 Fa-Jie Chen
Fa-Jie Chen Zhenguo Hua1†
Zhenguo Hua1† Jianhui Chen
Jianhui Chen Yuanzhi Xia
Yuanzhi Xia- 1College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, China
- 2Department of Chemistry, University of Illinois at Chicago, Chicago, IL, United States
Herein, we report an efficient method for the synthesis of (Z)-β-halovinyl ketones through a one-pot Sonogashira coupling and hydrohalogenation reaction promoted by palladium-copper catalyst and Brønsted acid. The ynone intermediates are generated in situ from readily available acid chlorides and terminal alkynes at room temperature, which are directly converted to (Z)-β-halovinyl ketones by treating with triflic acid. This method avoids the use of an external halogen source and features broad substrate scope, high yield, and good to excellent stereoselectivity.
Introduction
The palladium and copper-catalyzed cross-coupling reaction of terminal alkynes with aryl or vinyl halides, also known as Sonogashira reaction, has emerged as a powerful method for the synthesis of substituted alkynes in recent decades (Sonogashira et al., 1975; Sonogashira, 2002). The scope of this reaction has been extended to acid halides, which reacts with terminal alkynes smoothly at room temperature to provide ynones in good yield (Tohda et al., 1977; Eckhardt and Fu, 2003). The electron-deficient ynones are good Michael acceptors and react with various nucleophiles to form vinyl ketones (such as enaminone and β-ketoenolether) and heterocycles (such as pyrimidine) (Karpov and Müller, 2003a,b). Ynones are also used as useful precursors for the synthesis of β-halovinyl ketones (Pohland and Benson, 1966; Goossen et al., 2009).
β-Halovinyl ketones are important building blocks in organic synthesis and biochemical processes, and they have been widely used for the constructions of heterocycles such as chromenones, furans, pyrazole, and pyridines (Kim et al., 2012, 2017; Kim and Oh, 2014, 2015, 2017, 2020; Koo et al., 2019a). The methods to form β-halovinyl ketones from ynones generally involve regioselective hydrohalogenation of electron-deficient carbon-carbon triple bond. In these cases, a variety of halogen sources have been employed, including HCl, SnCl4, AlBr3, TMSCl, and LiBr (Scheme 1A) (Kundu and Chaudhuri, 1991; Shchukin and Vasilyev, 2008; Yang et al., 2011; Semenova et al., 2013; Yan et al., 2015; Zeng et al., 2017; Zhang et al., 2017). However, those methods suffer from relatively low stereoselectivity, which limits their synthetic applications. Recently, Xu and co-workers reported an atom-economical method for the regio- and stereoselective hydrohalogenation of ynones and ynamides using DMPU/HX (X = Br or Cl) reagents. Paixão and co-workers reported the regioselective synthesis of (Z)-β-halo α,β-unsaturated carbonyl systems via combination of halotrimethylsilane and tetrafluoroboric acid (da Silva et al., 2019).
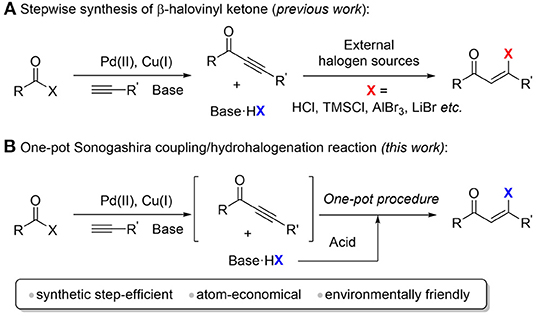
Scheme 1. Synthesis of β-halovinyl ketones. (A) Stepwise synthesis of β-halovinyl ketone (previous work). (B) One-pot sonogashira coupling/hydrohalogenation reaction (this work).
Ynones used in these hydrohalogenation methods are usually not commercially available. One of the common ways to prepare the precursor ynones is the Sonogashira cross-coupling reaction as mentioned above (Kokubo et al., 1996; Hua et al., 2005; Kashiwabara et al., 2005, 2008; Iwai et al., 2009, 2012; Kashiwabara and Tanaka, 2011). This hydrohalogenation strategy for β-halovinyl ketone synthesis requires a multi-step synthetic procedure (Scheme 1A). Moreover, in the starting material preparation step, at least one equivalent of chemical waste hydrohalide salt (for example, triethylamine hydrochloride) will be generated, which is commonly removed in the workup procedure. From the viewpoint of atom-economy and green chemistry, we envisioned that the chemical waste hydrohalide salt can be reused as the halogen source for the sequential hydrohalogenation to form β-halovinyl ketones by the one-pot treatment of strong Brønsted acid, thus avoiding the use of an external halogen source. Compared to the stepwise methods, this strategy is more step- and atom-economical and environment friendly.
Similar strategies have been developing relying on transition-metal (rhodium and iridium catalyst) (Kokubo et al., 1996; Hua et al., 2005; Kashiwabara et al., 2005, 2008; Goossen et al., 2009; Iwai et al., 2009, 2012; Kashiwabara and Tanaka, 2011), or Lewis acid (Price and Pappalardo, 1950; Benson and Pohland, 1964; Martens et al., 1975; Zhou et al., 2006; Wang et al., 2010; Hosseini-Sarvari and Mardaneh, 2011; Gandeepan et al., 2012; Koo et al., 2019b). However, these developed methods usually require relatively harsh reaction conditions, such as elevated temperature, long reaction time, use of expensive ligands or catalysts. To the best of our knowledge, the efficient synthesis of β-halovinyl ketones at room temperature through the cascade of the Sonogashira coupling and hydrohalogenation has not been reported.
Herein, we developed a one-pot synthesis of (Z)-β-halovinyl ketone via a Sonogashira coupling and hydrohalogenation sequence at room temperature (Scheme 1B). The ynone intermediates are generated in situ from the palladium and copper catalyzed cross coupling of acid halides and terminal alkynes, which are hydrohalogenated to afford (Z)-β-halovinyl ketone using side product hydrohalide salt as a halogen source. This method shows good stereoselectivity, high yield, and broad substrate scope.
