 Magdalena Rapp1*
Magdalena Rapp1* Klaudia Margas-Musielak1Patrycja Kaczmarek1Agnieszka Witkowska1
Klaudia Margas-Musielak1Patrycja Kaczmarek1Agnieszka Witkowska1 Tomasz Cytlak1,2
Tomasz Cytlak1,2 Tomasz Siodła1Henryk Koroniak1
Tomasz Siodła1Henryk Koroniak1- 1Faculty of Chemistry, Adam Mickiewicz University in Poznań, Poznań, Poland
- 2Centre for Advanced Technologies Adam Mickiewicz University in Poznań, Poznań, Poland
The synthesis of the stable surrogates of an important amino acid (R)-4-amino-3-hydroxybutyric acid (GABOB) such as substituted hydroxy aminophosphonic acids bearing a quaternary stereogenic center is presented. Highly diastereoselective formations of fluorinated spiroepoxy alkylphosphonate or related tertiary carbon-containing oxiranes from β-keto phosphonates possessing methyl, phenyl, or cyclohexenyl substituents, are reported. Stereoselective acid-promoted epoxide opening by bromide or azide followed by reduction/protection afforded tertiary bromides or N-Boc derivatives of β-amino-γ-hydroxy alkylphosphonates in most cases, while the reactions of oxiranes with different amines yielded their β-hydroxy-γ-amino regioisomers. Surprisingly, during the synthesis of amino phosphonic acids, we observe that the acid-induced rearrangement proceeded in a high diastereospecific manner, leading finally to substituted β-hydroxy-γ-aminoalkylphosphonic acids.
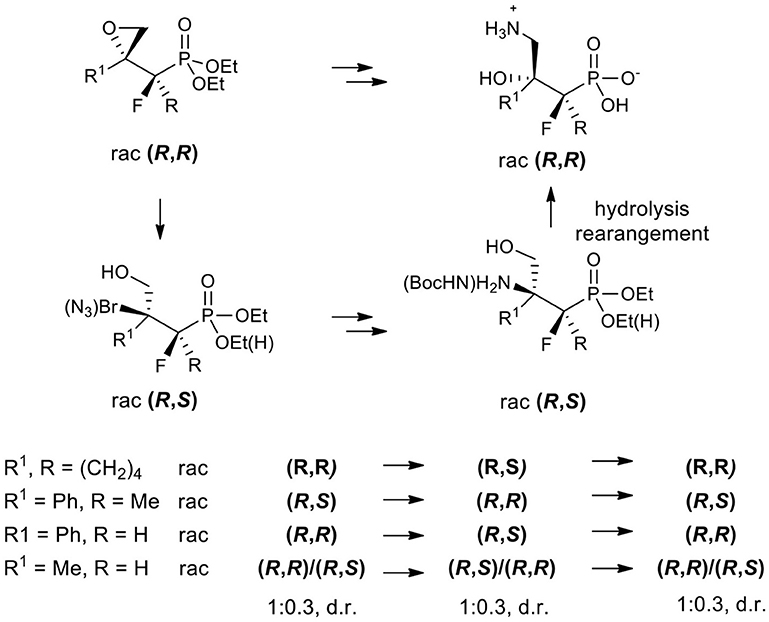
Graphical Abstract. Diastereoselective transformations of fluorinated epoxyalkylphosphonates.
Introduction
Aminophosphonates can be considered as good amino acid surrogates when the tetrahedral phosphonic acid moiety corresponds to the planar carboxylic group (Kukhar and Hudson, 2000; Kafarski and Lejczak, 2001; Palacios et al., 2005; Ordóñez et al., 2009, 2015; Naydenova et al., 2010; Orsini et al., 2010; Kafarski, 2020). The C–P bond is stable in different biochemical and thermal conditions and resists photochemical decomposition (Fields, 1999; Horsman and Zechel, 2017). For these reasons, in medicinal as well as in organic chemistry the synthesis and applications of α-, β-, or γ-amino phosphonates are of special interest (Kafarski and Lejczak, 1991; Fields, 1999; Foss et al., 2007; Mucha et al., 2011; Bera et al., 2013; Gu et al., 2018; Kafarski, 2020). Among them, phosphorus-containing amino alcohols frequently exhibit various biological interactions (Patel et al., 1990; Drag et al., 2013; Mandadapu et al., 2013). As a representative example (R)-2-amino-1-hydroxyethylphosphonic acid (HO-AEP), has been recognized as a component of the protozoal plasma membrane as well as in marine invertebrates and microorganisms (Korn et al., 1973; Watanabe et al., 2001), while FR-33289 with antibiotic properties was first isolated from Streptomyces rubellomurinus subsp., indigoferus (Iguchi et al., 1980). To compare, the obtained various 2-amino-1-hydroxy-alkanephosphonate dipeptides have been found as potent, non-covalent organophosphonate inhibitors of cathepsin C, papain, cathepsin B, and cathepsin K (Drag et al., 2013), renin (Patel et al., 1990), or norovirus (Mandadapu et al., 2013). Furthermore, the α-fluorinated δ-hydroxy-γ-aminophosphonates have been applied as the inhibitors of neutral Sphingomyelinase (N-SMase) (Yokomatsu et al., 2003; Figure 1).
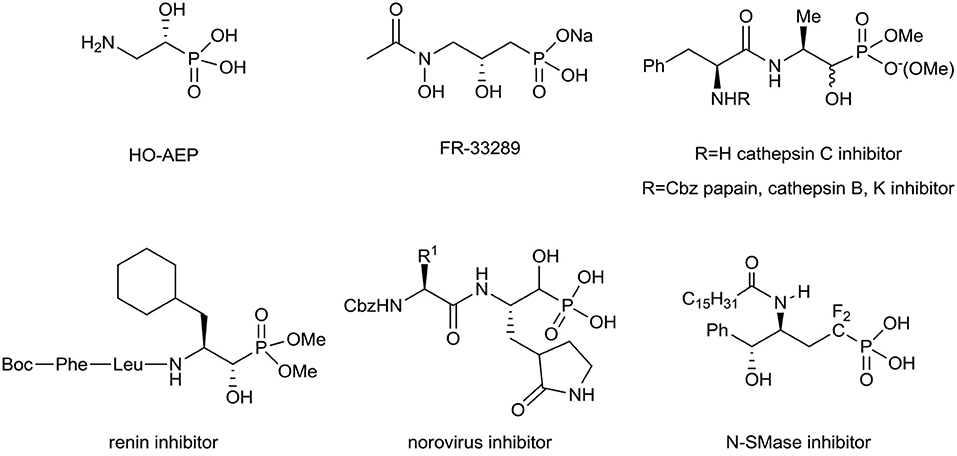
Figure 1. Structures of different bioactive amino alcohol derivatives of alkylphosphonates.
In medicinal chemistry, the introduction of a phosphonate group to biologically significant compounds is a frequently applied method to modify its physicochemical properties that could play an important role in a biological environment. The group of biologically important amino alcohols includes the attractive target 4-amino-3-hydroxybutyric acid (GABOB). (R)-(–)-γ-Amino-β-hydroxybutanoic acid (L-GABOB), also known as (R)-(–)-β-hydroxy-γ-aminobutanoic acid, is an important amino acid acting as an agonist of neurotransmitter γ-aminobutyric acid (GABA) (Roberts et al., 1981; Falch et al., 1986; Kristiansen and Fjalland, 1991). It is used in numerous illness treatments including epilepsy therapy (De Maio and Pasquariello, 1963; Banfi et al., 1983; García-Flores and Farías, 1997; Melis et al., 2014). Among several GABA and GABOB analogs the γ-amino-β-hydroxy phosphonic acid (P-GABOB) and its organophosphorus derivatives constitute an interesting group of compounds (Dingwall, 1983; Yuan et al., 2002; Ordóñez et al., 2003, 2010; Wang et al., 2003; Wróblewski and Hałajewska-Wosik, 2003; Tadeusiak, 2004; Figure 2).
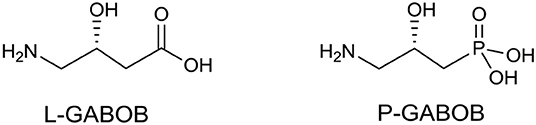
Figure 2. Structures of (R)-β-hydroxy-γ-aminobutanoic acid (L-GABOB) and its organophosphorus analogs P-GABOB.
Their syntheses have been successfully accomplished by the transformation of chiral precursors (Ordóñez et al., 2003; Wróblewski and Hałajewska-Wosik, 2003; Tadeusiak, 2004), the application of racemic starting materials (Dingwall, 1983; Ordóñez et al., 2003), and kinetic resolution (Wang et al., 2003; Wróblewski and Hałajewska-Wosik, 2003) or biocatalysis (Tadeusiak, 2004). Some GABA analogs are known as GABA receptor agonists and are used as drugs, e.g., β-phenyl-γ-aminobutanoic acid (phenibut) (Lapin, 2001), β-(4-chlorophenyl)-γ-aminobutyric acid (baclofen) (Brogden et al., 1974; Leggio et al., 2010), or 1-(aminomethyl)cyclohexane acetic acid (gabapentin) (Goa and Sorkin, 1993; Smith et al., 2016). On the other hand, two enantiomers of monofluorinated analogs of GABOB (with a fluorine instead of hydroxyl group) have been synthesized in order to determine the conformation of GABA when binding to specific protein receptors (Clift et al., 2007; Deniau et al., 2007; Yamamoto et al., 2011). In these types of compounds, due to a charge-dipole interaction between fluorine and a charged nitrogen atom, the gauche alignment F-C-C-N+ is preferred (Briggs et al., 2004). These properties revealed the distinct structural features of GABA binding sites to GABAA synopsis receptors comparing them to GABA-aminotransferase which may be of importance in Alzheimer's and Parkinson's diseases treatment (Clift et al., 2007; Deniau et al., 2007; Yamamoto et al., 2011). Analogously, in protonated vicinal fluorohydrine the gauche conformers are strongly favored lowering its energy (Clift et al., 2007; Deniau et al., 2007; Yamamoto et al., 2011). This effect has reinforced or destabilized chain conformations influencing binding affinities as it was reported for the fluorinated analogs of Indinavir (HIV protease inhibitor) (Myers et al., 2001). Other electronic and steric impacts mirroring an enzyme-substrate interaction have already been applied in medicinal chemistry (O'Hagan and Rzepa, 1997; Bégué and Bonnet-Delpon, 2008). What is more, electronegative fluorine increases the acidity of neighboring carboxylic acid (Koppel et al., 1994) or phosphonic acid (O'Hagan and Rzepa, 1997), and in the same manner, lowers the basicity of amines (Abraham et al., 1990), inducing a remarkable effect upon the physical and biochemical properties of fluorine-containing molecules. Similarly, the introduction of a phosphonate group to organic compounds modifies its physicochemical properties and could play an important role in the biological environment.
On the basis of a combination of reactivity and synthetic application of oxiranes in organic synthesis (Parker and Isaacs, 1959; Ready and Jacobsen, 2002; Azoulay et al., 2005; Wu and Xia, 2005; Padwaa and Murphree, 2006; Fustero et al., 2011; Singh et al., 2013; Zhao and Weix, 2014; Faiz and Zahoor, 2016), we decided to apply those three-membered heterocycles toward aminophosphonates. Recently we developed the method for the synthesis of two types of monofluorinated α, β-epoxyphosphonates with the vicinal and geminal arrangement of fluorine and phosphorus atoms via a Michaelis-Becker addition or by an intramolecular ring closure reaction (Rapp et al., 2015). Herein, we report our results concerning the diastereoselective synthesis and application of fluorinated epoxyalkylphosphonates toward α-fluoro γ-amino-β-hydroxybutanoic acid (GABOB) as well as β-amino-γ-hydroxyalkylphosphonic acid analogs. We expect that the interaction between fluorine and charged nitrogen or oxygen (gauche effect) in α-fluoro β,γ-amino alcohol derivatives of alkylphosphonates will play an important role and may be used in the future to reveal different binding sites when inhibiting certain enzymes.
Results and Discussion
The first aim of our study was the synthesis of α-fluoro-β,γ-epoxy alkylphosphonates as valuable intermediates for the construction of tertiary hydroxy- or aminophosphonates. We started from the convenient racemic diethyl α-fluoro-β-ketophosphonates bearing methyl, phenyl, or cyclohexyl substituents (compounds 1–4). The oxiranes 5–8 were received by the treatment of corresponding substrates with diazomethane. In the case of 1, the reaction led to the formation of oxiranes: 5 (93%, 3.3:1 d.r.), while from ketones 2–4 the epoxides 6–8 were obtained (yields from 90 to 95%) (Table 1).
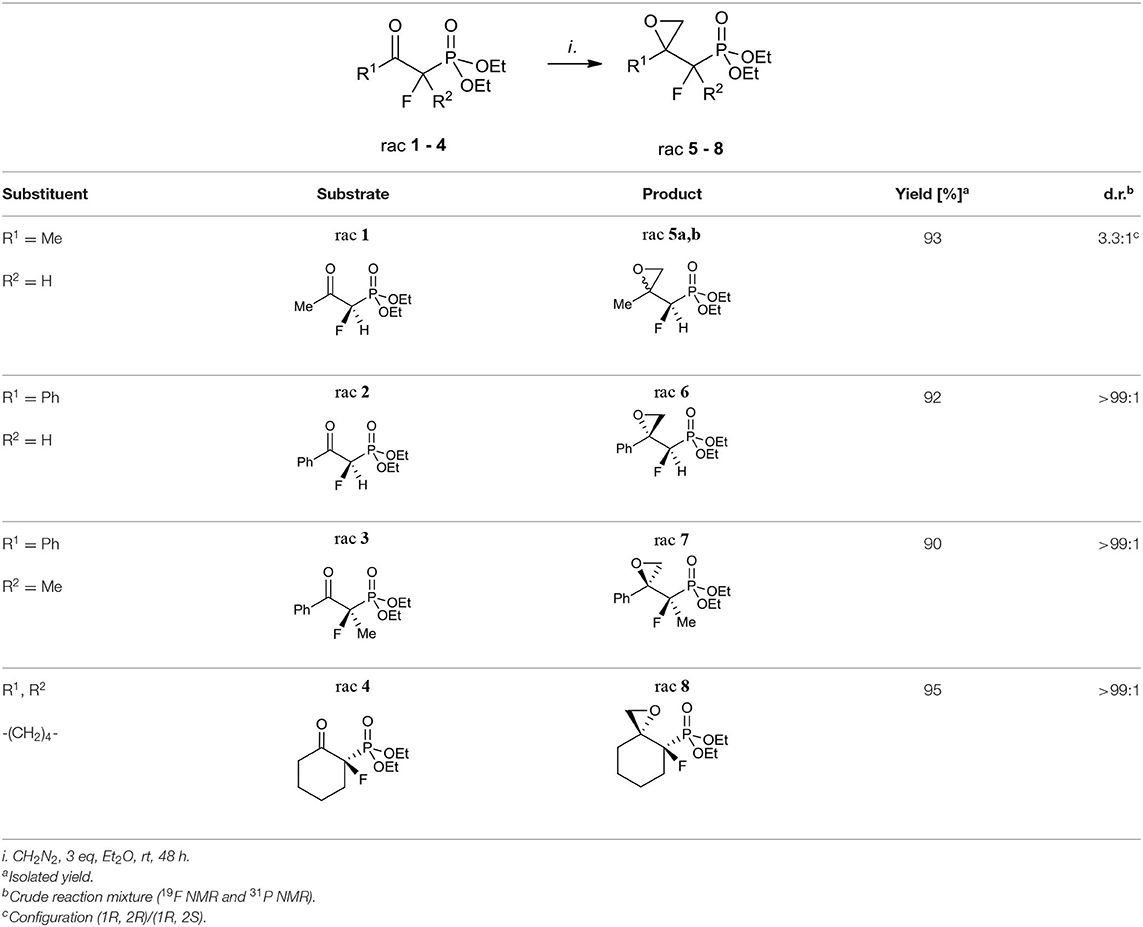
Table 1. Synthesis of fluorinated epoxy alkylphosphonates 5–8.
The diastereoselective addition of CH2N2 to acyclic β-ketophosphonate occurred contrary to the Felkin-Anh model with the bulky diethylphosphonate moiety as the biggest substituent perpendicular to C=O, with the medium–sized group R2 (e.g., F for 1, 2, and Me in the case of 3) and smallest substituents R1 (e.g., H for 1, 2, and F for 3), respectively (Sano et al., 2006) leading to 5a and 6–7 and could be explained by the addition to the carbonyl group as presented in Figure 3A. Thus, the steric interactions between the bulky diethylphosphonate moiety (perpendicular), fluorine, and other substituents and the approaching nucleophile are minimized. On the other hand, the equatorial attack and coordination of diazomethane with phosphonyl oxygen, in the six-membered chair-like transition state, led to compound 8 (Figure 3B). We also observed the influence of ketone substituents on diastereoselectivity. Thus, in the case of the addition to 2–4 [R1 = Ph or -(CH2)4-] the diastereomerically pure oxiranes 6–8 (>99:1 d.r. as determined by NMR) were formed, while the reaction of 1 (R1 = CH3, R2 = H) with diazomethane gave two diastereoisomers of 5 (5a:5b, 3.3:1, d.r.).
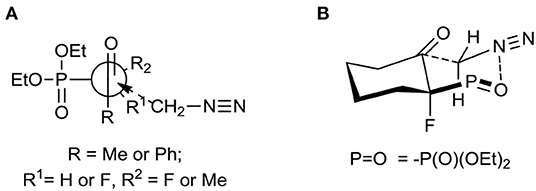
Figure 3. (A) Unhindered attack of CH2N2 to β-ketophosphonates 1–3 during formation of 5–7; (B) Proposed transition state leading to 8.
