original monolayer MoS2, N and P doped atoms can enhance the sensing sensitivity of monolayer MoS2 to O2 and NO gas molecules (Abbas et al., 2018). (Sahoo et al., 2016) have shown that the original monolayer MoS2 is inert to gas molecules such as NH3 and NO2, and the detection sensitivity of antisite doped with MoS2 for these gas molecules and other chemical substances is significantly improved (Sahoo et al., 2016). Luo et al. have shown that Al, Si and P doped atoms increase the orbital hybridization effect between NO2, NH3 molecules and monolayer MoS2, and promote the electron transfer. The doped monolayer MoS2 has better adsorption performance than the undoped monolayer MoS2 (Luo et al., 2016). Ma et al. have shown that when Au, Fe, CO and Ni are doped into the monolayer MoS2, the charge transfer occurs, the distance between the adsorbed molecule and the dopant is shortened, the adsorption energy is increased, and the gas sensitivity to H2, CO, NO and O2 is increased (Ma et al., 2016b; Kwak et al., 2018; Kwon et al., 2018; Wang et al., 2019a). Au doped monolayer MoS2 has high charge transfer and strong orbital hybridization ability. Doping Au atoms affect the electronic structure of MoS2 monolayer, thus improving the adsorption capacity, so that the adsorption structure of C2H6 and C2H4 molecules on Au doped MoS2 monolayer is relatively stable (Qian et al., 2020). In conclusion, although there are some reports on the surface activity of doped monolayer MoS2, the adsorption of formaldehyde on the surface of transition metal Zn doped MoS2 has not been confirmed. Therefore, the geometry, electronic structure and small molecule adsorption of different transition metal atom doped monolayer MoS2 system can be obtained by theoretical simulation method, which has guiding significance for the study of the unique gas sensing properties, adsorption properties and chemical activities of transition metal atom doped monolayer MoS2.
At present, toxic gas molecules are one of the main problems of environmental pollution. H2CO is a common toxic gas. H2CO is widely used in household materials (Chung et al., 2013; Salthammer, 2013). Long term exposure to H2CO will irritate the eyes and throat, make breathing difficult, and even pose a serious threat to the lungs. Therefore, it is of great significance to detect and control H2CO in home, residence and scientific research (Lara-Ibeas et al., 2020; Song et al., 2020; Zhou et al., 2020). Based on the first principle calculation, this paper studies the changes of the configuration, geometric stability and electronic structure of MoS2 substrate caused by the adsorption of H2CO on the original and Zn doped monolayer MoS2 gas after the S vacancy is filled with Zn dopant, and the adsorption energy, adsorption structure and charge transfer of gas molecules are analyzed. This study is helpful to find suitable chemical modification methods to improve the performance of MoS2 based gas sensors, and has important scientific significance and application prospects for the design of high-efficiency gas sensing materials.
Computing Method
All calculations were carried out using the first principle method, which was carried out by Vienna Ab-initio Simulation Package (VASP) and projector enhanced wave (PAW) method of DFT (Qian et al., 2019; Wang et al., 2019b). The configuration of transition metal Zn doped MoS2 monolayer was optimized. The generalized gradient approximation (GGA) and perdew-Burke-Ernzerhof (PBE) are used to calculate the exchange correlation energy. The C 2s2p, Zn 3d4s and O 2s2p states are regarded as valence electrons. In the process of geometric optimization, all internal coordinates are allowed to relax under a fixed lattice constant, and the energy cutoff for plane waves is set to 450 eV. The Brillouin domain integral uses 3 × 3 × 1 (Monkhorst and Pack, 1976) monkhorst pack (MP) grid and 0.1 eV Gaussian smear. The convergence criterion is 10−5 eV. When the force applied to the atom is less than 0.01 ev/Å, the optimized structure is obtained. The calculated lattice constant of MoS2 is 3.18 Å, which is well consistent with the experimental and theoretical value of 3.20 and 3.18 Å (Ataca and Ciraci, 2011; Le et al., 2014). Dispersive interactions are not included because we are concerned about the effects of chemical bonds and gas molecules on the electronic and magnetic properties of MoS2. This method has been used to study gas adsorption on atom doped MoS2 (Ma et al., 2016a). MoS2 cell is constructed as a 4 × 4 supercell, in which one S atom is replaced by a Zn doped atom, with a total of 48 atoms, including 16 Mo atoms, 31 S atoms and one Zn atom. In order to avoid the interaction between the MoS2 monolayer and its periodic image, a vacuum space of 18 Å was added perpendicular to the MoS2 layer. The binding energy (Eb) between metal atom and support is defined as Eb = Etot (MoS2-S) + Etot(M) − Etot (M + MoS2), where Etot (M + MoS2), Etot (MoS2-S) and Etot (M) are the total energy of M/MoS2 catalyst, energy of MoS2 vacancy S-based carrier and the energy of single metal atom, respectively. Positive values indicate that the reaction gives off heat. In addition, the adsorption energy (Eads) was calculated to describe the interaction strength between gas and gas/MoS2 catalyst. According to the formula Eads = Etot (M + MoS2) + Etot (gas) − Etot (gas-M/MoS2), Etot (M + MoS2), Etot (gas) and Etot (gas-M/MoS2) are the energy of M/MoS2 catalyst, the energy of gas and the total energy of adsorption system respectively. According to this definition, positive adsorption energy represents exothermic adsorption. The density of states (DOS) is calculated by using the K point 5 × 5 × 1 with higher density. The results of DOS were analyzed by P4vasp. Bader charge (Henkelman et al., 2006) was used to analyze the charge transfer. The 3D visualization program Vesta (Momma and Izumi, 2011) was used to visualize all molecular structures, and the electron density difference was obtained to analyze the electron transfer direction. The electron density difference is defined as △ρ = ρgas-M/MoS2 − ρM/MoS2 − ρgas, where ρgas-M/MoS2, ρM/MoS2 and ρgas represent the electron density of adsorption system, M/MoS2 catalyst and gas, respectively. The PAW results based on VASP processing and the COHP diagram of LOBSTER 4.0.0 (Local Orbital Basis Suite Towards Electronic-Structure Reconstruction) (Maintz et al., 2016) were used to analyze the bonding.