Results and Discussion
We commenced our study with the synthesis of β-chlorovinyl ketone 3a through a cross-coupling reaction of phenylacetylene 1 with benzoyl chloride 2 under the catalysis of PdCl2(PPh3)3 and CuI followed by Brønsted acid treatment (Table 1). After systematic optimization of the reaction conditions, we found that the best reaction conditions: 1.0 equivalent of phenylacetylene reacts with 1.3 equivalent of benzoyl chloride in the presence of 2 mol% of PdCl2(PPh3)2, 4 mol% of CuI, and 1.2 equivalent of triethylamine in 1,2-dichloroethane (0.4 M) at room temperature for 10 min, then treat the reaction mixture with 1.5 equivalent of triflic acid for 4 h at room temperature. The stereoselectivity for this transformation is up to 91/9 (Z/E) and (Z)-β-chlorovinyl ketone 3a was obtained in 87% yield (Table 1, entry 14). When triflic acid was replaced with weaker Brønsted acids, such as trifluoroacetic acid (entry 4), acetic acid (entry 5), and benzoic acid (entry 6), chlorovinyl ketone 3a was not generated. This result indicates the crucial role of triflic acid to react with triethylamine hydrochloride to release hydrochloride acid, which acts as the chloride source for the following hydrochlorination step. Other solvents such as ethanol (entry 9) and acetonitrile (entry 10) are not effective for this transformation. Extra co-solvent for the hydrohalogenation step is not necessary. Shorten the reaction time of Sonogashira coupling step to 2 min results in a decrement of the yield (65%) and stereoselectivity (86/14, Z/E).
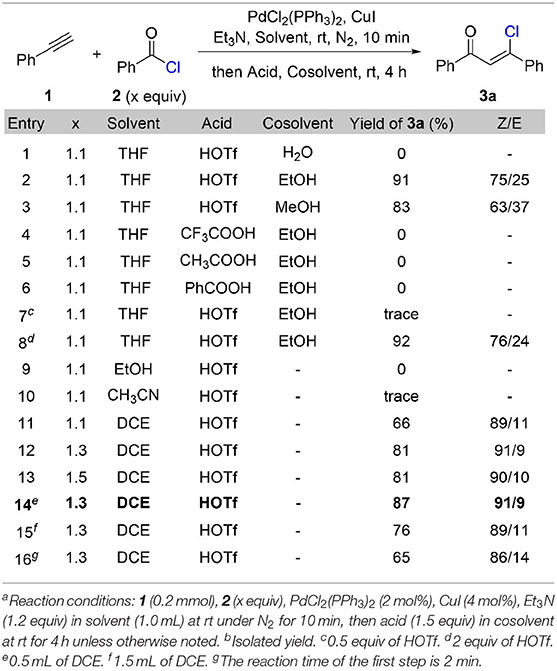
Table 1. Optimization of reaction conditionsa,b.
With the optimized reaction conditions in hand, we started to explore the substrate scope of this protocol. As showed in Table 2, a wide range of terminal alkynes were suitable in this transformation. Both electron-withdrawing groups, such as Br (3e), Cl (3b, 3c), F (3d) and electron-donating groups, such as Me (3f), OMe (3g) on the aromatic rings are well-tolerated, giving the (Z)-β-chlorovinyl ketone in good to high yield as well as good stereoselectivity. An alkyne bearing a thiophene moiety is also a good coupling partner, giving β-heterocycle-substituted unsaturated ketone 3i in 75% yield with a 92/8 ratio of Z/E isomers. The Z/E ratio of the product can be further increased up to 99/1 when sterically hindered alkynes were used (3l). The low yields were obtained for hydroxyl group-containing products (3h and 3j). It is likely because the hydroxyl group is acid sensitive. Replacement with a less sterically hindered linear 1-hexyne results in a dramatic decrease of the stereoselectivity of the ketone 3k to 41/59 (Z/E), indicating a bulky substituent at the alkyne moiety is important for high stereoselectivity. Trimethylsilyl-containing chlorovinyl ketone 3m was obtained in 63% yield and 89/11 (Z/E) stereoselectivity.
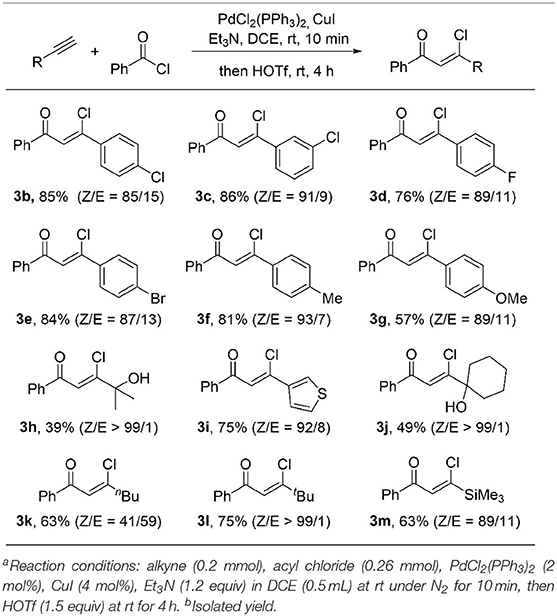
Table 2. Substrate scope of alkynesa,b.
Encouraged by the broad substrate scope of alkynes, we further examined the generality of the acyl chloride counterpart under standard reaction conditions (Table 3). A variety of benzoic acid chlorides substituted with either electron-donating groups (such as Me, OMe) or electron-withdrawing groups (such as F, Cl, Br) react with phenylacetylene and 1-hexyne smoothly, giving β-chlorovinyl ketone in good to excellent yield and high stereoselectivity. Inclusion of a strong electron-withdrawing CN group results 4l in only 27% yield, while the 4m could be formed in 60% yield when this was replaced by CF3. The Z/E ratio of the product is up to 99/1 in most cases. Ketone 4n was obtained in 82% yield from the reaction of aliphatic acid chloride. Thiophene containing product 4o was also obtained in 77% yield and excellent Z/E selectivity (>99/1).