The assignment of the relative configuration of spiroepoxide or related tertiary carbon-containing oxiranes can be troublesome and difficult to achieve. However, the structure and stereochemistry of compounds 5–8 were confirmed by NMR spectra, analysis of nOe effect between H-H (1D or 2D NOESY), and H-F correlations (2D or 1D HOESY). Thus, the 2D NOESY and 2D H-F HOESY experiments registered for both diastereoisomers 5a and 5b, indicated correlations between one of the diastereotopic protons of the oxirane ring and proton derived from the CHFP moiety as well as with fluorine. At the same time, the nOe effects between the remaining oxirane proton and methyl group as well as its long-range interactions “W-pathway” with fluorine (4JFH 5.6/5.8 Hz, in the case of major 5a) (Dolbier, 2009) or phosphorous atoms (4JHP 2.1 Hz, for minor 5b) (Zymańczyk-Duda et al., 1995) indicated stereochemistry rac (1R, 2R) for major 5a and rac (1R, 2S) in the case of minor 5b. At the same time, the correlation of signals derived from fluorine and the methyl group for 5a compared to the weak nOe effect for F-Me in the case of 5b additionally confirm the assigned configurations (Scheme 1). Additionally, the slightly different enhancements in nOe effects for the CHF proton and fluorine with phenyl as well as one of the protons of the oxirane ring were detected in the NOESY and HOESY spectra of 6. The second epoxide proton was involved in the “W-pathway” (4JFH 5.6Hz). In the case of 7, similar interactions between the methyl group and fluorine (2D NOESY and 2D H-F HOESY) together with one of the oxirane protons were observed. These analyses in comparison with spectral properties of 5a, b allow us to assign the configuration for 6 as rac (1R, 2R) and 7 as rac (1R, 2S). The analysis of the nOe effects (2D NOESY, H-F HOESY) for spiroepoxide 8 revealed the correlations of epoxide protons C2HH with axial proton C5H and equatorial C7H of cyclohexane, while the nOe effects between fluorine and the closer proton of oxirane as well as the contacts between fluorine and the axial protons of the cyclohexane ring (C6H and C8H), as presented in Scheme 1, indicated configuration (3R, 4R) of 8, and were the most informative in the stereochemical assignment. Additionally in the 13C NMR spectra of 5–8, the typical CFP(O)(OEt)2 chemical shifts and values of 1JC−P are from 166 to 172 Hz, while 1JC−F varying from 185 to 191 Hz, according to literature, were observed (Dolbier, 2009). Interestingly, the coupling of fluorine and phosphorus to the carbon atom is readily observable in 13C NMR and allowed us to distinguish the proximity of each carbon atom to these heteroatoms (Gorenstein, 1984; Hesse et al., 1997; Dolbier, 2009). At the same time, values of coupling constants 2JC−P were <9 Hz while analogous 2JC−F appeared usually around 20 Hz (with an exception of 12 Hz for 6). Moreover, 3JC−F and 3JC−P were c.a. 6–8 Hz and 2–3 Hz for 5a and 6–7 while for cyclohexane 8 3J was 7–10 and 2 Hz, respectively. The spectral properties of the obtained compounds (IR, 1H NMR) as well as chemical shift values in the 13C NMR spectra are consistent with the data reported for epoxyalkylphosphonates (Griffin and Kundu, 1969; Wróblewski and Bak-Sypień, 2007) as well as with spiroepoxides (Zanardi et al., 2016).
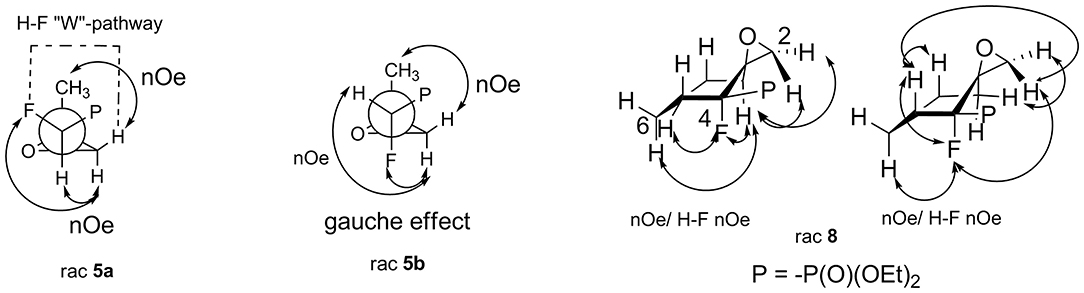
Scheme 1. Nuclear Overhauser effects (nOe effects) and diagnostic coupling constants in oxiranes 5a,b and 8.
The oxiranes 5a and 5b exist in [D] chloroform predominantly as their gauche conformers (respectively to the C1-C2 bond) as can be judged from the analysis of nOe interactions as well as W-pathway effects. This conclusion was further supported by the comparison of relative energies calculated for each conformers of 5a, b (Tables 2, 3). In the case of compound 5a, the DFT conformation analysis showed the preference of conformation 1 (Table 2—conformations 1Y and 1Z have the lowest energies). We also observed the influence of geometries of the bulky diethoxyphosphoryl group and the relative energies of calculated conformers. In the case of compound 5b, the conformation analysis suggested that the most stable were two conformers 2Y and 3X (Table 3), while from the NMR spectra conformation 3 seemed to be the major one. Such an inconsistency may be related to the fact that DFT calculations were performed “in vacuum” and do not reflect any interactions with solvent.
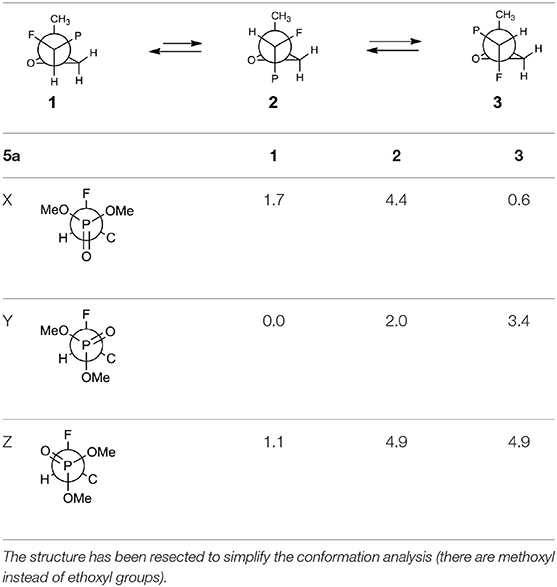
Table 2. The geometries and relative energies [kcal/mol] of nine energy minima for the compound 5a.
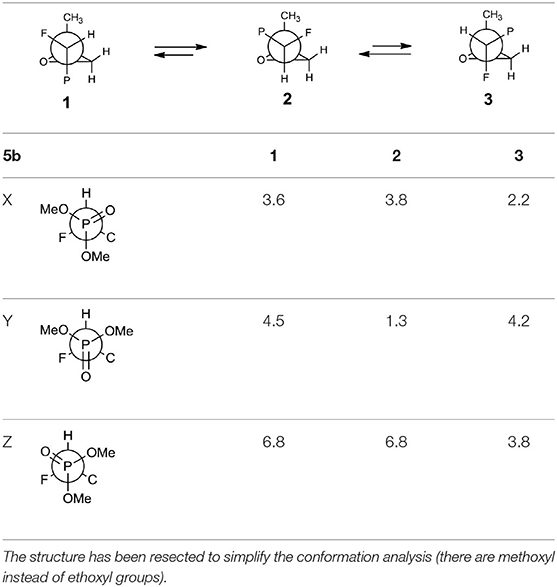
Table 3. The geometries and relative energies [kcal/mol] of nine energy minima for the compound 5b.
Stereoselective addition of a nucleophile to α-fluorophosphonoacetates approaching opposite to the phosphonates trajectory was already reported and applied in tandem reduction-olefination (Sano et al., 2006). Moreover, the similar diastereoselectivity of the diazomethane addition and its coordination in the case of the reaction of chiral α-alkyl/aryl substituted β-keto (Rs)-sulfoxides were presented by Sorochinsky and Soloshonok (2010). Also, the addition of the Grignard reagent to (2-oxocyclohexyl)phosphonate resulted in a hydroxy phosphonate with a cyclohexane ring possessing both OH and phosphonate groups in cis geometry as the major isomer [cis/trans, 95:5, respectively] (Lentsch and Wiemer, 1999). To compare, the additions of diazomethane or ethyl(iodomethyl)zinc to non-fluorinated β-ketophosphonates such as 2-oxo-2-phenylethyl- or 2-oxopropyl phosphonates were already reported. Thus, the chain extensions yielding γ-ketophosphonates were observed (Arbuzov et al., 1963; Verbicky and Zercher, 2000).
Considering the existing literature on the different reactions of epoxides, we decided to use them as building blocks in the synthesis of fluorinated β,γ-amino alcohol derivatives of alkylphosphonates. The first approach involves the application of amines to the epoxide ring opening with further transformations. The second strategy is based on the use of azides followed by the reduction and phosphonates hydrolysis. The results concerning the applied reaction in the case of compounds 5–8 with different amines are presented in Table 4.
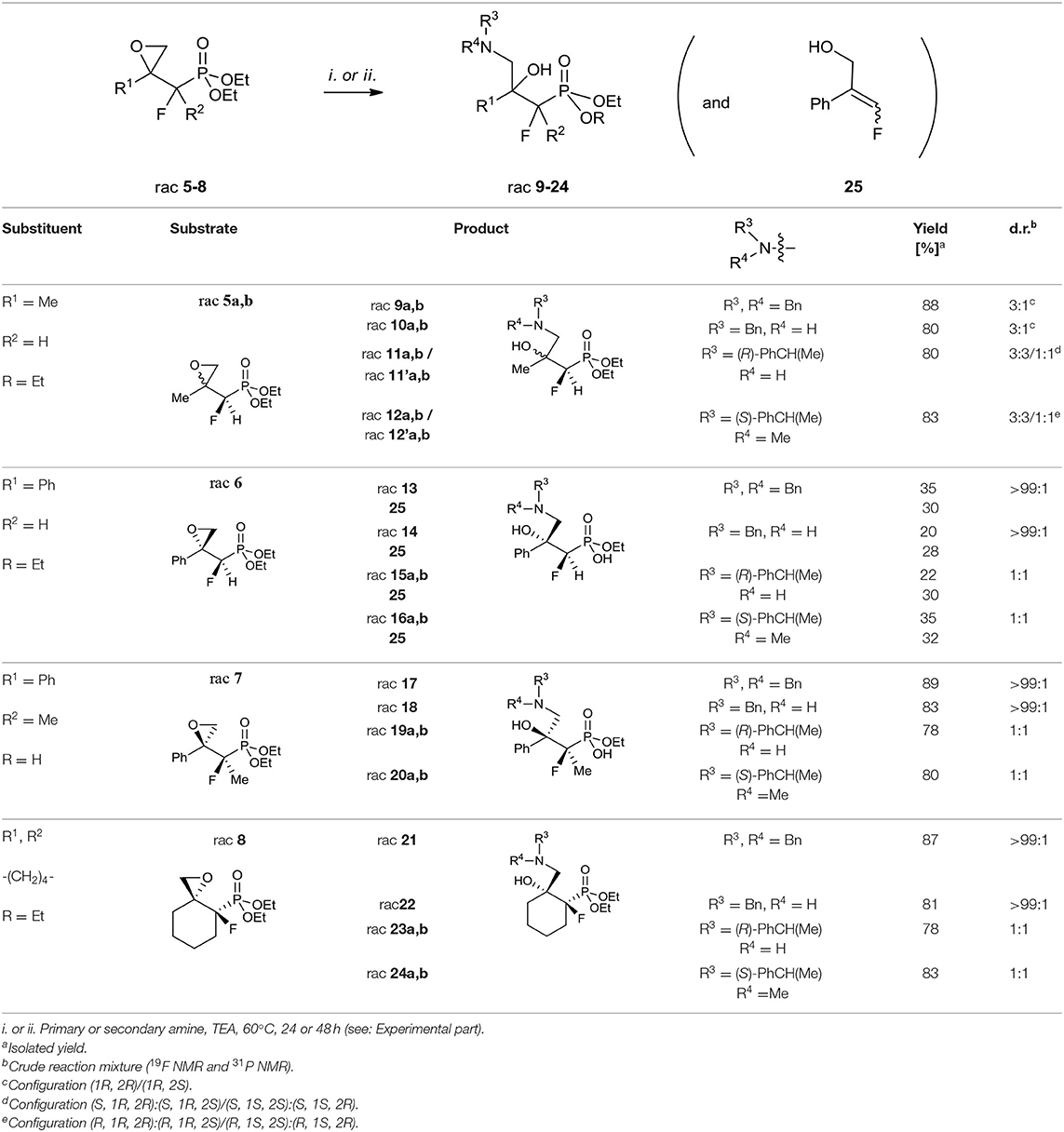
Table 4. Synthesis of γ-amino-β-hydroxy alkylphosphonates 9–24 and 25.
The use of secondary amines [dibenzylamine (Bn2NH), N,α-(R)-dimethylbenzylamine (R)-PhCH(Me)NH(Me)] or primary amines [benzylamine (BnNH2), or (S)-methylbenzylamine [(S)-PhCH(Me)NH2] with the addition of triethylamine (TEA) led to oxirane opening from the unhindered side to give one regioisomer of γ-amino-β-alcohols 9–24. The applications of secondary amines gave products with higher yields than with primary amines. Moreover, the reactions with chiral (S)-methylbenzylamine and N,α-(R)-dimethylbenzylamine gave 11a, b/11'a, b or 12a, b/12'a, b, respectively, as a mixture of diastereoisomers [3:3/1:1, d.r.]. To compare, we observed the formation of two stereoisomers 15–16 and 19–20, and 23–24 [1:1, d.r.] in the case of reaction with 6–8, as judged by NMR (Table 4). However, to open oxirane 7 with amines, an extended reaction time was necessary, yielding phosphonate monoesters 17–20. Base-mediated hydrolysis of dialkyl phosphonates to their monoalkyl phosphonic acids salts was already reported (Westheimer et al., 1988). Usually this reaction requires drastic experimental conditions (high pH and prolongated heating time). To compare, in the case of the reaction of oxirane 6 with amine, the mixtures of unreacted 6, appropriate amino alcohols 13–16 and 25 with poor yields were obtained. The addition of Bn2NH to epoxide 6 was studied in detail by 19F and 31P NMR. Thus, reaction of 6 with one equivalent of Bn2NH led to 13 with low yields (18%), while 35% yield was obtained when applying five equiv. of dibenzylamine and TEA (5 equiv.). During the experiment we observed formation of 25 (McDonald et al., 1985; Huleatt et al., 2015) as a mixture of isomers E/Z: 28:1 (Table 4). Apparently, due to acidic α-proton abstraction (by amide formed from primary or secondary amine and TEA-both amines are necessary), followed by E1cb elimination leading to epoxide-ring opening with subsequent intramolecular phosphonate rearrangement, the phosphate was formed. This compound during acidic work-up transformed into allylic alcohols 25 (Scheme 2).
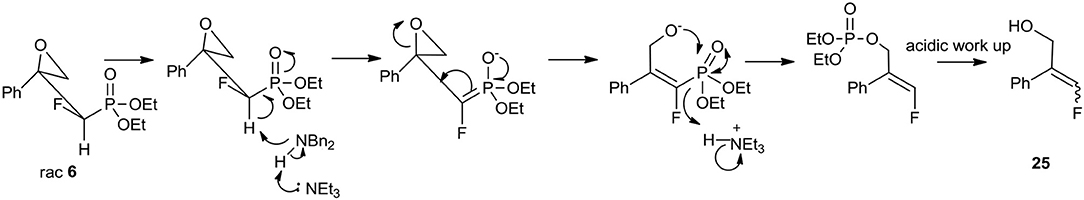
Scheme 2. Mechanism of transformation of 6 to 25 during reaction with Bn2NH and TEA.
The E1cb reactions occurring with syn or anti orientation were already applied in the stereoselective olefin syntheses (Clayden et al., 2008). Moreover, the saponification and elimination of a mixtures of syn and anti diastereoisomers (1:1) of β-hydroksyphosphonates led to the E/Z mixture of olefins (1:1) or vinyl phosphonates, while application of pure anti isomer caused formation of Z stereoisomer via oxaphosphetane, exclusively (Reichwein and Pagenkopf, 2003). Also, the hydrolysis of the C–P bond under basic conditions yielding phosphonate/phosphate conversion has been already reported (McKenna and Shen, 1981; Piettre and Cabanas, 1996; Beier et al., 2008).
Taking into account the low yields of amino alcohols 13–16, difficulties in the purification of the decomposing mixture of 6, 25, and amino alcohols formed from 6 (19F NMR, 31P NMR) the use of the phenyl series toward GABOB analogs synthesis was abandoned.