Results and Discussion
Properties of Zn Doped Monolayer MoS2 (Zn-MoS2)
Seen from Supplementary Figure S1A, nine possible adsorption sites were considered for H2CO adsorb on the original MoS2 surface, where TS, TMo1/TMo2, H1/H2 and B1/B2/B3 represent the top of S and Mo atoms, hexagonal ring center and bridge sites. The TSV represent the top site of Zn doped the S vacancy. As depicted in Supplementary Figure S1B, the band structure and total density of states (TDOS) of the original monolayer MoS2 show that the original MoS2 monolayer is a non-magnetic semiconductor with a direct band gap of 1.74 eV, which is well consistent with the results reported in the literature value of 1.74 eV (Dimple et al., 2017). The calculated binding energy of Zn doping on MoS2 surface is 0.5 eV, which indicates that this structure is easy to form under thermodynamic equilibrium conditions due to the exothermic process. As shown in Figure 1B, the charge accumulation and loss can be observed in both Zn and Mo atoms, which means that chemical bonds are formed between Zn and Mo atoms in the ZnMo3 clusters, which can be used as a new surface single cluster catalyst (SCC) (Ma et al., 2018) to study the adsorption performance of H2CO molecules.
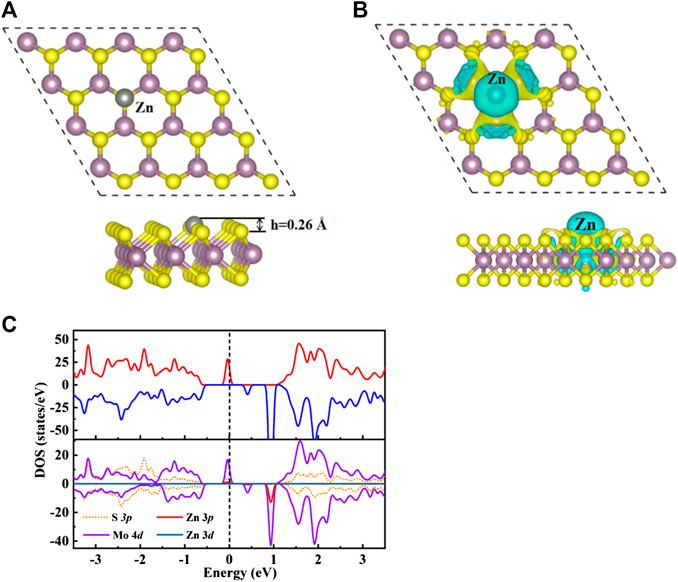
FIGURE 1. (A) Optimize the top view (top) and side view (bottom) structure of Zn-MoS2. Zn atoms are silver spheres. (B) The charge density difference diagram of Zn-MoS2. The yellow area indicates charge accumulation and the cyan area indicates charge loss. (C) TDOS (upper panels) and PDOS (lower panels) of Zn-MoS2 are projected on 3p orbital (red curve) and 3d orbital (dark blue curve) of Zn, 4d orbital (purple curve) of Mo atom and 3p orbit (orange dotted line) of S atom. The positive and negative values of DOS indicate spin up and spin down, respectively. The Fermi level is marked with a black dotted line.
For the average bond length between Zn dopant and adjacent Mo atoms, the calculated average bond length of Zn-Mo is 2.65 Å, which is larger than that of S-Mo bond in original monolayer MoS2 with the value of 2.41 Å. As exhibited in Figure 1A, accordingly the expansion of Zn-Mo bond relative to S-Mo bond makes the doped Zn atom protrude 0.26 Å above the S-plane. The electron transfer between dopant and MoS2 is calculated by Bader charge analysis. The trend of charge transfer is consistent with that of element electronegativity (Allred, 1961). The paulin electronegativity of Mo is 2.16, which is greater than that of Zn (1.65). Accordingly, Zn dopant loses electrons and carries a positive charge of 0.35 e. The charge distribution of Zn-MoS2 can be confirmed from Figure 1B. It is easy to see that Zn atoms are surrounded by cyan, which indicates that Zn atoms lose electrons. In addition, the magnetic moment of MoS2 is produced by Zn doping, the total magnetic moment of the single-layer MoS2 is 2.00 µB, only 0.1µB is located on the doped Zn atom, which indicates that the magnetic moment of the system mainly comes from Mo atom. In order to further understand the electronic and magnetic properties of Zn-MoS2, the total DOS (TDOS) and projected DOS (PDOS) of Zn-MoS2 spin polarization are given in Figure 1C. It can be seen from Figure 1C that an asymmetric DOS peak appears near the Fermi level, which is obviously different from the perfect MoS2 monolayer. This is consistent with the fact that the Zn-MoS2 system is paramagnetic (2.0 µb). Compared with MoS2 monolayer Supplementary Figure S1B, the spin down channel of Zn-MoS2 maintains the zero gap semiconductor characteristics of MoS2, but the spin up channel presents a non-zero density of states near Fermi level which indicates that the Zn-MoS2 system is semimetallic. Seen from the PDOS shown in Figure 1C, the degree of spin asymmetry of the 4d orbit of Mo is greater than that of the 3d orbit of Zn. This is consistent with the fact that the magnetic moment is mainly located on Mo atoms near Zn doping. In addition, since the asymmetric DOS peak is dominated by the 4d orbit of Mo, it can be expected that the spin charge density is mainly distributed on Mo atoms. Therefore, as shown in Figure 1B, the spin charge density of Zn-MoS2 system is mainly concentrated around Mo atom. In addition, Figure 1C also shows that near the Fermi level, the 3p state of Zn atom is hybridized with the 4d orbital of Mo, indicating the interaction between metal atom and S-vacancy.
Adsorption of H2CO on Original MoS2 Monolayer
In order to obtain a stable configuration, various possible initial adsorption structures were considered Supplementary Figure S1A. The interaction between H2CO and original MoS2 is very weak, and the stable configuration obtained belongs to physical adsorption. In this work, only the configuration with the largest and most stable adsorption energy is discussed. The adsorption energy, charge transfer and other related parameters are shown in Table 1, and the geometric electronic structure is shown in Figure 2.