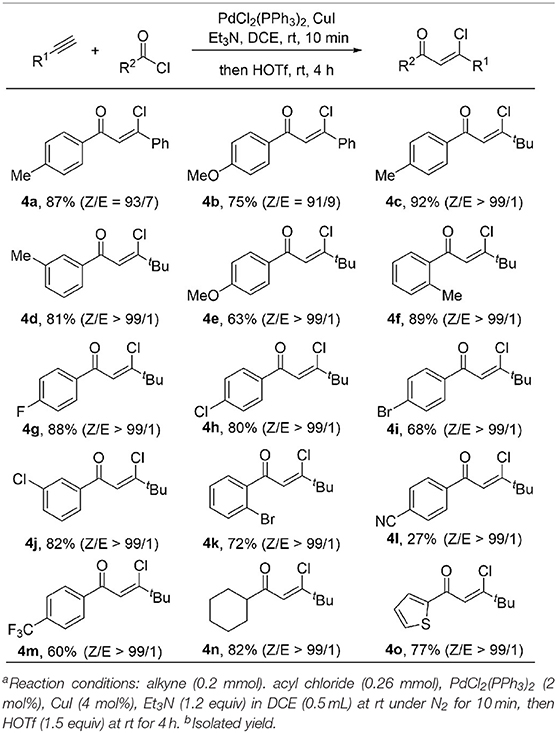
Table 3. Substrate scope of acyl chloridea,b.
To demonstrate the application of this method in organic synthesis, a gram scale experiment was carried out (Scheme 2A). We were delighted to find that the reaction of tert-butylacetylene 5 (10 mmol) with benzoic acid chloride 2 gave ketone 3l in gram-scale with high yield and stereoselectivity. As acid chlorides have been proved to be efficient for this transformation, we then turned our attention to the reactivity of acyl bromide with benzoic acid bromide (Scheme 2B), which gave rise to the β-bromovinyl ketone 6 in satisfactory yield (70%) with excellent Z/E ratio (99/1). However, reaction of 5 with benzoic acid iodide resulted a complex mixture.
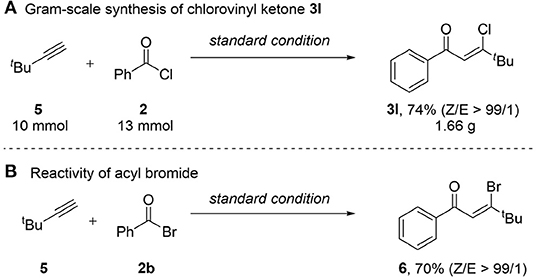
Scheme 2. Synthetic application of the protocol. (A) Gram-scale synthesis of chlorovinyl ketone 3I. (B) Reactivity of acyl bromide.
Based on the experimental results and precedents (Chinchilla and Nájera, 2007), a plausible mechanism for this sequential Sonogashira reaction/hydrohalogenation reaction was proposed (Scheme 3). Firstly, palladium precatalyst PdCl2(PPh3)2 forms a reactive Pd(0) species (Amatore and Jutand, 2000), which undergoes oxidative addition with acyl chloride to produce intermediate A. Secondly, the transmetallation of intermediate A with copper acetylide C, which is formed from terminal alkyne via complexation and deprotonation, gives intermediate D. Sequently, reductive elimination of D affords ynone and regenerated Pd(0) species. For the hydrohalogenation step, halogen source HX (such as HCl) is generated from the reaction of triflic acid with triethylammonium chloride formed in Sonogashira coupling step. The Michael addition of the halide source to the ynone E generates an allenyl intermediate G, which tautomerizes to the final product β-halovinyl ketone 3.
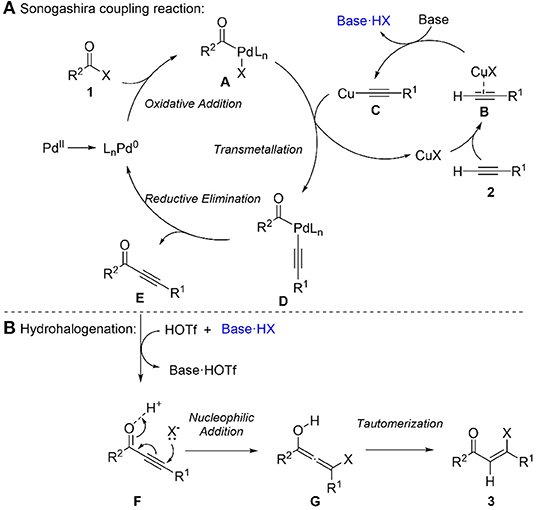
Scheme 3. Proposed mechanism. (A) sonogashira coupling reaction. (B) hydrohalogenation.
Conclusion
We have developed an efficient method for the synthesis of (Z)-β-halovinyl ketones from readily available terminal alkynes and acid chlorides through sequential Sonogashira coupling/hydrohalogenation reaction. This method features simple operations, high yield, and excellent stereoselectivity in most cases. It can be easily scaled up to a gram scale. The broad substrate scopes of both alkyne and acyl chloride show its potential application in organic synthesis.
Experimental Section
General Information
Unless otherwise noted, all chemicals were purchased from J&K, Energy-Chemical or Tansoole, and used as received. All reactions were carried out using oven-dried glassware and magnetic stirring under N2. The product was monitored and tracked by TLC (iodine, potassium permanganate, and other color reagents will be used if necessary). The product was extracted and filtered with 100–200 mesh silica gel and separated and purified with chromatography silica gel column or chromatography silica gel plate (specification of silica gel is 300–400 mesh silica gel). 1H NMR spectrum and 13C NMR spectrum were determined by Bruker-400 instrument or Bruker-500 instrument with TMS as internal standard and deuterium chloroform as solvent. 1H NMR chemical shifts were referenced to tetramethylsilane signal (0 ppm), 13C NMR chemical shifts were referenced to the solvent resonance (77.00 ppm, CDCl3). The following abbreviations (or combinations thereof) were used to explain multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet, br = broad, q = quadruplet. The melting point of solid compounds was determined by X-5 micro melting point tester.
Synthesis of β-Halovinyl Ketones
PdCl2(PPh3)2 (2.8 mg, 0.02 equiv.), CuI (1.5 mg, 0.04 equiv.), Et3N (33.3 μL, 1.2 equiv.), alkynes (0.2 mmol), acid halides (0.26 mmol), and DCE (0.5 mL) were added sequentially under N2 to a reaction tube (10 mL), equipped with a magnetic stir bar. Then the resulting mixture was stirred at room temperature for 10 min. HOTf (26.5 μL, 1.5 equiv.) was added to the reaction, which was stirred at room temperature for 4 h. The mixture was filtered through silica gel and the filtrate was concentrated. The residue was purified by flash column chromatography to afford the desired product.
