 Hui Li1
Hui Li1 Ryutaro Komatsu2Jihane Hankache3
Ryutaro Komatsu2Jihane Hankache3 Hisahiro Sasabe2Latevi Max Lawson Daku3Bilal Özen1Songjie Chen1Jürg Hauser1Andreas Hauser3
Hisahiro Sasabe2Latevi Max Lawson Daku3Bilal Özen1Songjie Chen1Jürg Hauser1Andreas Hauser3 Silvio Decurtins1Junji Kido2*
Silvio Decurtins1Junji Kido2* Shi-Xia Liu1*
Shi-Xia Liu1*- 1Department of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Bern, Switzerland
- 2Department of Organic Device Engineering Research Center for Organic Electronics, Yamagata University, Yamagata, Japan
- 3Department of Physical Chemistry, University of Geneva, Geneva, Switzerland
A series of bis(triphenylamine)benzodifuran chromophores have been synthesized and fully characterised. Starting from suitably functionalized benzodifuran (BDF) precursors, two triphenylamine (TPA) moieties are symmetrically coupled to a central BDF unit either at 4,8-positions through double bonds (1) and single bonds (2) respectively, or at 2,6-positions through double bonds (3). Their electronic absorption and photoluminescence properties as well as redox behaviour have been investigated in detail, indicating that the π-extended conjugation via vinyl linkers in 1 and 3 leads to comparatively strong electronic interactions between the relevant redox moieties TPA and BDF. Due to intriguing electronic properties and structural planarity, 3a has been applied as a dopant emitter in organic light-emitting diodes. A yellowish-green OLED exhibits a high external quantum efficiency (EQE) of 6.2%, thus exceeding the theoretical upper limit most likely due to energy transfer from an interface exciplex to an emissive layer and/or favorable horizontal orientation.
Introduction
Benzodifuran (BDF) derivatives are of particular interest and relevance for applications as organic semiconductors due to their structural and electronic features. The symmetry and planarity as well as π-extended conjugation can enhance electron delocalization and intermolecular interactions and thus improve charge mobility. After recent explorations of facile and efficient synthetic methodologies, BDF derivatives, including small molecules and polymers, have been widely used for high performance organic electronics such as organic photovoltaics (Li et al., 2010; Huo et al., 2012a; Huo et al., 2012b; Li et al., 2012; Aeschi et al., 2013; Li et al., 2013b; Kularatne et al., 2013; Gao et al., 2020), organic field effect transistors (OFETs) (Huang et al., 2014; Wang et al., 2016; Zhang et al., 2017; Wang et al., 2018), organic light-emitting diodes (OLEDs) (Tsuji et al., 2007; Tsuji et al., 2009; Mitsui et al., 2012) and single-molecule devices (Li et al., 2014; Huang et al., 2015; Xiang et al., 2015; Baghernejad et al., 2020). It has been demonstrated that extended π-conjugated BDF derivatives suitably functionalised with either pyridine (Yi et al., 2010), or pyrene and anthracene (Keller et al., 2011) or triphenyl amine (TPA) (Faurie et al., 2018) termini show very strong fluorescence emission with luminescence quantum yields ΦF of up to 0.98. Inspired by these results we have explored new chromophores for high performance OLED applications, utilising the synergetic effect of the BDF core and two TPA substituents linked either through single bonds or double bonds along the long and the short axes (Scheme 1).
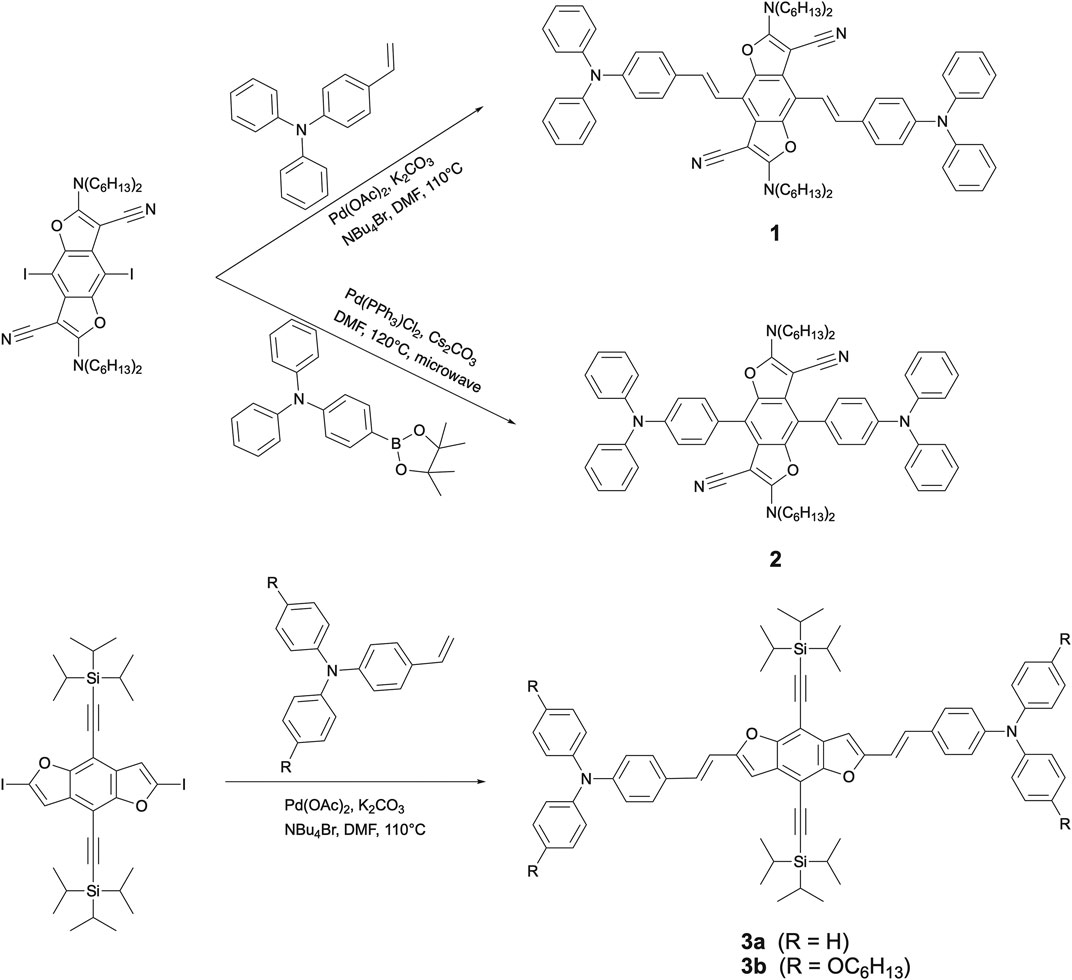
SCHEME 1. Synthetic routes to triads 1, 2, and 3.