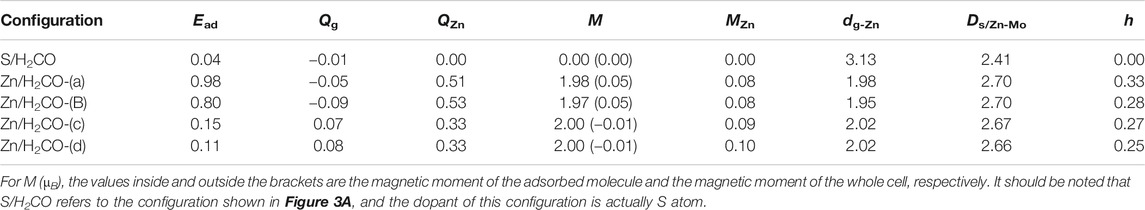
TABLE 1. Parameters of stable configuration of H2CO adsorbed on original and Zn doped monolayer MoS2: adsorption energy (Ead in eV), charge obtained by H2CO (Qg in e), charge obtained by Zn (QZn in e), magnetic moment of Zn atom (MZn in µB), nearest distance between adsorbed molecule and Zn atom (dg-Zn in Å), average distance between Zn atom and its adjacent molybdenum atom (ds/Zn-Mo in Å), The height of Zn atom relative to S plane (h in Å).
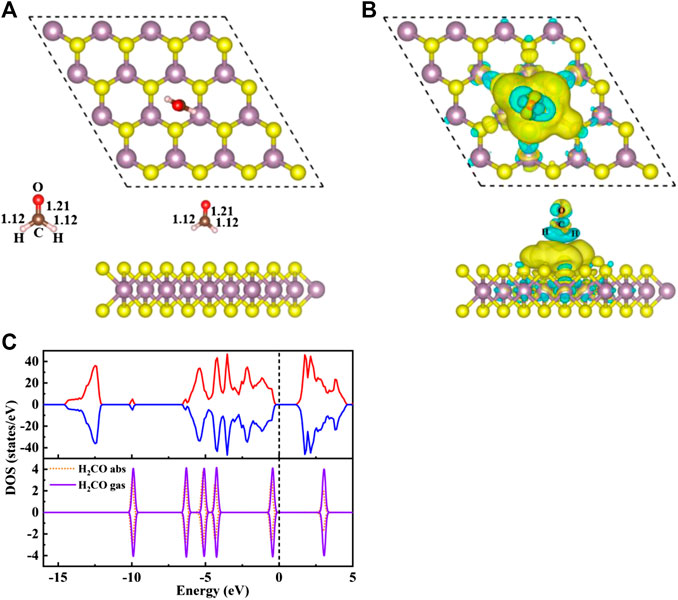
FIGURE 2. (A) The top view (top) and side view (bottom) structure of S/H2CO, and the left side shows the structure of H2CO in the gas phase. (B) Charge density difference diagram of S/H2CO. The yellow area indicates charge accumulation and the cyan area indicates charge loss. (C) The spin polarized TDOS of S/H2CO (upper panels) and the projected density of states (PDOS) of the original monolayer MoS2 for H2CO adsorption (lower panels): Orange dotted line, H2CO in the adsorption state; purple curve, H2CO in the gas phase. The positive and negative values of DOS indicate spin up and spin down, respectively. The Fermi level is marked with a black dotted line.
Seen from Figure 2A, the adsorption energy of H2CO on the original monolayer MoS2 is 0.04 eV. The adsorption of H2CO is almost perpendicular to the plane, and the H-end is downward. The nearest distance between the molecule and the substrate is 3.13 Å. Due to the small adsorption energy, the interaction between the adsorbed molecules and the substrate is weak, and the geometric structure of the adsorbed molecules is almost undisturbed. The C-O bond length is about 1.21 Å, and C-H bond length is about 1.12 Å, which is the same as that of H2CO bond in gas phase Figure 2A. The position of S atom under the adsorbed H2CO does not change, and the bond length between S and Mo is still 2.41 Å. This result can also be confirmed by Figure 2B where H2CO is almost surrounded by cyan, and only 0.01 e was obtained from the original monolayer MoS2. In order to further understand the interaction between the adsorbed H2CO and the original monolayer MoS2, we also calculated the TDOS of S/H2CO structure and the DOS of H2CO before and after adsorption, as shown in Figure 2C. After the adsorption of H2CO, we can see that there is no induced impurity state, and the band gap has no obvious change. Just because of the introduction of molecules, the single peak increases, indicating that the adsorption almost does not change the electrical properties of the original monolayer MoS2, It is well consistent with the literature report (Ma et al., 2016b). In other words, the original monolayer MoS2 is not sensitive to H2CO, which further proves that the adsorption of H2CO on the original monolayer MoS2 belongs to physical adsorption.
Adsorption of H2CO on Zn Doped Monolayer MoS2 (Zn/H2CO)
Finally, four stable configurations of H2CO adsorption structure on Zn-MoS2 were obtained, including Zn/H2CO-(a), Zn/H2CO-(b), Zn/H2CO-(c) and Zn/H2CO-(d). The structure parameters are shown in Figure 3, and the other related parameters are shown in Table 1.
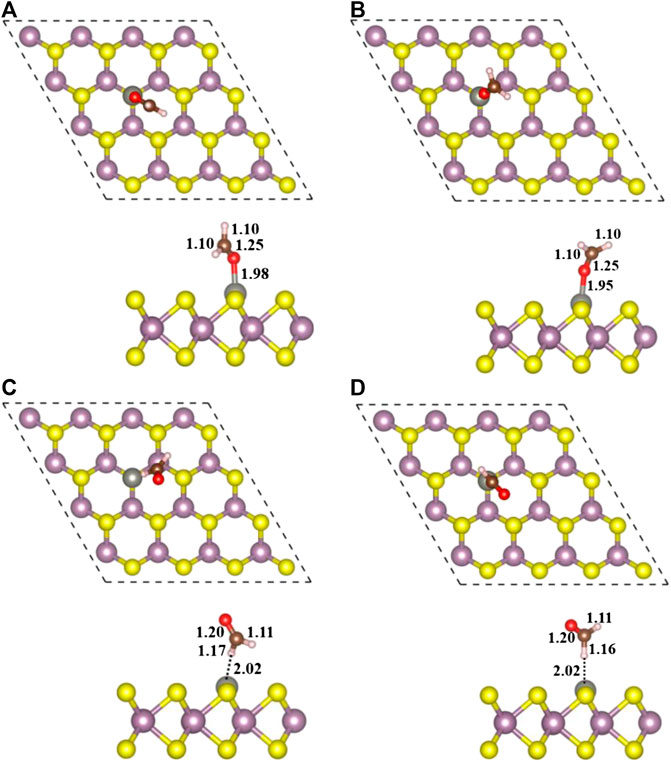
FIGURE 3. Top view (top) and side view (bottom) of geometric structures of Zn/H2CO-(A), Zn/H2CO-(B), Zn/H2CO-(C) and Zn/H2CO-(D) configurations are shown in (A–D), respectively.