Gram-Scale Synthesis of β-Chlorovinyl Ketone 3l
To a round bottom flask (100 mL), equipped with a magnetic stir bar, was added PdCl2(PPh3)2 (140.4 mg, 0.02 equiv.), CuI (76.2 mg, 0.04 equiv.), Et3N (1.7 mL, 1.2 equiv.), 3,3-dimethyl-1-butyne (1.2 mL, 10 mmol), benzoyl chloride (1.5 mL, 13 mmol), and 25 mL of DCE sequentially under N2 and then the resulting mixture was stirred at room temperature for 10 min. HOTf (1.3 mL, 1.5 equiv.) was added to the reaction, stirred at room temperature for 4 h. The mixture was filtered through silica gel and the filtrate was concentrated. EtOAc (30 mL) was added and the reaction mixture was washed with NaHCO3 (10 mL), water (2 × 10 mL), brine (10 mL) to remove excess acid. The organic layer was dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by flash column chromatography (silica gel, petroleum ether/ethyl acetate = 20/1) to afford the desired product 3l as a pale yellow oil (1.66 g, 74%).
Characterization Data
(Z)-3-Chloro-1,3-diphenylprop-2-en-1-one (3a): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 41.8 mg (87% yield) of 3a (Z/E = 91/9) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.0 Hz, 2H), 7.77–7.75 (m, 2H), 7.61–7.58 (m, 1H), 7.53–7.42 (m, 5H), 7.36 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 189.9, 143.3, 137.8, 137.3, 133.3, 130.5, 128.7, 128.7, 127.2, 121.5.
(Z)-3-Chloro-3-(4-chlorophenyl)-1-phenylprop-2-en-1-one (3b): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 45.7 mg (85% yield) of 3b (Z/E = 85/15) as a pale yellow solid; m.p. 54–57°C; 1H NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.0 Hz, 2H), 7.69 (d, J = 8.0 Hz, 2H), 7.61–7.59 (m, 1H), 7.51–7.48 (m, 2H), 7.41 (d, J = 8.0 Hz, 2H), 7.33 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 189.7, 141.8, 137.5, 136.7, 135.7, 133.4, 128.9, 128.7, 128.6, 128.4, 121.8.
(Z)-3-Chloro−3-(3-chlorophenyl)-1-phenylprop-2-en-1-one (3c): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 44.2 mg (86% yield) of 3c (Z/E = 91/9) as a pale yellow solid; m.p. 84-86 °C; 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 7.5 Hz, 2H), 7.74 (s, 1H), 7.64 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 7.0 Hz, 1H), 7.52–7.49 (m, 2H), 7.44 (d, J = 8.0 Hz, 1H), 7.41–7.37 (m, 1H), 7.33 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 189.7, 141.2, 139.0, 137.4, 134.8, 133.5, 130.5, 129.9, 128.8, 128.7, 127.3, 125.3, 122.6. HRMS (ESI): m/z calcd. for C15H10Cl2O [M + Na]+ 299.0001, found 299.0002.
(Z)-3-Chloro-3-(4-fluorophenyl)-1-phenylprop-2-en-1-one (3d): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 39.5 mg (76% yield) of 3d (Z/E = 89/11) as a pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 8.00 (d, J = 5.5 Hz, 2H), 7.77–7.75 (m, 2H), 7.64–7.56 (m, 1H), 7.55–7.46 (m, 2H), 7.31 (s, 1H), 7.15–7.12 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 189.7, 164.0 (d, J = 250.0 Hz), 142.1, 137.6, 133.5 (d, J = 3.8 Hz), 133.4, 129.2 (d, J = 8.8 Hz), 128.7, 128.6, 121.4, 115.7 (d, J = 21.3 Hz).
(Z)-3-(4-Bromophenyl)-3-chloro-1-phenylprop-2-en-1-one (3e): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 53.9 mg (84% yield) of 3e (Z/E = 87/13) as a pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 2H), 7.66-7.54 (m, 5H), 7.51–7.48 (m, 2H), 7.33 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 189.7, 141.9, 137.5, 136.2, 133.4, 131.9, 128.7, 128.6, 128.6, 125.0, 121.9.
(Z)-3-Chloro-1-phenyl-3-(p-tolyl)prop-2-en-1-one (3f): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 40.9 mg (81% yield) of 3f (Z/E = 93/7) as a pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 2H), 7.66 (d, J = 8.0 Hz, 2H), 7.59–7.56 (m, 1H), 7.50–7.47 (m, 2H), 7.35 (s, 1H), 7.24 (d, J = 8.0 Hz, 2H), 2.40 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 189.8, 143.6, 141.0, 137.9, 134.5, 133.2, 129.3, 128.6, 128.6, 127.1, 120.4, 21.3.
(Z)-3-Chloro-3-(4-methoxyphenyl)-1-phenylprop-2-en-1-one (3g): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 80/1 as the eluent to give 32 mg (57% yield) of 3g (Z/E = 89/11) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 7.2 Hz, 2H), 7.73 (d, J = 8.8 Hz, 2H), 7.60–7.56 (m, 1H), 7.51–7.47 (m, 2H), 7.32 (s, 1H), 6.95 (d, J = 9.2 Hz, 2H), 3.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.6, 161.6, 143.8, 138.2, 133.1, 129.7, 128.8, 128.6, 128.5, 119.2, 114.0, 55.5.
(Z)-3-Chloro-4-hydroxy-4-methyl-1-phenylpent-2-en-1-one (3h): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 10/1 as the eluent to give 24.9 mg (39% yield) of 3h (Z/E > 99/1) as a pale yellow solid; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 7.2 Hz, 2H), 7.58–7.54 (m, 1H), 7.51–7.47 (m, 2H), 5.97 (s, 1H), 1.49 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 207.0, 183.4, 132.6, 129.2, 128.8, 127.1, 98.6, 89.0, 23.1. HRMS (ESI): m/z calcd. for C12H13ClO2 [M + Na]+ 225.0677, found 225.0657.
