 Simone Di Micco1
Simone Di Micco1 Stefania Terracciano2
Stefania Terracciano2 Dafne Ruggiero2
Dafne Ruggiero2 Marianna Potenza2
Marianna Potenza2 Maria C. Vaccaro2Katrin Fischer3
Maria C. Vaccaro2Katrin Fischer3 Oliver Werz3Ines Bruno2*
Oliver Werz3Ines Bruno2* Giuseppe Bifulco2*
Giuseppe Bifulco2*- 1European Biomedical Research Institute of Salerno (EBRIS), Salerno, Italy
- 2Dipartimento di Farmacia, University degli Studi di Salerno, Fisciano, Italy
- 3Institute of Pharmacy, Friedrich-Schiller-University Jena, Jena, Germany
We report the implementation of our in silico/synthesis pipeline by targeting the glutathione-dependent enzyme mPGES-1, a valuable macromolecular target in both cancer therapy and inflammation therapy. Specifically, by using a virtual fragment screening approach of aromatic bromides, straightforwardly modifiable by the Suzuki-Miyaura reaction, we identified 3-phenylpropanoic acid and 2-(thiophen-2-yl)acetic acid to be suitable chemical platforms to develop tighter mPGES-1 inhibitors. Among these, compounds 1c and 2c showed selective inhibitory activity against mPGES-1 in the low micromolar range in accordance with molecular modeling calculations. Moreover, 1c and 2c exhibited interesting IC50 values on A549 cell lines compared to CAY10526, selected as reference compound. The most promising compound 2c induced the cycle arrest in the G0/G1 phase at 24 h of exposure, whereas at 48 and 72 h, it caused an increase of subG0/G1 fraction, suggesting an apoptosis/necrosis effect.
Introduction
Recently, we reported the successful identification of new mPGES-1 (Microsomal prostaglandin E synthase-1) inhibitors endowed with 1-fluoro-2,4-dinitro-biphenyl scaffold (Di Micco et al., 2018), acting in the low micromolar range in cell-free assays. Our hit discovery strategy was based on the virtual screening of commercially available fragments featuring aryl halide moieties, which represent the basic partners for the Suzuki-Miyaura reactions (Miyaura and Suzuki, 1995), a very suitable synthetic strategy leading to platforms highly prone to further chemical modifications (Giordano et al., 2018b). The obtained encouraging results prompted us to expand the compounds collection aiming to improve the affinity toward the biological target to find new potential drug candidates. mPGES-1 is an inducible transmembrane enzyme, belonging to the MAPEG [membrane-associated proteins in eicosanoid and glutathione (GSH) metabolism] protein family (Jakobsson et al., 2000). This glutathione-dependent enzyme catalyzes the last step of the biosynthesis of prostaglandin E2 (PGE2), resulting particularly upregulated in inflammation (Gerstmeier et al., 2019), in tumors (Nakanishi et al., 2010), and other pathologies such as Alzheimer's disease, arthritis, burn injury, atherosclerosis (Akitake et al., 2013). Besides the involvement of mPGES-1 in inflammation-related disorders, its biosynthetic product, PGE2, has been demonstrated to affect cancer cell proliferation and tumor development in several animal models. In recent years, mPGES-1 has been gaining great attention as a strategic therapeutic target, being the inducible terminal synthase in the production of PGE2. Indeed, its inhibition selectively affects the PGE2 upregulated by pathological conditions, while keeping the tissue levels of other important prostanoid molecules (Trebino et al., 2003), which are responsible for key cellular physiological functions. This special feature allows circumventing the typical gastrointestinal and cardiovascular side effects of COX-1/2 and selective COX-2 inhibitors (Di Micco et al., 2016). Despite the great interest in this biological target and the many efforts focused on the development of potential mPGES-1 binders (Figure 1), so far, no mPGES-1 inhibitors for clinical use have reached the market (Bergqvista et al., 2020). MK-886 is the first synthetic mPGES-1 inhibitor structurally based on an indole-carboxylic acid scaffold (Mancini et al., 2001), which led to some phenanthrene imidazole derivatives such as MF63 (Figure 1; Psarra et al., 2017), but no clinical data are available. Among the benzoxazole piperidine carboxamides, PF4693627 (Figure 1) showed good pharmacodynamic and pharmacokinetic properties, and it has been proposed for use in clinical studies for the treatment of rheumatoid arthritis and osteoarthritis (Arhancet et al., 2013). Some of the proposed inhibitors reached Phase 1, such as the imidazole derivative LY3023703 and the substituted pyrimidine GRC 27864 (Figure 1; Chini et al., 2020). Overall, the structural differences in mouse and human mPGES-1 hamper the development of candidates, both working in preclinical and clinical stages (Bergqvista et al., 2020). Indeed, different research groups from academia and pharma companies have introduced in their drug discovery pipeline biological assays on mouse/rat cell lines (Kim et al., 2016; Ding et al., 2018; Larsson et al., 2019) and in vivo experiments on guinea pig or human mPGES-1 knock-in mice (Xu et al., 2008; Bergqvista et al., 2020). Furthermore, the high plasma protein binding of lead structures also represents a big deal in drug advancement (Koeberle et al., 2016; Di Francesco et al., 2020). Thus, the discovery of new promising chemotypes, hopefully endowed with cross-species activity, for clinical practice is highly requested. In this scenario, many efforts have been made by our research group in order to gain insights into the structural requirements needed to assure an optimal interaction between a ligand and the biological target under investigation (Di Micco et al., 2016, 2018; Gerstmeier et al., 2019; Chini et al., 2020). Different chemical entities, with a promising anti-MPGES-1 activity, have been identified and are currently under further investigation to deepen their biological and pharmacological profile.
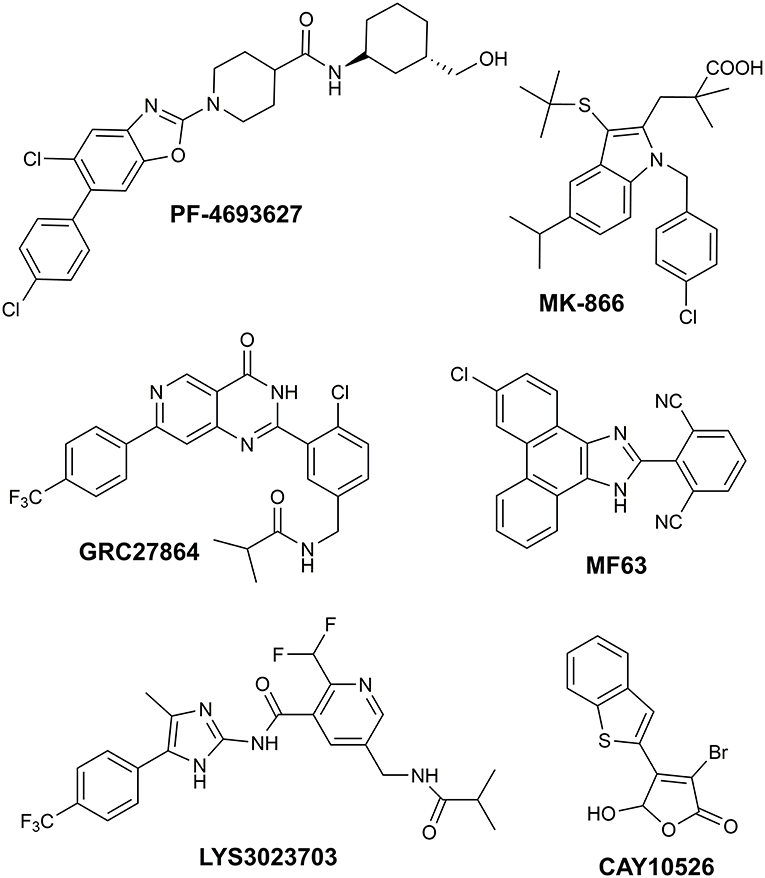
Figure 1. Molecular structure of known mPGES-1 inhibitors.
However, among the several drug discovery strategies available to perform this structure–activity relationship task, one approach is to expand the chemical diversity of the potential ligands, by developing several compound collections to gather as much information as possible. Thereby, following our fragment-based approach, we succeeded in identifying two very promising novel chemotypes whose antiproliferative effect is strongly related to their ability to inhibit mPGES-1, which was not demonstrated in the precedent contribution (Di Micco et al., 2018). Moreover, we implemented our in silico workflow by widening the input aryl bromides and inserting further filters to identify new potential molecular seeds to build the lead compounds. The implemented approach allowed for the wide exploration of the chemical diversity, resulting in the identification of two starting fragments that are structurally different, unlike the previous work (Di Micco et al., 2018).
Materials and Methods
Computational Details
The 3D structures of 1a-f and 2a-f were built by means of Build Panel of Maestro (version 11) and refined by applying the following: the OPLS3 force field (Harder et al., 2016), Polak–Ribier conjugate gradient algorithm (maximum derivative <0.001 kcal/mol), and the GB/SA (generalized Born/surface area) (Still et al., 1990) solvent treatment for the presence of H2O. Then, 1a-f and 2a-f were processed with LigPrep (2017), generating all possible tautomers, stereoisomers, and protonation states at a pH of 7.0 ± 1.0. As for the protein model, the x-ray structure of mPGES-1 (PDB ID: 4AL0) was employed and processed by means of Protein Preparation Wizard (2017) and Sastry et al. (2013): missing side chains and loops were checked; bond order was assigned and all hydrogens were added; and residue alternate positions were checked considering the A conformation. The charges of the side chains were attributed according to their pKa at pH 7.0 and the hydrogen bond network was refined through the optimize option. All water molecules were deleted. Glide (2017) and Friesner et al. (2006) was used for molecular docking calculations. The docking methodology was previously validated (Di Micco et al., 2018) by docking two co-crystallized low-size molecules (Giordano et al., 2018a; Di Micco et al., 2019) endowed with a molecular size similar to that of screened fragments. We sized the inner and outer receptor grid boxes to be 10 and 17 Å, respectively, and centered between chain A and C on the x-, y-, and z-coordinates: 3.04, 21.16, −7.75, respectively. For each step of our workflow, firstly we used Standard Precision (SP), applying default parameters and generating one pose per ligand. Secondly, the generated poses from the SP calculations were employed as input conformations for two rounds of predictions in the Extra Precision (XP) Glide mode, considering the halogen atoms to be the acceptor and donor of bonds. The ligands were treated as flexible, allowing the sole trans conformation for amide bonds, also sampling the nitrogen inversion and ring conformations (with an energy cutoff of 2.5 kcal/mol). The enhanced sampling mode was applied, maintaining 10,000 poses/ligand for the initial step of docking and selecting 1,000 poses per ligand for energy minimization. For each ligand, 1,000 maximum output structures were kept by using 0.15 as the partial charge cutoff and 0.8 as the scaling factor for van der Waals radii. Post-docking optimization was performed on docked conformations, taking into account 100 maximum number of poses and applying 0.5 kcal/mol as a cutoff for rejecting obtained minimized poses. The following energy contributions were accounted for: aromatic H and halogen bonds (as donor and acceptor); the reward of intramolecular H-bonds; and Epik state penalty. Maestro (version 11) was employed for docking outcome analysis and for the generation of figures.