OLEDs have attracted significant attention because of their promising applications in flexible large-scale flat-panel displays and their potential for low cost fabrication (Baldo et al., 2000; Wu et al., 2004; Burn et al., 2007; de Sousa et al., 2020; Xu et al., 2021). The characteristic organic semiconducting materials in OLED devices will realise good flexibility over a large-area fabrication and high-performance optical and electrical properties. As one of the most important parameters for the device, the external quantum efficiency (EQE) of fluorescent OLEDs is determined by several factors, including the charge balance of the holes and the electrons, the singlet exciton ratio, the photoluminescence quantum efficiency (PLQE) and the light-outcoupling efficiency (Thejo Kalyani and Dhoble, 2012; Poriel and Rault-Berthelot, 2020). With the limit of the singlet exciton ratio of 25% and the upper theoretical limit of the EQE of 5%, the great challenge has indeed been to improve the EQE of fluorescent OLED devices. In recent studies, the charge balance of holes and electrons has been investigated for elucidating how to affect and maximise the EQE (Pu et al., 2012; Wei et al., 2018; Teng et al., 2020). Although the application of exciplex formation for tuning emission colors (Cheng et al., 2005) or for obtaining white OLEDs (Kalinowski et al., 2007; Tong et al., 2007) has been pursued, there are only few reports concerning the enhancement of the EQE by energy transfer from exciplexes in OLED devices (Park et al., 2011; Park et al., 2013; Kim & Kim, 2019). In this paper, we describe the synthesis, redox and photophysical properties of BDF-TPA triads, 1–3 (Scheme 1). Among them, 3a holds the promise of a better OLED performance in term of intriguing electronic properties and structural planarity. Therefore, only 3a-based OLED devices have been investigated in detail. The yellow-greenish OLED device using 1 wt% 3a-doped 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP, Figure 1) as an emissive layer shows a low turn-on voltage of 3.2 V and an EQE of 6.2%, which is 35% higher than the theoretical EQE upper limit. This is very probably due to energy transfer from an exciplex formed between an electron transporting layer and the host materials to the emissive layer.
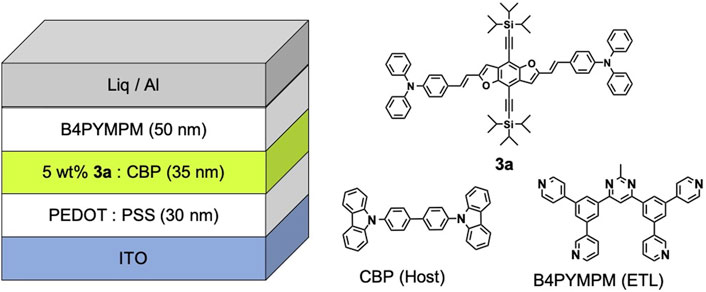
FIGURE 1. The configuration of the OLED device and the chemical structures of 3a, CBP and B4PyMPM.
Experimental
Materials and Methods
2,6-Bis(N,N-dihexylamino)-4,8-diiodobenzo[1,2-b:4,5-b′] difuran-3,7-dicarbonitrile (Yi et al., 2010) and 4,8-bis(triisopropylsilylethynyl)-2,6-diiodobenzo[1,2-b:4,5-b′]difuran (Huang et al., 2015) were previously reported. 4-(4,4,5,5-Tetramethyl-[1,3,2]dioxaborolane)phenyl -N,N-diphenylamine (Fan et al., 2012), N-phenyl-N-(4-vinylphenyl)aniline (Zhou et al., 2011) and 4-(hexyloxy)-N-[4-(hexyloxy)phenyl]-N-(4-vinylphenyl)aniline (Ko et al., 2012) were synthesized according to the literature procedures. All the chemicals and solvents were purchased from commercial sources and were used without further purification, unless specially mentioned. All the reactions were carried out, unless mentioned, under normal laboratory conditions in air. Microwave reactions were conducted using the Biotage Initiator Eight EXP microwave apparatus and the corresponding vials. The reaction was performed in a glass vial (capacity 20 ml) sealed with a septum, under magnetic stirring. The temperature of the reaction mixture was monitored using a calibrated infrared temperature control mounted under the reaction vial. 1H NMR spectra were obtained on a Bruker AC 300 spectrometer operating at 300 MHz. Chemical shift in ppm is quoted relative to residual solvent signals calibrated as follows: δ(CDCl3) = 7.26 ppm, δ(CD2Cl2) = 5.32 ppm. Multiplicities in the 1H NMR spectra are described as: s = singlet, d = doublet, t = triplet, m = multiplet; coupling constants are reported in Hz. Mass spectra were recorded either with ESI (electrospray ionization) on a Thermo Scientific LTQ Orbitrap XL in the positive mode or with Matrix assisted Laser desorption ionization, coupled to a Time-of-Flight analyzer (MALDI-TOF) on a Ultraflex spectrometer.