(Z)-3-Chloro-1-phenyl-3-(thiophen-3-yl)prop-2-en-1-one (3i): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 37.4 mg (75% yield) of 3i (Z/E = 92/8) as pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 7.2 Hz, 2H), 7.85–7.83 (m, 1H), 7.60–7.57 (m, 1H), 7.47-7.51 (m, 2H), 7.43–7.41 (m, 1H), 7.41–7.39 (m, 1H), 7.37 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 189.6, 139.3, 138.0, 137.9, 133.2, 128.7, 128.5, 127.7, 127.1, 125.0, 119.3. HRMS (ESI): m/z calcd. for C13H9ClOS [M + Na]+ 270.9955, found 270.9939.
(Z)-3-Chloro-3-(1-hydroxycyclohexyl)-1-phenylprop-2-en-1-one (3j): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 10/1 as the eluent to give 26.0 mg (49% yield) of 3j (Z/E > 99/1) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 7.2 Hz, 2H), 7.58–7.54 (m, 1H), 7.51–7.46 (m, 2H), 5.98 (s, 1H), 1.97–1.52 (m, 10H); 13C NMR (100 MHz, CDCl3) δ 206.9, 183.4, 132.5, 129.3, 128.8, 127.1, 99.2, 90.8, 31.9, 24.5, 21.9. HRMS (ESI): m/z calcd. for C15H17ClO2 [M + H]+ 265.0990, found 265.1009.
(Z)-3-Chloro-1-phenylhept-2-en-1-one (3k): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 28.4 mg (63% yield) of 3k (Z/E = 41/59) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 7.0 Hz, 2H), 7.59–7.55 (m, 1H), 7.49–7.45 (m, 2H), 6.82 (s, 1H), 2.55 (t, J = 8.0 Hz, 2H), 1.72–1.65 (m, 2H), 1.47–1.37 (m, 2H), 0.97 (t, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 188.6, 157.8, 138.1, 133.0, 128.7, 128.3, 123.3, 36.2, 29.9, 22.1, 13.8.
(Z)-3-Chloro-4,4-dimethyl-1-phenylpent-2-en-1-one (3l): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 33.0 mg (75% yield) of 3l (Z/E > 99/1) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 7.2 Hz, 2H), 7.59-7.55 (m, 1H), 7.49–7.45 (m, 2H), 6.72 (s, 1H), 1.28 (s, 9H); 13C NMR (100.0 MHz, CDCl3) δ 191.6, 154.6, 137.3, 133.2, 128.8, 128.6, 119.5, 39.9, 28.7.
(Z)-3-Chloro-1-phenyl-3-(trimethylsilyl)prop-2-en-1-one (3m): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 30.4 mg (63% yield) of 3m (Z/E = 89/11) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 7.2 Hz, 2H), 7.61–7.57 (m, 1H), 7.50–7.47 (m, 2H), 7.04 (s, 1H), 0.31 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.4, 148.6, 136.7, 133.5, 133.4, 128.9, 128.7,−2.5. HRMS (ESI): m/z calcd. for C12H15ClOSi [M + Na]+ 261.0473, found 261.0477.
(Z)-3-Chloro-3-phenyl-1-(p-tolyl)prop-2-en-1-one (4a): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 46.6 mg (87% yield) of 4a (Z/E = 93/7) as a pale yellow solid; m.p.: 71–73°C; 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.0 Hz, 2H), 7.81–7.73 (m, 2H), 7.45–7.43 (m, 3H), 7.33 (s, 1H), 7.29 (d, J = 8.0 Hz, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.6, 144.3, 142.6, 137.3, 135.1, 130.4, 129.4, 128.8, 128.6, 127.1, 121.8, 21.7.
(Z)-3-Chloro-1-(4-methoxyphenyl)-3-phenylprop-2-en-1-one1 (4b): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 100/1 as the eluent to give 40.5 mg (75% yield) of 4b (Z/E = 91/9) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.8 Hz, 2H), 7.76–7.74 (m, 2H), 7.45–7.43 (m, 3H), 7.29 (s, 1H), 6.96 (d, J = 8.8 Hz, 2H), 3.88 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 188.7, 163.8, 141.9, 137.3, 131.1, 130.5, 130.3, 128.6, 127.0, 122.0, 113.9, 55.5.
(Z)-3-Chloro-4,4-dimethyl-1-(p-tolyl)pent-2-en-1-one (4c): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 50/1 as the eluent to give 40.7 mg (92% yield) of 4c (Z/E > 99/1) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.83 (d, J = 8.4 Hz, 2H), 7.27–7.25 (m, 2H), 6.68 (s, 1H), 2.42 (s, 3H), 1.32 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 191.4, 153.9, 144.2, 134.8, 129.3, 129.0, 119.7, 39.8, 28.7, 21.7. HRMS (ESI): m/z calcd. for C14H17ClO [M+H]+ 237.1041, Found 237.1048.
(Z)-3-chloro-4,4-dimethyl-1-(m-tolyl)pent-2-en-1-one (4d): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 38.8 mg (81% yield) of 4d (Z/E > 99/1) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.74 (s, 1H), 7.70 (d, J = 7.2 Hz, 1H), 7.44–7.29 (m, 2H), 6.71 (s, 1H), 2.41 (s, 3H), 1.32 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 191.8, 154.4, 138.4, 137.3, 134.0, 129.1, 128.4, 126.1, 119.6, 39.8, 28.6, 21.3. HRMS (ESI): m/z calcd. for C14H17ClO [M+H]+ 237.1041, found 237.1053.
(Z)-3-Chloro-1-(4-methoxyphenyl)-4,4-dimethylpent-2-en-1-one (4e): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 30/1 as the eluent to give 31.1 mg (63% yield) of 4e (Z/E > 99/1) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.91 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 8.8 Hz, 2H), 6.63 (s, 1H), 3.87 (s, 2H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 190.6, 163.7, 153.2, 131.3, 130.2, 119.9, 113.8, 55.5, 39.7, 28.7. HRMS (ESI): m/z calcd. for C14H17ClO2 [M + Na]+ 275.0809, found 275.0805.
(Z)-3-Chloro-4,4-dimethyl-1-(o-tolyl)pent-2-en-1-one (4f): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 30/1 as the eluent to give 41 mg (89% yield) of 4f (Z/E > 99/1) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.57 (d, J = 7.2 Hz, 1H), 7.40–7.36 (m, 1H), 7.27–7.24 (m, 2H), 6.61 (s, 1H), 2.53 (s, 3H), 1.29 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 194.6, 154.8, 138.4, 138.1, 131.7, 131.4, 129.5, 125.6, 121.8, 39.9, 28.6, 20.9. HRMS (ESI): m/z calcd. for C14H17ClO [M + Na]+ 259.0860, found 259.0858.