Chemistry
All commercially available starting materials were purchased from Sigma-Aldrich and used as received. All solvents used for the synthesis were of HPLC grade and purchased from Sigma-Aldrich. The NMR spectra (1H, 13C) were recorded using the Bruker Avance 400, 500, 600 MHz instruments (Billerica, MA, United States), T = 298 K. The compounds were dissolved in 0.5 mL of DMSO-d6, CD3OD, CDCl3 (Sigma-Aldrich, 99.8 Atom %D). Coupling constants (J) are reported in Hertz, and the chemical shifts are expressed in ppm on the delta (δ) scale relative to the solvent peak as the internal reference. Multiplicities are reported as follows: s, singlet; d, doublet; t, triplet; m, multiplet; dd, doublet of doublet; ddd, doublet of doublet of doublet. Mass spectrometry experiments were performed using an HPLC–MS system Q-ToF Premiere instrument (Waters, Co., Milford, MA, United States) equipped with an ESI source and Waters pump system. The reactions were monitored on silica gel 60 F254 plates (Merck, Darmstadt, Germany) and the spots were visualized under UV light (λ = 254, 365 nm). Analytical and semi-preparative reversed-phase HPLC was performed on Agilent Technologies 1200 Series high-performance liquid chromatography using a Nucleodur, C8 reversed-phase column (100 mm × 2 mm, 4 μM, 80 Å, flow rate = 1 mL/min; 250 mm × 10 mm, 4 μM, 80 Å, flow rate = 4 mL/min, respectively, Phenomenex®). The binary solvent system (A/B) was as follows: 0.1% TFA in water (A) and 0.1% TFA in CH3CN (B). The absorbance was detected at 220/240 nm. The purity of all the tested compounds (>98%) was determined by HPLC analysis.
General Procedure for the Suzuki-Miyaura Reaction
First, 3-(2-bromophenyl)propanoic acid (1) or 2-(4-bromothiophen-2-yl)acetic acid (2) (1.0 equiv.), the appropriate substituted boronic acid (a-f) (1.2 equiv.), K2CO3 (2 equiv.), and Pd(PPh3)4 (0.05 equiv.) were placed in a 25 mL round-bottom flask equipped with a stir bar in the air. The flask was evacuated and refilled with N2 five times. A mixture of dioxane/H2O (3.0 and 1.5 mL; rate 2:1) was placed in another 25 mL round-bottom flask, which was also evacuated and refilled with N2 five times. Finally, the dioxane/H2O mixture was added using a syringe in the flask with the powders. The reaction was fitted with a condenser, immersed in an oil bath, and refluxed overnight. The reaction mixture was cooled, diluted with acidified water (20 mL), and filtered. The filtrate was washed with CH3OH and CHCl3 (20 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure. The desired compounds 1a-1f and 2a-2f were confirmed by analytical RP-HPLC (Nucleodur, C8 reversed-phase column: 100 mm × 2 mm, 4 μM, 80 Å, flow rate = 1 mL/min). HPLC purification was performed by semi-preparative reversed-phase HPLC (Nucleodur, C8 reversed-phase column: 250 mm × 10 mm, 4 μM, 80 Å, flow rate = 4 mL/min). The final products were obtained with high purity (>98%) as detected by HPLC analysis and were fully characterized by electrospray ionization mass spectrometry (ESI-MS) and NMR spectra.
3-(4′-(4-methylphenylsulfonamido)-[1,1′-biphenyl]-2-yl)propanoic Acid (1a)
Compound 1a was obtained by following the general procedure as a brown solid (30% yield after HPLC). RP-HPLC tR = 31.8 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CD3OD): δ = 7.55 (d, J = 8.2 Hz, 2H), 7.20 (d, J = 8.0 Hz, 2H), 7.16–7.13 (m, 2H), 7.10 (ddd, J = 8.8, 5.3, 3.7 Hz, 1H), δ 7.06–7.01 (m, 4H), 7.00–6.97 (m, 1H), 2.74 (t, J = 7.9 Hz, 2H), 2.26 (s, 3H), 2.24 (t, J = 7.9 Hz, 2H). 13C NMR (100 MHz, CD3OD): δ = 173.5, 143.6 (2C), 141.1, 138.0, 137.7, 136.7, 129.7, 129.4 (2C), 129.2 (2C), 128.8, 127.3, 126.85 (2C), 126.0, 120.9 (2C), 34.5, 28.0, 20.0. ESI-MS: calculated for C22H21NO4S 395.47; found m/z = 394.93 [M-H]−.
3-(3′-(4-methylphenylsulfonamido)-[1,1′-biphenyl]-2-yl)propanoic Acid (1b)
Compound 1b was obtained by following the general procedure as a dark brown solid (60% yield after HPLC). RP-HPLC tR = 35.3 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CD3OD): δ = 7.67–7.64 (m, 2H), 7.32–7.20 (m, 6H), 7.10–6.99 (m, 4H), 2.80 (t, J = 7.9 Hz, 2H), 2.39 (s, 3H), 2.32 (t, J = 7.9 Hz, 2H). 13C NMR (100 MHz, CD3OD): δ = 173.4, 143.6, 142.5, 141.2, 137.6, 137.5, 136.6, 129.5, 129.2 (2C), 128.9, 128.7, 127.5, 126.9 (2C), 126.0, 125.2, 121.9, 119.9, 34.6, 27.9, 20.0. ESI-MS: calculated for C22H21NO4S 395.47; found m/z = 394.12 [M-H]−.
3-(3′-((2-chlorobenzyl)oxy)-[1,1′-biphenyl]-2-yl)propanoic Acid (1c)
Compound 1c was obtained by following the general procedure as a yellow solid (65% after HPLC). RP-HPLC tR = 40.9 min, gradient condition: from 5% B to 100% B in 60 min, Flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CD3OD): δ = 7.60–7.55 (m, 1H), 7.48–7.39 (m, 1H), δ 7.37–7.26 (m, 5H), 7.23 (t, J = 4.6 Hz, 1H), 7.17 (d, J = 7.1 Hz, 1H), 7.04–6.97 (m, 1H), 6.91 (d, J = 7.1 Hz, 2H), 5.21 (s, 2H), 2.88 (t, J = 7.9 Hz, 2H), 2,38 (t, J = 7.9 Hz, 2H). 13C NMR (125 MHz, CD3OD): δ = 175.4, 158.4, 143.1, 141.6, 137.9, 134.7, 132.6, 129.6, 129.1, 129.1 (2C), 129.0, 128.8, 127.3, 126.8, 125.8, 121.8, 115.4, 113.3, 66.9, 34.9, 28.1. ESI-MS: calculated for C22H19ClO3 366.84; found m/z = 367.09 [M+H]+.
3-(3′-benzoyl-[1,1′-biphenyl]-2-yl)propanoic Acid (1d)
Compound 1d was obtained by following the general procedure as a dark brown solid (45% after HPLC). RP-HPLC tR = 36.5 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CD3OD): δ = 7.91–7.80 (m, 4H), 7.68 (dd, J = 8.4, 6.3 Hz, 1H), 7.57 (t, J = 7.7, 2H), 7.51 (dd, J = 8.3, 3.1 Hz, 2H), 7.41–7.33 (m, 3H), 7.24 (d, J = 7.2 Hz, 1H), 2.96 (t, J = 7.9 Hz, 2H), 2.45 (t, J = 7.9 Hz, 2H). 13C NMR (150 MHz, CD3OD): δ = 196.9, 175.0, 146.3, 140.9, 137.8, 137.5, 136.0, 132.4, 129.7 (2C), 129.6 (2C), 129.5, 129.0 (2C), 128.9, 128.1 (2C), 127.9, 126.1, 34.7, 28.0. ESI-MS: calculated for C22H18O3 330.38; found m/z = 331.09 [M+H]+.
3-(4′-(phenoxymethyl)-[1,1′-biphenyl]-2-yl)propanoic Acid (1e)
Compound 1e was obtained by following the general procedure as a red solid (65% after HPLC). RP-HPLC tR = 39.7 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CD3OD): δ = 7.43–7.37 (m, 2H), 7.23–7.11 (m, 7H), 7.05 (d, J = 7.4 Hz, 1H), 6.91 (d, J = 8.0 Hz, 2H), 6.83 (t, J = 7.4 Hz, 1H), 4.75 (s, 2H), 2.79 (t, J = 8.0 Hz, 2H), 2.28 (t, J = 8.0 Hz, 2H). 13C NMR (100 MHz, CD3OD): δ = 173.5, 158.8, 141.6, 141.2, 137.7, 136.2, 129.8, 129.1 (2C), 128.9 (2C), 128.8, 127.3, 127.1 (2C), 126.0, 120.6, 114.6 (2C), 69.3, 34.5, 28.0. ESI-MS: calculated for C22H20O3; found 332.39 m/z = 372.08 [M+K]+.
3-(3′-(benzyloxy)-[1,1′-biphenyl]-2-yl)propanoic Acid (1f)
Compound 1f was obtained by following the general procedure as a dark green solid (60% after HPLC). RP-HPLC tR = 39.3 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CD3OD): δ = 7.33 (d, J = 7.5 Hz, 2H), 7.25 (t, J = 7.4 Hz, 2H), 7.22–7.16 (m, 4H), 7.15–7.09 (m, 1H), 7.04 (d, J = 7.6 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.81–6.74 (m, 2H), 4.75 (s, 2H), 2.75 (t, J = 8.0 Hz, 2H), 2.25 (t, J = 8.0 Hz, 2H). 13C NMR (100 MHz, CD3OD): δ = 175.4, 158.6, 143.0, 141.7, 137.8, 137.3, 129.6, 128.9, 128.7, 128.1 (2C), 127.4, 127.2, 127.1 (2C), 126.0, 121.5, 115.4, 113.4, 69.6, 34.9, 28.1. ESI-MS: calculated for C22H20O3; found 332.39 m/z = 333.17 [M+H]+.