Synthetic Procedures
Synthesis of 1
A mixture of 2,6-bis(N,N-dihexylamino)-4,8-diiodobenzo[1,2-b:4,5-b′]difuran-3,7-dicarbonitrile (317.2 mg, 0.384 mmol), N-phenyl-N-(4-vinylphenyl)aniline (260 mg, 0.96 mmol), K2CO3 (132.6 mg, 0.96 mmol), NBu4Br (123.7 mg, 0.384 mmol), Pd(OAc)2 (17 mg, 0.076 mmol) and anhydrous DMF (20 ml) was bubbled with N2 for 10 min and then the resulting solution was heated at 110°C overnight in an inert atmosphere. The volatile was evaporated by rotavapor and the residue was subjected to column chromatography on silica gel, eluting with a gradient of CH2Cl2 and hexane (1:1 to 1:0) to afford an orange crystalline product (273.7 mg, 64%). 1H NMR (300 MHz, CDCl3) δ 7.89 (d, J = 16.4 Hz, 2H), 7.43 (m, 6H), 7.26 (m, 8H), 7.12 (m, 8H), 7.03 (m, 8H), 3.63 (m, 8H), 1.80 (m, 8H), 1.36 (m, 24H), 0.87 (t, J = 6.9 Hz, 12H). ESI-MS: Calc. for C76H84N6O2 [M]+ 1112.67, found: 1112.66; [M]2 + 556.34, found: 556.33.
Synthesis of 2
A mixture of 4-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolane) phenyl-N,N-diphenylamine (317 mg, 1 mmol), 2,6-bis(N,N-dihexylamino)-4,8-diiodobenzo[1,2-b:4,5-b′]difuran-3,7-di- carbonitrile (330 mg, 0.4 mmol), Pd(PPh3)2Cl2 (140 mg, 0.2 mmol) and Cs2CO3 (326 mg, 1 mmol) in DMF (6 ml)/H2O (1 ml) was added to a 20 ml microwave vial. The reactor was sealed and purged with Ar for 20 min. The resulting mixture was subjected to microwave irradiation by prestirring for 1 min and reacted at 120°C for 60 min. The resulting mixture was poured into water (10 ml) and extracted with CH2Cl2 (20 ml). The volatile was evaporated by rotavapor, and the residue was subjected to column chromatography on silica gel, eluting with 1:1 CH2Cl2/hexane, to afford a light-yellow solid product (301 mg, 71%). 1H NMR (300 MHz, CDCl3) δ: 7.45 (d, J = 8.7 Hz, 4H), 7.23 (m, 8 H), 7.17 (m, 12H), 7.04 (m, 4H), 3.47 (t, J = 7.74 Hz, 8H), 1.66 (m, 8H), 1.27 (m, 24H), 0.85 (m, 12H). 13C NMR (75.5 MHz, CDCl3) δ: 129.21, 124.70, 47.86, 31.59, 31.20, 26.56, 25.91, 22.68, and 14.03. MALDI-TOF MS: Calcd. for C72H80N6O2 [M]+ 1060.63, found: 1060.75.
Synthesis of 3a
A mixture of N-phenyl-N-(4-vinylphenyl)aniline (339 mg, 1.25 mmol), 4,8-bis(triisopropylsilylethynyl)-2,6-diiodobenzo[1,2-b:4,5-b′]difuran (385 mg, 0.5 mmol), anhydrous potassium carbonate (172 mg, 1.25 mmol), tetrabutylammonium bromide (161 mg, 0.5 mmol), and Pd(OAc)2 (22 mg, 0.01 mmol) was added into anhydrous DMF (30 ml) under N2 atmosphere. The resulting mixture was stirred at 110°C for 4 h, and then cooled to room temperature, followed by pouring into water (400 ml) with stirring. The mixture was extracted with CH2Cl2, and the organic phase was combined and washed with brine. After drying with anhydrous MgSO4, the solvent was removed. The crude product was purified by column chromatography (silica gel) using hexanes/dichloromethane (4:1) as eluent to obtain the product as an orange powder (250 mg). Yield 47%; 1H NMR (300 MHz, CD2Cl2) δ 7.42 (d, J = 8.7 Hz, 4H), 7.32 (m, 10H), 7.13 (m, 16H), 6.97 (d, J = 15.9 Hz, 2H), 6.82 (s, 2H), 1.25 (s, 42H); 13C NMR (75.5 MHz, CDCl3) δ 156.99, 152.48, 148.42, 147.53, 131.07, 130.34, 129.54, 127.92, 125.11, 123.66, 122.96, 113.98, 104.62, 100.96, 99.28, 98.28, 18.77, and 11.61; MALDI-TOF MS: Calcd. for C72H76N2O2Si2 [M]+ 1056.54, found: 1056.54. HRMS: Calcd. for C72H76N2O2Si2 [M]+ 1056.5445, found: 1056.5448.
Synthesis of 3b
Following the same synthetic procedure for 3a, N-phenyl-N-(4-vinylphenyl)aniline was replaced by 4-(hexyloxy)-N-(4-(hexyloxy)phenyl)-N-(4-vinylphenyl)aniline (590 mg, 1.25 mmol). The reaction mixture was stirred under 110°C for 6 h. After cooling down to room temperature, the mixture was poured into water and extracted with CH2Cl2. The organic phase was evaporated and purified on silica gel by flash chromatography using a gradient of hexanes:CH2Cl2 (1:0 to 1.5:1). The pure product was isolated as a red powder (0.36 g). Yield 49%; 1H NMR (300 MHz, CDCl3) δ 7.32 (d, J = 8.6 Hz, 4H), 7.07 (d, J = 7.4 Hz, 8H), 6.85 (d, J = 8.8 Hz, 14H), 3.95 (t, J = 6.5 Hz, 8H), 1.79 (m, 8H), 1.37 (m, 24H), 1.26 (s, 42H) 0.92 (t, J = 7.0 Hz, 12H); ESI-MS: Calcd. for C96H124N2O6Si2 [M]+ 1456.90, found: 1456.90; [M]2 + 728.45, found: 728.45.