(Z)-3-Chloro-1-(4-fluorophenyl)-4,4-dimethylpent-2-en-1-one (4g): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 42.4 mg (88% yield) of 4g (Z/E > 99/1) as a pale yellow solid; m.p. 54–55°C; 1H NMR (400 MHz, CDCl3) δ 7.97–7.93 (m, 2H), 7.17–7.12 (m, 2H), 6.65 (s, 1H), 1.31 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 190.2, 165.9 (d, J = 254 Hz), 154.7, 133.7 (d, J = 2.8 Hz), 131.5 (d, J = 9.4 Hz), 119.3, 115.8 (d, J = 22.0 Hz), 39.9, 28.6. HRMS (ESI): m/z calcd. for C13H14ClFO [M + Na]+ 263.0609, found 263.0613.
(Z)-3-Chloro-1-(4-chlorophenyl)-4,4-dimethylpent-2-en-1-one (4h): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 40.9 mg (80% yield) of 4h (Z/E > 99/1) as a pale yellow solid; m.p. 64–66°C; 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 8.4 Hz, 2H), 7.48 (d, J = 8.4 Hz, 2H), 6.70 (s, 1H), 1.36 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 190.4, 155.2, 139.7, 135.6, 130.2, 128.9, 119.0, 39.9, 28.6. HRMS (ESI): m/z calcd. for C13H14Cl2O [M + Na]+ 279.0314, found 279.0327.
(Z)-1-(4-Bromophenyl)-3-chloro-4,4-dimethylpent-2-en-1-one (4i): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 41 mg (68% yield) of 4i (Z/E > 99/1) as a pale yellow solid; m.p. 68–69°C; 1H NMR (400 MHz, CDCl3) δ 7.77 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 6.65 (s, 1H), 1.31 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 190.6, 155.3, 136.1, 131.9, 130.3, 128.4, 119.0, 40.0, 28.6. HRMS (ESI): m/z calcd. for C13H14BrClO [M + Na]+ 322.9809, found 322.9795.
(Z)-3-Chloro-1-(3-chlorophenyl)-4,4-dimethylpent-2-en-1-one (4j): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 42.4 mg (82% yield) of 4j (Z/E > 99/1) as a pale yellow solid; m.p. 55–56°C; 1H NMR (400 MHz, CDCl3) δ 7.88–7.87 (m, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.55–7.53 (m, 2H), 7.43–7.39 (m, 1H), 6.68 (s, 1H), 1.31 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 190.2, 167.1, 164.6, 154.7, 131.5, 131.4, 119.3, 115.9, 115.6, 39.9, 28.6. HRMS (ESI): m/z calcd. for C13H14Cl2O [M + Na]+ 279.0314, found 279.0323.
(Z)-1-(2-Bromophenyl)-3-chloro-4,4-dimethylpent-2-en-1-one (4k): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 43.5 mg (72% yield) of 4k (Z/E > 99/1) as a pale yellow solid; m.p. 64–66°C; 1H NMR (400 MHz, CDCl3) δ 7.59–7.61 (m, 1H), 7.49–7.46 (m, 1H), 7.40-7.36 (m, 1H), 7.33–7.29 (m, 1H), 6.68 (s, 1H), 1.27 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 192.5, 157.4, 141.6, 133.5, 131.8, 129.9, 127.5, 121.2, 119.5, 40.3, 28.5. HRMS (ESI): m/z calcd. for C13H14BrClO [M + Na]+ 322.9809, found 322.9804.
(Z)-4-(3-chloro-3-phenylacryloyl)benzonitrile (4l): Prepared according to the general procedure. The crude reaction mixture was purified by prep-TLC using PE/EA = 10/1 as the eluent to give 14.3 mg (27% yield) of 4l (Z/E > 99/1) as a yellow solid; m. p. 108.8–110.6°C; 1H NMR (500 MHz, CDCl3) δ 8.08 (d, J = 8.4 Hz, 2H), 7.84–7.73 (m, 4H), 7.54–7.44 (m, 3H), 7.33 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 188.3, 145.8, 141.0, 136.9, 132.6, 131.3, 129.0, 128.8, 127.3, 120.2, 117.9, 116.4.
(Z)-3-chloro-3-phenyl-1-(4-(trifluoromethyl)phenyl)prop-2-en-1-one (4m): Prepared according to the general procedure. The crude reaction mixture was purified by prep-TLC using PE/EA = 10/1 as the eluent to give 37.4 mg (60% yield) of 4m (Z/E > 99/1) as a pale yellow solid; m.p. 80.7–81.7°C; 1H NMR (500 MHz, CDCl3) δ 8.09 (d, J = 8.0 Hz, 2H), 7.83–7.68 (m, 4H), 7.53–7.40 (m, 3H), 7.35 (s, 1H); 13C NMR (126 MHz, CDCl3) δ 188.7, 145.1, 140.6,137.1, 134.5 (q, J = 32.8 Hz), 131.0, 128.9, 128.8, 127.3, 125.8 (q, J = 3.8 Hz), 123.6 (q, J = 273.4 Hz), 120.6.
(Z)-3-Chloro-1-cyclohexyl-4,4-dimethylpent-2-en-1-one (4n): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 35.9 mg (82% yield) of 4n (Z/E > 99/1) as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 6.31 (s, 1H), 2.56–2.50 (m, 1H), 1.87–1.75 (m, 6H), 1.43-1.25 (m, 4H), 1.23 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 203.1, 154.6, 120.3, 51.3, 40.0, 28.7, 28.3, 25.8, 25.6. HRMS (ESI): m/z calcd. for C13H21ClO [M + Na]+ 251.1173, found 251.1174.
(Z)-3-Chloro-4,4-dimethyl-1-(thiophen-2-yl)pent-2-en-1-one (4o): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 34.3 mg (77% yield) of 4o (Z/E > 99/1) as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.72–7.58 (m, 2H), 7.15–7.13 (m, 1H), 6.76 (s, 1H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 182.8, 156.0, 145.0, 134.1, 132.4, 128.1, 118.4, 40.1, 28.6. HRMS (ESI): m/z calcd. for C11H13ClOS [M + Na]+ 251.0268, found 251.0274.