The analysis of the NMR spectra of 9–24 confirmed the formation of aminohydroxyphosphonates possessing the tertiary hydroxyl group. Thus, the signals of the tertiary carbon atom (C-OH) were located around δ: 72–78 (2JC−F 19–21 Hz) while signals of CH2NR3R4 appeared at δ: 53–59 similarly to tertiary alcohols (Duangdee et al., 2012) and N-benzyl or trityl protected amines (Verbruggen et al., 1996; Wróblewski and Hałajewska-Wosik, 2003). The stereochemistry of the obtained amino alcohol was a consequence of configuration in starting epoxides and was distinguished by spectroscopic data analysis. The examination of the 1H NMR spectra combined with 2D NOESY and 1D H-F HOESY correlations for amino alcohols 9a, b confirmed the configurations as rac (1R, 2R/1R, 2S). Thus, the nOe interactions between the proton as well as fluorine (CHFP) and both oxirane protons in major 9a were observed, while the different enhancements of nOe effects for the CHF proton and fluorine and analogous epoxide protons in the case of minor 9b were noticeable. By analogy, the stereochemistry of one diastereoisomer of 17 as rac (1R, 2S) and tentatively as rac (1R, 2R) for 13 has been assigned. The analysis of nOe effects (2D NOESY, 1D H-F HOESY) for 21 showed, similar to those found on the spectra of 8, the correlations of the methylene (CH2OH) protons with both protons C3H (slightly different nOe effect) of cyclohexane, while the nOe effects between fluorine and both CH2OH protons as well as the contacts between fluorine and axial protons of the cyclohexane ring (C3H and C5H) and vicinal C6 protons indicated configuration (3R,4R) of 21, in the mixture with the predominant conformation. The 2-hydroxyalkylphosphonates are known to exist in CDCl3 in major “frozen” conformation where intramolecular hydrogen bonding between hydroxyl hydrogen and phosphonate oxygen allows them to form a six-membered ring in chair conformation and increase its stability (Zymańczyk-Duda et al., 1995; Genov et al., 1998; Gancarz et al., 2000; Wróblewski and Hałajewska-Wosik, 2002). In the case of cyclohexane derivatives, amino alcohols 21–24 can adopt a conformation similar to trans-decalin as presented in Figure 4.
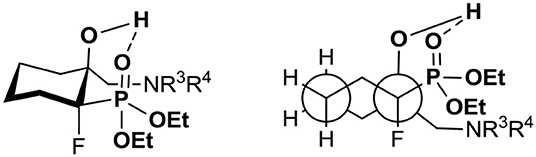
Figure 4. The proposed predominant conformation of γ-amino-β-hydroxyphosphonates 21–24.
The proposed predominant conformation of compounds 21–24 based on the values of 3JC−P 3–5 Hz in a signal derived from the CH2N group in the 13C NMR spectra indicate gauche conformation (regarding phosphonate moiety, P-Cγ). To compare, the other Cγ carbon atoms of the cyclohexane ring are in anti-arrangement as can be demonstrated by 3JC−P 8–9 Hz visible in signals appearing around δ: 32–35 and 19–20 ppm (Wróblewski and Hałajewska-Wosik, 2002).
The obtained regiochemistry is in agreement with the stereochemical outcome of the ring opening of other terminal epoxides at the least hindered position (Parker and Isaacs, 1959; Azoulay et al., 2005; Wu and Xia, 2005; Padwaa and Murphree, 2006; Fustero et al., 2011; Singh et al., 2013). The analogous reaction of optically active (S)-2,3-epoxypropylphosphonate with Bn2NH almost exclusively gave one (S)-enantiomer of amino alcohol (Wróblewski and Hałajewska-Wosik, 2002). Similar reactions of 2,3-oxiranepropylphosphonates with N-tritylamine as well as N-benzhydrylamine were already applied for organophosphorus analog of (R)-4-amino-3-hydroxybutyric acid (L-GABOB) synthesis (Wróblewski and Hałajewska-Wosik, 2003). Moreover, the reaction of dimenthyl-(S)-2,3-oxiranopropylophosphonate with dibenzylamine serve for the synthesis of the non-fluorinated analog of P-GABOB (Nesterov and Kolodiazhnyi, 2007). An alternative method of oxirane 6 opening with aqueous ammonia as conveniently applied in the case of the reaction with epoxy vinyl phosphonate (Cristau et al., 2000) gave a complicated mixture of products decomposing during column chromatography.
Next, the synthesis of γ-amino-β-hydroxy alkylphosphonic acids was performed. Thus, the hydrogenation with concomitant introduction of the Boc group (Pd//C, Boc2O, Table 5, method i.) of dibenzyl derivatives 9, 17, and 21 were applied resulting in the N-Boc (N-tert-butoxycarbonyl)-protected phosphonates 26 and 28. This step, necessary to avoid decomposition of the amino alcohol, was already used in the synthesis of organophosphorus analogs of GABOB (Wróblewski and Hałajewska-Wosik, 2003). On the other hand, due to the presence of one free acidic group (R = H), the debenzylation of 17 to get 27 required only hydrogen and a palladium catalyst as reagents (Table 5, method ii.). The examination of the 1H NMR spectra combined with 1D NOE difference spectrometry and the 1D H-F HOESY spectra for amino alcohols 27 indicated the nOe interactions between methyl protons and one of the diastereotopic epoxide protons at the same time without nOe effect with a phenyl group, while in the case of fluorine spectra (1D HOESY) the interactions for both oxirane rings were detected. Taking into account the predominant “frozen” conformation where intramolecular hydrogen bonding between hydroxyl hydrogen and phosphonate oxygen were formed, the configuration of 27 as rac (2R, 3S) was assigned, additionally confirming the stereochemistry of starting oxirane 7. In the last step, acidic hydrolysis of 26–28 (conc HCl, reflux, 2–3 days) followed by propylene oxide treatment (Wróblewski and Hałajewska-Wosik, 2003; Table 5, method iii.) gave target compounds as salts 29–31.
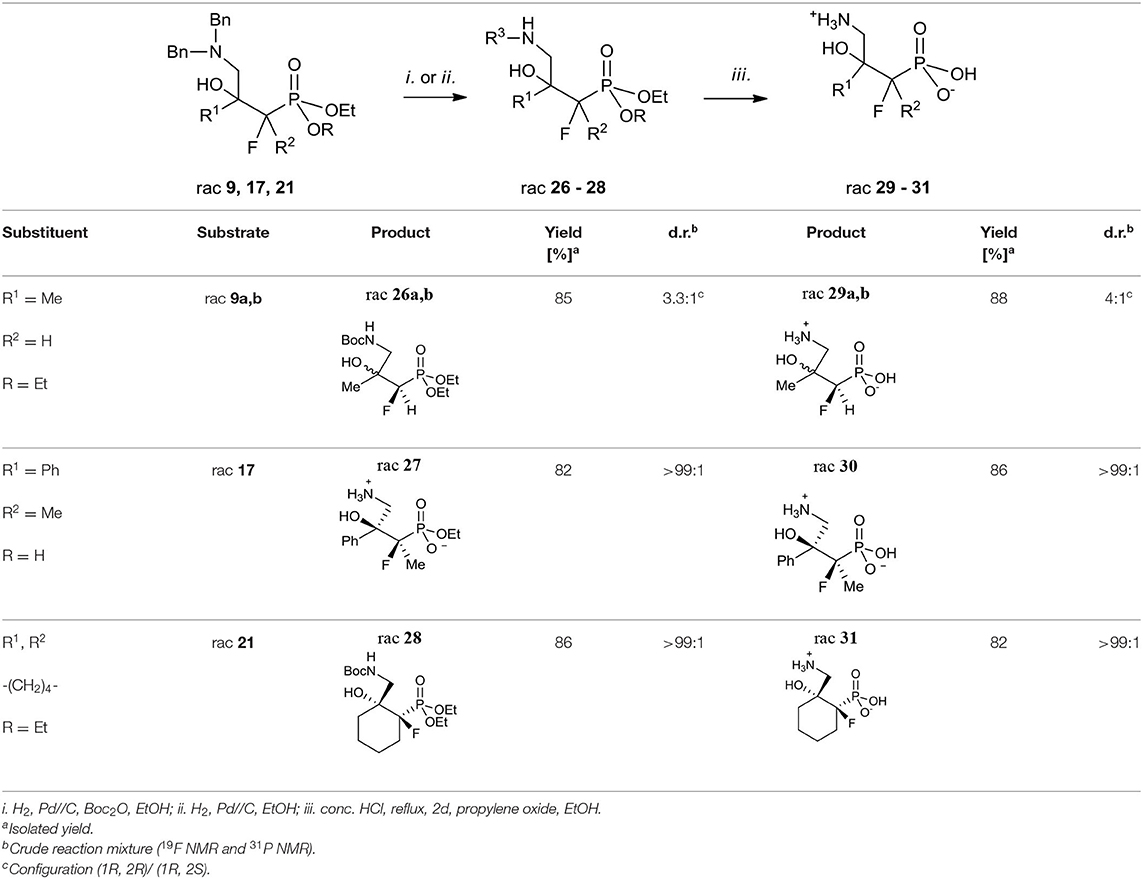
Table 5. Synthesis of fluorinated γ-amino-β-hydroxy alkylphosphonic acids 29–31.
In the case of substrate 26a, b, two diastereoisomers of 29a, b (4:1, d.r.) were formed, while the reaction of 27–28 gave the single diastereoisomers of 30–31 (>99:1, d.r.). The configurations in the obtained γ-amino-β-alcohols 29–31 resulted from the stereochemistry of starting amino alcohols and as rac (1R, 2R) for major 29a, as rac (1R, 2S) for minor 29b, and as rac (2R, 3S) for 30 were assigned (Table 5). Additionally, the analysis of one- and two-dimensional NMR spectra allowed us to confirm the structure of 31 as rac (1R, 2R). Importantly, in the acidic conditions, we did not observe rearrangement or racemization, but we noticed that compounds were contaminated with elimination products as can be judged by NMR.
Our strategy for the synthesis of β-amino-γ-hydroxy phosphonates relied on the construction of tertiary amines via bromides or azides.
Considering oxiranes as useful intermediates for the syntheses of halogenohydrines, we decided to react obtained epoxides 5–8 with hydrogen bromide (generated from AcBr and MeOH). The reactions led to vicinal bromohydrines 32–35 with yields from 86% for 33 to 94% for 34 (method i. Table 6).
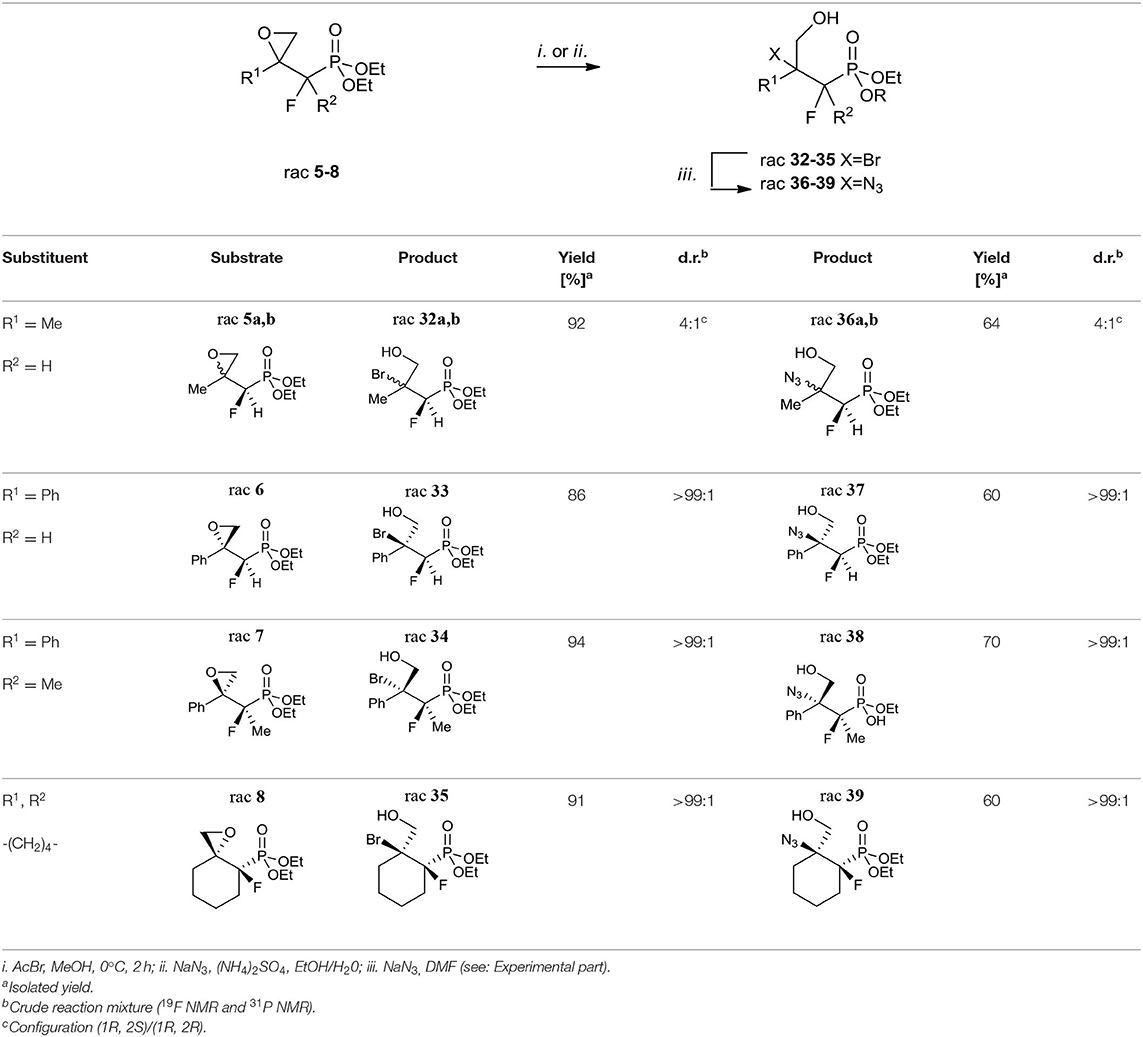
Table 6. Synthesis of fluorinated β-bromo- or β-azido-γ-hydroxy- alkylphosphonates 32–35 or 36–39.
As a result of the halide-induced oxirane opening, the only one regioisomer, tertiary bromide was observed in all cases, even in the case of derivatives of styrene and more substituted terminal cyclohexane oxiranes. The diastereoselectivity of the bromohydrine formations was analogous to those observed for the synthesis of compounds 5–8. We observed the formation of both diastereoisomers of bromohydrine in the case of compound 32 (32a:32b, 4:1, d.r.), while single diastereoisomers (>99:1, d.r.) were obtained in the case of 33–35. The analysis of the 2D NOESY and 1D H-F HOESY difference spectra of 32a indicated that fluorine interacts with only one of the diastereotopic protons of CH2OH, while its geminal proton derived from the CHF moiety had a slightly different nOe effect with both methylene protons. To compare, in minor 32b the nuclei of the CHF group correlated with both protons of the hydroxymethyl group indicating an opposite tendency compared to 9a, b, and configurations rac (1R, 2S) for major 32a and rac (1R, 2S) for minor 32b were assigned. Moreover, the different nOe effects (1D NOE, 1D HOESY) with both methylene protons of CH2OH and the fluorine atom as well as the Me group indicated a similarity to interactions characterizing minor 32b. To compare, the correlation between one of the methylene protons derived from CH2OH with C3 and axial C4&C6 together with a different enhancement of signals of CHHOH with fluorine observed on HOESY spectra suggested the axial arrangement of both fluorine and hydroxymethyl moiety together with the bulky equatorial phosphonate group as a predominant conformer of 35 with (1R, 2S) configuration. By analogy, the stereochemistry of one diastereoisomer of 33 as rac (1R, 2S) and 34 as rac (2R, 3R) were assigned. The additional diagnostic signals confirming tertiary bromide formation appeared in the 13C NMR spectra. Thus, signals of atom C-2 (Br) in the spectra of 32–35 were located around δ: 72–78 (2JC−F 18–21 Hz), while in starting oxiranes C-2 signals appeared in the range δ: 55–63, and were slightly shifted compared to tertiary bromides such as 2-bromo-2-phenylpropane (Atack et al., 2014). Altogether, this allows us to suggest that the introduction of bromide via (protonated) oxirane ring-opening occurred through an SN2-like (mixed SN1/SN2) mechanism with the nucleophilic attack on a more substituted carbon atom. Usually, in the case of acid-mediated opening of derivatives of styrene oxide and more substituted terminal cyclohexane oxirane the formation of the mixture of regioisomers (with the predominance of a more substituted product) or partial racemization through carbocation takes place (Parker and Isaacs, 1959; Costantino et al., 1982; Bell and Ciaccio, 1986; Lin and Whalen, 1994; Padwaa and Murphree, 2006; Singh et al., 2007; Morton et al., 2009).