Electrochemistry
Cyclic voltammetry measurements in CH2Cl2 (10−4 M) were performed at room temperature under Ar with a three-electrode cell, using 0.1 M Bu4NPF6 as supporting electrolyte, an Ag/AgCl electrode containing 2 M LiCl (in ethanol) as reference electrode, a glassy carbon electrode as counter electrode, and a Pt disk as working electrode at a scan rate of 100 mV/s.
Spectroscopic Measurements
High quality absorption spectra of all compounds in CH2Cl2 were recorded in 1 cm quartz cells with a Cary 5000 dual beam spectrometer at concentrations of 1 × 10−5 M. PL and excitation spectra in CH2Cl2 of the same solutions were recorded with a Fluorolog 3 spectrometer.
Spectroelectrochemical measurement were performed using an optically transparent thin layer cell (d = 0.7 mm) from Specac, which was placed inside the Cary 5000 spectrometer, and the electrochemical potential was applied by using a potentiostat in three electrode configuration. All potentials are given versus the silver wire pseudo-reference electrode.
Computational Details
DFT (Hohenberg and Kohn, 1964; Kohn and Sham, 1965) and TD-DFT (Casida and Chong, 1995) based calculations on 1 were performed with the B3LYP functional (summer 1994; Becke, 1993) and the DZVP2 basis (Godbout et al., 1992) set using the NWChem program package (Aprà et al., 2020). Optimization calculations were performed using tight convergence criteria and the subsequent TDDFT electronic calculations were performed within the Tamm-Dancoff approximation (Hirata and Head-Gordon, 1999).
Crystallography
A crystal of 2 was mounted with Apiezon on a glass needle and used for X-ray structure determination at −100°C. All measurements were made on an Oxford Diffraction SuperNova area-detector diffractometer (2010) using mirror optics monochromated Mo Kα radiation (λ = 0.71073 Å). The unit cell constants and an orientation matrix for data collection were obtained from a least-squares refinement of the setting angles of 5,150 reflections in the range 2.10° < θ < 25.30°. A total of 1,300 frames were collected using ω scans, 120 s exposure time and a rotation angle of 0.5° per frame, and a crystal-detector distance of 50.0 mm.
Data reduction was performed using the (CrysAlisPro, 2010) program. The intensities were corrected for Lorentz and polarization effects, and an absorption correction based on the multi-scan method using SCALE3 ABSPACK in (CrysAlisPro, 2010) was applied. Data collection and refinement parameters are given in Supplementary Table 1.
The structure was solved by direct methods using SUPERFLIP (Palatinus and Chapuis, 2007), which revealed the positions of all non-hydrogen atoms of the title compound. The non-hydrogen atoms were refined anisotropically. All H-atoms but the ones from water were placed in geometrically calculated positions and refined using a riding model where each H-atom was assigned a fixed isotropic displacement parameter with a value equal to 1.2 Ueq of its parent atom (1.5 Ueq for the ammonium groups).
Refinement of the structure was carried out on F2 using full-matrix least-squares procedures, which minimized the function Σw(Fo2 – Fc2)2. The weighting scheme was based on counting statistics and included a factor to downweight the intense reflections. All calculations were performed using the SHELXL-97 (Sheldrick, 2008) program.
Device Fabrication
A solution processed OLED was fabricated with a structure of [ITO (130 nm)/PEDOT:PSS (40 nm)/3a (1 and 5 wt%) doped CBP (30 nm)/B4PYMPM (50 nm)/Liq (3 nm)/Al (100 nm)]. A 40 nm of PEDOT:PSS (CH8000) film as a hole injection layer (HIL) was spin-coated on an ITO substrate and annealed at 200°C in air. Then, a 30 nm of 1 or 5 wt% 3a-doped CBP film as an emitting layer (EML) was spin-coated from a THF solution (5 mg ml−1) and annealed at 60°C for 10 min B4PYMPM as an electron-transporting layer (ETL) and Liq/Al as a cathode were thermally evaporated under vacuum. The current density (J) – luminance (L) – voltage (V) characteristics of the OLEDs were measured by a Keithley source meter 2,400 and a Konica Minolta CS-200, respectively. Electroluminescence (EL) spectra were taken by an optical multichannel analyzer, Hamamatsu PMA 11. The angular dependence of luminous intensity was measured using a Keithley source measure unit 2,400 and a Minolta CS2000. External quantum efficiencies were calculated from the front luminance, current density and EL spectrum.
Results and Discussion
Synthesis and Characterization
The synthetic routes to triads 1, 2, and 3, in which two TPA moieties are symmetrically coupled to a central BDF unit, are outlined in Scheme 1. Starting from 2,6-bis(N,N-dihexylamino)-4,8-diiodobenzo[1,2-b:4,5-b′]difuran-3,7-dicarbonitrile (Yi et al., 2010), the target compounds 1 and 2 were prepared in good yields via palladium-catalyzed Heck and Suzuki cross-coupling reactions with the corresponding TPA precursors, respectively. Similarly, triads 3 were readily obtained from 4,8-bis(triisopropylsilylethynyl)-2,6-diiodobenzo[1,2-b:4,5-b′]difuran in reasonable yields by reactions with 2.5 equivalents of the corresponding TPA derivatives under standard Heck cross-coupling conditions. The structural planarity and π-extended conjugation between redox active TPA and BDF units through vinyl spacers are the same in both 3a and 3b, hence they show equivalent electronic properties. It is, however, envisaged that 3a has a strong tendency to aggregate in solutions. For ease of electrochemical and optical characterization as demonstrated in the following, 3b was therefore prepared by introducing several hexyloxy substituents on the TPA moieties. All of these BDF-TPA triads have been characterized by NMR and Mass spectroscopy. Compound 2 crystallizes as solvate-free colorless needles in the monoclinic space group P21/n. In one unit cell, there are two independent molecules situated on a crystallographic center of symmetry. As illustrated in Figure 2, the BDF core is planar, and the dihedral angles formed between the phenyl rings directly linked to BDF core and the BDF unit is 56.4°. There are no exceptional geometrical features, and all bond lengths and angles are within the expected range; they compare well with reported structures of BDF and TPA derivatives. There are no strong intermolecular interactions such as π-π interactions because of the steric effects of the two TPA groups.