2-(4-(4-(4-methylphenylsulfonamido)phenyl)thiophen-2-yl)acetic Acid (2a)
Compound 2a was obtained by following the general procedure as a brown solid (55% after HPLC). RP-HPLC tR = 39.3 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CDCl3): δ = 7.67 (d, J = 7.9 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 7.27–7.21 (m, 3H), 7.09 (d, J = 8.1 Hz, 2H), 6.63 (s, 1H), 3.92 (s, 2H), 2.40 (s, 3H). 13C NMR (100 MHz, CD3OD): δ = 172.7, 143.6, 140.8, 136.8, 136.7, 136.5, 132.4, 129.1 (2C), 126.9 (2C), 126.3 (2C), 125.5, 121.3 (2C), 119.00, 34.7, 20.0. ESI-MS: calculated for C19H17NO4S2; found 387.47 m/z = 388.10 [M+H]+.
2-(4-(3-(4-methylphenylsulfonamido)phenyl)thiophen-2-yl)acetic Acid (2b)
Compound 2b was obtained by following the general procedure as a dark brown solid (40% after HPLC). RP-HPLC tR = 26.9 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CDCl3): δ = 7.59 (d, J = 8.0 Hz, 2H), 7.25–7.12 (m, 5H), 7.10 (s, 1H), 6.89 (d, J = 7.8 Hz, 1H), 6.55 (s, 1H), 3.83 (s, 2H), 2.30 (s, 3H). 13C NMR (100 MHz, CD3OD): δ = 172.7, 143.7, 140.9, 138.1, 137.0, 136.8, 136.6, 129.2 (2C), 129.1, 126.9 (2C), 125.5, 122.0, 119.7, 119.3, 118.4, 34.6, 20.0. ESI-MS: calculated for C19H17NO4S2; found 387.47 m/z = 388.08 [M+H]+.
2-(4-(3-((2-chlorobenzyl)oxy)phenyl)thiophen-2-yl)acetic Acid (2c)
Compound 2c was obtained by following the general procedure as a dark brown solid (45% after HPLC). RP-HPLC tR = 27.9 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, DMSO-d6): δ = 7.76 (d, J = 1.5 Hz, 1H), 7.66–7.62 (m, 1H), 7.55–7.51 (m, 1H), 7.42–7.39 (m, 3H), 7.36–7.31 (m, 2H), 7.30–7.26 (m, 1H), 6.95 (dd, J = 8.0, 2.5 Hz, 1H), 5.22 (s, 2H), 3.85 (s, 2H). 13C NMR (150 MHz, CD3OD): δ = 171.0, 157.9, 145.1, 136.5, 135.2, 134.8, 132.6, 129.0, 128.9, 128.6, 128.3, 128.1, 126.7, 126.4, 125.8, 116.6, 115.9, 66.7, 39.0. ESI-MS: calculated for C19H15ClO3S; found 358.84 m/z = 359.29 [M+H]−.
2-(4-(4-benzoylphenyl)thiophen-2-yl)acetic Acid (2d)
Compound 2d was obtained by following the general procedure as a dark brown solid (40% after HPLC). RP-HPLC tR = 35.3 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CD3OD): δ = 7.85–7.78 (m, 6H), 7.74–7.72 (m, 1H), 7.67 (t, J = 7.5 Hz, 1H), 7.56 (t, J = 7.6 Hz, 2H), 7.43 (s, 1H), 3.96 (s, 2H). 13C NMR (100 MHz, CD3OD): δ = 196.6, 171.2, 140.4, 139.9, 137.7, 136.9, 135.6, 132.3, 130.5 (2C), 129.5 (2C), 128.1 (2C), 125.7, 125.6 (2C), 121.7, 34.5. ESI-MS: calculated for C19H14O3S; found 322.38 m/z = 321.07 [M-H]+.
2-(4-(4-(phenoxymethyl)phenyl)thiophen-2-yl)acetic Acid (2e)
Compound 2e was obtained by following the general procedure as a dark brown solid (55% after HPLC). RP-HPLC tR = 37.8 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CD3OD): δ = 7.56–7.52 (m, 2H), 7.40 (s, 1H), 7.36 (d, J = 7.7 Hz, 2H), 7.22 (s, 1H), 7.17 (t, J = 8.0 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 6.83 (t, J = 7.7 Hz, 1H), 4.97 (s, 2H), 3.76 (s, 2H). 13C NMR (100 MHz, CD3OD): δ = 179.3, 160.0, 145.5, 141.9, 141.9, 137.7, 129.6 (2C), 128.8 (2C), 128.2, 128.8 (2C), 127.7, 121.7, 115.8 (2C), 70.4, 40.3. ESI-MS: calculated for C19H16O3S; found 324.39 m/z = 325.08 [M-H]−.
2-(4-(3-(benzyloxy)phenyl)thiophen-2-yl)acetic Acid (2f)
Compound 2f was obtained by following the general procedure as a dark brown solid (40% after HPLC). RP-HPLC tR = 36.3 min, gradient condition: from 5% B to 100% B in 60 min, flow rate of 4 mL/min, λ = 240 nm. 1H NMR (400 MHz, CDCl3): δ = 7.38 (d, J = 7.0 Hz, 2H), 7.32 (t, J = 7.4 Hz, 2H), 7.28–7.24 (m, 2H), 7.21–7.16 (m, 2H), 7.11–7.07 (m, 2H), 6.83 (dd, J = 8.3, 2.5 Hz, 1H), 5.03 (s, 2H), 3.84 (s, 2H). 13C NMR (150 MHz, CD3OD): δ = 172.9, 159.2, 141.5, 137.4, 137.2, 136.7, 129.4, 128.1 (2C), 127.4, 127.2 (2C), 125.7, 119.5, 118.5, 113.1, 112.4, 69.6, 34.4. ESI-MS: calculated for C19H16O3S; found 324.39 m/z = 325.20 [M-H]−.
Biological Assay for mPGES-1 Activity
mPGES-1 was obtained by differential centrifugation from interleukin (IL)-1β-stimulated A549 cell microsomes (Koeberle et al., 2008). Firstly, the A549 cells were stimulated by IL-1β (1 ng/mL) for 48 h; then, the cells were harvested and sonicated. The obtained homogenate underwent differential centrifugation at 10,000 × g for 10 min, whereas the supernatant was centrifuged at 174,000 × g for 1 h at 4°C. The microsomal fraction (pellet) was resuspended in 1 mL of homogenization buffer (0.1 M potassium phosphate buffer, pH 7.4, 60 μg/mL soybean trypsin inhibitor, 1 mM phenylmethanesulfonyl fluoride, 250 mM sucrose, 2.5 mM glutathione, 1 μg/mL leupeptin), and the total protein concentration was assessed. The microsomes were resuspended in potassium phosphate buffer (0.1 M, pH 7.4) containing 2.5 mM of glutathione. Compounds 1a-1f and 2a-2f or dimethyl sulfoxide (DMSO; as vehicle control) were added, and 15 min later, the reaction (4°C, 100 μL total volume) started following the addition of 20 μM of PGH2. After 1 min, 100 μL of stop solution (40 mM FeCl2, 80 mM citric acid, and 10 μM 11β-PGE2) were dropped in at 4°C. PGE2 was firstly separated by solid-phase extraction and then analyzed by RP-HPLC as previously described (Koeberle et al., 2008).
Cell Line
Human lung carcinoma A549 cell lines were purchased from Cell Application Inc., Sigma-Aldrich, and Merck (Darmstadt, Germany) and maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, United States) in a 5% CO2 humid atmosphere. To ensure logarithmic growth, the cells were subcultured every 2 days. The cell line was tested for mycoplasma using PCR analysis.
Inhibition of PGE2 Production in Cells
The A549 cells were plated at 10,000 cells/well in DMEM with 10% FBS medium in a 96-well plate. After overnight incubation, the cells were treated with 10 μM of compounds 1c and 2c in DMEM 1% FBS and IL-1β (10 ng/mL) to upregulate the expression of PG synthases.
After 24 h, the amount of PGE2 released in the supernatant was evaluated by using a commercially available enzyme immunoassay kit (Prostaglandin E2 EIA kit Monoclonal, Cayman Chemical Company, Ann Arbor, MI, United States).
Activity Assays of Isolated COX-1 and COX-2
Compounds 1c and 2c were submitted for the evaluation of their activity on cyclooxygenases (COXs), specifically using isolated ovine COX-1 and recombinant human COX-2. The COXs were diluted to a final concentration of 50 U/mL for COX-1 and 20 U/mL for COX-2 in Tris buffer (100 mM, pH = 8) supplemented with glutathione (5 mM), EDTA (100 μM) and hemoglobin (5 μM) and pre-incubated with vehicle (0.1% DMSO) or test compounds (at 10 μM) over 5 min at room temperature. Then, the temperature of the samples was increased up to 37°C over 1 min, and the reactions were triggered by adding arachidonic acid to a final concentration of 5 μM for COX-1 and 2 μM for COX-2. After 5 min, 1 mL of ice-cold methanol was added and the reactions were blocked on ice. Internal PGB1 standard (183 ng) and 530 μL of acidified phosphate-buffered saline (PBS) were added for the purification step, and solid-phase extraction was performed using C18 RP-columns (100 mg, UCT, Bristol, PA, United States) as reported previously (Albert et al., 2002). COX product formation was calculated by analyzing 12- hydroxyheptadecatrienoic acid (12-HHT) formation by means of RP-HPLC (Albert et al., 2002).