Similarly, the reactions of oxiranes 5–8 with sodium azides [NaN3, (NH4)2SO4, EtOH/H20] (Głowacka, 2009) were conducted to give azidohydrine 36–39 as the single regioisomers with yields from 60% for 37 and 39 to 70% for 38 and diastereoselectivity from (4:1, d.r.) for 36 to the sole product in the case of 37–39 (>99:1, d.r; Table 6 method ii.). The application of ammonium chloride (Głowacka, 2009) instead of (NH4)2SO4 resulted in the formation of a mixture of appropriate chlorides and hydrolysis products. Additionally, it is worth noting that the introduction of azide in the case of 7 had to be conducted for 2 days, and gave monoester 38 (R = H). The analysis of IR (band at 2,100 cm−1) (Hesse et al., 1997) as well as the 13C NMR spectra of 36–39 confirmed the formation of tertiary azides. Thus, the signals of (C-OH) carbon atoms were located around δ: 73–79 (3JC−P 19–21 Hz) while signals of CN3 appeared at δ: 57, similarly to 2-azido-2-phenylpropan-1-ol (Prasad et al., 2015). Although 3-hydroxyphosphonates 36–39 can rotate freely around the P-C1 and C1-C2 bonds, we would like to emphasize that in [D] chloroform they exist predominantly as their anti-conformers (arrangement of phosphonate and CH2OH moieties regarding C1-C2 bonds) as can be judged from the large (18–21 Hz) values of their 3JC−P couplings together with the analysis of the NOESY and H-F HOESY spectra. In the case of major 36a, similar interactions between the proton of the CHF group and fluorine (2D NOESY and 2D H-F HOESY) with both methylene CH2OH protons were observed, while in the case of the minor isomer 36b the different enhancements of analogous signals due to nOe effects were detected, similarly to interactions in 32a, b. In the case of 37, we observed nOe effects (1D NOESY, 1D HOESY) related to 36a nOe effects, while the interactions between fluorine and other protons in the 38 spectra corresponded to these detected for 36b, as expected. Moreover, the different nOe effects (1D NOE, 1D HOESY) with both methylene protons of CH2OH and fluorine as well as the Me group indicated a similarity to interactions characterizing minor 32b. Analogously to 35, the correlation between one of the methylene protons derived from CH2OH with both C3 protons and axial C4 together with a different enhancement of signals of CHHOH with fluorine and the interaction of F with axial C3&C5 protons observed on the HOESY spectra of 39 indicated the (1R, 2S) configuration. These results are analogical to the stereochemistry determined for bromides 32–35 (Table 6). These observations confirmed that the oxirane ring-opening happened with inversion of configuration (SN2-like mechanism) and substitution occurred at the tertiary carbon atom in each case.
A conventional way to get the vicinal azido alcohols involves epoxides ring-opening (Benedetti et al., 1998; Fringuelli et al., 1999; Amantini et al., 2002; Badalassi et al., 2004; Nesterov and Kolodiazhnyi, 2007; Głowacka, 2009) or displacement of halohydrins (Draper, 1983; Iacazio and Réglier, 2005) under acidic/basic conditions with the application of different azide sources as well as chemoselectivity by the reduction of α-azido ketones (Rao et al., 1992; Ordóñez et al., 2003; Ankati et al., 2008). As an example, the opening of diethyl (S)-2,3-oxiranepropylphosphonate with sodium azide in the presence of (NH4)2SO4 resulted in the formation of single regioisomer (R)-3-azido-2-hydroxypropylphosphonate as well (Głowacka, 2009). To compare, the alternative reactions of bromohydrines 32–35 with sodium azide in dimethylformamide as a solvent (Table 6, method iii.) gave the mixture of azidohydrines 36–37, 39, or 38′ (R = Et) as major compounds, as well as appropriate starting oxiranes 5–8 (ratio: see Experimental part). To explain the observed diastereoselectivity, the reaction of bromide 32a, b (4:1, d.r.) with NaN3 in DMF was carried out. As a result, the azide 36a, b (3:1, d.r.; δ = 17.6:18.6 in 31P NMR) and epoxide 5a, b (14:1, d.r.; δ 14.1:14.9 in 31P NMR) with a ratio for 36/5 as 5:1, were formed. Apparently, the formation of carbocation took place. Stereochemistry of the addition of azide to carbocation (like in carbonyl compound) was a consequence of the steric hindrance on the adjacent stereogenic center allowing them to attack from opposite the fluorine side (path a, Scheme 3). Moreover, partial racemization leading to both diastereoisomers of azide and formation of both diastereoisomeric epoxides as a result of the attack of a lone-pair nucleophile such as the hydroxyl group from the alternative side (path b, Scheme 3) supported the proposed course of the reaction.
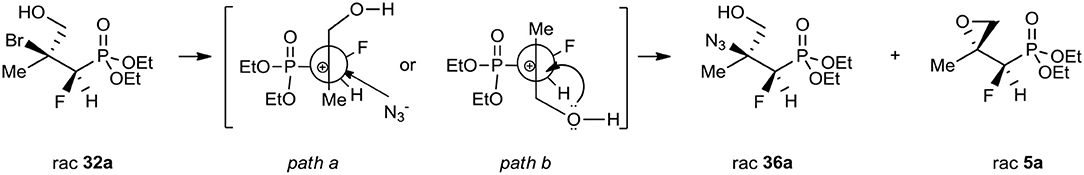
Scheme 3. The preferred trajectory of nucleophilic attack during transformation of 32a.
Next, the obtained azidohydrines were applied to afford the vicinal β-amino-γ-hydroxy phosphonates. The reduction of azides 36–39 followed by protection of the nitrogen group [H2, Pd//C, (Boc2O) method i. or ii. Table 7] led to the N-Boc derivatives 40–42 or compound 27. Subsequent acidic hydrolysis (conc HCl, reflux, 2–3 days, propylene oxide) (Wróblewski and Hałajewska-Wosik, 2003; Table 7, method iii.) surprisingly gave γ-amino-β-hydroxy phosphonates 29–31, and 43 instead of their β-amino-γ-hydroxy regioisomers. Analysis of spectra confirmed that as the products compounds 29a, b as a mixture (4:1, d.r.) and 30–31 as well as 43 [with tentatively assigned stereochemistry (1R,2S)] as single diastereoisomers were obtained.
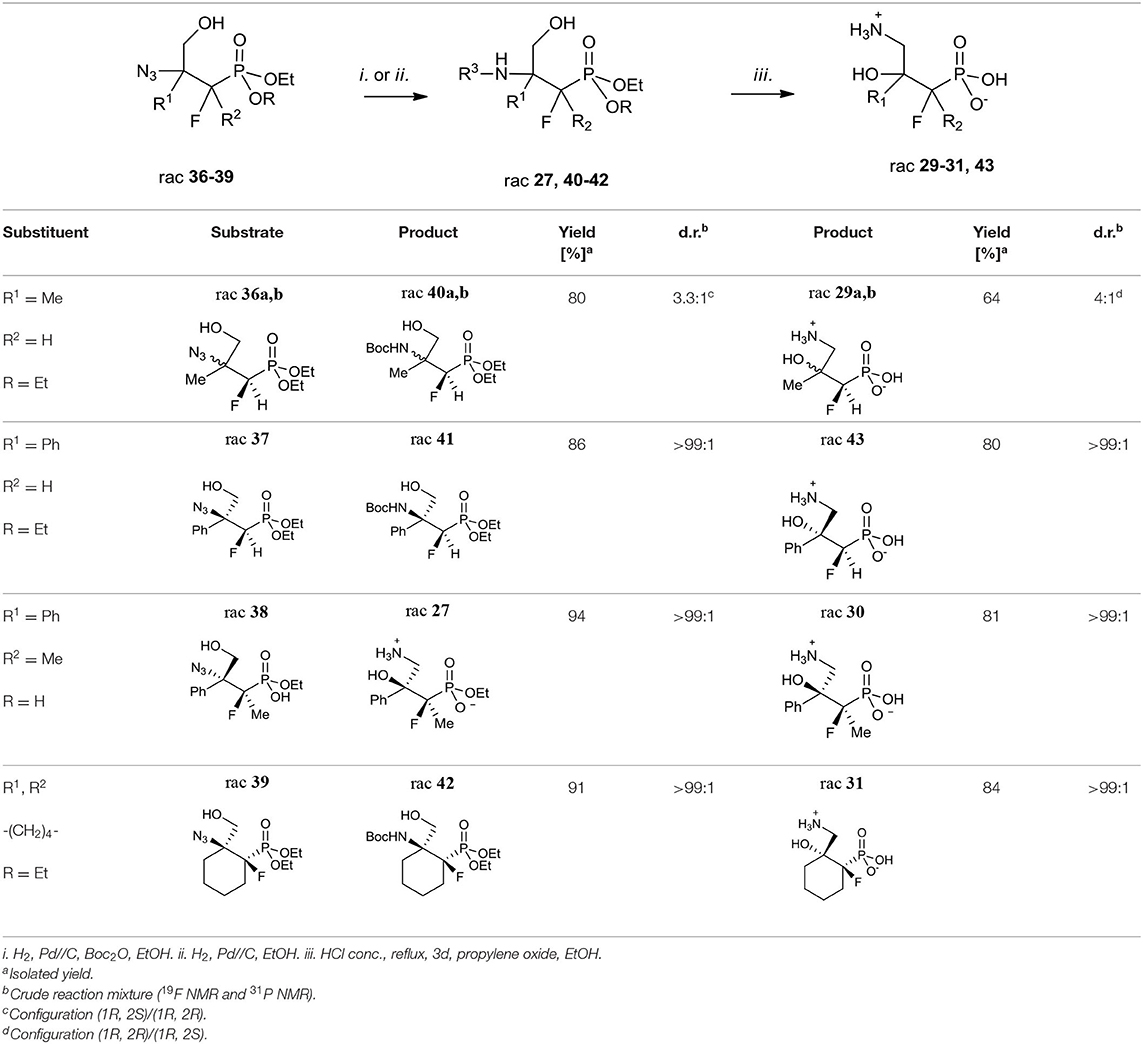
Table 7. Synthesis of fluorinated γ-amino-β-hydroxy alkylphosphonic acids 29–31 and 43.
Apparently, during acid treatment the protonation of a more basic amino group takes place. Next, due to the attack of the neighboring group from the opposite side, the protonated oxirane is formed. Subsequent oxirane ring-opening by ammonia leads to rearrangement products such as γ-amino-β-hydroxy alkylphosphonic acid (Scheme 4).
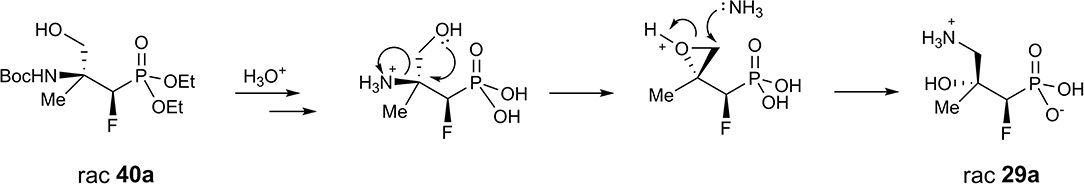
Scheme 4. Proposed mechanism of the rearrangement of 40a to 29a during acid-mediated hydrolysis.
Moreover, the formation of compound 27 supports the proposed mechanism. We already observed the participation of the neighboring group (substituted amines) during deoxyfluorination of α-hydroxy-β-aminophosphonate derivatives of amino acids, leading to β-fluoro-α-aminophosphonates (Kazmierczak and Koroniak, 2012; Kaczmarek et al., 2018).
Conclusion
The diastereoselective reaction of monofluorinated β-ketophosphonates bearing methyl, phenyl, or cycloalkane substituents with diazomethane lead to oxiranes possessing a tertiary carbon atom. The heteronucleophiles-induced ring-opening, followed by acidic hydrolysis allowed us to obtain designed γ-amino-β-hydroxy alkylphosphonic acids. Moreover, epoxide ring-opening with HBr or NaN3 yielded substituted tertiary β-bromohydrines or β-azidoalcohol phosphonate derivatives. Subsequent azide reduction and acidic hydrolysis resulted in the formation of γ-amino-β-hydroxy alkylphosphonic acids, as the rearrangement products. Interestingly, in the cases of phenyl and cyclohexane derivatives, we observed excellent regio- and diastereoselectivity in the reactions.
We expect that our aminophosphonates and the observed rearrangement will found an application for drug design. To verify our assumptions concerning interactions of fluorine with neighboring groups in aminophosphonates, further syntheses will be carried out. We believe our methods will find tremendous application in the synthesis of biologically active molecules and useful intermediates.
Materials and Methods
General Information
1H NMR, 13C NMR, 19F NMR, and 31P NMR spectra were performed on Bruker ASCEND 400 (400 MHz), Bruker ASCEND 600 (600 MHz), and Varian Mercury (300 MHz) spectrometers, as is noted. The 2D and 1D selective NMR spectra (1D NOESY and 1D H-F HOESY) were recorded on Bruker ASCEND 600 (600 MHz) or Bruker ASCEND 400 (400 MHz) spectrometers. Chemical shifts of 1H NMR were expressed in parts per million downfield from tetramethylsilane (TMS) as an internal standard (δ = 0) in CDCl3. Chemical shifts of 13C NMR were expressed in parts per million downfield and upfield from CDCl3 as an internal standard (δ 77.16) or CD3OD (δ 49.00) or CF3COOD (δ 164.2) or traces of solvent. Chemical shifts of 19F NMR were expressed in parts per million upfield from CFCl3. The ethereal solution of diazomethane was prepared as described (Vogel et al., 1989). Compounds 1 (Radwan-Olszewska et al., 2011), 2 (Cox et al., 2005), 3 (Hamashima et al., 2005), and 4 (Kim, 2005) were prepared as described. The NMR data for 25 (McDonald et al., 1985; Huleatt et al., 2015) was in good agreement. For more information see Supplementary Materials.
Theoretical Calculations
The quantum mechanical calculations of potential energy under vacuum at the M06/6-31+G** (Hehre et al., 1986; Zhao and Truhlar, 2008) level of theory were performed using the GAUSSIAN09 program (Frisch et al., 2013), in order to systematically search for possible conformations. The vibrational frequencies were calculated using the same method, and then their positivity was applied to confirm that each of the calculated structures corresponded to a minimum on the potential energy surface. To simplify the calculations, the ethoxyl substituents were replaced with methoxyl substituents for which several conformations were calculated with the aim of choosing the global minimum-energy structure.
Procedures
Procedure for Oxirane 5–8 Preparation
The ethereal solution of diazomethane was cooled in an ice-bath [distilled from the mixture of diazald (964 mg, 4.5 mmol)] in diethyl ether (15 mL) and KOH (252 mg, 4.5 mmol) [dissolved in EtOH (4.5 mL); yield 65–70%] (Vogel et al., 1989) was quickly cooled in an ice-bath and carefully added to the flask containing β-ketophosphonates 1–4 (1.5 mmol). Next, the tightly closed reaction mixture (with glass stopper) was stirred for 48 h at room temperature (monitored by NMR). The solvent was evaporated and the residue was purified by column chromatography to give compounds 5–8.
rac Diethyl ((R)-fluoro((R)-2-methyloxiran-2-yl)methyl)phosphonate (rac 5a), major isomer: Isolated as a mixture with 5b, which could not be separated by the chromatography techniques employed in this study transparent oil (315 mg, 93%, 3.3:1 d.r.): 1H NMR (600 MHz, CDCl3) δ = 4.29 (dd, J = 45.8, 7.8 Hz, 1H, CHF), 4.16–4.06 (m, 4H, OCH2), 2.78 (“d,” J = 4.4 Hz, 1H, CHH), 2.66 (dd, J = 5.8, 4.4 Hz, 1H, CHH), 1.41 (s, 3H, CH3), 1.25 (t, J = 7.3 Hz, 3H, OCH2CH3), 1.24 (td, J = 7.3 Hz, 3H, OCH2CH3). 13C NMR (151 MHz, CDCl3) δ = 92.23 (dd, J = 187.7, 166.2 Hz, CFP), 63.47 (d, J = 6.8 Hz, OCH2), 62.97 (d, J = 6.6 Hz, OCH2), 55.28 (dd, J = 22.4, 9.2 Hz, C(OC)Me), 50.62 (d, J = 7.2 Hz, CH2), 16.36 (d, J = 3.7 Hz, CH3), 16.27 (d, J = 5.5 Hz, OCH2CH3), 16.29 (d, J = 5.5 Hz, OCH2CH3). 19F NMR (565 MHz, CDCl3) δ = −213.83 (ddd, J = 79.8, 45.8, 5.6 Hz). 31P{/1H} NMR (243 MHz, CDCl3) δ = 14.12 (d, J = 79.8 Hz). 31P NMR (122 MHz, CDCl3) δ = 14.12 (dh, J = 79.8, 7.9 Hz). MS (EI) m/z = 227.1 [M+H] + IR (film): ν = 2,985, 2,934, 1,445, 1,368, 1,256, 1,163, 1,017, and 971 cm−1.
rac Diethyl ((R)-fluoro((S)-2-methyloxiran-2-yl)methyl)phosphonate (rac 5b), minor isomer: 1H NMR (600 MHz, CDCl3) δ = 4.52 (dd, J = 45.9, 5.7 Hz, 1H, CHF), 4.16–4.06 (m, 4H, OCH2), 2.92 (d, J = 4.8 Hz, 1H, CHH), 2.58 (dd, J = 4.8, 2.1 Hz, 1H, CHH), 1.40 (d, J = 1.5 Hz, 3H, CH3), 1.26 (t, J = 7.3 Hz, 3H, OCH2CH3), 1.24 (t, J = 7.3 Hz, 3H, OCH2CH3). 13C NMR (151 MHz, CDCl3) δ = 88.75 (dd, J = 187.6, 166.2 Hz, CFP), 63.47 (d, J = 6.8 Hz, OCH2), 62.97 (d, J = 6.6 Hz, OCH2), 54.56 (dd, J = 21.9, 5.3 Hz, C(OC)Me), 51.43 (t, J = 4.9 Hz, CH2), 17.86 (t, J = 1.9 Hz, CH3), 16.31 (d, J = 4.9 Hz, OCH2CH3), 16.26 (d, J = 5.1 Hz, OCH2CH3). 19F NMR (565 MHz, CDCl3) δ = −214.21 (dd, J = 74.0, 45.9 Hz). 31P{/1H} NMR (243 MHz, CDCl3) δ = 14.96 (d, J = 74.4 Hz). 31P NMR (122 MHz, CDCl3) δ = 14.96 (br dh, J = 74.2, 7.4 Hz).