Configuration of the Adsorbed Zn/H2CO-(a) and Zn/H2CO-(b)
As shown in Figure 3A,B, the adsorption energies of Zn/H2CO-(a) and Zn/H2CO-(b) are 0.98 and 0.80 eV, respectively. The distance between the O atoms of the adsorbed molecule and Zn dopant is 1.98 and 1.95 Å, respectively, indicating that the molecule has strong adsorbed ability. The interaction between the adsorbed H2CO and the substrate makes the geometry of H2CO change obviously, the C-O bond length of H2CO increased by 0.04 Å and the C-H bond length decreased by 0.02 Å. The average bond length of Zn-Mo on the substrate Zn-MoS2 increases from 2.41 to 2.70 Å, and the distance between the dopant Zn and the S-plane is 0.33 and 0.28 Å larger than that of the substrate Zn-MoS2, respectively. In addition, Bader charge analysis showed that H2CO was the electron acceptor of Zn/H2CO-(a) and Zn/H2CO-(b), and the electron transfer between Zn-MoS2 substrate and H2CO was 0.05 and 0.09 e, respectively, this is mainly due to the doping of Zn atoms (0.51 and 0.53 e), it may be due to the difference of electronegativity between O (3.44) and Zn (1.65).
The difference of charge density of the final configuration of Zn/H2CO-(a) and Zn/H2CO-(b) is shown in Figures 4A,B to further understand the interaction between H2CO molecule and Zn doped monolayer MoS2. The yellow area is the electron accumulation area, and the cyan area is the electron consumption area. As shown in Figure 4A, the electron transfer is not only located on the C and O atoms of H2CO adsorption, but also on the O-Zn bond, which is consistent with the strong adsorption capacity of H2CO. In addition, the loss of electrons on the C-O bond leads to the weakening of the C-O bond, which makes the O atom protruding above the S plane chemically active to other molecules, including H2CO itself. For the adsorption of Zn/H2CO-(b), it can be found that the adsorption behavior is similar to that of Zn/H2CO-(a), as shown in Figure 4B, which will not be further discussed.
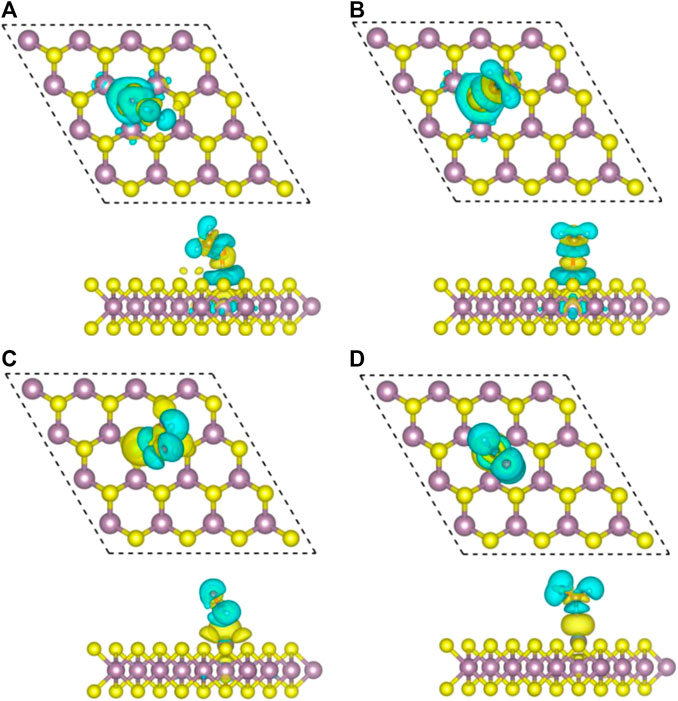
FIGURE 4. Top view (top) and side view (bottom) of charge density differences of final configurations of Zn/H2CO-(A), Zn/H2CO-(B), Zn/H2CO-(C) and Zn/H2CO-(D) are shown in (A–D), respectively. Yellow and cyan represent regions of electron accumulation and depletion, respectively.
In order to further understand the adsorption behavior of H2CO on Zn-MoS2 surface. Figures 5A–D displayed the spin-polarized total densities of states (TDOS) (upper panels) and corresponding DOS projected on 3d states of Zn atom, adsorbed H2CO gas molecules and the isolated H2CO gas molecules (lower panels) after H2CO adsorption on Zn-embedded monolayer MoS2.
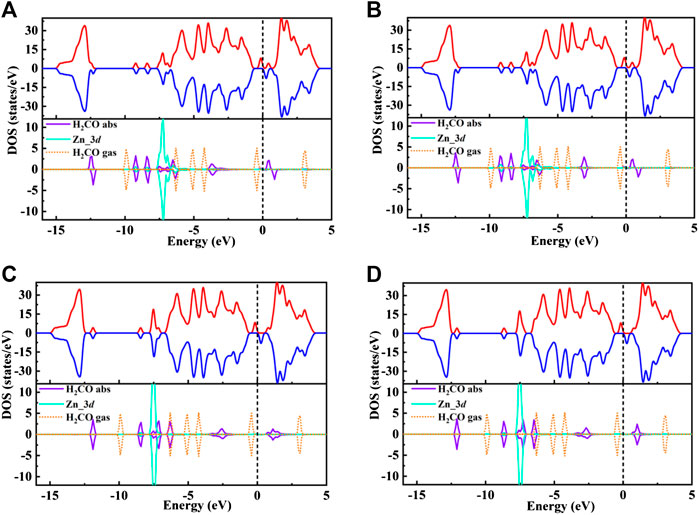
FIGURE 5. (A–D) shows the total spin polarized density of states (TDOS) of Zn/H2CO-(A), Zn/H2CO-(B), Zn/H2CO-(C) and Zn/H2CO-(D) systems (upper panels), and the projected density of states (PDOS) of H2CO adsorbed by Zn-MoS2(lower panels): Purple curve, H2CO in adsorption state; orange dotted line, H2CO in gas phase; blue curve, d-projected PDOS for Zn atom. The positive and negative values of DOS indicate spin up and spin down, respectively. The Fermi level is marked with a black dotted line.