(Z)-3-Bromo-4,4-dimethyl-1-phenylpent-2-en-1-one (6): Prepared according to the general procedure. The crude reaction mixture was purified by flash column chromatography using PE/EA = 20/1 as the eluent to give 39.2 mg (70% yield) of 6 (Z/E > 99/1) as a pale yellow solid; m.p. 82–84°C; 1H NMR (400 MHz, CDCl3) δ 7.92 (d, J = 7.2 Hz, 2H), 7.60–7.56 (m, 1H), 7.50–7.46 (m, 2H), 6.91 (s, 1H), 1.31 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 192.6, 147.5, 136.6, 133.4, 129.0, 128.6, 123.2, 40.5, 29.3.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
YX designed the research. F-JC and ZH carried out the experiments. All authors contributed to the manuscript.
Funding
Financial support was provided by NSFC (Nos. 21801191, 21572163, and 21873074) and the Wenzhou Science & Technology Bureau (G20180016).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.621545/full#supplementary-material
References
Amatore, C., and Jutand, A. (2000). Anionic Pd(0) and Pd(II) intermediates in palladium-catalyzed heck and cross-coupling reactions. Acc. Chem. Res. 33, 314–321. doi: 10.1021/ar980063a
Benson, W. R., and Pohland, A. E. (1964). trans-β-Chlorovinyl Ketones and trans-(β-Acylvinyl)trimethylammonium Chlorides1,2. J. Org. Chem. 29, 385–391. doi: 10.1021/jo01025a033
Chinchilla, R., and Nájera, C. (2007). The sonogashira reaction: a booming methodology in synthetic organic chemistry. Chem. Rev. 107, 874–992. doi: 10.1021/cr050992x
da Silva, V. A. F., da Silva, G. P., Matsuo, B. T., Ali, A., Davis, R. L., Zukerman-Schpector, J., et al. (2019). Synthesis of (Z)-β-Halo α,β-unsaturated carbonyl systems via the combination of halotrimethylsilane and tetrafluoroboric acid. Org. Biomol. Chem. 17, 519–526. doi: 10.1039/C8OB02110D
Eckhardt, M., and Fu, G. C. (2003). The first applications of carbene ligands in cross-couplings of alkyl electrophiles: sonogashira reactions of unactivated alkyl bromides and iodides. J. Am. Chem. Soc. 125, 13642–13643. doi: 10.1021/ja038177r
Gandeepan, P., Parthasarathy, K., Su, T.-H., and Cheng, C.-H. (2012). Iron-catalyzed synthesis of β-chlorovinyl and α,β-alkynyl ketones from terminal and silylated alkynes with acid chlorides. Adv. Synth. Catal. 354, 457–468. doi: 10.1002/adsc.201100670
Goossen, L. J., Rodriguez, N., and Goossen, K. (2009). Stereoselective synthesis of β-chlorovinyl ketones and arenes by the catalytic addition of acid chlorides to alkynes. Angew. Chem. Int. Ed. 48, 9592–9594. doi: 10.1002/anie.200904615
Hosseini-Sarvari, M., and Mardaneh, Z. (2011). Selective and CO-retentive addition reactions of acid chlorides to terminal alkynes in synthesis of β-chloro-α, β-unsaturated ketones using ZnO. Bull. Chem. Soc. Jpn. 84, 778–782. doi: 10.1246/bcsj.20110040
Hua, R., Onozawa, S.-Y., and Tanaka, M. (2005). Rhodium-catalyzed nondecarbonylative addition reaction of ClCOCOOC2H5 to alkynes. Chem. Eur. J. 11, 3621–3630. doi: 10.1002/chem.200401279
Iwai, T., Fujihara, T., Terao, J., and Tsuji, Y. (2009). Iridium-catalyzed addition of acid chlorides to terminal alkynes. J. Am. Chem. Soc. 131, 6668–6669. doi: 10.1021/ja901778y
Iwai, T., Fujihara, T., Terao, J., and Tsuji, Y. (2012). Iridium-catalyzed addition of aroyl chlorides and aliphatic acid chlorides to terminal alkynes. J. Am. Chem. Soc. 134, 1268–1274. doi: 10.1021/ja209679c
Karpov, A. S., and Müller, T. J. J. (2003a). Straightforward novel one-pot enaminone and pyrimidine syntheses by coupling-addition-cyclocondensation sequences. Synthesis 2003, 2815–2826. doi: 10.1055/s-2003-42480
Karpov, A. S., and Müller, T. J. J. (2003b). New entry to a three-component pyrimidine synthesis by TMS–ynones via sonogashira coupling. Org. Lett. 5, 3451–3454. doi: 10.1021/ol035212q
Kashiwabara, T., Fuse, K., Hua, R., and Tanaka, M. (2008). Rhodium-complex-catalyzed addition reactions of chloroacetyl chlorides to alkynes. Org. Lett. 10, 5469–5472. doi: 10.1021/ol802260w
Kashiwabara, T., Kataoka, K., Hua, R., Shimada, S., and Tanaka, M. (2005). CO-retentive addition reactions of fluorinated acid chlorides to alkynes. Org. Lett. 7, 2241–2244. doi: 10.1021/ol050632w
Kashiwabara, T., and Tanaka, M. (2011). Rhodium-catalyzed addition of α-keto acid chlorides with terminal alkynes. Adv. Synth. Catal. 353, 1485–1490. doi: 10.1002/adsc.201100108
Kim, H. Y., Li, J.-Y., and Oh, K. (2012). Studies on elimination pathways of β-halovinyl ketones leading to allenyl and propargyl ketones and furans under the action of mild bases. J. Org. Chem. 77, 11132–11145. doi: 10.1021/jo302253c
Kim, H. Y., and Oh, K. (2014). A facile access to 4-substituted-2-naphthols via a tandem friedel–crafts reaction: a β-Chlorovinyl Ketone Pathway. Org. Lett. 16, 5934–5936. doi: 10.1021/ol502951v
Kim, H. Y., and Oh, K. (2015). 1,3-dienones and 2H-Pyran-2-ones from Soft α-vinyl enolization of β-chlorovinyl ketones: defined roles of brönsted and lewis base. Org. Lett. 17, 6254–6257. doi: 10.1021/acs.orglett.5b03265