rac Diethyl ((R)-fluoro((R)-2-phenyloxiran-2-yl)methyl)phosphonate (rac 6), transparent oil (398 mg, 92%): 1H NMR (300 MHz, CDCl3) δ = 7.53–7.48 (m, 2H, Ph), 7.39–7.32 (m, 3H, Ph), 5.05 (dd, J = 45.4, 8.1 Hz, 1H, CHF), 4.19–4.04 (m, 2H, OCH2), 4.04–3.93 (m, 2H, OCH2), 3.40 (d, J = 5.2 Hz, 1H, CHH), 2.93 (td, J = 5.6, 0.7 Hz 1H, CHH), 1.26 (td, J = 7.1, 0.5 Hz, 3H, OCH2CH3), 1.15 (td, J = 7.1, 0.6 Hz, 3H, OCH2CH3). 13C NMR (75 MHz, CDCl3) δ = 136.45 (d, J = 1.9 Hz, Ph), 128.98, 128.54, 127.40 (3 × s, Ph), 90.75 (dd, J = 190.9 Hz, 168.1 Hz, CFP), 63.19 (dd, J = 12.5, 6.6 Hz, C(OC)Ph), 62.61 (d, J = 6.5 Hz, OCH2), 51.80 (dd, J = 7.9, 2.9 Hz, CH2), 16.19 (d, J = 6.0 Hz, OCH2CH3), 16.18 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (282 MHz, CDCl3) δ = −210.19 (ddd, J = 78.3, 45.5, 5.5 Hz). 31P{/1H} NMR (121 MHz, CDCl3) δ = 12.79 (d, J = 78.3 Hz). 31P NMR (122 MHz, CDCl3) δ = 12.73 (dp, J = 78.4, 7.9 Hz). MS (EI) m/z = 288.0 [M+]. IR (film): ν = 2,985, 2,927, 1,642, 1,447, 1,391, 1,371, 1,258, 1,161, 1,017, and 974 cm−1.
rac Diethyl ((R)-1-fluoro-1-((S)-2-phenyloxiran-2-yl)ethyl)phosphonate (rac 7), transparent oil (408 mg, 90%): 1H NMR (400 MHz, CDCl3) δ = 7.54–7.49 (m, 2H, Ph), 7.39–7.28 (m, 3H, Ph), 4.17–4.08 (m, 2H, OCH2), 4.05 (“quint,” J = 7.0 Hz, 2H, OCH2), 3.39 (dd, J = 5.1, 0.8 Hz, 1H, CHH), 2.88 (dd, J = 5.1, 2.7 Hz, 1H, CHH), 1.62 (dd, J = 24.7, 14.0 Hz, 3H, CH3), 1.23 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.18 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 135.95 (t, J = 1.8 Hz, Ph), 129.13 (d, J = 1.4 Hz, Ph), 128.35, 127.61 (2 × s, Ph), 95.91 (dd, J = 184.9, 171.7 Hz, CFP), 63.25 (d, J = 7.2 Hz, OCH2), 63.23 (dd, J = 7.0, 1.7 Hz, OCH2), 61.87 (dd, J = 23.4, 8.3 Hz, C(OC)Ph), 50.84 (dd, J = 6.2, 2.7 Hz, CH2), 19.26 (dd, J = 22.5, 2.2 Hz, CH3), 16.25 (t, J = 5.8 Hz, OCH2CH3), 16.19 (t, J = 5.8 Hz, OCH2CH3). 19F NMR (376 MHz, CDCl3) δ = −170.39 (dqd, J = 89.9, 24.9, 2.8 Hz). 31P{/1H} NMR (162 MHz, CDCl3) δ = 16.46 (d, J = 89.2 Hz). 31P NMR (122 MHz, CDCl3) δ = 16.47 (dqd, J = 89.6, 14.2, 7.1 Hz). MS (EI) m/z = 303.2 [M+H] +.
rac Diethyl ((3R,4R)-4-fluoro-1-oxaspiro[2.5]octan-4-yl)phosphonate (rac 8), transparent oil (379 mg, 95%): 1H NMR (400 MHz, CDCl3) δ = 4.29–4.24 (m, 2H, OCH2CH3), 4.24–4.19 (m, 2H, OCH2CH3), 3.30–3.27 (m, 1H, CHH, C2H), 2.53 (q, J = 4.7 Hz, CHH, C2H), 2.45–2.37 (m, 1H, C5H), 2.32–2.26 (m, 1H, C8H), 1.88–1.79 (m, 2H, C5H&C6H), 1.82–1.73 (m, 2H, C6H&C7H), 1.56–1.50 (m, 2H, C7H&C8H), 1.34 (t, J = 7.0 Hz, 3H, OCH2CH3), 1.33 (t, J = 7.0 Hz, 3H, OCH2CH3). 1H{/19F} NMR (400 MHz, CDCl3) δ = 4.23 (quint, J = 7.2 Hz, 2H), 4.16 (quint, J = 7.2 Hz, 2H), 3.24 (d, J = 5.3 Hz, 1H), 2.49 (d, J = 4.9 Hz, 1H), 2.45–2.35 (m, 1H), 2.26 (m, 1H), 1.88–1.79 (m, 1H), 1.81 (br d, J = 10.0 Hz, 1H), 1.75–1.63 (m, 2H), 1.54–1.48 (m, 2H), 1.34 (t, J = 7.0 Hz, 3H), 1.33 (t, J = 7.0 Hz, 3H). 1H{/31P} NMR (400 MHz, CDCl3) δ = 4.23 (q, J = 7.1 Hz, 2H), 4.16 (q, J = 7.1 Hz, 2H), 3.24 (d, J = 5.3 Hz, 1H), 2.48 (t, J = 4.9 Hz, 1H), 2.45–2.37 (m, 1H), 2.32–2.26 (m, 1H), 1.88–1.79 (m, 1H), 1.81 (br dd, J = 13.0, 10.0 Hz, 1H), 1.75–1.63 (m, 2H), 1.54–1.48 (m, 2H), 1.33 (t, J = 7.0 Hz, 3H), 1.32 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ = 93.47 (dd, J = 191.1, 168.8 Hz, CFP, C4), 63.19 (d, J = 6.5 Hz, OCH2), 62.94 (d, J = 7.2 Hz, OCH2), 59.95 (d, J = 20.2 Hz, C(OC)C), 50.25 (dd, J = 10.3, 7.1 Hz, CH2O), 33.72 (dd, J = 18.8, 2.3 Hz, C5), 31.58 (d, J = 1.4 Hz, C8), 24.12 (s, C7), 21.64 (dd, J = 8.3, 1.7 Hz, C6), 16.30 (d, J = 6.3 Hz, OCH2CH3), 16.24 (d, J = 6.4 Hz, OCH2CH3). 19F NMR (376 MHz, CDCl3) δ = −173.04 (br d, J = 90.3 Hz). 31P{/1H} NMR (162 MHz, CDCl3) δ = 16.88 (d, J = 88.4 Hz). 31P NMR (122 MHz, CDCl3) δ = 16.89 (ddq, J = 87.6, 24.5, 8.2 Hz). MS (EI) m/z = 268.1 [M+H]+.
General Procedure (Procedure A) for Oxiranes 5, 7–8 Opening by Secondary or Primary Amine
To the mixture of secondary or primary amine (0.48 mmol) and triethylamine (56 μL, 40 mg, 0.4 mmol) dissolved in EtOH (2 mL), epoxides 5–8 (0.4 mmol) were added. Next, the reaction mixture was heated in an oil bath at 60°C for 24–60 h (monitoring by TLC). Then, the reaction mixture was evaporated and purified by flash column chromatography (1 cm layer of silica gel) with chloroform CHCl3 → 5% MeOH/CHCl3 (v:v) to give appropriate amino alcohols 9–12 and 17–24. For the spectroscopic properties of compounds 10–12, 18–20, and 22–24 see Supplementary Materials.
General Procedure (Procedure B) for Oxirane 6 Opening by Secondary or Primary Amine
To the mixture of secondary or primary amine (2 mmol) and triethylamine (279 μL, 200 mg, 2 mmol) dissolved in EtOH (2 mL), epoxide 6 (0.4 mmol) was added. Next, the reaction mixture was heated in an oil bath at 60°C for 24–60 h (monitoring by TLC). Then the reaction mixture was diluted with CH2Cl2 (20 mL) and aqueous HCl (1 M, 10 mL) was added. The mixture was extracted with CH2Cl2 (3 ×20 mL). The combined extracts were washed with aqueous sodium bicarbonate, brine, dried over Na2SO4, filtrated, and concentrated under reduced pressure. The residue was purified by flash column chromatography (1 cm layer of silica gel) with CHCl3 → 5% MeOH/CHCl3 (v:v) to give a mixture of 6, appropriate amino alcohols 13–16, and allylic alcohol 25. For spectroscopic properties of compounds 14–16 see Supplementary Materials.
rac Diethyl ((1R,2R)-3-(dibenzylamino)-1-fluoro-2-hydroxy-2-methylpropyl)phosphonate (rac 9a), procedure A (Bn2NH, TEA). Major isomer: isolated as a mixture with 9b, which could not be separated by the chromatography techniques employed in this study; transparent oil (145 mg, 85%, 3:1 d.r.): 1H NMR (300 MHz, CDCl3) δ = 7.35–7.31 (m, 10H, Ph), 4.54 (dd, J = 44.9, 4.9 Hz, 1H, CHF), 4.26–4.06 (m, 4H, OCH2), 3.86 (d, J = 13.9 Hz, 2H, CHHPh), 3.54 (d, J = 13.7 Hz, 2H, CHHPh), 2.86–2.82 (m, 1H, CHH), 2.79 (dd, J =14.1, 2.0 Hz, 1H, CHH), 1.36–1.33 (m, 3H, CH3), 1.33–1.30 (m, 3H, OCH2CH3), 1.30–1.28 (m, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 140.34, 128.51, 128.27, 127.06 (4 × s, Ph), 91.79 (dd, J = 188.3, 162.5 Hz, CFP), 73.64 (dd, J = 17.9, 2.2 Hz, COH), 64.01 (dd, J = 6.9, 1.9 Hz, OCH2), 62.65 (d, J = 6.8 Hz, OCH2), 59.79 (d, J = 3.9 Hz, CH2Ph), 59.05 (dd, J = 7.9, 4.6 Hz CH2N), 23.08 (CH3), 16.51 (d, J = 5.3 Hz, OCH2CH3), 16.46 (d, J = 5.3 Hz, OCH2CH3). 19F NMR (283 MHz, CDCl3) δ = −213.28 (dd, J = 78.5, 44.8 Hz). 31P{/1H} NMR (122 MHz, CDCl3) δ = 16.81 (d, J = 78.6 Hz). IR (film): ν = 3,331, 3,061, 2,980, 2,925, 2,850, 2,797, 1,643, 1,604, 1,494, 1,453, 1,366, 1,258, 1,056, 1,028, and 976 cm−1 GC–MS m/z = 424 [M+H]+ tR = 14.60 min.
rac Diethyl ((1R,2S)-3-(dibenzylamino)-1-fluoro-2-hydroxy-2-methylpropyl)phosphonate (rac 9b), minor isomer: 1H NMR (300 MHz, CDCl3) δ = 7.35–7.31 (m, 10H, Ph), 4.87 (dd, J = 44.7, 2.6 Hz, 1H, CHF), 4.26–4.06 (m, 4H, OCH2), 3.85 (d, J = 13.9 Hz, 2H, CHHPh), 3.65 (d, J = 13.7 Hz, 2H, CHHPh), 2.88 (dt, J = 13.4, 1.4 Hz, 1H, CHH), 2.61 (ddd, J = 14.2, 3.1, 2.0 Hz, 1H, CHH), 1.33–1.30 (m, 3H, CH3), 1.30–1.28 (m, 3H, OCH2CH3), 1.28–1.26 (m, 3H, OCH2CH3). 19F NMR (283 MHz, CDCl3) δ = −209.25 (ddd, J = 71.4, 44.8, 2.0 Hz). 31P{/1H} NMR (122 MHz, CDCl3) δ = 18.21 (d, J = 71.5 Hz).
rac Diethyl ((1R,2R)-3-(dibenzylamino)-1-fluoro-2-hydroxy-2-phenylpropyl phosphonate (rac 13), procedure B (Bn2NH, TEA). Isolated as a mixture with 6 and 25, which could not be separated by the chromatography techniques employed in this study, (6/13/25 crude ratio: 20/40/40, NMR) transparent oil (rac 13 68 mg, 35%; 25 18 mg, 30%): 1H NMR (300 MHz, CDCl3): δ = 7.20–7.51 (m, 15H, Ph), 5.1 (s, 1H, OH), 4.79 (dd, J = 45.0, 5.9 Hz, 1H, CHF), 4.0–4.15 (m, 2H, OCH2), 3.7–3.8 (m, 2H, OCH2), 3.59 (d, J = 12.7 Hz, 1H, CHH), 3.37 (d, J = 13.5 Hz, 2H, CHHPh), 3.32 (dd, J = 13.6, 1.8 Hz, 1H, CHHPh), 3.12 (dd, J = 13.6, 1.2 Hz, 1H, CHH), 1.23 (td, J = 7.1, 0.5 Hz, 3H, OCH2CH3), 1.02 (td, J = 7.1, 0.4 Hz, 3H, OCH2CH3). 19F NMR: δ = −212.15 (dd, J = 81.8, 44.9 Hz). 31P{/1H} NMR: δ = 15.96 (d, J = 81.9 Hz). EI–MS m/z = 396.2 [M-Bn+2H]+.
rac Ethyl hydrogen ((2R,3S)-4-(dibenzylamino)-2-fluoro-3-hydroxy-3-phenylbutan-2-yl)phosphonate (rac 17), procedure A (Bn2NH, TEA), reaction time 60 h; white solid (132 mg, 89%): 1H NMR (400 MHz, CDCl3) δ = 7.62–7.53 (m, 1H, Ph), 7.42–7.31 (m, 12H, Ph), 7.32–7.26 (m, 2H, Ph), 4.12–4.04 (m, 2H, OCH2CH3), 3.85 (s, 4H, NCH2Ph), 3.43 (d, J = 5.1 Hz, 1H, CHH), 2.93 (dd, J = 5.2, 2.7 Hz, 1H, CHH), 1.67 (dd, J = 24.7, 14.0 Hz, 3H, CH3), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 139.58 (s, Ph), 136.01 (d, J = 1.8 Hz, Ph), 129.17, 128.45, 128.34, 127.66, 127.12 (4 × s, Ph) 95.97 (dd, J = 185.0, 171.6 Hz, CFP), 63.29 (dd, J = 7.0, 2.4 Hz, OCH2), 61.91 (dd, J = 23.5, 8.3 Hz, COH), 52.84 (s, CH2Ph), 50.91 (dd, J = 6.2, 2.7 Hz, CN), 19.32 (dd, J = 22.7, 2.1 Hz, CH3), 16.27 (t, J = 5.9 Hz, OCH2CH3). 19F NMR (376 MHz, CDCl3) δ = −170.28 (dqd, J = 89.4, 24.8, 2.5 Hz). 31P{/1H} NMR (162 MHz, CDCl3) δ = 16.51 (d, J = 90.1 Hz). MS(ESI) C26H32FNO4P [M+H]+ calc. 472.21, found 472.26.
rac Diethyl ((1R,2R)-2-((dibenzylamino)methyl)-1-fluoro-2-hydroxycyclohexyl) phosphonate (rac 21), procedure A (Bn2NH, TEA), slightly yellow oil (166 mg, 87%): 1H NMR (400 MHz, CDCl3): δ = 7.26–7.21 (m, 10H, Ph), 4.06–3.94 (m, 4H, OCH2), 3.94 (d, J = 14.3 Hz, 2H, NCH2Ph), 3.51 (d, J = 13.6 Hz, 2H, NCH2Ph), 3.31 (d, J = 14.0 Hz, 1H, CHH), 2.79 (d, J = 14.1 Hz, 1H, CHH), 2.18–2.10 (m, 1H, C6H), 2.15–2.01 (m, 2H, C6&3H), 1.92–1.86 (m, 1H, C3H), 1.70–1.65 (br s, 1H, OH), 1.68–1.64 (m, 1H, C4H), 1.54–1.46 (m, 2H, C4&5H), 1.46–1.40 (m, 2H, C4&5H), 1.40–1.36 (m, 1H, C3H), 1.30 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.26 (t, J = 7.3 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3): δ = 139.51, 129.15, 128.47, 127.02 (4 × s, Ph), 97.37 (dd, J = 190.1, 161.7 Hz, CFP, C1), 73.60 (dd, J = 21.6, 2.2 Hz, COH, C2), 63.18 (d, J = 6.4 Hz, OCH2), 63.16 (d, J = 6.4 Hz, OCH2), 60.01 (s, CH2Ph), 55.77–55.69 (m, CN), 32.37 (dd, J = 8.1, 0.6 Hz, CH2, C3), 29.11 (dd, J = 20.2, 2.2 Hz, CH2, C6), 20.21 (s, CH2, C4), 20.05 (dd, J = 9.1, 3.1 Hz, CH2, C5), 16.48 (d, J = 5.9 Hz, OCH2CH3), 16.47 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (377 MHz, CDCl3): δ = −180.09 (br s). 31P{/1H} NMR (162 MHz, CDCl3): δ = 20.09 (d, J = 83.6 Hz). IR (film): ν = 3,688, 3,609, 3,054, 2,985, 2,871, 1,456, 1,368, 1,263, and 1,027 cm−1. GC–MS (EI) m/z = 374.2 [M-Bn+2H]+, tR = 18.32 min.