The TDOS of Zn/H2CO-(a) and Zn/H2CO-(b) systems are shown in Figures 5A,B. Compared with Zn-MoS2, due to the hybridization of Zn atoms and H2CO molecules, the charge is transferred from the matrix to the adsorbed H2CO molecule, resulting in a new DOS peak at CBM for the TDOS of Zn/H2CO-(a) and Zn/H2CO-(b) systems. It can be observed that the occupied state PDOS of the adsorbed H2CO molecule Figures 5A,B, below is much lower panels than Fermi level, and the induced impurity state is produced, which reveals the reaction between Zn-MoS2 monolayer and H2CO molecule. For Zn/H2CO-(a) and Zn/H2CO-(b) configurations, the molecular orbitals of adsorbed H2CO are delocalized relative to the isolated H2CO in the gas phase. The 3d orbital of Zn atom is coupled with H2CO in the range of 9.60∼0.00 eV. The interaction between H2CO molecule and Zn atom leads to charge transfer. Zn/H2CO-(a) and Zn/H2CO-(b) structures have higher adsorption energy, which indicates that the adsorption is chemisorption, which is suitable for gas removal under H2CO adsorption. The results show that, compared with undoped MoS2, doping Zn atoms in the defective monolayer MoS2 is beneficial to the adsorption of H2CO. We use crystal orbital Hamiltonian populations (COHP) to analyze the interaction between different O-Zn/H-Zn bonds by means of a pair of atomic orbital bonding or antibonding states (van Santen and Tranca, 2016; Steinberg and Dronskowski, 2018; Tsuji and Yoshizawa, 2018). Moreover, through the integration of COHP (ICOHP), we can get a precise value of bond strength. The positive and negative values of ICOHP represent the anti bonding state and bonding state respectively. The smaller the ICOHP value, the stronger the bond strength. Figures 6A-D shows the -COHP curve and the integral (ICOHP) value of the O-Zn and H-Zn bonds in the structured Zn/H2CO-(a) and Zn/H2CO-(b) systems and the Zn/H2CO-(c) and Zn/H2CO-(d) systems, respectively. It can be seen from Figures 6A,B that when H2CO is adsorbed to Zn-MoS2, there are a small amount of O-Zn bonding orbitals (-COHP values) at Fermi level, and the orbitals above Fermi level belong to anti bonding orbitals. In other words, the O-Zn bond is enhanced after H2CO adsorption, which is consistent with the increase of C-O distance (Figures 4A,B). The O atoms in the two adsorption systems have strong binding with Zn atoms (ICOHP value is small), which also shows that these phases are stable.
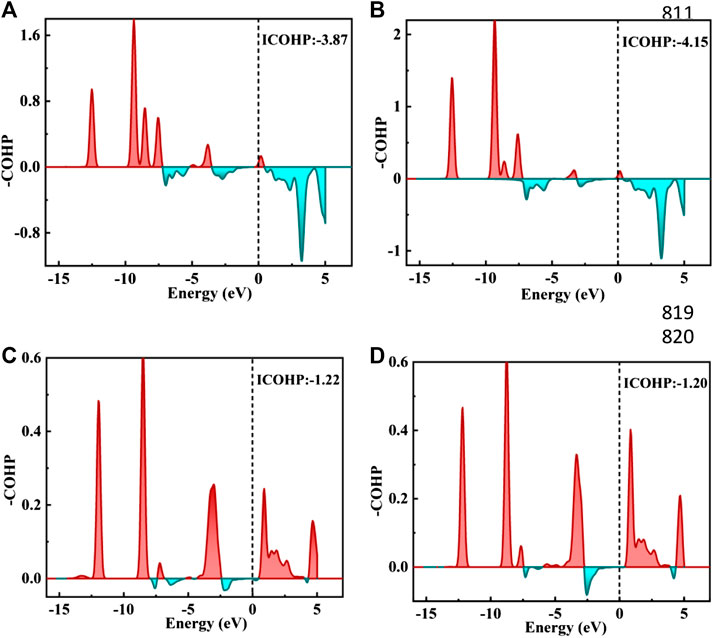
FIGURE 6. (A–D) figure shows the negative crystal orbital Hamilton density (-COHP) on the Zn/H2CO-(A) and Zn/H2CO-(B) system O-Zn bond and the Zn/H2CO-(C) and Zn/H2CO-(D) system H-Zn bond, respectively. The red filled area is the O-Zn/H-Zn bonding area, and the green filled area is the O-Zn/H-Zn antibonding. Fermi energy level marked with black dotted line.
Configuration Adsorption of Zn/H2CO-(c) and Zn/H2CO-(d)
As shown in Figures 3C,D the Zn/H2CO-(c) and Zn/H2CO-(d) configurations with adsorption energies of 0.15 and 0.11 eV, respectively, are much less stable than those of Zn/H2CO-(a) and Zn/H2CO-(b), and the H atoms point to the doped Zn atoms. Different from the configurations of Zn/H2CO-(a) and Zn/H2CO-(b), the adsorption energies are also very different, similar to the adsorption on the original monolayer MoS2. Therefore, the orientation of H atom or O atom in H2CO can be used to determine the adsorption energy. At the same time, in Figures 3C,D the interaction between the adsorbed molecules and the substrate is also different due to the different orientations of hydrogen atoms, i.e., the angle of C-H-Zn is different. In the two structures, the distance between H and Zn atoms is 2.02 Å.
Therefore, the strong covalent bond or ionic bond between the two atoms can be eliminated. The weak electrostatic attraction of negatively charged H atom (0.06 e) and positively charged Zn atom (0.33 e) should be combined, which is consistent with the weak adsorption of Zn/H2CO-(c) and Zn/H2CO-(d) configurations.
Compared with H2CO in the gas phase, the geometry and substrate of the adsorbed state are only slightly changed. The C-O bond of H2CO decreased by 0.01 Å. In addition, for Zn/H2CO-(c) and Zn/H2CO-(d), there is a small electron transfer (0.07 and 0.08 e) between the adsorbed H2CO and the substrate, and the molecule has a small magnetic moment of 0.01 µB. The adsorption of Zn/H2CO-(c) and Zn/H2CO-(d) is weaker than that of Zn/H2CO-(a) and Zn/H2CO-(b), indicating that the probability of occurrence of the latter two configurations is much higher than that of the former two. For the adsorption of Zn/H2CO-(c) and Zn/H2CO-(d) configurations, the charge density difference diagram shows that there is almost no electron accumulation between the adsorbed molecule H2CO and the Zn doped MoS2 monolayer (Figures 4C,D). It is further confirmed that the adsorption capacity of H2CO on the substrate is weak, which is consistent with the small adsorption energy and large distance mentioned above.