Cell Viability Assay
Cell viability was determined by MTT conversion assay. The cells were seeded in triplicate in 96 well-plates and incubated with increasing concentrations (between 5 and 100 μM) of compounds 1c, 2c and CAY10526 (Cayman Chemical Company), or DMSO 0.1% (v/v) for the 24, 48, or 72 h in DMEM. Following the treatment, 20 μL of MTT solution (5 mg/mL in PBS) [3-4,5-dimethyldiazol-2-yl]-2,5-diphenyl tetrazolium bromide reagent (Sigma-Aldrich), was added, and the cells were incubated for an additional 3 h at 37°C. The formazan crystals thus formed were dissolved in 100 μL of buffer containing 50% (v/v) N,N-dimethylformamide, 20% SDS (sodium dodecyl sulfate) (pH 4.5). The IC50 values were defined as the compound concentration resulting in 50% inhibition of cell survival compared to control cells treated with DMSO. The absorbance was measured at 570 nm with a Multiskan™ GO Microplate Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States).
Cell-Cycle Progression Analysis
The A549 cells were treated with compound 2c (5 or 10 μM) or DMSO for 24, 48, or 72 h in DMEM with 10% FBS. After each treatment, the cells were harvested and incubated with propidium iodide (PI) solution (0.1% sodium citrate, 0.1% Triton X-100, and 50 mg/mL of PI) for 30 min at 4°C. For each sample, 10,000 events were recorded using FACScalibur flow cytometry (Becton Dickinson, San Jose, CA, United States), and the proportion of the cells in each cell cycle phase was calculated using the ModFit LT software (BD). Necrosis/apoptosis cell fraction was quantified as the proportion of cells with hypodiploid DNA (subG0/G1 peak) using the CellQuest software (Becton Dickinson). The data reported were obtained by two different experiments with similar results, performed in duplicate, and the statistical significance of results was examined using two-way ANOVA with Bonferroni post-test analysis.
Statistical Analysis
The obtained data are expressed as mean ± SEM of single determinations performed in independent experiments on different days. The IC50 values were graphically calculated from averaged measurements at five different concentrations of the compounds by means of the GraphPad Prism 4.0 software (San Diego, CA, United States). One-way ANOVA followed by Tukey-Kramer post-hoc test for multiple comparisons was applied for statistical evaluation of the data. A p value of <0.05 (*) was considered significant.
Results
Molecular Modeling
We adopted a virtual fragment screening approach (Botta et al., 2015; Giordano et al., 2018a, 2019) starting from aromatic bromides, straightforwardly modifiable by the Suzuki-Miyaura reaction, as successfully reported in a recent contribution of the authors (Di Micco et al., 2018). We focused on aromatic fragments as starting molecular scaffold, based on our previous observation that in the 3D protein structure, different aromatic residues, such as Phe44, His53, and Tyr130, flank the mPGES-1 binding site (Lauro et al., 2016). In the present work, we updated and enlarged the collection of commercially available (Sigma Aldrich + Otava Chemicals) aromatic bromide fragments, following an implemented in silico workflow, which is sketched in Figure 2.
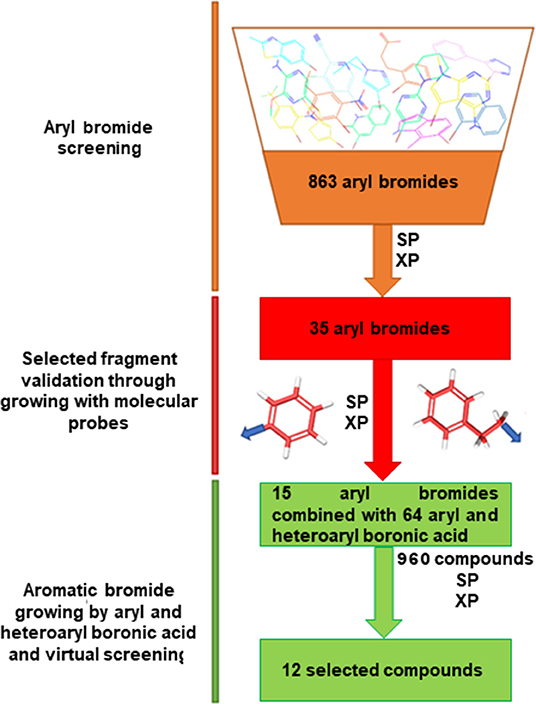
Figure 2. Fragment-based approach workflow.
The built library of 863 fragments was virtually screened into the mPGES-1 catalytic site by means of the Glide software (Friesner et al., 2004, 2006; Halgren et al., 2004; Glide, 2017), which is a very useful tool in fragment-based drug design (Loving et al., 2009; Sándor et al., 2010; Good et al., 2012; Vass and Keseru, 2013; Lauro et al., 2016; Di Micco et al., 2018). As for the protein model, we employed the high-resolution crystal structure (1.16 Å) of the enzyme linked to GSH (PDB ID: 4AL0) (Sjçgren et al., 2013), as no significant structural changes upon inhibitor binding were observed by comparing the different reported experimental structures (Li et al., 2014; Luz et al., 2015; Kuklish et al., 2016; Schiffler et al., 2016; Partridge et al., 2017) of mPGES-1. In our analysis of docked fragments into the catalytic cavity, we particularly paid attention to the bromine atom orientation, representing the functionalization point according to the synthetic strategy used. The docking outcomes suggested 35 potential molecular seeds that can be employed for chemical modifications, all showing to make interactions with residues Phe44, Asp49, Arg52, His53, Arg126, and Ser127. These fragment candidates were expanded by substituting the bromine with both an aryl and a phenylethyl moiety as molecular probes mimicking the boronic acid partners into Suzuki reactions, to evaluate their influence on the binding mode of the parent molecular seeds (Figure 2). The so-built 70 small molecules were subjected to virtual screening, selecting 15 potential aromatic bromides as electrophile partners in the Suzuki-Miyaura reaction. The docked poses of the selected fragments showed that the bromine atom points toward a 9 Å cavity delimited by the transmembrane helixes (Sjçgren et al., 2013; Di Micco et al., 2016, 2018), constituted by the residues A:Tyr28, A:Ala31, A:Ile32, A:Gln36, C:Ser127, C:Tyr130, C:Thr131, C:Gln134, C:Leu135, and C:Cys137. Hence, we combined the selected fragments with 64 boronic acids endowed with two aromatic rings to cover the unoccupied 9-Å pocket and to establish π-π interactions, van der Waals contacts, and H-bonds with the macromolecular counterparts. These structural considerations are fully supported by x-ray crystallography (Sjçgren et al., 2013; Li et al., 2014; Luz et al., 2015; Kuklish et al., 2016; Schiffler et al., 2016; Partridge et al., 2017), showing that the inhibitors bound to mPGES-1 give π-π interactions with Tyr28 and Tyr130 and van der Waals contacts with the other residues outlining the transmembrane cavity. The 960 generated compounds were screened by molecular docking and the obtained outcomes narrowed down the library to, at least, two small collections of compounds 1a-1f and 2a-2f (Figure 2), which are helpful for grasping a basic structure–activity profile.
The two sets of compounds 1a-f and 2a-f contain as basic units, respectively, a 3-biphenyl-2-yl propionic acid and a 4-phenyl-(thiophen-2-yl)acetic acid, derived from the starting aromatic bromide fragments coupled with six different boronic acids (Figure 3). The analysis of the docked poses of 1a-f and 2a-f (Figures 4, 5) showed that all the compounds fill equivalent spaces of the catalytic pocket, making a similar pattern of interactions with the protein counterparts (Table 1). In detail, they establish H-bonds with Arg52, His53, and Ser127, whereas ring A gives a π-cation with Arg126 and an aromatic H-bond with Asp49 (Figures 4, 5). It is worth noting that the contacts with residues Arg52, His53, and Ser127 have been already shown by co-crystallized binders of mPGES-1 (Li et al., 2014; Luz et al., 2015; Kuklish et al., 2016; Schiffler et al., 2016; Partridge et al., 2017). The 3-phenylpropanoic acid (1a-f) and the 2-(thiophen-2-yl)acetic acid (2a-f) portions also establish van der Waals contacts with Gly35, Leu39, Phe44, Arg52, His53, Ala123, and Pro124 (Figures 4, 5). The central aromatic ring gives π-π interactions with Tyr130 and contacts with Gly35, Ser127, Tyr130, and Thr131 (Figures 4, 5). The ring C is engaged in π-π interactions with Tyr130 and gives van der Waals bonds with Tyr28, Ile32, Thr131, and Gln134 (Figures 4, 5). Compounds 1d and 2d, compared to their congeners, do not give contacts with Tyr28 and Gln134 (Figures 4, 5). The sulphonamide groups in 1a, 2a, and 2b donate an H-bond to Thr131 (Figures 4A, 5A,B), whereas 1b accepts an H-bond from Tyr130 (Figure 4B). The oxymethylene group of 1c, 1e, 1f, 2c, 2e, and 2f is hydrogen-bonded to the side chain of Tyr130 (Figures 4C,E,F, 5C,E,F). In this extensive network of contacts, the halogen bond, established by the chlorine atom present on ring C of 1c and 2c, could be responsible for the higher affinity with the protein model and accordingly for the higher biological activity manifested.