General Procedure (Procedure C) for Hydrogenation and N-Boc Protection of γ-Amino-β-Hydroxyphosphonates
A solution of N, N-dibenzyl-protected hydroxyphosphonate (0.3 mmol) in absolute EtOH (2 mL) containing Boc2O (98 mg, 0.45 mmol) was hydrogenated over 10% Pd-C (30 mg) under atmospheric pressure for 48 h. Then, the catalyst was filtrated through Celite with MeOH, the solution was concentrated on vacuum, and purified by flash column chromatography CHCl3 → 5% MeOH/CHCl3 (v:v) (1 cm layer of silica gel), to give appropriate N-Boc-protected amino hydroxyphosphonate.
General Procedure (Procedure D) for Hydrogenation of Monoesters of Hydroxyalkylphosphonic Acids
A solution of monoester of N, N-dibenzyl-protected or azido-hydroxyphosphonic acids 17 or 38 (0.3 mmol) in absolute EtOH (2 mL) was hydrogenated over 10% Pd-C (30 mg) under atmospheric pressure for 48 h. Then, the catalyst was filtrated through Celite with MeOH, the solution was concentrated on vacuum, and purified by flash column chromatography CHCl3 → 5% MeOH/CHCl3 (v:v) (1 cm layer of silica gel) to give monoester of amino hydroxyphosphonic acid 27. For spectroscopic properties of compounds 26–28 (procedures C, D) see Supplementary Materials.
General Procedure (Procedure E) for Acidic Hydrolysis of Hydroxyphosphonates
To the appropriated hydroxyphosphonates 26–28 and 40–42 (0.25 mmol), aqueous HCl (12 M, 2 ml) was added. Next, the mixture was refluxed for 48 h, then evaporated and co-evaporated with ethanol (3 × 4 mL). To the obtained residue, ethanol (1 mL) was added, and the mixture was treated dropwise with propylene oxide (to pH 7). The residue was filtered off, evaporated, dissolved in methanol, and evaporated to give 29–31, or 43 as white amorphous, low-soluble (in polar solvents at rt) powder, which decomposed before melting.
rac Hydrogen ((1R,2R)-3-ammonio-1-fluoro-2-hydroxy-2-methylpropyl)phosphonate (rac 29a), procedure E, major isomer. Isolated as a mixture with 29b, which could not be separated, white solid (41 mg, 88%, 3:1, d.r.): 1H NMR (300 MHz, D2O) δ = 4.55 (dd, J = 44.6, 6.4 Hz, 1H, CHFP), 3.22 (d, J = 13.1 Hz, 1H, CHH), 3.10 (d, J = 13.1 Hz, 1H, CHH), 1.38 (p, J = 2.5 Hz, 3H, CH3). 13C NMR (101 MHz, D2O) δ = 93.50 (dd, J = 181.4, 152.5 Hz, CFP), 70.62 (d, J = 18.8 Hz, COH), 45.70 (d, J = 11.1 Hz, CN), 20.81 (t, J = 3.5 Hz, CH3). 19F NMR (283 MHz, D2O) δ = −209.12 (dd, J = 69.6, 45.9 Hz). 31P{/1H} NMR (122 MHz, D2O) δ = 9.95 (d, J = 69.2 Hz). IR (film): ν = 3,349, 2,980, 2,933, 1,712, 1,518, 1,392, 1,367, 1,167, 1,029, and 974 HRMS calc for C4H12FNO4P 188.0488; found 188.0483 [M+H]+.
rac Hydrogen ((1R,2S)-3-ammonio-1-fluoro-2-hydroxy-2-methylpropyl)phosphonate (rac 29b), minor isomer: 19F NMR (283 MHz, D2O) δ = −207.23 (dd, J = 69.1, 44.9 Hz).
31P{/1H} NMR (122 MHz, D2O) δ = 9.39 (d, J = 67.1 Hz).
rac Hydrogen ((2R,3S)-4-ammonio-2-fluoro-3-hydroxy-3-phenylbutan-2-yl)phosphonate (rac 30), procedure E, slightly cream-colored solid (57 mg, 86%): 1H NMR (300 MHz, D2O) δ = 7.47–7.39 (m, 2H, Ph), 7.34–7.24 (m, 3H, Ph), 3.78 (d, J = 13.3 Hz, 1H, CHH), 3.55 (d, J = 13.4 Hz, 1H, CHH), 1.16 (ddd, J = 25.5, 12.7, 1.4 Hz, 3H, CH3). 13C NMR (76 MHz, D2O) δ = 137.21 (d, J = 7.2 Hz, Ph), 128.84 (s, Ph), 128.74 (s, Ph), 127.27 (d, J = 2.8 Hz, Ph), 97.36 (dd, J = 186.4, 152.2 Hz, CFP), 76.79 (d, J = 19.5 Hz, COH), 45.73 (s, CN), 18.56 (d, J = 21.4 Hz, CH3). 13C NMR (101 MHz, CF3COOD) δ = 136.94 (d, J = 8.4 Hz, Ph), 132.28, 131.59, 128.30 (3 × s, Ph), 98.66 (dd, J = 188.3, 165.2 Hz, CFP), 79.81 (d, J = 21.3 Hz), 48.83 (s, CN), 19.70 (d, J = 21.3 Hz, CH3). 19F NMR (283 MHz, D2O) δ = −169.07 (dtd, J = 74.5, 30.1, 22.5 Hz). 31P{/1H} NMR (122 MHz, D2O) δ = 14.78 (d, J = 77.7 Hz). IR (film): ν = 3,182 (br), 2,938, 2,868, 1,447, 1,404, 1,205, 1,044, and 940 cm−1. HRMS calc for C10H16FNO4P 264.0801; found 264.0799 [M+H]+.
rac Hydrogen ((1R,2R)-2-(ammoniomethyl)-1-fluoro-2-hydroxycyclohexyl)phosphonate (rac 31), procedure E, white solid (47 mg, 82%). Alternatively, compound rac 31 can be obtain from rac 42 by procedure E (45 mg, 81%): 1H NMR (401 MHz, CD3OD) δ = 3.45 (d, J = 13.2 Hz, 1H, CHHOH), 2.91 (d, J = 12.8 Hz, 1H, CHHOH), 2.26–2.00 (m, 2H, CH2), 2.23–1.63 (m, 2H, CH2), 1.64–1.49 (m, 4H, CH2). 13C NMR (101 MHz, D2O) δ = 96.14 (dd, J = 183.2, 153.7 Hz, CFP, C1), 70.96 (dd, J = 22.9, 2.1 Hz, COH, C2), 45.80 (d, J = 2.0 Hz, CN), 31.46 (d, J = 6.5 Hz, C3), 28.73 (dd, J = 20.0, 2.9 Hz, C6), 19.47 (s, C4), 19.21 (dd, J = 8.3, 3.3 Hz, C5). 13C NMR (101 MHz, CF3COOD) δ = 97.06 (dd, J = 181.1, 169.0 Hz, CFP, C1), 74.21 (d, J = 24.5 Hz, COH, C2), 48.14 (CN), 35.48 (C3), 30.19 (d, J = 19.9 Hz, C6), 21.25 (C4), 20.52 (C5). 19F NMR (283 MHz, D2O) δ = −178.24 (dd, J = 74.2, 43.6 Hz). 19F NMR (283 MHz, CD3OD) δ = −179.99 (dd, J = 75.7, 47.1 Hz). 31P{/1H} NMR (122 MHz, D2O) δ = 14.95 (d, J = 77.1 Hz). 31P{/1H} NMR (122 MHz, CD3OD) δ = 16.18 (d, J = 75.9 Hz). IR (film): ν = 3,185 (br), 2,936, 2,865, 1,454, 1,338, 1,147, 1,052, 1,033, 1,012, and 956 HRMS calc for C7H16FNO4P 228.0801; found 228.0797 [M+H]+.
General Procedure (Procedure F) for Oxiranes 5–8 Opening by HBr
After being dissolved in chloroform (1.5 mL), acethyl bromide (44 μL, 74 mg, 0.6 mmol) and methanol (24 μL, 19 mg, 0.6 mmol) were mixed at 0°C and the reaction mixture was stirred for 20 min. Then, oxiranes 5–8 (0.5 mmol) in chloroform (1.5 mL) were added, and stirring was continued at 0°C for 2 h. Next, the crude reaction mixture was extracted with CH2Cl2 (NaHCO3aq/brine), dried with anhydrous Na2SO4, and evaporated to give bromohydrines 32–35. Flash column chromatography CHCl3 → 5% MeOH/CHCl3 (v:v) (1 cm layer of silica gel) gave compounds with lower yields. For spectroscopic properties of compounds 32–35 see Supplementary Materials.
General Procedure (Procedure G) for Oxiranes 5–8 Opening by Sodium Azide
To the dissolved mixture of EtOH:H2O (8:1, v:v) oxiranes 5–8 (0.5 mmol), sodium azide (150 mg, 2.3 mmol) and ammonium sulfate (132 mg, 1 mmol) were added. Then, the obtained mixture was refluxed for 24/48 h (monitored by TLC). Next, the crude reaction mixture was extracted with CH2Cl2 (NaHCO3aq/brine), dried (Na2SO4), evaporated, and purified by flash column chromatography CHCl3 → 5% MeOH/CHCl3 (v:v) (1 cm layer of silica gel) to give compounds 36–39.
General Procedure (Procedure H) for Reaction of Bromides 32–35 With Sodium Azide
Bromides 32–35 (0.3 mmol) were dissolved in DMF (2 mL), then sodium azide (91 mg, 1.4 mmol) was added. Then, the obtained mixture was stirred for 48 h (monitored by TLC). Next, the crude reaction mixture was extracted with CH2Cl2 (NaHCO3aq/brine), dried (Na2SO4), evaporated, and purified by flash column chromatography to give the mixture of appropriate azides 36–39 and oxiranes 5–8 which could not be separated by the chromatography techniques employed in this study (31P NMR).
rac Diethyl ((1R,2S)-2-azido-1-fluoro-3-hydroxy-2-methylpropyl)phosphonate (rac 36a), major isomer. Procedure G (24 h): isolated as a mixture with 36b, which could not be separated by the chromatography techniques employed in this study; slightly yellow oil (86 mg, 64%, 4:1, d.r.). Procedure H gave a mixture 36a, b (3:1, d.r.) / 5a, b (5:1, d.r.) with ratio 1.5:1. 1H NMR (300 MHz, CDCl3) δ = 4.67 (dd, J = 45.1, 5.8 Hz, 1H, CHF), 4.33–4.17 (m, 4H, OCH2), 3.78 (br s, 1H, OH), 3.49 (d, J = 12.1, 1.8 Hz, 1H, CHH), 3.45 (d, J = 12.4 Hz, 1H, CHH), 1.42–1.39 (m, 6H, CH3, OCH2CH3), 1.39–1.38 (m, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 90.18 (dd, J = 188.4, 163.7 Hz, CFP), 73.30 (dd, J = 18.3, 2.4 Hz, COH), 64.30 (dd, J = 6.8, 2.0 Hz, OCH2), 62.83 (d, J = 7.0 Hz, OCH2CH3), 56.59 (dd, J = 7.9, 5.6 Hz, CN), 21.95 (t, J = 3.3 Hz, CH3), 16.27 (d, J = 6.0 Hz, OCH2CH3), 16.26 (d, J = 6.0 Hz, OCH2CH3). 19F NMR (376 MHz, CDCl3) δ = −213.89 (dd, J = 76.9, 44.8 Hz). 31P{/1H} NMR (243 MHz, CDCl3) δ = 17.57 (d, J = 77.5 Hz). IR (film): ν = 3,368, 2,985, 2,923, 2,853, 2,103, 1,237, 1,163, 1,023, 973, and 549 cm−1. HRMS (EI-MS) calcd for C8H18FN3O4P [M]+: 270.1019, found: 270.1033.
rac Diethyl ((1R,2R)-2-azido-1-fluoro-3-hydroxy-2-methylpropyl)phosphonate (rac 36b), minor isomer: 1H NMR (300 MHz, CDCl3) δ = 4.82 (dd, J = 44.9, 3.6 Hz, 1H, CHF), 4.33–4.17 (m, 2H, OCH2), 4.13–4.08 (m, 2H, OCH2), 3.85 (br s, 1H, OH), 3.41 (dt, J = 12.7, 2.0 Hz, 1H, CHH), 3.36 (d, J = 12.6, 2.3 Hz, 1H, CHH), 1.39–1.38 (m, 3H, CH3), 1.38–1.32 (m, 6H, OCH2CH3). 19F NMR (376 MHz, CDCl3) δ = −210.73 (dd, J = 71.7, 44.6 Hz). 31P{/1H} NMR (243 MHz, CDCl3) δ = 18.60 (d, J = 71.5 Hz).
rac Diethyl ((1R,2S)-2-azido-1-fluoro-3-hydroxy-2-phenylpropyl)phosphonate (rac 37), procedure G (24 h): transparent oil (99 mg, 60%). Procedure H gave a mixture 37/ 6 with a ratio of 3.8:1. 1H NMR (300 MHz, CDCl3) δ = 7.57–7.34 (m, 5H, Ph), 5.07 (dd, J = 45.5, 6.6 Hz, 1H, CHF), 4.22 (“quint,” J = 7.6 Hz, 2H, OCH2), 3.74 (ddd, J = 12.3, 2.4, 0.8 Hz, 1H, CHH), 3.69 (dd, J = 12.5, 1.2 Hz, 1H, CHH), 3.75–3.64 (m, 1H, OCHHCH3), 3.36–3.26 (m, 1H, OCHH), 1.33 (t, J = 7.0 Hz, 3H, OCH2CH3), 0.90 (t, J = 7.1 Hz, 3H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 138.70 (d, J = 4.7 Hz, Ph), 128.36, 128.32, 126.02 (3 × s, Ph), 89.55 (dd, J = 191.8, 166.0 Hz, CFP), 76.86 (s, COH masked by CHCl3), 64.44 (dd, J = 6.6, 2.5 Hz, OCH2), 62.44 (d, J = 6.9 Hz, OCH2), 57.33 (dd, J = 12.1, 5.5 Hz, CN), 16.33 (d, J = 5.8 Hz, OCH2CH3), 15.77 (d, J = 6.1 Hz, OCH2CH3). 19F NMR (282 MHz, CDCl3) δ = −214.83 (dd, J = 82.2, 45.5 Hz). 31P{/1H} NMR (121 MHz, CDCl3) δ = 16.29 (d, J = 82.2 Hz). IR (film): ν = 3,369, 2,985, 2,933, 2,102, 1,530, 1,520, 1,231, 1,162, 1,020, 976, and 702 cm−1. MS (EI) m/z = 332.2 [M+H]+.
rac Ethyl hydrogen ((2R,3R)-3-azido-2-fluoro-4-hydroxy-3-phenylbutan-2-yl)phosphonate (rac 38) and rac diethyl ((2R,3R)-3-azido-2-fluoro-4-hydroxy-3-phenylbutan-2-yl)phosphonate (rac 38′), procedure G (48 h, evaporation, flush column chromatography 95:5 → 9:1 CHCl3/MeOH, v:v) gave 38 Compound 38 had: 1H NMR (300 MHz, DMSO-d6) δ = 7.51–7.19 (m, 6H, Ph, OH), 4.33 (d, J = 13.2 Hz, 1H, CHH), 3.89 (quintet, J = 7.0 Hz, 2H, OCH2), 3.70 (dd, J = 13.2, 2.6 Hz, 1H, CHH), 1.16 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.04 (dd, J = 24.9, 11.4 Hz, 3H, CH3). 13C NMR (76 MHz, DMSO-d6) δ = 140.65 (d, J = 10.0 Hz, Ph), 127.34 (s, Ph), 127.11 (d, J = 3.0 Hz, Ph), 126.73 (s, Ph), 96.99 (dd, J = 190.6, 144.3 Hz, CFP), 79.04 (dd, J = 19.7, 5.0 Hz, COH), 60.85 (d, J = 5.9 Hz, OCH2), 56.93 (s, CH2), 19.19 (dd, J = 21.5, 4.0 Hz, CH3), 16.94 (d, J = 5.6 Hz, OCH2CH3). 19F NMR (283 MHz, DMSO-d6) δ = −170.77 (dq, J = 68.9, 24.8 Hz). 31P{/1H} NMR (122 MHz, DMSO-d6) δ = 12.43–14.36 (m). MS (ESI) calc for C12H16FNO4P− [M-N2]− 288.081 obtained 288.09. Compound 38': 19F NMR (376 MHz, CDCl3) δ = −171.86 (dq, J = 84.3, 25.0 Hz). 31P{/1H} NMR (162 MHz, CDCl3) δ = 19.55 (d, J = 84.1 Hz).
rac Diethyl ((1R,2S)-2-azido-1-fluoro-2-(hydroxymethyl)cyclohexyl)phosphonate (rac 39), procedure G (24 h): transparent oil (93 mg, 60%). Procedure H gave a mixture 39/8 with a ratio of 1:1.1H NMR (400 MHz, CDCl3) δ = 4.31–4.18 (m, 4H, OCH2), 3.82 (d, J = 12.9 Hz, 1H, CHH), 3.34 (dd, J = 12.9, 2.0 Hz, 1H, CHH), 2.27–2.13 (m, 1H, C6H), 2.12–2.04 (m, 1H, C6H), 2.04–1.92 (m, 1H, C3H), 1.78–1.65 (m, 1H, C4H), 1.65–1.55 (m, 2H, C3&C5H), 1.55–1.50 (m, 2H, C4&C5H), 1.39 (dt, J = 7.0,1.1 Hz, 6H, OCH2CH3). 13C NMR (101 MHz, CDCl3) δ = 95.9 (dd, J = 193.1, 160.5 Hz, CFP, C1), 73.6 (dd, J = 21.1, 2.4 Hz, COH), 64.16 (d, J = 7.2 Hz, OCH2), 63.98(d, J = 7.9, 1.7 Hz, OCH2), 57.7 (d, J = 1.7 Hz, CN, C2), 30.0 (d, J = 8.5 Hz, CH2, C3), 28.3 (dd, J = 20.5, 2.8 Hz, CH2, C6), 19.4 (s, CH2, C4), 19.6 (dd, J = 10.1, 2.5 Hz, CH2, C5), 16.5 (d, J = 5.7 Hz, OCH2CH3), 16.4 (d, J = 5.9 Hz, OCH2CH3). 19F NMR (377 MHz, CDCl3) δ = −180.95 (dd, J = 78.2, 43.4 Hz). 31P{/1H} NMR (162 MHz, CDCl3) δ = 19.51 (d, J = 79.8 Hz). IR (film): ν = 3,364, 2,938, 2,869, 2,102, 1,234, 1,165, 1,023, and 973 cm−1. MS (EI) m/z = 309.2 [M]+.