Further analyzing the adsorption behavior of H2CO on the Zn-MoS2 surface, it can be seen from Figures 5C,D above that the TDOS of the Zn/H2CO-(c) and Zn/H2CO-(d) systems do not show induced impurity state near the Fermi level, and the DOS curves of the adsorbed H2CO of the Zn/H2CO-(c) and Zn/H2CO-(d) systems overlap slightly with the 3d orbitals of the Zn atoms (Figures 5C,D, below), shows that the interaction between H2CO and Zn-MoS2 is weak, the bonding between H-Zn atoms is weak, and the ICOHP negative value is large (Figures 6C,D), resulting in only 0.07 and 0.08 e being transferred from the adsorbed H2CO molecule to the final substance. The H-Zn strength in Zn/H2CO-(c) system is higher than that in Zn/H2CO-(d) system, indicating that Zn/H2CO-(c) system is more stable than Zn/H2CO-(d) system. This conclusion is consistent with the previous calculation of adsorption energy (Table 1). In terms of magnetic properties, the total magnetic moment of the whole adsorption system did not change after adsorption of H2CO by Zn-MoS2 (Zn/H2CO-(c) and Zn/H2CO-(d) were 2.00 µB), which was consistent with the small adsorption energy of Zn/H2CO-(c) and Zn/H2CO-(d).
Conclusion
In conclusion, according to the first principles calculations, we have studied the effects of Zn doping S vacancy on the electronic structure, magnetic properties and chemical activity of monolayer MoS2. The calculation of binding energy shows that Zn atoms are closely bound to S-defects, which is mainly due to the hybridization between dopant atoms and their nearest Mo atoms. Finally, a new single cluster catalyst on ZnMo3 surface is formed, which improves the selectivity of MoS2 surface. By embedding Zn atoms, the magnetic properties of MoS2 monolayer can be adjusted and the spin magnetic moment of Zn-MoS2 is 2.00 µB. The electronic properties of MoS2 are also changed by the impurity states induced in the band gap. The effects of H2CO adsorption and doping on the chemical activity of MoS2 monolayers were further investigated. It was found that the H-end downward adsorption of H2CO in the original monolayer MoS2 and Zn doped monolayer MoS2 was very weak, and the electronic structure of the two substrates changed little after adsorption, indicating that the two systems were not sensitive to H2CO.When the O atom in H2CO molecule faces to the substrate, the adsorption capacity is strong, and the adsorbed H2CO is effectively activated. DOS analysis showed that the electronic structure of Zn-MoS2 could be changed by introducing impurities in the band gap when the O-terminal of H2CO molecule was adsorbed downward. At the same time, the magnetic properties of Zn-MoS2 system are also adjusted. The COHP diagram shows that Zn atoms are strongly bonded with O atoms. This study shows that Zn doping is a promising method to optimize the electronic structure, magnetic properties and chemical activity of MoS2, which provides a promising way to improve the electronic properties of MoS2 materials.
Author Contributions
CH: conceptualization, methodology, funding acquisition, supervision, project administration. GZ: formal analysis. JH: software. TX: software, methodology. HL: writing—original draft, investigation, formal analysis, methodology. LF: supervision, funding acquisition, project administration. GD: formal analysis. HY: software.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Funding
This study was funded by the Natural Science Foundation of China (Nos. 21603109 and 21876104), the Henan Joint Fund of the National Natural Science Foundation of China (No. U1404216), Transformation of Scientific and Technological Achievements Programs of Higher Education Institutions in Shanxi (No. 2020CG032), Cultivation Plan of Young Scientific Researchers in Higher Education Institutions of Shanxi Province.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.605311/full#supplementary-material.
References
Abbas, H. G., Debela, T. T., Hussain, S., and Hussain, I. (2018). Inorganic molecule (O2, NO) adsorption on nitrogen- and phosphorus-doped MoS2 monolayer using first principle calculations. RSC Adv. 8 (67), 38656–38666. doi:10.1039/c8ra07638c
Abbasi, A. (2019). Adsorption of phenol, hydrazine and thiophene on stanene monolayers: a computational investigation. Synth. Met. 247, 26–36. doi:10.1016/j.synthmet.2018.11.012
Abbasi, A., and Sardroodi, J. J. (2018a). Exploration of sensing of nitrogen dioxide and ozone molecules using novel TiO2/Stanene heterostructures employing DFT calculations. Appl. Surf. Sci. 442, 368–381. doi:10.1016/j.apsusc.2018.02.183
Abbasi, A., and Sardroodi, J. J. (2018b). Investigation of the adsorption of ozone molecules on TiO2/WSe2 nanocomposites by DFT computations: applications to gas sensor devices. Appl. Surf. Sci. 436, 27–41. doi:10.1016/j.apsusc.2017.12.010
Abbasi, A., and Sardroodi, J. J. (2019). The adsorption of sulfur trioxide and ozone molecules on stanene nanosheets investigated by DFT: applications to gas sensor devices. Phys. E Low-Dimens. Syst. Nanostruct. 108, 382–390. doi:10.1016/j.physe.2018.05.004
Allred, A. L. (1961). Electronegativity values from thermochemical data. J. Inorg. Nucl. Chem. 17 (3-4), 215–221. doi:10.1016/0022-1902-5