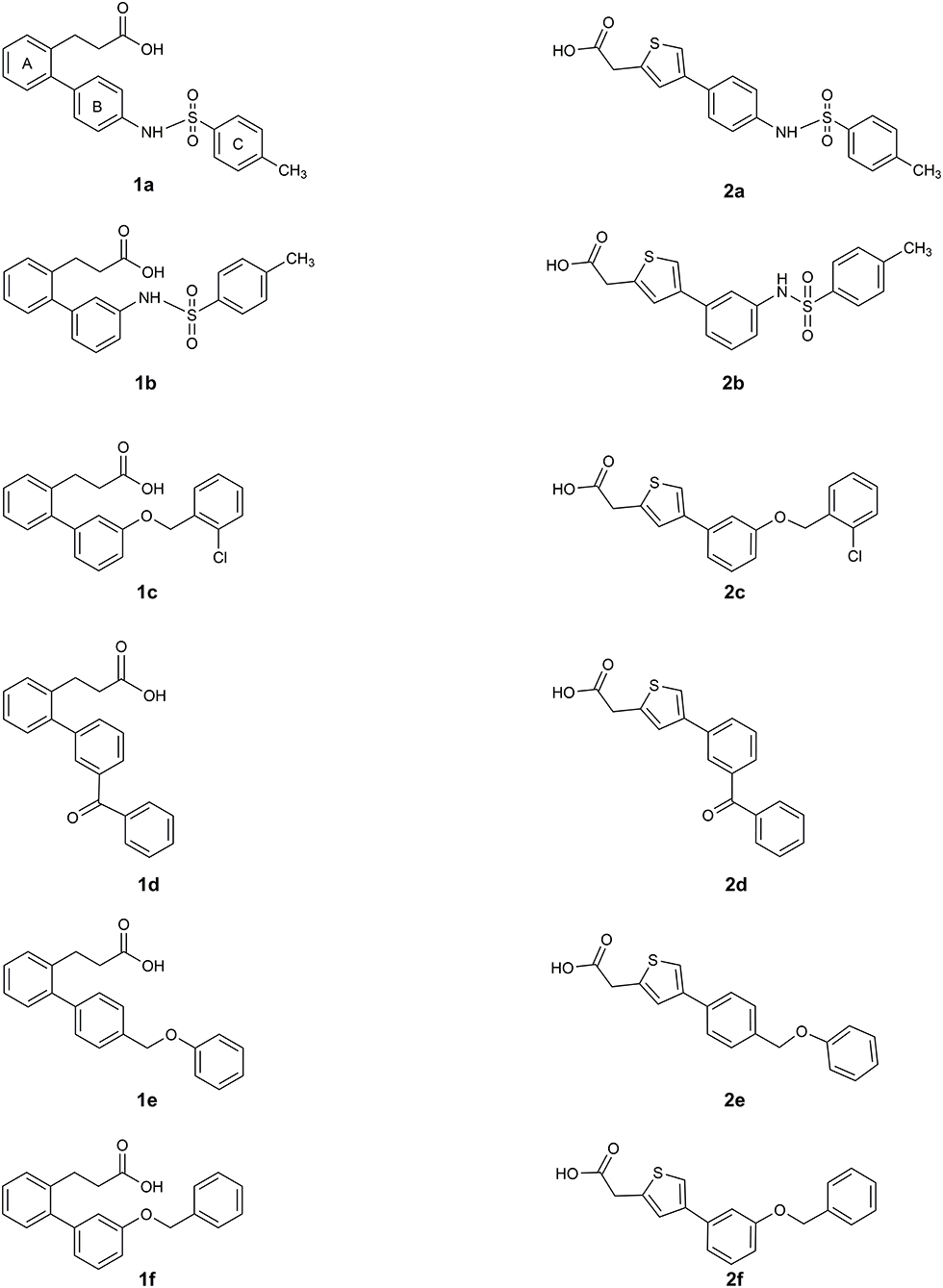
Figure 3. Molecular structure of compounds 1a-f and 2a-f.
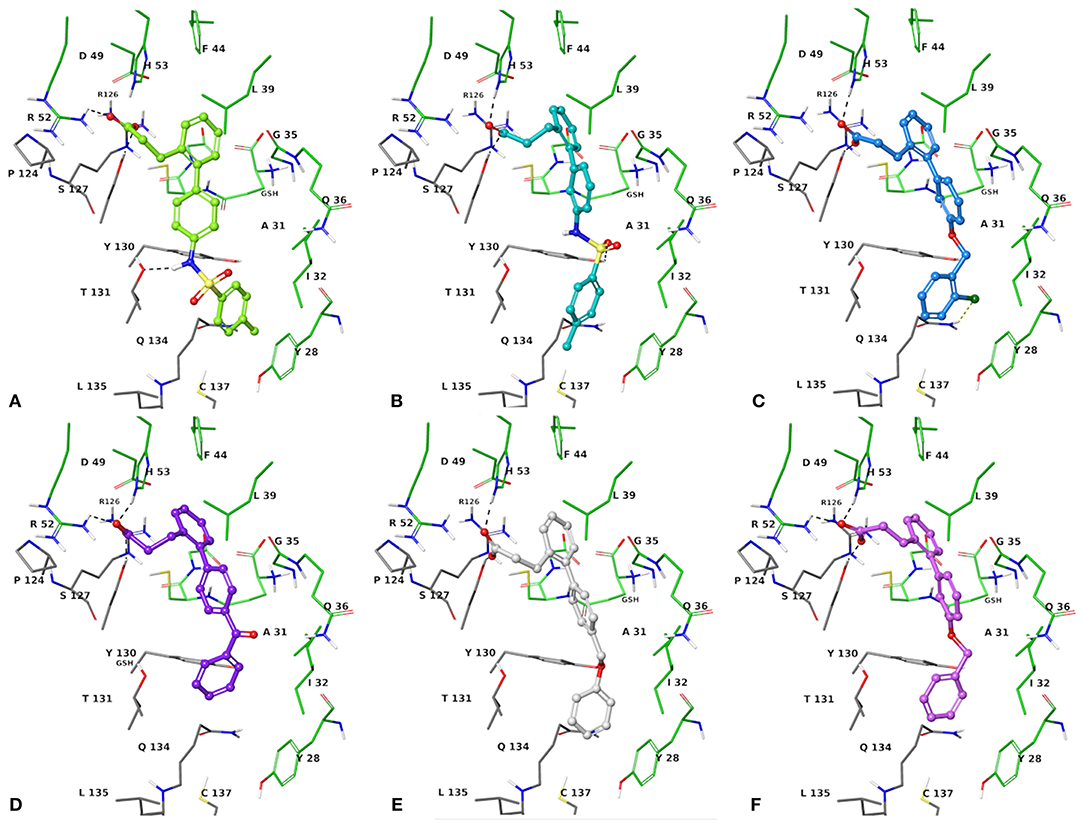
Figure 4. Three-dimensional model of the interactions given by 1a (A), 1b (B), 1c (C), 1d (D), 1e (E), and 1f (F) with mPGES-1. The protein is depicted by tube [colored: C, gray (chain A) and green (chain B); polar H, white; N, dark-blue; O, red; S, yellow]. The GSH is sketched in the faded green tube. The small molecules are represented by sticks (yellow-green for 1a, teal for 1b, azure for 1c, violet for 1d, platinum for 1e, faded plum for 1f) and balls (colored: C, as for the sticks; polar H, white; N, dark-blue; O, red). The dashed black and yellow lines indicate the hydrogen and halogen bonds, respectively, between ligand and protein.
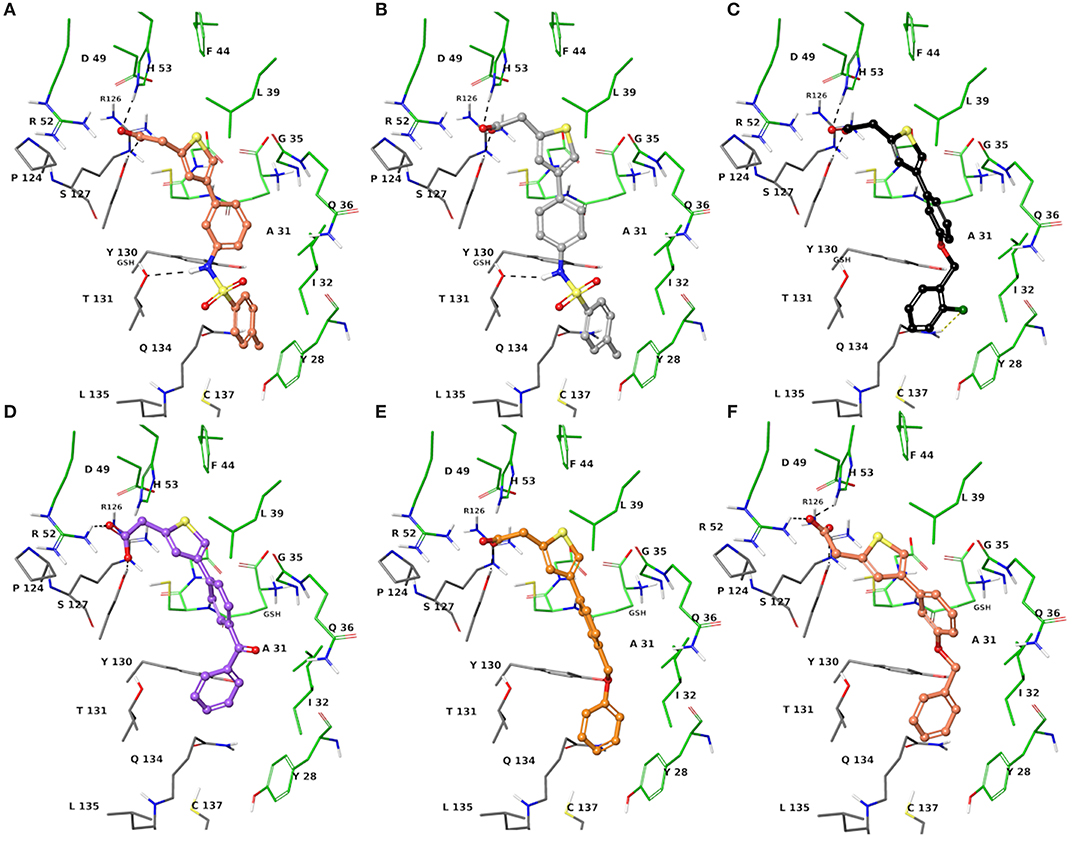
Figure 5. Three-dimensional model of the interactions given by 2a (A), 2b (B), 2c (C), 2d (D), 2e (E), and 2f (F) with mPGES-1. The protein is depicted by the tube [colored: C, gray (chain A) and green (chain B); polar H, white; N, dark-blue; O, red; S, yellow]. The GSH is sketched in the faded green tube. The small molecules are represented by sticks (faded salmon for 2a, light gray for 2b, black for 2c, faded violet for 2d, orange for 2e, faded red-orange for 2f) and balls (colored: C, as for the sticks; polar H, white; N, dark-blue; O, red). The dashed black and yellow lines indicate the hydrogen and halogen bonds, respectively, between ligand and protein.
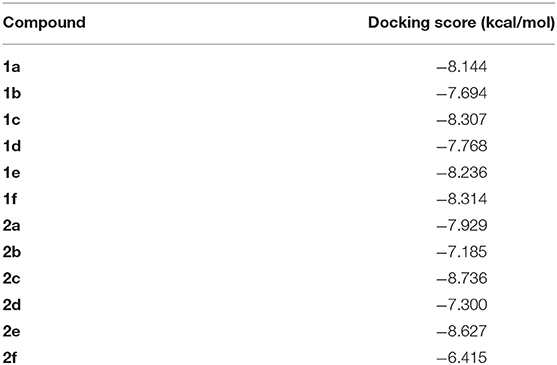
Table 1. Docking scores for 1a-f and 2a-f.