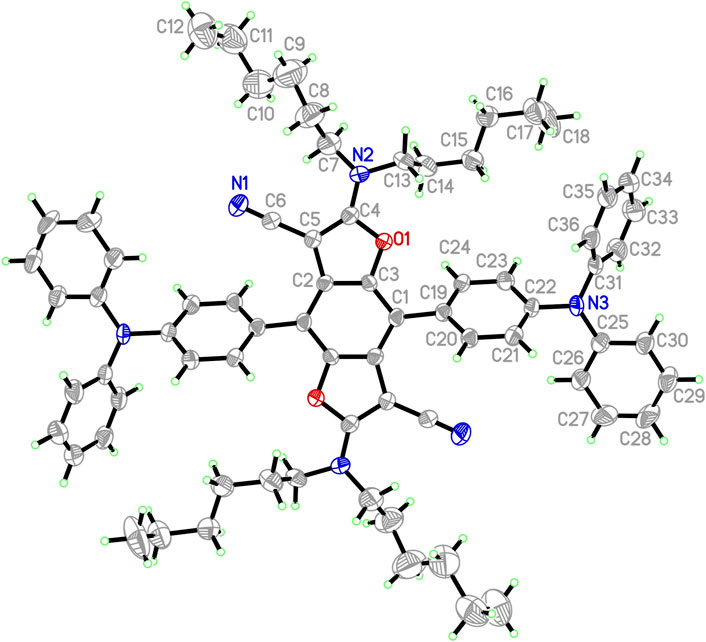
FIGURE 2. ORTEP representation of one independent molecule 2 (50% probability ellipsoids; H-atoms given arbitrary displacement parameters for clarity).
Electrochemical Properties
The redox properties of triads 1−3 were investigated by cyclic voltammetry (CV) in CH2Cl2. As depicted in Figure 3, triad 1 undergoes four distinct, reversible single-electron oxidation processes with the corresponding redox potentials of 0.69, 0.82, 1.20, and 1.35 V against an Ag/AgCl reference electrode. For non-conjugated 2, three reversible single-electron oxidation waves appear at very similar but slightly higher potentials of 0.73, 1.00, and 1.24 V compared to 1, indicative of the lack of strong electronic communications between BDF and TPA units. In contrast, 3b shows likewise three reversible oxidation waves at very similar but slightly lower potentials of 0.55, 1.08, and 1.38 V, owing to the presence of electron-donating hexyloxy substituents and extended π-conjugation. Two electrons are likely involved during the first oxidation process. For 3a, it is impossible to obtain well-resolved redox peaks, presumably due to strong aggregation of the π-extended BDF skeleton without the presence of hexyloxy groups. Both BDF and TPA subunits are electron donating groups with oxidation potentials very close to each other. Consequently, the sequence of oxidation steps for such triads is not obvious.
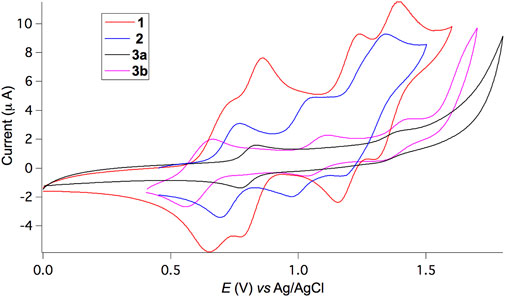
FIGURE 3. Cyclic voltammograms versus Ag/AgCl of 1, 2, and 3 in CH2Cl2 and 0.1 M Bu4NPF6 supporting electrolyte at a scan rate of 100 mV/s.
Furthermore, the conjugated double bonds of the linkers in 1 and 3 lead to comparatively strong electronic interactions between the relevant redox moieties and the description of the oxidation processes as being localised on a given fragment may not be adequate. In this case, charge-transfer processes are expected to be more complex. Spectroelectrochemistry has been used to unravel the sequence of events (vide infra).
Absorption and Photoluminescence Properties in Solution
Figure 4A shows UV-vis absorption, photoluminescence (PL) and excitation spectra of 1 in CH2Cl2 at room temperature. The absorption spectrum consists of a broad structured band with maxima at 20,600, 22,000, and 23,500 cm−1, and a broad unstructured band centered at 32,800 cm−1. On excitation at 430 nm (23,000 cm−1) compound 1 shows an intense, only slightly structured PL band centered at 19,500 cm−1 (530 nm). The perfect superposition of the absorption spectrum with the excitation spectrum indicates that the PL is indeed intrinsic to the molecular triad and not due to impurities. The absorption and PL spectra do not have a mirror-image relationship. This is due to the contribution of several electronic transitions to the absorption band. The PL lifetime as measured in deoxygenated solution at room temperature upon pulsed excitation at 395 nm is 3.2(1) ns (see Figure 4A, inset), and the quantum efficiency is 0.77, giving a radiative lifetime of 4.2 ns.