Kim, H. Y., and Oh, K. (2017). A one-pot synthesis of pyranone and pyrrole derivatives from β-chlorovinyl ketones via direct conjugate addition approach. Org. Lett. 19, 4904–4907. doi: 10.1021/acs.orglett.7b02381
Kim, H. Y., and Oh, K. (2020). Divergent reaction pathways of β-chlorovinyl ketones: microwave-assisted thermal nazarov cyclization versus cycloisomerization via soft vinyl enolization. Adv. Synth. Catal. 362, 1391–1398. doi: 10.1002/adsc.201901399
Kim, H. Y., Song, E., and Oh, K. (2017). Unified approach to (thio)chromenones via one-pot friedel–crafts acylation/cyclization: distinctive mechanistic pathways of β-chlorovinyl ketones. Org. Lett. 19, 312–315. doi: 10.1021/acs.orglett.6b03348
Kokubo, K., Matsumasa, K., Miura, M., and Nomura, M. (1996). Rhodium-catalyzed reaction of aroyl chlorides with alkynes. J. Org. Chem. 61, 6941–6946. doi: 10.1021/jo960915p
Koo, H., Kim, H. Y., and Oh, K. (2019a). Continuous flow synthesis of isoxazoles via vinyl azides from friedel–crafts acylation of alkynes: a modulated troubleshooting optimization approach. Org. Lett. 21, 10063–10068. doi: 10.1021/acs.orglett.9b04010
Koo, H., Kim, H. Y., and Oh, K. (2019b). (E)-Selective friedel–crafts acylation of alkynes to β-chlorovinyl ketones: defying isomerizations in batch reactions by flow chemistry approaches. Org. Chem. Front. 6, 1868–1872. doi: 10.1039/C9QO00217K
Kundu, N. G., and Chaudhuri, L. N. (1991). Synthesis of dimethoxypyrimidines and uracils with novel C-5 substituents. J. Chem. Soc. Perkin Trans. 1, 1677–1682. doi: 10.1039/p19910001677
Martens, H., Janssens, F., and Hoornaert, G. (1975). Stereochemistry and mechanism of acylation of acetylenes. Tetrahedron 31, 177–183. doi: 10.1016/0040-4020(75)85014-9
Pohland, A. E., and Benson, W. R. (1966). β-Chlorovinyl ketones. Chem. Rev. 66, 161–197. doi: 10.1021/cr60240a003
Price, C. C., and Pappalardo, J. A. (1950). The synthesis of some α,β-unsaturated aldehydes1. J. Am. Chem. Soc. 72, 2613–2615. doi: 10.1021/ja01162a074
Semenova, I. S., Yarovenko, V. N., Levchenko, K. S., and Krayushkin, M. M. (2013). Synthesis of 1,3-thioxoketones from salicylaldehyde. Russ. Chem. Bull. 62, 1022–1025. doi: 10.1007/s11172-013-0135-9
Shchukin, A. O., and Vasilyev, A. V. (2008). Different reactivities of acetylene carbonyl compounds under the catalysis by Bronsted superacids and Lewis acids. Appl. Catal. A Gen. 336, 140–147. doi: 10.1016/j.apcata.2007.07.035
Sonogashira, K. (2002). Development of Pd–Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 653, 46–49. doi: 10.1016/S0022-328X(02)01158-0
Sonogashira, K., Tohda, Y., and Hagihara, N. (1975). A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 16, 4467–4470. doi: 10.1016/S0040-4039(00)91094-3
Tohda, Y., Sonogashira, K., and Hagihara, N. (1977). A convenient synthesis of 1-Alkynyl ketones and 2-alkynamides. Synthesis 1977, 777–778. doi: 10.1055/s-1977-24574
Wang, B., Wang, S., Li, P., and Wang, L. (2010). Iron-catalyzed regio- and stereoselective addition of acid chlorides to alkynes. Chem. Commun. 46, 5891–5893. doi: 10.1039/c0cc01182g
Yan, W., Li, Z., and Kishi, Y. (2015). Selective activation/coupling of polyhalogenated nucleophiles in Ni/Cr-mediated reactions: synthesis of C1–C19 building block of halichondrin Bs. J. Am. Chem. Soc. 137, 6219–6225. doi: 10.1021/jacs.5b03498
Yang, F., Ji, K.-G., Ali, S., and Liang, Y.-M. (2011). Sc(OTf)3-Catalyzed synthesis of indoles and SnCl4-mediated regioselective hydrochlorination of 5-(Arylamino)pent-3-yn-2-ones. J. Org. Chem. 76, 8329–8335. doi: 10.1021/jo201514q
Zeng, X., Lu, Z., Liu, S., Hammond, G. B., and Xu, B. (2017). Metal-free, regio-, and stereo-controlled hydrochlorination and hydrobromination of ynones and ynamides. J. Org. Chem. 82, 13179–13187. doi: 10.1021/acs.joc.7b02257
Zhang, J., Liu, L., Duan, J., Gu, L., Chen, B., Sun, T., et al. (2017). Stereoselective one-pot sequential dehydrochlorination/trans-hydrofluorination reaction of β-Chloro-α,β-unsaturated aldehydes or ketones: facile access to (Z)-β-Fluoro-β-arylenals/β-Fluoro-β-arylenones. Adv. Synth. Catal. 359, 4348–4358. doi: 10.1002/adsc.201700981
Keywords: Sonogashira coupling, one-pot synthesis, β-halovinyl ketone, hydrohalogenation, ynones
Citation: Chen F-J, Hua Z, Chen J, Chen J, Lee D and Xia Y (2021) One-Pot Synthesis of (Z)-β-Halovinyl Ketones via the Cascade of Sonogashira Coupling and Hydrohalogenation. Front. Chem. 8:621545. doi: 10.3389/fchem.2020.621545
Received: 26 October 2020; Accepted: 03 December 2020;
Published: 22 April 2021.
Edited by:
Guigen Li, Texas Tech University, United StatesReviewed by:
Wanqing Wu, South China University of Technology, ChinaSrinath Pashikanti, Idaho State University, United States
Copyright © 2021 Chen, Hua, Chen, Chen, Lee and Xia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daesung Lee, ZHN1bmdsZWVAdWljLmVkdQ==; Yuanzhi Xia, eHl6QHd6dS5lZHUuY24=
†These authors have contributed equally to this work