Chemical Synthesis
The synthesis of compounds 1a-f and 2a-f was accomplished by the Suzuki-Miyaura cross-coupling reaction of 3-(2-bromo-phenyl)-propionic acid 1 or (4-bromo-thiophen-2-yl)-acetic acid 2 with selected arylboronic acids/esters (a-f), bearing various substitution patterns, as reported in Scheme 1. By means of the conventional Suzuki reaction conditions, the designed compounds 1a-f and 2a-f were easily prepared with good and moderate yields, respectively, and under mild conditions (see Experimental section) (Miyaura and Suzuki, 1995; Di Micco et al., 2018; Campeau and Hazari, 2019).
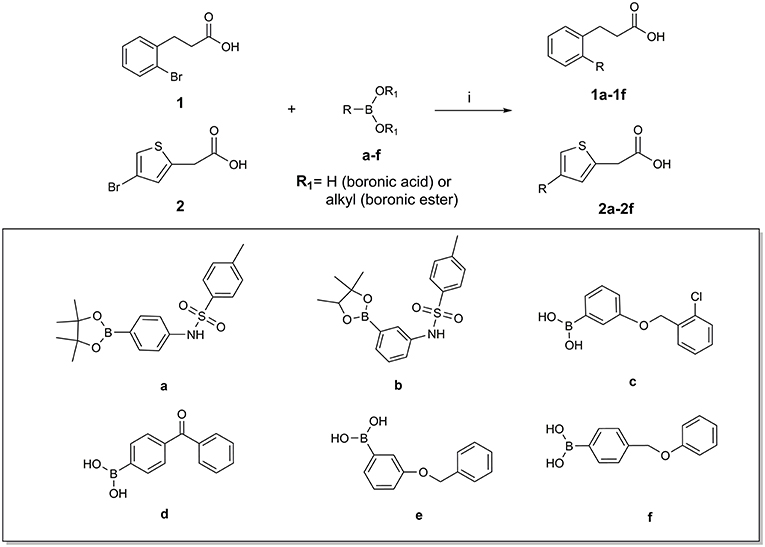
Scheme 1. Synthetic procedure: (i) Pd(PPh3)4, K2CO3, dioxane/H2O (2:1), reflux, overnight.
Biological Investigation
The synthesized compounds (1a-f and 2a-f) were investigated for inhibition of human mPGES-1, derived from microsomes of the IL-1β-stimulated A549 cell line by means of cell-free assay. All compounds were tested at 10 μM and the residual mPGES-1 activity was detected as shown in Figure 6.
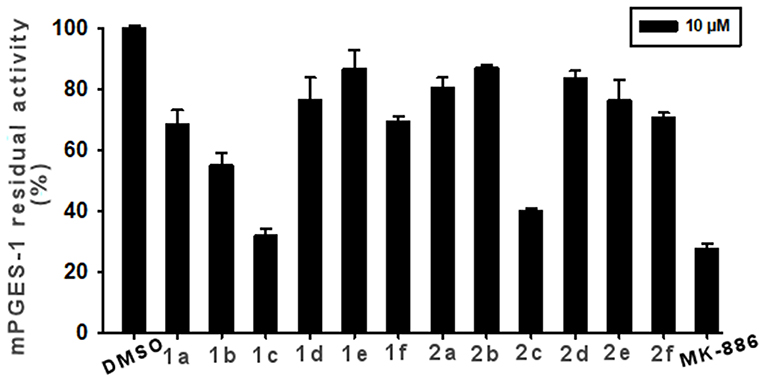
Figure 6. Inhibition of mPGES-1 by compounds 1a-f, 2a-f, and MK-886 (10 μM) in a cell-free enzyme activity assay. The percentage of residual mPGES-1 activity is calculated with respect to the control. Data are given as mean ± SEM, n = 3.
The experiments showed that 1c and 2c presented the highest inhibitory activity (>60%, Figure 6). Compound 1b also appreciably decreased enzyme activity by 42%. Lower enzyme modulation (20–25%) was observed for 1a, 1f, 2e, and 2f, whereas the remaining analogs (1d, 1e, 2a, 2b, and 2d) presented an inhibitory activity of ≤ 20%. Notably, these experimental data are in good qualitative agreement with the theoretical outcomes. Indeed, the chlorine, atom present on ring C and its specific spatial position on ring B, contributes to the increase in binding affinity for the target enzyme.
We further proceeded to investigate the inhibitory activity of 1c and 2c by evaluating their IC50 values vs. mPGES-1 through the same assay. Comparable IC50 values in the low micromolar range were observed for compounds 1c (IC50 = 3.4 ± 0.5 μM) and 2c (IC50 = 5.9 ± 1.0 μM).
Moreover, we investigated the effects of compounds 1c and 2c on PGE2 production in A549 cells, which were incubated with IL-1β and the two selected compounds (10 μM) for 24 h. Both the tested molecules considerably reduced cytokine-induced PGE2 production (Figure 7).
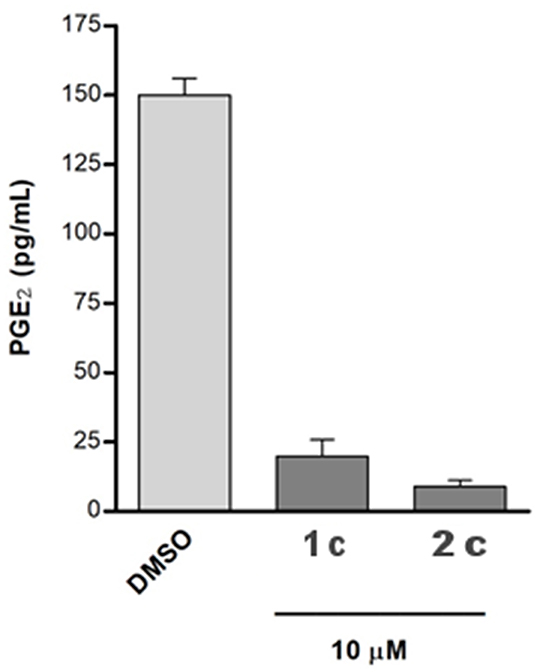
Figure 7. The A549 cells were incubated for 24 h with 10 μM of compounds in conditioned medium (1% FBS and 10 ng/mL IL-1β). The PGE2 released into medium was quantified using a specific ELISA kit assay. The results were compared with that of the control cells IL-1β stimulated and treated with chemical vehicle (DMSO) and are expressed as mean ± SEM (pg/mL) of two different experiments.
To assess the selectivity of the compounds, we investigated the impact of 1c and 2c on both COX isoforms by means of cell-free assays to exclude possible effects on other prostanoid formations. Specifically, the enzymes were incubated with 1c and 2c at 10 μM or vehicle (0.1% DMSO) and the 12-HHT formation was quantified. Interestingly, the compounds did not compromise the activity of COXs (Figure 8), thus confirming their selective inhibition against mPGES-1, which is known to be functionally linked to the inducible isoform COX-2 on the arachidonic acid cascade.
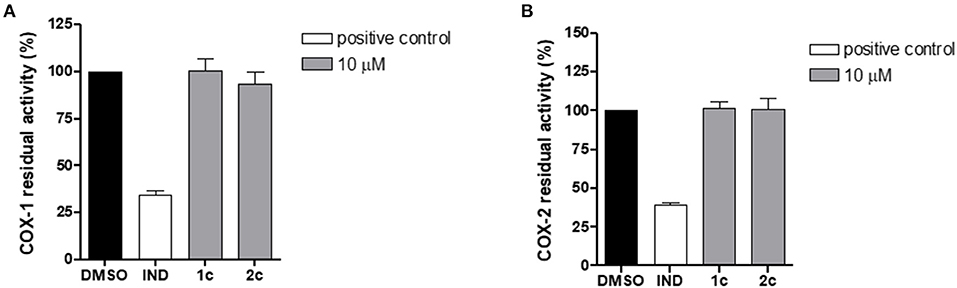
Figure 8. Residual activity of COX-1 (A) and COX-2 (B) after incubation with compounds 1c, 2c, and indometacin (10 μM) or vehicle (0.1% DMSO). Data are expressed as a percentage of control (100%), n = 3.
Cytotoxic Activity and Cell Cycle
Microsomal prostaglandin E synthase-1 is highly expressed and strongly associated with signaling pathways in different types of malignant cells. As suggested in previous studies, the decreasing of mPGES-1 activity may inhibit cell proliferation, induce apoptosis, and cell cycle arrest.
In order to evaluate the potential anticancer properties of the mPGES-1 inhibitors, we tested the antiproliferative or cytotoxic effect of compounds 1c and 2c (5–100 μM) in human A549 cells (Hanaka et al., 2009; Maeng et al., 2014) after exposure for 24, 48, and 72 h, determining cell viability by MTT proliferation assay. The compound CAY10526, a selective modulator of mPGES-1 expression (Guerrero et al., 2007), was used as control in our experiments and reproduced the previously reported IC50 values well (Chini et al., 2020). Moreover, the IC50 values at 48 and 72 h, obtained for CAY10526, were in agreement with the concentrations used to evaluate the interference of the PGE2 production levels in A549 cells (Maeng et al., 2014). As shown in Table 2, 1c and 2c inhibited the cancer cell vitality in the A549 cell line, exhibiting good cytotoxic activity compared to the known inhibitor (CAY10526). Since compound 2c showed the best IC50 values after 48 and 72 h of exposure, it was selected for further analysis.
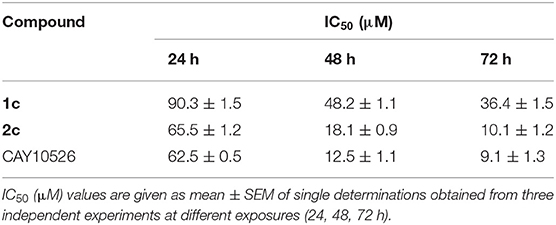
Table 2. Cell viability assay of mPGES-1 inhibitors in the A549 cancer cell line.