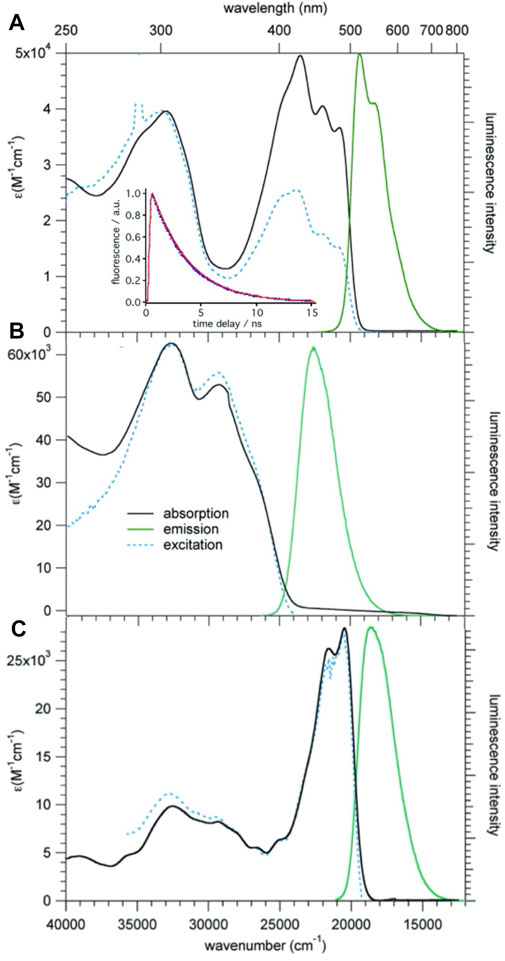
FIGURE 4. Absorption, emission, and excitation spectra of 1 (A), 2 (B), and 3b (C), c = 10−5 M in CH2Cl2 solution at room temperature; for the emission and excitation spectra of 1 and 3b, λex = 430 nm and λem = 550 nm, respectively, for 2 the corresponding values are 320 and 450 nm, respectively. Inset: luminescence decay of 1, experimental: blue, single exponential fit: red.
The assignment of the lowest energy absorption bands can be based on TD-DFT calculations performed on 1 using the B3LYP functional and the DZVP2 basis set implemented in the NWChem program package (Aprà et al., 2020). In particular the lowest energy S0 → S1 transition calculated at 20,700 cm−1 (483 nm), has very high oscillator strength and corresponds with 98% to a one-electron transition during which the electron is promoted from the highest occupied MO (HOMO) to the lowest unoccupied MO (LUMO). Both are extended over the whole molecule due to the conjugated double bonds linking the three moieties (Supplementary Figure 1). The lowest energy excitation is thus best identified as π-π* transition with at most a partial charge localisation on the BDF core in the excited state. The calculated Franck-Condon energy of this transition is in very good agreement with the lowest energy band in the experimental spectrum, and the optical HOMO-LUMO gap according to DFT is computed at 2.82 eV = 22,750 cm−1. Above the first intense band, several weaker transitions follow, giving rise to the observed structure in the lowest energy band. The next higher energy transitions with large oscillator strength are the S0 → S7 and S0 → S8 transitions calculated at around 30,000 cm−1 (330 nm), that is, close to the higher energy band in the experimental absorption spectrum. The overall agreement between the experimental and the calculated absorption spectrum of 1 is very good.
Figures 4B,C show the absorption emission and excitation spectra for compounds 2 and 3b. The lowest energy transition of 3b is at very similar energies as for 1 and the luminescence quantum efficiency is with 0.75 also quite similar. For 2 the lowest energy absorption band is at substantially higher energy. This is attributed to the fact that without the vinyl linker, the phenyl ring of the TPA linked to BDF is no longer co-planar to the latter, and that therefore the π-system is less extended. This observation is consistent with sizable dihedral angles as shown in its single crystal structure. The corresponding luminescence is likewise blue-shifted and the quantum efficiency is 0.48.
Spectroelectrochemistry
The CV of 1 inside the spectroelectrochemical cell (Supplementary Figure 2) shows the same four oxidation waves as the high-quality voltammogram. For the Ag wire pseudo-reference electrode used in the spectroelectrochemical cell, the onset of the oxidation occurs at approximately 100 mV lower potential than for the Ag/AgCl reference used for recording the high-quality CV.
Figure 5 shows the spectral evolution of 1 with an applied positive potential vs. the silver wire pseudo reference electrode. The spectrum before applying any potential consists of the abovementioned structured band with a maximum 23,250 cm−1 (430 nm) and no absorption in the visible range below 20,000 cm−1 and in the NIR. At an applied potential of 650 mV (Figure 5A) a broad unstructured band appears rapidly in the NIR with a maximum at around 6,000 cm−1 (1650 nm) and a weaker shoulder on the low-energy side, that is at 4,200 cm−1 (2,400 nm, Supplementary Figure 3 showing the spectra on a wavelength scale for better visibility of this band). Concomitantly, a second, more structured feature appears in the region of 16,660 – 11,800 cm−1 (600 – 850 nm) and the intensity of the original band structure at around 23,000 cm−1 nm decreases rapidly. Upon prolonged application of 650 mV a second broad band and more intense band centred at 8,000 cm−1 (1250 nm) starts to appear slowly on the high-energy side of the band at 6,000 cm−1. On applying a potential of 850 mV (Figure 5B) to a fresh solution, this second band, in turn, increases rapidly at the expense of the band at 6,000 cm−1. At the same time the shoulder at 4,200 cm−1 disappears again. Finally, application of 1200 mV (Figure 5C) to yet again a fresh solution results in a very rapid appearance of the spectrum obtained at 850 mV followed by a decrease of the intense band at 8,000 cm−1 to zero intensity and the appearance of strong bands at 13,000 cm−1 (770 nm) and 21,000 cm−1 (475 nm). Clearly, the oxidation up to a potential of 1200 mV occurs in three distinct steps. The corresponding most likely spectra of the three species extracted from Figure 5 are shown in Figure 6A and Supplementary Figure 4. The spectrum appearing at a potential of 650 mV corresponds to the one-electron oxidised form 1+. Even though the HOMO is delocalised, the interaction of the oxidised molecule with the weakly polar solvent will quickly result in the localisation of the positive charge on one of the subunits with the inherently lowest oxidation potential, that is, on one of TPA units. In the absorption spectrum following the first oxidation, the intense absorption band at 6,000 cm−1 (1650 nm) corresponds therefore to BDF to TPA+ charge-transfer transition. The weaker band at 4,200 cm−1 (2,400 nm) can be associated with a photo-induced hole transfer from the TPA+ to the neutral TPA, that is an intervalence transition (IV-CT) (Lambert et al., 2002). On the second oxidation step, the intense band at 6,000 cm−1 loses its intensity at the expense of new even more intense band at 8,000 cm−1 (1250 nm). This is due to the oxidation of the second TPA, which results in the shift to higher energy and an increase in intensity of the BDF to TPA+ charge-transfer transition. At the same time this also explains the disappearance of the band associated with an intervalence transition. In the third oxidation process, BDF is oxidised to BDF•+ and the BDF to TPA+ absorption band is fully replaced by the new intense band at 13,000 cm−1 (770 nm). The nature of this transition is not entirely clear, it could be a delocalised SOMO to LUMO transition or BDF+-related transitions (Shukla et al., 2008; Keller et al., 2011).