Ataca, C., and Ciraci, S. (2011). Functionalization of single-layer MoS2 honeycomb structures. J. Phys. Chem. C 115 (27), 13303–13311. doi:10.1021/jp2000442
Chen, D., Tang, J., Zhang, X., Li, Y., and Liu, H. (2019). Detecting decompositions of sulfur hexafluoride using MoS2 monolayer as gas sensor. IEEE Sensor. J. 19 (1), 39–46. doi:10.1109/jsen.2018.2876637
Chung, P. R., Tzeng, C. T., Ke, M. T., and Lee, C. Y. (2013). Formaldehyde gas sensors: a review. Sensors. 13 (4), 4468–4484. doi:10.3390/s130404468
Cui, H., Zhang, X., Zhang, G., and Tang, J. (2019). Pd-doped MoS2 monolayer: a promising candidate for DGA in transformer oil based on DFT method. Appl. Surf. Sci. 470, 1035–1042. doi:10.1016/j.apsusc.2018.11.230
Dimple, , Jena, N., and De Sarkar, A. (2017). Compressive strain induced enhancement in thermoelectric-power-factor in monolayer MoS2 nanosheet. J. Phys. Condens. Matter. 29 (22), 225501. doi:10.1088/1361-648X/aa6cbc
Guo, J., Tadesse Tsega, T., Ul Islam, I., Iqbal, A., Zai, J., and Qian, X. (2020). Fe doping promoted electrocatalytic N2 reduction reaction of 2H MoS2. Chin. Chem. Lett. 31(09): 2487–2490. doi:10.1016/j.cclet.2020.02.019
Guo, T., Wang, L., Sun, S., Wang, Y., Chen, X., Zhang, K., et al. (2019). Layered MoS2@graphene functionalized with nitrogen-doped graphene quantum dots as an enhanced electrochemical hydrogen evolution catalyst. Chin. Chem. Lett. 30 (6), 1253–1260. doi:10.1016/j.cclet.2019.02.009
Henkelman, G., Arnaldsson, A., and Jónsson, H. (2006). A fast and robust algorithm for Bader decomposition of charge density. Comput. Mater. Sci. 36 (3), 354–360. doi:10.1016/j.commatsci.2005.04.010
Kathiravan, D., Huang, B.-R., Saravanan, A., Prasannan, A., and Hong, P.-D. (2019). Highly enhanced hydrogen sensing properties of sericin-induced exfoliated MoS2 nanosheets at room temperature. Sensor. Actuator. B Chem. 279, 138–147. doi:10.1016/j.snb.2018.09.104
Kwak, I. H., Kwon, I. S., Abbas, H. G., Jung, G., Lee, Y., Park, J., et al. (2018). Stable methylammonium-intercalated 1T′-MoS2 for efficient electrocatalytic hydrogen evolution. J. Mater. Chem. 6 (14), 5613–5617. doi:10.1039/c8ta00700d
Kwon, I. S., Kwak, I. H., Abbas, H. G., Jung, G., Lee, Y., Park, J., et al. (2018). Intercalation of aromatic amine for the 2H-1T' phase transition of MoS2 by experiments and calculations. Nanoscale 10 (24), 11349–11356. doi:10.1039/c8nr02365d
Lara-Ibeas, I., Megías-Sayago, C., Louis, B., and Le Calvé, S. (2020). Adsorptive removal of gaseous formaldehyde at realistic concentrations. J. Environ. Chem. Eng. 8 (4), 103986. doi:10.1016/j.jece.2020.1
Le, D., Rawal, T. B., and Rahman, T. S. (2014). Single-layer MoS2 with sulfur vacancies: structure and catalytic application. J. Phys. Chem. C 118 (10), 5346–5351. doi:10.1021/jp411256g
Lolla, S., and Luo, X. (2020). Tuning the catalytic properties of monolayer MoS2 through doping and sulfur vacancies. Appl. Surf. Sci. 507, 144892. doi:10.1016/j.apsusc.2019.144892
Luo, H., Cao, Y., Zhou, J., Feng, J., Cao, J., and Guo, H. (2016). Adsorption of NO2, NH3 on monolayer MoS2 doped with Al, Si, and P: a first-principles study. Chem. Phys. Lett. 643, 27–33. doi:10.1016/j.cplett.2015.10.077
Ma, D., Ju, W., Li, T., Yang, G., He, C., Ma, B., et al. (2016a). Formaldehyde molecule adsorption on the doped monolayer MoS2: a first-principles study. Appl. Surf. Sci. 371, 180–188. doi:10.1016/j.apsusc.2016.02.230
Ma, D., Ju, W., Li, T., Zhang, X., He, C., Ma, B., et al. (2016b). The adsorption of CO and NO on the MoS2 monolayer doped with Au, Pt, Pd, or Ni: a first-principles study. Appl. Surf. Sci. 383, 98–105. doi:10.1016/j.apsusc.2016.04.171
Ma, D., Wang, Q., Li, T., He, C., Ma, B., Tang, Y., et al. (2016c). Repairing sulfur vacancies in the MoS2 monolayer by using CO, NO and NO2 molecules. J. Mater. Chem. C 4 (29), 7093–7101. doi:10.1039/c6tc01746k
Ma, X. L., Liu, J. C., Xiao, H., and Li, J. (2018). Surface single-cluster catalyst for N2-to-NH3 thermal conversion. J. Am. Chem. Soc. 140 (1), 46–49. doi:10.1021/jacs.7b10354
Maintz, S., Deringer, V. L., Tchougreeff, A. L., and Dronskowski, R. (2016). LOBSTER: a tool to extract chemical bonding from plane-wave based DFT. J. Comput. Chem. 37 (11), 1030–1035. doi:10.1002/jcc.24300
Momma, K., and Izumi, F. (2011). VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44 (6), 1272–1276. doi:10.1107/s0021889811038970
Monkhorst, H. J., and Pack, J. D. (1976). Special points for Brillouin-zone integrations. Phys. Rev. B 13 (12), 5188–5192. doi:10.1103/PhysRevB.13.5188
Pan, X., Ji, J., Zhang, N., and Xing, M. (2020). Research progress of graphene-based nanomaterials for the environmental remediation. Chin. Chem. Lett. 31 (6), 1462–1473. doi:10.1016/j.cclet.2019.10.002
Qian, G., Peng, Q., Zou, D., Wang, S., Yan, B., and Zhou, Q. (2020). First-principles insight into Au-doped MoS2 for sensing C2H6 and C2H4. Front. Mater. 7. doi:10.3389/fmats.2020.00022
Qian, H., Lu, W., Wei, X., Chen, W., and Deng, J. (2019). H2S and SO2 adsorption onPt-MoS2 adsorbent for partial discharge elimination: a DFT study. Results Phys. 12, 107–112. doi:10.1016/j.rinp.2018.11.035
Ren, J., Liu, H., Xue, Y., and Wang, L. (2019). Adsorption behavior of CH4 gas molecule on the MoX2(S, Se, Te) monolayer: the DFT study. Nanoscale Res. Lett. 14 (1), 293. doi:10.1186/s11671-019-3125-5