The cell cycle progression analysis of A549 cancer cells by flow cytometry was performed to explore the effect of compound 2c on the inhibition of cancer cell viability, using concentrations close to the best IC50 values (Table 2). The treatment with 2c displayed a modest cytostatic activity (Figure 9) after 24 h, and this effect was enhanced after 48 and 72 h with a considerable arrest in the G0/G1 phase. Moreover, the cell exposure with 10 μM of 2c for 48 and 72 h caused an increase in subG0/G1 fraction (about 8.0 ± 0.9% and 13.5 ± 0.5%, respectively), suggesting the occurrence of an apoptosis/necrosis event.
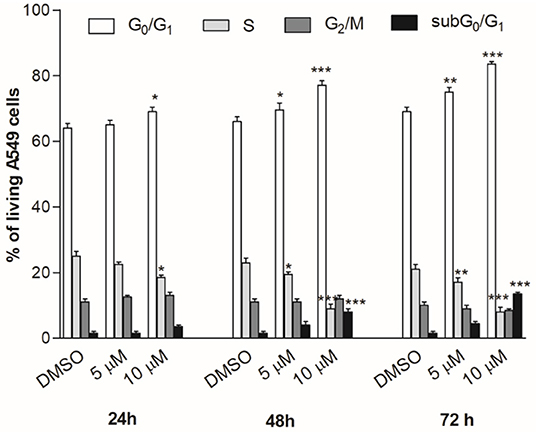
Figure 9. Cell cycle progression. A549 cancer cells were treated for 24, 48, or 72 h with compound 2c used in different concentrations. The percentage of cell cycle stages was analyzed by flow cytometry with DNA propidium iodide staining. Results are expressed as mean ± SEM of two independent experiments performed in triplicate (*P < 0.05, **P < 0.005, ***P < 0.001).
Discussion
By combining a virtual fragment-based approach and biological investigations in cell-free and cell-based assays, we succeeded in identifying the 4-phenyl-(thiophen-2-yl)acetic acid-based compound 2c as a novel lead compound worthy to progressively develop new-generation m-PGES-1 inhibitors with anti-tumoral and/or anti-inflammatory properties. Compound 2c showed low micromolar activity against mPGES-1 and did not affect COXs in cell-free assays. Considering the well-known side effects of commercially available COX inhibitors, the selectivity for mPGES-1 of compound 2c is a promising outcome that paves the way to further investigation. Interestingly, 2c demonstrated considerable inhibition of PGE2 production in A549 cells. On the same cell line, 2c showed good cytotoxic activity.
In particular, we observed that the biological activity profile of 2c at 24, 48, and 72 h is comparable to the reference compound, CAY10526. Interesting results were also obtained for 1c, which presents a selective inhibition activity of mPGES-1 in the low micromolar range. Moreover, 1c considerably inhibits the PGE2 biosynthesis in A549 cells similarly to 2c, and its cytotoxicity is lower than that of 2c. However, the 4-phenyl-(thiophen-2-yl)acetic acid-based scaffold could be used as the molecular framework for the rational design of new mPGES-1 inhibitors. The structure–activity relationships gathered in our investigations suggested that ring C and its meta position on ring B are responsible for the increase in the binding affinity toward mPGES-1. In particular, the presence of a chlorine atom on both the most active compounds against mPGES-1 (1c and 2c) highlights the advantage of a halogen bond for strengthening the ligand–protein complex formation. Furthermore, rings B and C greatly contribute to the complex line-up through π-π interactions, van der Waals contacts, and H-bonds with transmembrane residues. The aryl bromide-derived portion also contributes to intermolecular recognition by contacts with Arg52, His53, and Ser127. The experimental outcomes validated the implementation of our in silico pipeline, which uses phenyl and phenylethyl groups as molecular probes to mimic the boronic acid partner of Suzuki-Miyaura cross-coupling in order to better filter the aryl-bromides. Moreover, the enrichment of the input library collection of aryl bromides with respect to our previous work has increased the chance to explore the chemical diversity of selected molecular seeds to develop potential clinical candidates. In future works, the computer-aided approach defined in this contribution could be of great help to design tighter binders of mPGES-1 with anti-inflammatory and anti-cancer properties, and it could be adapted to different biological targets under investigation. We envisage an application toward other members of the membrane-associated proteins in eicosanoid and glutathione (MAPEG) metabolism superfamily, such as LTC4 and FLAP, aimed at a multitargeted approach against the prostaglandin pathway for safer therapeutical treatment. These outcomes could provide with further chances of disclosing an interesting hit to be directed toward further investigations and for adding another piece to tackle the hard challenge to develop clinical candidates. Thus, the identified lead compounds shall be tested against mouse cell lines (RAW 264.7 and NIH cells) to verify the cross-species activity, thus laying the foundation for successful in vivo tests.
Dedication
This work was dedicated to the memory of our dear colleague Prof. Maurizio Botta.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding authors.
Author Contributions
SD and GB: conceptualization, supervision, and project administration. SD: methodology, validation, and visualization. DR and ST: synthesis. MV, MP and KF: biological assays. SD and ST: formal analysis, investigation, and data curation. GB: resources and funding acquisition. SD, ST, IB, and GB: writing—original draft preparation. SD, ST, OW, IB, and GB: writing—review and editing. All authors have read and agreed on the published version of the manuscript.
Funding
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) (Grant IG 2018 - ID. 21397 project – P.I. GB); Fighting Cancer Resistance: Multidisciplinary Integrated Platform for a Technological Innovative Approach to Oncotherapies (Campania Oncotherapies).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JL declared a past co-authorship with one of the authors OW to the handling Editor.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.676631/full#supplementary-material
References
Akitake, Y., Nakatani, Y., Kamei, D., Hosokawa, M., Akatsu, H., Uematsu, S., et al. (2013). Microsomal prostaglandin E synthase-1 is induced in Alzheimer's disease and its deletion mitigates alzheimer's disease-like pathology in a mouse model. J. Neurosci. Res. 91, 909–919. doi: 10.1002/jnr.23217
Albert, D., Zundorf, I., Dingermann, T., Muller, W. E., Steinhilber, D., and Werz, O. (2002). Hyperforin is a dual inhibitor of cyclooxygenase-1 and 5-lipoxygenase. Biochem. Pharmacol. 64, 1767–1775. doi: 10.1016/S0006-2952(02)01387-4
Arhancet, G. B., Walker, D. P., Metz, S., Fobian, Y. M., Heasley, S. E., Carter, J. S., et al. (2013). Discovery and SAR of PF-4693627, a potent, selective and orally bioavailable mPGES-1 inhibitor for the potential treatment of inflammation. Bioorg. Med. Chem. Lett. 23, 1114–1119. doi: 10.1016/j.bmcl.2012.11.109
Bergqvista, F., Morgensternb, R., and Jakobsson, P.-J. (2020). A review on mPGES-1 inhibitors: From preclinical studies to clinical applications. Prostaglandins Other Lipid Mediators 147, 106383–106393. doi: 10.1016/j.prostaglandins.2019.106383
Botta, A., Sirignano, E., Popolo, A., Saturnino, C., Terracciano, S., Foglia, A., et al. (2015). Identification of lead compounds as inhibitors of STAT3: design, synthesis and bioactivity. Mol. Inform. 34, 689–697. doi: 10.1002/minf.201500043
Campeau, L.-C., and Hazari, N. (2019). Cross-coupling and related reactions: connecting past success to the development of new reactions for the future. Organometallics 38, 3–35. doi: 10.1021/acs.organomet.8b00720
Chini, M. G., Giordano, A., Potenza, M., Terracciano, S., Fischer, K., Vaccaro, M. C., et al. (2020). Targeting mPGES-1 by a combinatorial approach: identification of the aminobenzothiazole scaffold to suppress PGE2 levels. ACS Med. Chem. Lett. 11, 783–789. doi: 10.1021/acsmedchemlett.9b00618
Di Francesco, L., Bruno, A., Ricciotti, E., Tacconelli, S., Dovizio, M., Guillem-Llobat, P., et al. (2020). Pharmacological characterization of the microsomal prostaglandin E2Synthase-1 inhibitor AF3485 in vitro and in vivo. Front. Pharmacol. 11:374. doi: 10.3389/fphar.2020.00374
Di Micco, S., Masullo, M., Bandak, A. F., Berger, J. M., Riccio, R., Piacente, S., et al. (2019). Garcinol and related polyisoprenylated benzophenones as topoisomerase II inhibitors: biochemical and molecular modeling studies. J. Nat. Prod. 82, 2768–2779. doi: 10.1021/acs.jnatprod.9b00382
Di Micco, S., Spatafora, C., Cardullo, N., Riccio, R., Fischer, K., Pergola, C., et al. (2016). 2,3-Dihydrobenzofuran privileged structures as new bioinspired lead compounds for the design of mPGES-1 inhibitors. Bioorg. Med. Chem. 24, 820–826. doi: 10.1016/j.bmc.2016.01.002
Di Micco, Terracciano, S., Cantone, V., Fischer, K., Koeberle, A., Foglia, A., Riccio, R., et al. (2018). Discovery of new potent molecular entities able to inhibit mPGES-1. Eur. J. Med. Chem. 143, 1419–1427. doi: 10.1016/j.ejmech.2017.10.039
Ding, K., Zhou, Z., Hou, S., Yuan, Y., Zhou, S., Zheng, X., et al. (2018). Structure-based discovery of mPGES-1 inhibitors suitable for preclinical testing in wild-type mice as a new generation of anti-inflammatory drugs. Sci. Rep. 8:5205. doi: 10.1038/s41598-018-23482-4
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. doi: 10.1021/jm0306430
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., et al. (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196. doi: 10.1021/jm051256o