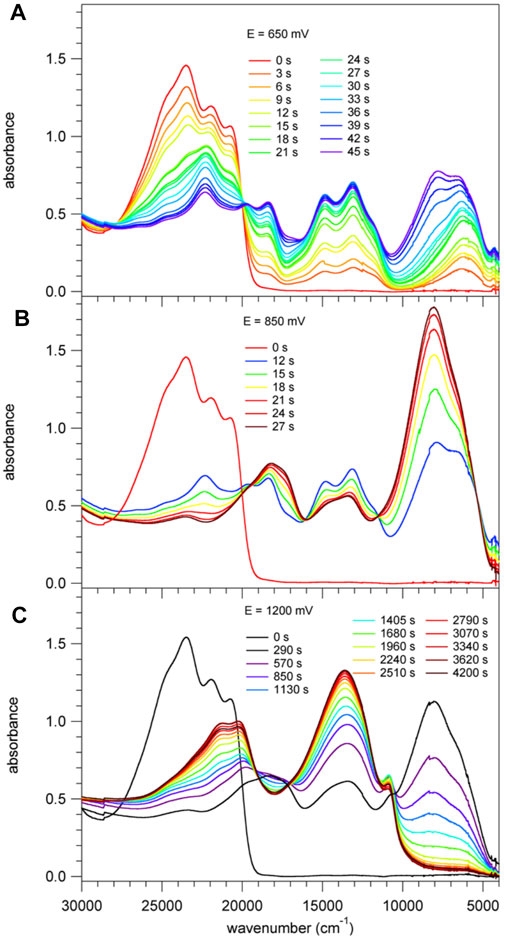
FIGURE 5. Absorption spectra of 1, c= 2 × 10−4 M in CH2Cl2 containing 0.1 M of TBAPF6. These spectra were recorded right after applying for a specific time a positive potential of (A) 650 mV, (B) 850 mV and (C) 1200 mV against an Ag wire pseudo reference electrode.
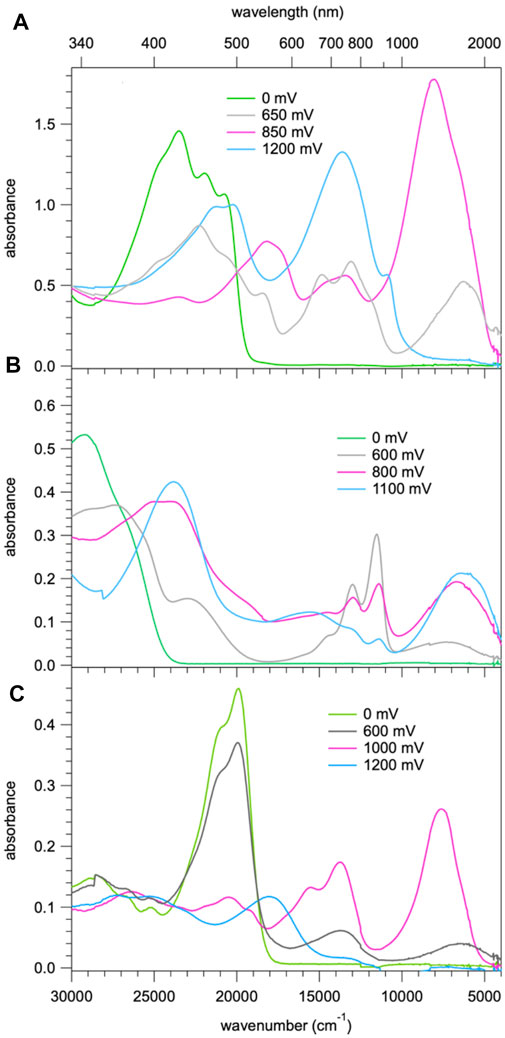
FIGURE 6. (A) Absorption spectra of 1 corresponding to the first three oxidation steps with the most probable spectra of the three species extracted from the spectra of Figure 4; (B) absorption spectra of 2 corresponding to the first three oxidation steps with the most probable spectra of the three species; (C) absorption spectra of 3b corresponding to the three species on sequential oxidation.
For compound 3b, the evolution of the spectrum on oxidation shows similar three steps as for compound 1. Figure 6C shows the most probable spectra of the species associated with the three oxidation steps (for the full evolution of the spectrum, see Supplementary Figure 5). As the potential shifts positively, a BDF to TPA+ charge-transfer transition around 5500 cm−1 initially appears and is subsequently shifted to 7,500 cm−1 which then completely disappears, concomitantly with the appearance of a new absorption band at 23,000 cm−1. In the last step, the BDF is oxidised to BDF+ species. The absence of an IV-CT transition indicates the two TPA units are oxidised simultaneously, which is in good agreement with its CV results.