Sahoo, M. P. K., Wang, J., Zhang, Y., Shimada, T., and Kitamura, T. (2016). Modulation of gas adsorption and magnetic properties of monolayer-MoS2 by antisite defect and strain. J. Phys. Chem. C 120 (26), 14113–14121. doi:10.1021/acs.jpcc.6b03284
Salthammer, T. (2013). Formaldehyde in the ambient atmosphere: from an indoor pollutant to an outdoor pollutant? Angew. Chem. Int. Ed. 52 (12), 3320–3327. doi:10.1002/anie.201205984
Sharma, A., Anu, , Khan, M. S., Husain, M., Khan, M. S., and Srivastava, A. (2018). Sensing of CO and NO on Cu-doped MoS2 monolayer-based single electron transistor: a first principles study. IEEE Sensor. J. 18 (7), 2853–2860. doi:10.1109/jsen.2018.2801865
Song, X., Dong, M., Li, Y., Wu, Y., Sun, Y., Yuan, G., et al. (2020). Formaldehyde adsorption effects of chlorine adatoms on lithium-decorated graphene: a DFT study. Chem. Phys. Lett. 761, 138085. doi:10.1016/j.cplett.2020.1380
Steinberg, S., and Dronskowski, R. (2018). The crystal orbital Hamilton population (COHP) method as a tool to visualize and analyze chemical bonding in intermetallic compounds. Crystals. 8 (5), 225. doi:10.3390/cryst8050225
Tan, J., Hu, J., Ren, J., Peng, J., Liu, C., Song, Y., et al. (2020). Fast response speed of mechanically exfoliated MoS2 modified by PbS in detecting NO2. Chin. Chem. Lett. 31 (8), 2103–2108. doi:10.1016/j.cclet.2020.03.060
Tsuji, Y., and Yoshizawa, K. (2018). Adsorption and activation of methane on the (110) surface of rutile-type metal dioxides. J. Phys. Chem. C 122 (27), 15359–15381. doi:10.1021/acs.jpcc.8b03184
van Santen, R. A., and Tranca, I. (2016). How molecular is the chemisorptive bond? Phys. Chem. Chem. Phys. 18 (31), 20868–20894. doi:10.1039/c6cp01394e
Wang, J., Zhou, Q., Lu, Z., Wei, Z., and Zeng, W. (2019a). Gas sensing performances and mechanism at atomic level of Au-MoS2 microspheres. Appl. Surf. Sci. 490, 124–136. doi:10.1016/j.apsusc.2019.06.075
Wang, J., Zhou, Q., and Zeng, W. (2019b). Competitive adsorption of SF6 decompositions on Ni-doped ZnO (100) surface: computational and experimental study. Appl. Surf. Sci. 479, 185–197. doi:10.1016/j.apsusc.2019.01.255
Wang, T., Gao, D., Zhuo, J., Zhu, Z., Papakonstantinou, P., Li, Y., et al. (2013). Size-dependent enhancement of electrocatalytic oxygen-reduction and hydrogen-evolution performance of MoS2 particles. Chemistry 19 (36), 11939–11948. doi:10.1002/chem.201301406
Wanno, B., and Tabtimsai, C. (2014). A DFT investigation of CO adsorption on VIIIB transition metal- doped graphene sheets. Superlattice. Microst. 67, 110–117. doi:10.1016/j.spmi.2013.12.025
Wu, D., Lou, Z., Wang, Y., Yao, Z., Xu, T., Shi, Z., et al. (2018). Photovoltaic high-performance broadband photodetector based on MoS2/Si nanowire array heterojunction. Sol. Energy Mater. Sol. Cell. 182, 272–280. doi:10.1016/j.solmat.2018.03.017
Xu, M., Liang, T., Shi, M., and Chen, H. (2013). Graphene-like two-dimensional materials. Chem. Rev. 113 (5), 3766–3798. doi:10.1021/cr300263a
Yu, L., El-Damak, D., Radhakrishna, U., Ling, X., Zubair, A., Lin, Y., et al. (2016). Design, modeling, and fabrication of chemical vapor deposition grown MoS2 circuits with E-mode FETs for large-area electronics. Nano Lett. 16 (10), 6349–6356. doi:10.1021/acs.nanolett.6b02739
Zhang, D., Jiang, C., and Wu, J. (2018). Layer-by-layer assembled In2O3 nanocubes/flower-like MoS2 nanofilm for room temperature formaldehyde sensing. Sensor. Actuator. B Chem. 273, 176–184. doi:10.1016/j.snb
Zhang, D., Sun, Y. E., Jiang, C., and Zhang, Y. (2017). Room temperature hydrogen gas sensor based on palladium decorated tin oxide/molybdenum disulfide ternary hybrid via hydrothermal route. Sensor. Actuator. B Chem. 242, 15–24. doi:10.1016/j.snb.2016.11.005
Zhang, Y., Zeng, W., and Li, Y. (2019). Porous MoS2 microspheres decorated with Cu2O nanoparticles for ammonia sensing property. Mater. Lett. 241, 223–226. doi:10.1016/j.matlet.2019.01.130
Zheng, J., Song, D., Chen, H., Xu, J., Alharbi, N. S., Hayat, T., et al. (2020). Enhanced peroxidase-like activity of hierarchical MoS2-decorated N-doped carbon nanotubes with synergetic effect for colorimetric detection of H2O2 and ascorbic acid. Chin. Chem. Lett. 31 (5), 1109–1113. doi:10.1016/j.cclet.2019.09.037
Zhou, J., Liu, G., Jiang, Q., Zhao, W., Ao, Z., and An, T. (2020). Density functional theory calculations on single atomic catalysis: Ti-decorated Ti3C2O2 monolayer (MXene) for HCHO oxidation. Chin. J. Catal. 41 (10), 1633–1644. doi:10.1016/s1872-2067(20)63571-9
Keywords: first-principles calculation, monolayer MoS2, H2CO, adsorption energy, gas sensitivity
Citation: Li H, Fu L, He C, Huo J, Yang H, Xie T, Zhao G and Dong G (2021) Formaldehyde Molecules Adsorption on Zn Doped Monolayer MoS2: A First-Principles Calculation. Front. Chem. 8:605311. doi: 10.3389/fchem.2020.605311
Received: 11 September 2020; Accepted: 17 December 2020;
Published: 16 April 2021.
Edited by:
Jinfeng Dong, Wuhan University, ChinaCopyright © 2021 Li, Fu, He, Huo, Yang, Xie, Zhao and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaozheng He, aGVjejIwMTlAeGF0dS5lZHUuY24=; Guozheng Zhao, emhhb2d1b3poZW5nQHN4bnUuZWR1LmNu; Guohui Dong, ZG9uZ2d1b2h1aUBzdXN0LmVkdS5jbg==
†These authors have contributed equally to this work