General Procedure (Procedure I) for Hydrogenation and N-Boc Protection of β-Azido-γ-Hydroxyphosphonates
A solution of azidohydroxyphosphonates 36–37 and 39 (0.3 mmol) in absolute EtOH (2 mL) containing Boc2O (98 mg, 0.45 mmol) was hydrogenated over 10% Pd-C (30 mg) under atmospheric pressure for 48 h. Then, the catalyst was filtrated through Celite with MeOH, the solution was concentrated on vacuum, and purified by flash column chromatography CHCl3 → 5% MeOH/CHCl3 (v:v) (1 cm layer of silica gel) to give appropriate N-Boc-protected amino hydroxyphosphonate. For spectroscopic properties of compounds 40–42 see Supplementary Materials.
rac Hydrogen ((1R,2R)-2-ammonio-1-fluoro-3-hydroxy-2-phenylpropyl)phosphonate (rac 43), procedure E, white solid (52 mg, 84%): 1H NMR (300 MHz, D2O) δ = 7.53–7.49 (m, 2H, Ph), 7.44–7.35 (m, 3H, Ph), 5.05 (dd, J = 44.9, 6.1 Hz, 1H, CHF), 3.66 (br d, J = 13.0 Hz, 1H, CHH), 3.38 (d, J = 13.2 Hz, 1H, CHH). 13C NMR (101 MHz, D2O) δ = 137.17, 128.45, 128.33, 125.44 (4 × s, Ph), 92.05 (dd, J = 185.1, 153.3 Hz, CFP), 74.23 (d, J = 18.2 Hz, COH), 46.22 (s, CN). 31P{/1H} NMR (122 MHz, D2O) δ = 9.65 (d, J = 71.2 Hz). 19F NMR (283 MHz, D2O) δ = −207.56 (dd, J = 71.3, 45.0 Hz). IR (film): ν = 3,391, 3,146, 3,050, 2,937, 2,864, 1,454, 1,410, 1,042, and 955 cm−1. HRMS calc for C9H14FNO4P+ 250.0639; found 250.0643 [M+H]+.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
MR and KM-M carried out chemical synthesis, characterization, and manuscript writing. AW carried out chemical synthesis and compounds characterization. PK participated to structure determination, manuscript writing, and revision. TC performed part of the NMR experiments and contributed to the structure determination, manuscript writing, and revision. TS carried out the DFT calculations. MR and HK designed and managed the study. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by Grant No. HARMONIA/2017/M/ST5/00437 from the Polish National Science Centre (NCN).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Apollo Scientific company (UK) for the supply of Selectfluor® and the Bruker Team (R. Kerssebaum and A. Pagelot) for help with the 1D HOESY experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.613633/full#supplementary-material
References
Abraham, M. H., Grellier, P. L., Prior, D. V., Morris, J. J., and Taylor, P. J. (1990). Hydrogen bonding. Part 10. A scale of solute hydrogen-bond basicity using log K values for complexation in tetrachloromethane. J. Chem. Soc. Perkin Trans. 2:521. doi: 10.1039/p29900000521
Amantini, D., Fringuelli, F., Piermatti, O., Tortoioli, S., and Vaccaro, L. (2002). Nucleophilic ring opening of 1, 2-epoxides in aqueous medium. ARKIVOC 2002, 293–311. doi: 10.3998/ark.5550190.0003.b26
Ankati, H., Yang, Y., Zhu, D., Biehl, E. R., and Hua, L. (2008). Synthesis of optically pure 2-azido-1-arylethanols with isolated enzymes and conversion to triazole-containing β-blocker analogues employing click chemistry. J. Org. Chem. 73, 6433–6436. doi: 10.1021/jo8009616
Arbuzov, B. A., Vinogradova, V. S., Polezhaeva, N. A., and Shamsutdinova, A. K. (1963). β-Keto phosphonic esters - communication 11. Action of diazomethane on diethyl acetyl- and benzoylphosphonates. Bull. Acad. Sci. USSR Div. Chem. Sci. 12, 604–610. doi: 10.1007/BF00843950
Atack, T. C., Lecker, R. M., and Cook, S. P. (2014). Iron-catalyzed borylation of alkyl electrophiles. J. Am. Chem. Soc. 136, 9521–9523. doi: 10.1021/ja505199u
Azoulay, S., Manabe, K., and Kobayashi, S. (2005). Catalytic asymmetric ring opening of meso-epoxides with aromatic amines in water. Org. Lett. 7, 4593–4595. doi: 10.1021/ol051546z
Badalassi, F., Klein, G., Crotti, P., and Reymond, J.-L. (2004). Fluorescence assay and screening of epoxide opening by nucleophiles. Eur. J. Org. Chem. 2557–2566. doi: 10.1002/ejoc.200400024
Banfi, S., Fonio, W., Allievi, E., and Raimondo, S. (1983). Experimental chronic epilepsy in rats: a screening method for antiepileptic drugs. Pharmacol. Res. Commun. 15, 553–558. doi: 10.1016/S0031-6989(83)80072-1
Bégué, J.-P., and Bonnet-Delpon, D. (2008). Bioorganic and Medicinal Chemistry of Fluorine, 1st Edn. New Jersey, NJ: Wiley and Sons.
Beier, P., Alexandrova, A. V., Zibinsky, M., and Prakash, G. K. S. (2008). Nucleophilic difluoromethylation and difluoromethylenation of aldehydes and ketones using diethyl difluoromethylphosphonate. Tetrahedron 64, 10977–10985. doi: 10.1016/j.tet.2008.10.006
Bell, T. W., and Ciaccio, J. A. (1986). Conversion of epoxides to bromohydrins by B-bromobis (dimethylamino) borane. Tetrahedron Lett. 27, 827–830. doi: 10.1016/S0040-4039(00)84111-8
Benedetti F. Berti F. and S. Norbedo (1998). Regio-and stereoselective ring opening of 2, 3-epoxyalcohols with diethylaluminium azide. Tetrahedron Lett. 39, 7971–7974. doi: 10.1016/S0040-4039(98)01733-X
Bera, K., Nadkarni, D., and Namboothiri, I. N. N. (2013). Asymmetric synthesis of γ-aminophosphonates: the bio-isosteric analogs of γ-aminobutyric acid. J. Chem. Sci. 125, 443–465. doi: 10.1007/s12039-013-0418-6
Briggs, C. R., Allen, M. J., O'Hagan, D., Tozer, D. J., Slawin, A. M. Z., Goeta, A. E., et al. (2004). The observation of a large gauche preference when 2-fluoroethylamine and 2-fluoroethanol become protonated. Org. Biomol. Chem. 2, 732–740. doi: 10.1039/b312188g
Brogden, R. N., Speight, T. M., and Avery, G. S. (1974). Baclofen: a preliminary report of its pharmacological properties and therapeutic efficacy in spasticity. Drugs 8, 1–14. doi: 10.2165/00003495-197408010-00001
Clayden, J., Greeves, N., Warren, S., and Wothers, P. (2008). Organic Chemistry. Oxford: University Press, 814–818.
Clift, M. D., Ji, H., Deniau, G. P., O'Hagan, D., and Silverman, R. B. (2007). Enantiomers of 4-amino-3-fluorobutanoic acid as substrates for γ-aminobutyric acid aminotransferase. Conformational probes for GABA binding. Biochemistry 46, 13819–13828. doi: 10.1021/bi701249q
Costantino, P., Crotti, P., Ferretti, M., and Macchia, F. (1982). Stereochemical evidence for aryl participation in the ring opening of oxiranes. Ring-opening reactions of 1-benzyl-1, 2-epoxycyclohexane under acidic conditions. J. Org. Chem. 47, 2917–2923. doi: 10.1021/jo00136a023
Cox, R. J., Gibson, J. S., and Hadfield, A. T. (2005). Design, synthesis and analysis of inhibitors of bacterial aspartate semialdehyde dehydrogenase. ChemBioChem 6, 2255–2260. doi: 10.1002/cbic.200500172
Cristau, H.-J., Pirat, J.-L., Drag, M., and Kafarski, P. (2000). Regio-and stereoselective synthesis of 2-amino-1-hydroxy-2-aryl ethylphosphonic esters. Tetrahedron Lett. 41, 9781–9785. doi: 10.1016/S0040-4039(00)01722-6
De Maio, D., and Pasquariello, G. (1963). Gamma-amino-beta-hydroxybutyric acid (GABOB) and brain serotonin. Psychopharmacologia 5, 84–86. doi: 10.1007/BF00405577
Deniau, G., Slawin, A. M. Z., Lebl, T., Chorki, F., Issberner, J. P., van Mourik, T., et al. (2007). Synthesis, conformation and biological evaluation of the enantiomers of 3-fluoro-γ-aminobutyric acid ((R)- and (S)-3F-GABA): an analogue of the neurotransmitter GABA. ChemBioChem 8, 2265–2274. doi: 10.1002/cbic.200700371
Dingwall, J. G. (1983). New carboxyphosphonic and phosphinic acid structures of technical and biological interest. Phosph. Sulf. Silicon Relat. Elem. 18, 353–356. doi: 10.1080/03086648308076038
Dolbier, W. R. Jr. (2009). Guide to Fluorine NMR for Organic Chemists. New Jersey, NJ: Wiley Interscience.
Drag, M., Wieczerzak, E., Pawełczak, M., Berlicki, Ł., Grzonka, Z., and Kafarski, P. (2013). Toward very potent, non-covalent organophosphonate inhibitors of cathepsin C and related enzymes by 2-amino-1-hydroxy-alkanephosphonates dipeptides. Biochimie 95, 1640–1649. doi: 10.1016/j.biochi.2013.05.006
Draper, R. W. (1983). Synthesis of steroidal azides. Part 1. Stereospecific vicinal azidohydroxylation of steroidal olefins. J. Chem. Soc. Perkin Trans. 1, 2781–2786. doi: 10.1039/P19830002781
Duangdee, N., Harnying, W., Rulli, G., Neudörfl, J.-M., Gröger, H., and Berkessel, A. (2012). Highly enantioselective organocatalytic trifluoromethyl carbinol synthesis—a caveat on reaction times and product isolation. J. Am. Chem. Soc. 134, 11196–11205. doi: 10.1021/ja302511t
Faiz, S., and Zahoor, A. F. (2016). Ring opening of epoxides with C-nucleophiles. Mol. Divers. 20, 969–987. doi: 10.1007/s11030-016-9686-7
Falch, E., Hegegaard, A., Nielsen, L., Jensen, B. R., Hjeds, H., and Krogsgaard-Larsen, P. (1986). Comparative stereostructure-activity studies on GABAA and GABAB receptor sites and GABA uptake using rat brain membrane preparations. J. Neurochem. 47, 898–903. doi: 10.1111/j.1471-4159.1986.tb00695.x
Fields, S. C. (1999). Synthesis of natural products containing a C-P bond. Tetrahedron 55, 12237–12273. doi: 10.1016/S0040-4020(99)00701-2
Foss, F. W. Jr., Snyder, A. H., Davis, M. D., Rouse, M., Okusa, M. D., Lynch, K. R., et al. (2007). Synthesis and biological evaluation of γ-aminophosphonates as potent, subtype-selective sphingosine 1-phosphate receptor agonists and antagonists. Bioorg. Med. Chem. 15, 663–677. doi: 10.1016/j.bmc.2006.10.060
Fringuelli, F., Piermatti, O., Pizzo, F, and Vaccaro, L. (1999). Ring opening of epoxides with sodium azide in water. A regioselective pH-controlled reaction. J. Org. Chem. 64, 6094–6096. doi: 10.1021/jo990368i
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2013). Gaussian 09, Revision D.01. Wallingford, CT: Gaussian Inc.
Fustero, S., Cuñat, A. C., Flores, S., Báez, C., Oliver, J., Cynamon, M., et al. (2011). Design, synthesis, and biological evaluation of novel fluorinated ethanolamines. Chem. Eur. J. 17, 14772–14784. doi: 10.1002/chem.201102078
Gancarz, R., Latajka, R., Małuszek, B., Zymańczyk-Duda, E., and Kafarski, P. (2000). Hydrogen bonding in diethyl 2-hydroxypropylphosphonate–an example of equilibrium between intramolecular and intermolecular bonding. Magn. Reson. Chem. 38, 197–200. doi: 10.1002/(SICI)1097-458X(200003)38:3<197::AID-MRC620>3.0.CO;2-J
García-Flores, E., and Farías, R. (1997). γ-Amino-β-hydroxybutyric acid as add-on therapy in adult patients with severe focal epilepsy. Stereotact. Funct. Neurosurg. 69(1–4 Pt 2), 243–246. doi: 10.1159/000099882;
Genov, D. G., Kresinski, R. A., and Tebby, J. C. (1998). Conformational analysis of 2-substituted alkylphosphoryl compounds. 4. Substituent effect on the nature of the hydrogen bonding in (2-hydroxyalkyl)phosphoryl systems. J. Org. Chem. 63, 2574–2585. doi: 10.1021/jo972036o
Głowacka, I. E. (2009). Synthesis of enantiomerically pure diethyl (R)-and (S)-2-hydroxy-3-(1, 2,3-triazol-1-yl) propylphosphonates. Tetrahedron Asymmetry 20, 2270–2278. doi: 10.1016/j.tetasy.2009.09.005
Goa, K. L., and Sorkin, E. M. (1993). Gabapentin. A review of its pharmacological properties and clinical potential in epilepsy. Drugs 46, 409–427. doi: 10.2165/00003495-199346030-00007
Gorenstein, D. G. (Ed.). (1984). Phosphorus-31 NMR. Principles and Applications. Orlando, FL: Academic Press Inc.
Griffin, C. E., and Kundu, S. K. (1969). Phosphonic acids and esters. XX. Preparation and ring opening reaction of alfa-, beta-, gamma-epoxyalkylphosphonates. The proton magnetic resonance spectra of vicinally substituted ethyl- and propyl-phosphonates. J. Org. Chem. 34, 1532–1539. doi: 10.1021/jo01258a002
Gu, Z., Zhou, J., Jiang, G.-F., and Zhou, Y.-G. (2018). Synthesis of chiral γ-aminophosphonates through the organocatalytic hydrophosphonylation of azadienes with phosphites. Org. Chem. Front. 5, 1148–1151. doi: 10.1039/C7QO01158J
Hamashima, Y., Suzuki, T., Shimura, Y., Shimizu, T., Umebayashi, N., Tamura, T., et al. (2005). An efficient catalytic enantioselective fluorination of β-ketophosphonates using chiral palladium complexes. Tetrahedron Lett. 46, 1447–1450. doi: 10.1016/j.tetlet.2005.01.018
Hehre, W. J., Radom, L., and Schleyer, P. V. R. (1986). Ab Initio Molecular Orbital Theory. New York, NY: Wiley.
Hesse, M., Meier, H., and Zeeh, B. (1997). Spectroscopic Methods in Organic Chemistry. New York, NY: Thieme.