Gerstmeier, J., Kretzer, C., Di Micco, S., Miek, L., Butschek, H., Cantone, V., et al. (2019). Novel benzoxanthene lignans that favorably modulate lipid mediator biosynthesis: a promising pharmacological strategy for anti-inflammatory therapy. Biochem. Pharmacol. 165, 263–274. doi: 10.1016/j.bcp.2019.03.003
Giordano, A., del Gaudio, F., Johansson, C., Riccio, R., Oppermann, U., and Di Micco, S. (2018a). Virtual fragment screening identification of a novel Quinoline-5,8-dicarboxylic acid derivative as selective JMJD3 inhibitor. ChemMedChem 13, 1160–1164. doi: 10.1002/cmdc.201800198
Giordano, A., Forte, G., Massimo, L., Riccio, R., Bifulco, G., and Di Micco, S. (2018b). Discovery of new erbB4 inhibitors: repositioning an orphan chemical library by inverse virtual screening. Eur. J. Med. Chem. 152, 253–263. doi: 10.1016/j.ejmech.2018.04.018
Giordano, A., Forte, G., Terracciano, S., Russo, A., Sala, M., Scala, M. C., et al. (2019). Identification of the 2-benzoxazol-2-yl-phenol scaffold as new hit for JMJD3 inhibition. ACS Med. Chem. Lett. 10, 601–605. doi: 10.1021/acsmedchemlett.8b00589
Good, A. C., Liu, J., Hirth, B., Asmussen, G., Xiang, Y., Biemann, H.-P., et al. (2012). Implications of promiscuous pim-1 kinase fragment inhibitor hydrophobic interactions for fragment-based drug design. J. Med. Chem. 55, 2641–2648. doi: 10.1021/jm2014698
Guerrero, M. D., Aquino, M., Bruno, I., Terencio, M. C., Paya, M., Riccio, R., et al. (2007). Synthesis and pharmacological evaluation of a selected library of new potential anti-inflammatory agents bearing the γ-hydroxybutenolide scaffold: a new class of inhibitors of prostanoid production through the selective modulation of microsomal prostaglandin E synthase-1 expression. J. Med. Chem. 50, 2176–2184. doi: 10.1021/jm0700823
Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759. doi: 10.1021/jm030644s
Hanaka, H., Pawelzik, S.-C., Johnsen, J. I., Rakonjac, M., Terawaki, K., Rasmuson, A., et al. (2009). Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. PNAS 106, 18757–18762. doi: 10.1073/pnas.0910218106
Harder, E., Damm, W., Maple, J., Wu, C., Reboul, M., Xiang, J. Y., et al. (2016). OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 12, 281–296. doi: 10.1021/acs.jctc.5b00864
Jakobsson, P. J., Morgenstern, R., Mancini, J., Ford-Hutchinson, A., and Persson, B. (2000). Membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG). A widespread protein superfamily. Am. J. Respir. Crit. Care Med. 161, S20–S24. doi: 10.1164/ajrccm.161.supplement_1.ltta-5
Kim, M., Lee, S., Park, E. B., Kim, K. J., Lee, H. H., Shin, J. S., et al. (2016). Hit-to-lead optimization of phenylsulfonyl hydrazides for a potent suppressor of PGE2 production: synthesis, biological activity, and molecular docking study. Bioorg. Med. Chem. 26, 94–99. doi: 10.1016/j.bmcl.2015.11.024
Koeberle, A., Laufer, S. A., and Werz, O. (2016). Design and development of microsomal prostaglandin E2 synthase-1 inhibitors: challenges and future directions. J. Med. Chem. 59, 5970–5986. doi: 10.1021/acs.jmedchem.5b01750
Koeberle, A., Siemoneit, U., Bühring, U., Northoff, H., Laufer, S., Albrecht, W., et al. (2008). Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J. Pharmacol. Exp. Ther. 326, 975–982. doi: 10.1124/jpet.108.139444
Kuklish, S. L., Antonysamy, S., Bhattachar, S. N., Chandrasekhar, S., Fisher, M. J., Fretland, A. J., et al. (2016). Characterization of 3,3-dimethyl substituted N-aryl piperidines as potent microsomal prostaglandin E synthase-1 inhibitors. Bioorg. Med. Chem. Lett. 26, 4824–4828. doi: 10.1016/j.bmcl.2016.08.023
Larsson, K., Steinmetz, J., Bergqvist, F., Arefin, S., Spahiu, L., Wannberg, J., et al. (2019). Biological characterization of new inhibitors of microsomal PGE synthase-1 in preclinical models of inflammation and vascular tone. Br. J. Pharmacol. 176, 4625–4638. doi: 10.1111/bph.14827
Lauro, G., Tortorella, P., Bertamino, A., Ostacolo, C., Koeberle, A., Fischer, K., et al. (2016). Structure-based design of microsomal prostaglandin E2 synthase-1 (mPGES-1) inhibitors using a virtual fragment growing optimization scheme. ChemMedChem 11, 612–619. doi: 10.1002/cmdc.201500598
Li, D., Howe, N., Dukkipati, A., Shah, S. T., Bax, B. D., Edge, C., et al. (2014). Crystallizing membrane proteins in the lipidic mesophase. Experience with human prostaglandin E2 synthase 1 and an evolving strategy. Cryst. Growth Des. 14, 2034–2047. doi: 10.1021/cg500157x
Loving, K., Salam, N. K., and Sherman, W. (2009). Energetic analysis of fragment docking and application to structure-based pharmacophore hypothesis generation. J. Comput. Aided Mol. Des. 23, 541–554. doi: 10.1007/s10822-009-9268-1
Luz, J. G., Antonysamy, S., Kuklish, S. L., Condon, B., Lee, M. R., Allison, D., et al. (2015). Crystal structures of mPGES-1 inhibitor complexes form a basis for the rational design of potent analgesic and anti-inflammatory therapeutics. J. Med. Chem. 58, 4727–4737. doi: 10.1021/acs.jmedchem.5b00330
Maeng, H.-J., Lee, W.-J., Jin, Q.-R., Chang, J.-E., and Shim, W.-S. (2014). Upregulation of COX-2 in the lung cancer promotes overexpression of multidrug resistance protein 4 (MRP4) via PGE2-dependent pathway. Eur. J. Pharm. Sci. 62, 189–196. doi: 10.1016/j.ejps.2014.05.023
Mancini, J. A., Blood, K., Guay, J., Gordon, R., Claveau, D., Chan, C. C., et al. (2001). Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide induced pyresis and adjuvant-induced arthritis. J. Biol. Chem. 276, 4469–4475. doi: 10.1074/jbc.M006865200
Miyaura, N., and Suzuki, A. (1995). Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 95, 2457–2483. doi: 10.1021/cr00039a007
Nakanishi, M., Gokhale, V., Meuillet, E. J., and Rosenberg, D. W. (2010). mPGES-1 as a target for cancer suppression: a comprehensive invited review “Phospholipase A2 and lipid mediators”. Biochimie 92, 660–664. doi: 10.1016/j.biochi.2010.02.006
Partridge, K. M., Antonysamy, S., Bhattachar, S. N., Chandrasekhar, S., Fishera, M. J., Fretland, A., et al. (2017). Discovery and characterization of [(cyclopentyl)ethyl]benzoic acid inhibitors of microsomal prostaglandin E synthase-1. Bioorg. Med. Chem. Lett. 27, 1478–1483. doi: 10.1016/j.bmcl.2016.11.011
Protein Preparation Wizard (2017). Schrödinger Release 2017-1: Schrödinger Suite 2017-1. New York, NY: Epik, Schrödinger, LLC; Impact, Schrödinger, LLC; Prime, Schrödinger, LLC.
Psarra, A., Nikolaou, A., Kokotou, M. G., Limnios, D., and Kokotos, G. (2017). Microsomal prostaglandin E2 synthase-1 inhibitors: a patent review. Expert Opin. Ther. Pat. 27, 1047–1059. doi: 10.1080/13543776.2017.1344218
Sándor, M., Kiss, R., and Keseru, G. M. (2010). Virtual fragment docking by glide: a validation study on 190 protein-fragment complexes. J. Chem. Inf. Model. 50, 1165–1172. doi: 10.1021/ci1000407
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R., and Sherman, W. (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 27, 221–234. doi: 10.1007/s10822-013-9644-8
Schiffler, M. A., Antonysamy, S., Bhattachar, S. N., Campanale, K. M., Chandrasekhar, S., Condon, B., et al. (2016). Discovery and characterization of 2-acylaminoimidazole microsomal prostaglandin E synthase-1 inhibitors. J. Med. Chem. 59, 194–205. doi: 10.1021/acs.jmedchem.5b01249
Sjçgren, T., Nord, J., Ek, M., Johansson, P., Liu, G., and Geschwindner, S. (2013). Crystal structure of microsomal prostaglandin E2 synthase provides insight into diversity in the mapeg superfamily. Proc. Natl. Acad. Sci. U.S.A. 110, 3806–3811. doi: 10.1073/pnas.1218504110
Still, W. C., Tempczyk, A., Hawley, R. C., and Hendrickson, T. (1990). Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 112, 6127–6129. doi: 10.1021/ja00172a038
Trebino, C. E., Stock, J. L., Gibbons, C. P., Naiman, B. M., Wachtmann, T. S., Umland, J. P., et al. (2003). Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. U.S.A. 100, 9044–9049. doi: 10.1073/pnas.1332766100
Vass, M., and Keseru, M. (2013). Fragments to link. A multiple docking strategy for second site binders. Med. Chem. Commun. 4, 510–514. doi: 10.1039/C2MD20267K
Xu, D., Rowland, S. E., Clark, P., Giroux, A., Cote, B., Guiral, S., et al. (2008). MF63 [2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)-isophthalonitrile], a selective microsomal prostaglandin E synthase-1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J. Pharmacol. Exp. Ther. 326, 754–763. doi: 10.1124/jpet.108.138776
Keywords: fragment-based approach, Suzuki-Miyaura cross-coupling, mPGES-1 inhibitors, anti-inflammatory drugs, anticancer agents
Citation: Di Micco S, Terracciano S, Ruggiero D, Potenza M, Vaccaro MC, Fischer K, Werz O, Bruno I and Bifulco G (2021) Identification of 2-(thiophen-2-yl)acetic Acid-Based Lead Compound for mPGES-1 Inhibition. Front. Chem. 9:676631. doi: 10.3389/fchem.2021.676631
Received: 05 March 2021; Accepted: 07 April 2021;
Published: 07 May 2021.
Edited by:
Antonio Randazzo, University of Naples Federico II, ItalyReviewed by:
Clemens Zwergel, Sapienza University of Rome, ItalyJae Yeol Lee, Kyung Hee University, South Korea
Copyright © 2021 Di Micco, Terracciano, Ruggiero, Potenza, Vaccaro, Fischer, Werz, Bruno and Bifulco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Bifulco, YmlmdWxjb0B1bmlzYS5pdA==; Ines Bruno, YnJ1bm9pbkB1bmlzYS5pdA==