Horsman, G. P., and Zechel, D. L. (2017). Phosphonate biochemistry. Chem. Rev. 117, 5704–5783. doi: 10.1021/acs.chemrev.6b00536
Huleatt, P. B., Khoo, M. L., Chua, Y. Y., Tan, T. W., Liew, R. S., Balogh, B., et al. (2015). Novel arylalkenylpropargylamines as neuroprotective, potent, and selective monoamine oxidase B inhibitors for the treatment of Parkinson's disease. J. Med. Chem. 58, 1400–1419. doi: 10.1021/jm501722s
Iacazio, G., and Réglier, M. (2005). Chemo-enzymatic synthesis of all four diastereoisomers of 1-fluoro-2-amino-indane. Tetrahedron Asymmetry 16, 3633–3639. doi: 10.1016/j.tetasy.2005.09.015
Iguchi, E., Okuhara, M., Kohsaka, M., Aoki, H., and Imanaka, H. (1980). Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR-32863 and FR-33289. J. Antibiot. 33:24. doi: 10.7164/antibiotics.33.24
Kaczmarek, P., Rapp, M., and Koroniak, H. (2018). Pyrrolidine and oxazolidine ring transformations in proline and serine derivatives of α-hydroxyphosphonates induced by deoxyfluorinating reagents. RSC Adv. 8, 24444–24457. doi: 10.1039/C8RA05186K
Kafarski, P. (2020). “Phosphonates: their natural occurrence and physiological role,” in Contemporary Topics about Phosphorus in Biology and Materials, ed D. Churchill (London: IntechOpen), 557–577. doi: 10.5772/intechopen.87155
Kafarski, P., and Lejczak, B. (1991). Biological activity of aminophosphonic acids. Phosph. Sulf. Silicon Relat. Elem. 63, 193–215. doi: 10.1080/10426509108029443
Kafarski, P., and Lejczak, B. (2001). Aminophosphonic acids of potential medical importance. Curr. Med. Chem. Anti Cancer Agents 1, 301–312. doi: 10.2174/1568011013354543
Kazmierczak, M., and Koroniak, H. (2012). DAST mediated preparation of β-fluoro-α-aminophosphonates. J. Fluorine Chem. 139, 23–27., doi: 10.1016/j.jfluchem.2012.03.016
Kim S. M.;, Kim, H. R.;, and Kim, D. Y. (2005). Catalytic enantioselective fluorination and amination of β-keto phosphonates catalyzed by chiral palladium complexes. Org. Lett. 7, 2309–2311. doi: 10.1021/ol050413a
Koppel, I. A., Taft, R. W., Anvia, F., Zhu, S.-Z., Hu, L.-Q., Sung, K.-S., et al. (1994). The gas-phase acidities of very strong neutral bronsted acids. J. Am. Chem. Soc. 116, 3047–3057. doi: 10.1021/ja00086a038
Korn, E. D., Dearborn, D. G., Fales, H. M., and Sokoloski, E. A. (1973). Phosphonoglycan A major polysaccharide constituent of the amoeba plasma membrane contains 2-aminoethylphosphonic acid and 1-hydroxy-2-aminoethylphosphonic acid. J. Biol. Chem. 248, 2257–2259.
Kristiansen, U., and Fjalland, B. (1991). Ligand structural specificity of GABAA receptors in guinea pig ileum. Pharmacol. Toxicol. 68, 332–339. doi: 10.1111/j.1600-0773.1991.tb01249.x
Kukhar, V. P., and Hudson, H. R. (2000). Aminophosphonic and Aminophosphinic Acids. Chichester: John Wiley and Sons Ltd.
Lapin, I. (2001). Phenibut (beta-phenyl-GABA): a tranquilizer and nootropic drug. CNS Drug Rev. 7, 471–481. doi: 10.1111/j.1527-3458.2001.tb00211.x
Leggio, L., Garbutt, J. C., and Addolorato, G. (2010). Effectiveness and safety of baclofen in the treatment of alcohol dependent patients. CNS Neurol. Disord. Drug Targets 9, 33–44. doi: 10.2174/187152710790966614
Lentsch, L. M., and Wiemer, D. F. (1999). Addition of organometallic nucleophiles to β-keto phosphonates. J. Org. Chem. 64, 5205–5212. doi: 10.1021/jo990388k
Lin, B., and Whalen, D. L. (1994). Stereochemistry of the spontaneous, acid-catalyzed and base-catalyzed hydrolyses of styrene oxide. J. Org. Chem. 59, 1638–1641. doi: 10.1021/jo00086a012
Mandadapu, S. R., Gunnam, M. R., Galasiti Kankanamalage, A. C., Uy, R. A. Z., Alliston, K. R., Lushington, G. H., et al. (2013). Potent inhibition of norovirus by dipeptidyl α-hydroxyphosphonate transition state mimics. Bioorg. Med. Chem. Lett. 23, 5941–5944. doi: 10.1016/j.bmcl.2013.08.073
McDonald, I. A., Lacoste, J. M., Bey, P., Palfreyman, M. G., and Zreika, M. (1985). Enzyme-activated irreversible inhibitors of monoamine oxidase: phenylallylamine structure-activity relationships. J. Med. Chem. 28, 186–193. doi: 10.1021/jm00380a007
McKenna, C. E., and Shen, P. D. (1981). Fluorination of methanediphosphonate esters by perchloryl fluoride. Synthesis of fluoromethanediphosphonic acid and difluoromethanediphosphonic acid. J. Org. Chem. 46 4573–4576 doi: 10.1021/jo00335a053
Melis, G. B., Paoletti, A. M., Mais, V., Mastrapasqua, N. M., Strigini, F., Fruzzetti, F., et al. (2014). Dose-related effects of γ-amino β-hydroxy butyric acid (GABOB) infusion on growth hormone secretion in normal women. J. Endocrinol. Invest. 5, 101–106. doi: 10.1007/BF03350499
Morton, J. G. M., Kwon, L. D., Freeman, J. D., and Njardarson, J. T. (2009). An Adler–Becker oxidation approach to vinigrol. Tetrahedron Lett. 50, 1684–1686. doi: 10.1016/j.tetlet.2009.01.134
Mucha, A., Kafarski, P., and Berlicki, Ł. (2011). Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J. Med. Chem. 54, 5955-5980. doi: 10.1021/jm200587f
Myers, A. G., Barbay, J. K., and Zhong, B. (2001). Asymmetric synthesis of chiral organofluorine compounds: use of nonracemic fluoroiodoacetic acid as a practical electrophile and its application to the synthesis of monofluoro hydroxyethylene dipeptide isosteres within a novel series of HIV protease inhibitors. J. Am. Chem. Soc. 123, 7207–7219. doi: 10.1021/ja010113y
Naydenova, E. D., Todorov, P. T., and Troev, K. D. (2010). Recent synthesis of aminophosphonic acids as potential biological importance. Amino Acids 38, 23-30. doi: 10.1007/s00726-009-0254-7
Nesterov, V. V., and Kolodiazhnyi, O. I. (2007). Efficient method for the asymmetric reduction of α-and β-ketophosphonates. Tetrahedron 63, 6720–6731. doi: 10.1016/j.tet.2007.04.101
O'Hagan, D., and Rzepa, H. S. (1997). Some influences of fluorine in bioorganic chemistry. Chem. Commun. 1997, 645–652. doi: 10.1039/A604140J
Ordóñez, M., González-Morales, A., Ruiz, C., De la Cruz-Cordero, R., and Fernández-Zertuche, M. (2003). Preparation of (R)-and (S)-γ-amino-β-hydroxypropylphosphonic acid from glycine. Tetrahedron Asymmetry 14, 1775–1779. doi: 10.1016/S0957-4166(03)00371-9
Ordóñez, M., Labastida-Galván, V., and Lagunas-Rivera, S. (2010). Stereoselective synthesis of GABOB, carnitine and statine phosphonates analogues. Tetrahedron Asymmetry 21, 129–147. doi: 10.1016/j.tetasy.2009.12.022
Ordóñez, M., Rojas-Cabrera, H., and Cativiela, C. (2009). An overview of stereoselective synthesis of α-aminophosphonic acids and derivatives Tetrahedron 65, 17–49. doi: 10.1016/j.tet.2008.09.083
Ordóñez, M., Viveros-Ceballos, J. L., Cativiela, C., and Sayago, F. J. (2015). An update on the stereoselective synthesis of α-aminophosphonic acids and derivatives. Tetrahedron 71, 1745–1784. doi: 10.1016/j.tet.2015.01.029
Orsini, F., Sello, G., and Sisti, M. (2010). Aminophosphonic acids and derivatives. Synthesis and biological applications. Curr. Med. Chem. 17, 264-289. doi: 10.2174/092986710790149729
Padwaa, A., and Murphree, S. S. (2006). Epoxides and aziridines - a mini review. ARKIVOC 2006, 6–33. doi: 10.3998/ark.5550190.0007.302
Palacios, F., Alonso, C., and de los Santos, J. M. (2005). Synthesis of β-aminophosphonates and β-phosphinates. Chem. Rev. 105, 899–931. doi: 10.1021/cr040672y
Parker, R. E., and Isaacs, N. S. (1959). Mechanisms of epoxide reactions. Chem. Rev. 59, 737–799. doi: 10.1021/cr50028a006
Patel, D. V., Rielly-Gauvin, K., and Ryono, D. E. (1990). Preparation of peptidic α-hydroxy phosphonates a new class of transition state analog renin inhibitors. Tetrahedron Lett. 31, 5587–5590. doi: 10.1016/S0040-4039(00)97903-6
Piettre, S. R., and Cabanas, L. (1996). Reinvestigation of the Wadsworth-Emmons reaction involving lithium difluoromethylenephosphonate. Tetrahedron Lett. 37 5881–5884. doi: 10.1016/0040-4039(96)01267-1
Prasad, P. K., Reddi, R. N., and Sudalai, A. (2015). Oxidant controlled regio-and stereodivergent azidohydroxylation of alkenes via I2 catalysis. Chem. Commun. 51, 10276–10279. doi: 10.1039/C5CC02374B
Radwan-Olszewska, K., Palacios, F., and Kafarski, P. (2011). Selective Synthesis of α-Fluoro-β-keto-and α-Fluoro-β-aminophosphonates via electrophilic fluorination by selectfluor. J. Org. Chem. 76, 1170–1173. doi: 10.1021/jo102276y
Rao, A. V. R., Gurjar, M. K., and Kaiwar, V. (1992). Synthesis of optically pure 2-azido-1-arylethanols with isolated enzymes and conversion to triazole-containing β-blocker analogues employing click chemistry. Tetrahedron Asymmetry 3, 859–862.
Rapp, M., Margas-Musielak, K., and Koroniak, H. (2015). Synthesis and spectral properties of fluorinated α,β-epoxyphosphonates. J. Fluorine Chem. 179, 142–149. doi: 10.1016/j.jfluchem.2015.07.009
Ready, J. M., and Jacobsen, E. N. (2002). A practical oligomeric [(salen)Co] catalyst for asymmetric epoxide ring-opening reactions. Angew. Chem. Int. Ed. 41, 1373–1377. doi: 10.1002/1521-3773(20020415)41:8<1374::AID-ANIE1374>3.0.CO;2-8
Reichwein, J. F., and Pagenkopf, B. L. (2003). A new Horner–Wadsworth–Emmons type coupling reaction between nonstabilized β-hydroxy phosphonates and aldehydes or ketones. J. Am. Chem. Soc. 125, 1821–1824. doi: 10.1021/ja027658s
Roberts, E., Krause, D. N., Wong, E., and Mori, A. (1981). Different efficacies of D-and L- gamma-amino-beta-hydroxybutyric acids in GABA receptor and transport test systems. J. Neurosci. Methods 1, 132–140. doi: 10.1523/JNEUROSCI.01-02-00132.1981
Sano, S., Kuroda, Y., Saito, K., Ose, Y., and Nagao, Y. (2006). Tandem reduction–olefination of triethyl 2-acyl-2-fluoro-2-phosphonoacetates and a synthetic approach to Cbz-Gly-Ψ[(Z)-CFC]-Gly dipeptide isostere. Tetrahedron 62, 11881–11890. doi: 10.1016/j.tet.2006.09.096
Singh, G. S., Mollet, K., D'hooghe, M., and De Kimpe, N. (2013). Epihalohydrins in organic synthesis. Chem. Rev. 113, 1441–1498. doi: 10.1021/cr3003455
Singh, V., Sahu, P. K., Singh, R. B., and Mobin, S. M. (2007). Molecular Complexity from aromatics: a novel, stereoselective route to Tricyclo[5.2.2.01, 5]undecenones, Tricyclo[6.2.2.01, 6]Dodecenones, and [n.3.3]Propellanes. J. Org. Chem. 72, 10155–10165. doi: 10.1021/jo702168s
Smith, R. V., Havens, J. R., and Walsh, S. L. (2016). Gabapentin misuse, abuse and diversion: a systematic review. Addiction 111, 1160–1174. doi: 10.1111/add.13324
Sorochinsky, A. E., and Soloshonok, V. A. (2010). Asymmetric synthesis of fluorine-containing amines, amino alcohols, α-and β-amino acids mediated by chiral sulfinyl group. J. Fluorine Chem. 131, 127–139. doi: 10.1016/j.jfluchem.2009.09.015
Tadeusiak, E. J. (2004). Synthesis of phosphonic analogues of carnitine and γ-amino-β-hydroxybutyric acid. Bioorg. Chem. 32, 473–482. doi: 10.1016/j.bioorg.2004.05.009
Verbicky, C. A., and Zercher, C. K. (2000). Zinc-mediated chain extension of β-keto phosphonates. J. Org. Chem. 65:5615. doi: 10.1021/jo000343f
Verbruggen, C., Craecker, S. D., Rajan, P., Jiao, X.-Y., Borloo, M., Smith, K., et al. (1996). Phosphonic acid and phosphinic acid tripeptides as inhibitors of glutathionylspermidine synthetase. Bioorg. Med. Chem. Lett. 6, 253–258. doi: 10.1016/0960-894x(96)00001-7
Vogel, A. I., Furniss, B. S., Hannaford, A. J., Tatchell, A. R., and Smith, P. W. G. (1989). Vogel's Textbook of Practical Organic Chemistry, 5th Edn. London: Longman Scientific and Technical.
Wang, K., Zhang, Y., and Yuan, C. (2003). Enzymatic synthesis of phosphocarnitine, phosphogabob and fosfomycin. Org. Biomol. Chem. 1, 3564–3569. doi: 10.1039/B307638P
Watanabe, Y., Nakajima, M., Hoshino, T., Jayasimhulu, K., Brooks, E. E., and Kaneshiro, E. S. (2001). A novel sphingophosphonolipid head group 1-hydroxy-2-aminoethyl phosphonate in Bdellovibrio stolpii. Lipids 36, 513–519. doi: 10.1007/s11745-001-0751-3
Westheimer, F. H., Huang, S., and Covitz, F. (1988). Rates and mechanisms of hydrolysis of esters of phosphorous acid. J. Am. Chem. Soc. 110, 181–185. doi: 10.1021/ja00209a029
Wróblewski, A. E., and Bak-Sypień, I. I. (2007). New analogues of fosfomycin—synthesis of diethyl (1R,2R)- and (1S,2R)-1,2-epoxy-3-hydroxypropylphosphonates. Tetrahedron Asymmetry 18, 520–526. doi: 10.1016/j.tetasy.2007.02.006
Wróblewski, A. E., and Hałajewska-Wosik, A. (2002). An efficient synthesis of enantiomeric (S)-phosphocarnitine. Eur. J. Org. Chem. 2002, 2758–2763. doi: 10.1002/1099-0690(200208)2002:16<2758::AID-EJOC2758>3.0.CO;2-Q
Wróblewski, A. E., and Hałajewska-Wosik, A. (2003). An efficient synthesis of an enantiomerically pure phosphonate analogue of l-GABOB. Tetrahedron Asymmetry 14, 3359–3363. doi: 10.1016/j.tetasy.2003.09.006
Wu, J., and Xia, H.-G. (2005). Tertiary amines as highly efficient catalysts in the ring-opening reactions of epoxides with amines or thiols in H2O: expeditious approach to β-amino alcohols and β-aminothioethers. Green Chem. 7, 708–710. doi: 10.1039/B509288D
Yamamoto, I., Deniau, G. P., Gavande, N., Chebib, M., Johnston, G. A. R., and O'Hagan, D. (2011). Agonist responses of (R)-and (S)-3-fluoro-γ-aminobutyric acids suggest an enantiomeric fold for GABA binding to GABA C receptors. Chem. Commun. 47, 7956–7958. doi: 10.1039/C1CC12141C
Yokomatsu, T., Murano, T., Akiyama, T., Koizumi, J., Shibuya, S., Tsuji, Y., et al. (2003). Synthesis of non-competitive inhibitors of sphingomyelinases with significant activity. Bioorg. Med. Chem. Lett. 13:229. doi: 10.1016/S0960-894X(02)00888-0
Yuan, C.-Y., Wang, K., Li, J.-F., and Li, Z.-Y. (2002). Enzymatic synthesis of phosphocarnitine, phosphogabob and fosfomycin. Phosph. Sulf. Silicon Relat. Elem. 177:2391.
Zanardi, M. M., Suárez, A. G., and Sarotti, A. M. (2016). Determination of the relative configuration of terminal and spiroepoxides by computational methods. Advantages of the inclusion of unscaled data. J. Org. Chem. 82, 1873–1879. doi: 10.1021/acs.joc.6b02129
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06 functionals and 12 other functionals. Theor. Chem. Acc. 119:525. doi: 10.1007/s00214-007-0401-8
Zhao, Y., and Weix, D. J. (2014). Nickel-catalyzed regiodivergent opening of epoxides with aryl halides: co-catalysis controls regioselectivity. J. Am. Chem. Soc.136, 48–51. doi: 10.1021/ja410704d
Keywords: diastereoselectivity, quaternary stereogenic center, aminophosphonates, fluorinated phosphonates, aminoalcohols, epoxyalkylphosphonates
Citation: Rapp M, Margas-Musielak K, Kaczmarek P, Witkowska A, Cytlak T, Siodła T and Koroniak H (2021) Highly Diastereoselective Construction of Carbon– Heteroatom Quaternary Stereogenic Centers in the Synthesis of Analogs of Bioactive Compounds: From Monofluorinated Epoxyalkylphosphonates to α-Fluoro-, β-, or γ-Amino Alcohol Derivatives of Alkylphosphonates. Front. Chem. 9:613633. doi: 10.3389/fchem.2021.613633
Received: 09 October 2020; Accepted: 26 March 2021;
Published: 02 June 2021.
Edited by:
Petri Turhanen, University of Eastern Finland, FinlandReviewed by:
Luke Hunter, University of New South Wales, AustraliaMichele Mancinelli, University of Bologna, Italy
Copyright © 2021 Rapp, Margas-Musielak, Kaczmarek, Witkowska, Cytlak, Siodła and Koroniak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena Rapp, bWFnZHJhcHBAYW11LmVkdS5wbA==