For compound 2, the evolution on oxidation is somewhat different. The most likely spectra of intermediate species are given in Figure 6B (for the full evolution of the spectrum, see Supplementary Figure 6). Upon the first oxidation, two well-defined absorption bands at 13,000 cm−1 (770 nm) and 12,000 cm−1 (830 nm), and one broad band at 7,000 cm−1 (1430 nm) appear. The former underpins clearly the signature of the BDF+ radical species (Shukla et al., 2008; Keller et al., 2011) whereas the latter is very probably due to a TPA to BDF+ charge-transfer transition. As the oxidation proceeds, an IV-CT band around 6,500 cm−1 (1540 nm) is observed, indicative of the participation of one TPA unit in the process. During the third oxidation, the intrinsic absorption bands of the radical cation BDF•+ significantly decrease and a new intense band at 24,000 cm−1 (420 nm) appears, which might be attributed to the formation of BDF2+ dication species.
Photoluminescence in Solid Films and OLED Performance
The UV-vis absorption and PL spectra of 3a in toluene solution, a neat film and a 5 wt% 3a doped CBP film are depicted in Figure 7. Triad 3a shows intense optical absorptions over the UV-vis spectral part similar to 3b, with absorption onset energies at about 520 nm (19,200 cm−1) and 580 nm (17,200 cm−1) for solution and film, respectively. The λabs,max of the 3a film appears at a longer wavelength than that in solution, most likely due to the aggregation in the solid state. Moreover, 3a exhibits a strong orange emission with a PL quantum efficiency (PLQE) of 70% in toluene, comparable to 3b in CH2Cl2 and other BDF derivatives (Keller et al., 2011; Li et al., 2013a; Yi et al., 2010). A very small stokes shift of 22 nm (880 cm−1) is observed probably because of the rigid molecular structure. The optical band gap is determined to be 2.3 eV from the intersection of the absorption and PL spectra in a neat film. Taken together with CV data, the LUMO energy level is estimated to be −2.7 eV. The ionisation potential (IP) value of a 3a neat film was also determined by photoelectron yield spectrometry to be around 5.2 eV (Supplementary Figure 8 and Supplementary Table 3), which matches well with the HOMO energy level (5.0 eV) estimated from CV measurements. The same holds true for IP values and HOMO energy levels of triads 1 and 2. Although the 5 wt% 3a doped CBP film shows a similar absorption spectrum as the host molecule CBP in the film (Gong et al., 2012), no CBP-based emission at 400 nm but a strong PL from 3a chromophore was observed, indicating efficient energy transfer from host CBP to 3a. Additionally, PLQEs of 3a-doped CBP films were recorded to be around 60% for 1 wt% and 5 wt%, which are significantly higher than those of 5 wt% 1 and 2 doped CBP films (32 and 47%, respectively, Supplementary Table 3). Notably, PLQEs are greatly decreased with increase of 3a concentration (Supplementary Figure 7 and Supplementary Table 2) indicative of the strong aggregation of the π-extended BDF skeleton.
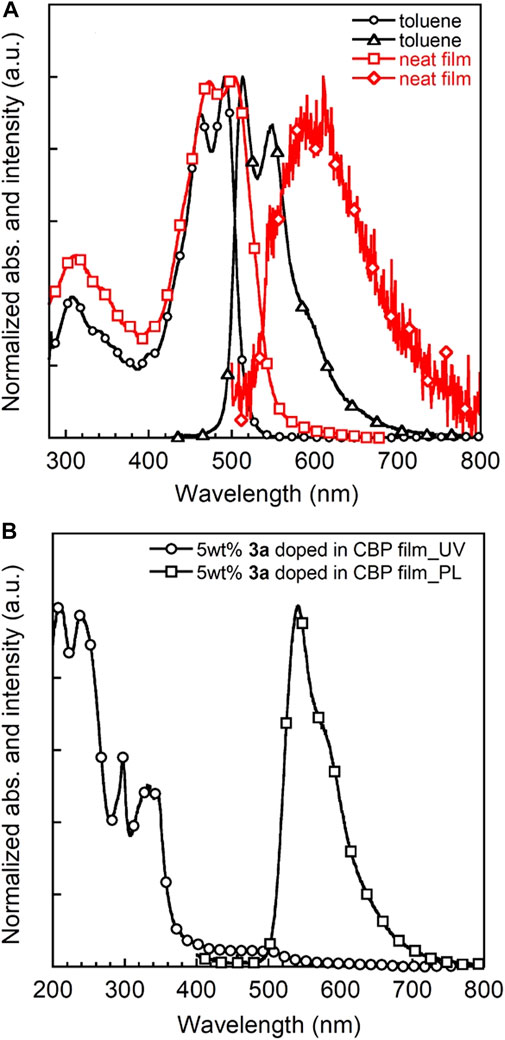
FIGURE 7. UV-vis and PL spectra of 3a in toluene (c = 1 × 10−5 M) and neat film (A) and 5 wt% 3a-doped CBP film (B).
To evaluate the device performance, two types of 3a based OLEDs were fabricated, (I) 1 wt% 3a-doped CBP and (II) 5wt% 3a-doped CBP as an emission layer (EML), respectively. The configuration of the OLED devices is designed as ITO (130 nm)/PEDOT:PSS (40 nm)/EML (30 nm)/bis-4,6-(3,5-di-4-pyridyl-phenyl)-2-methylpyrimidine (B4PYMPM) (50 nm)/Liq (3 nm)/Al (100 nm) (Figure 1). The current density–voltage–luminance (I–V–L) and electroluminescence (EL) spectra of two devices are illustrated in Figure 8, device characteristics (Supplementary Figure 9) are summarised in Table 1 and these data were calculated from Lambertian assumption.
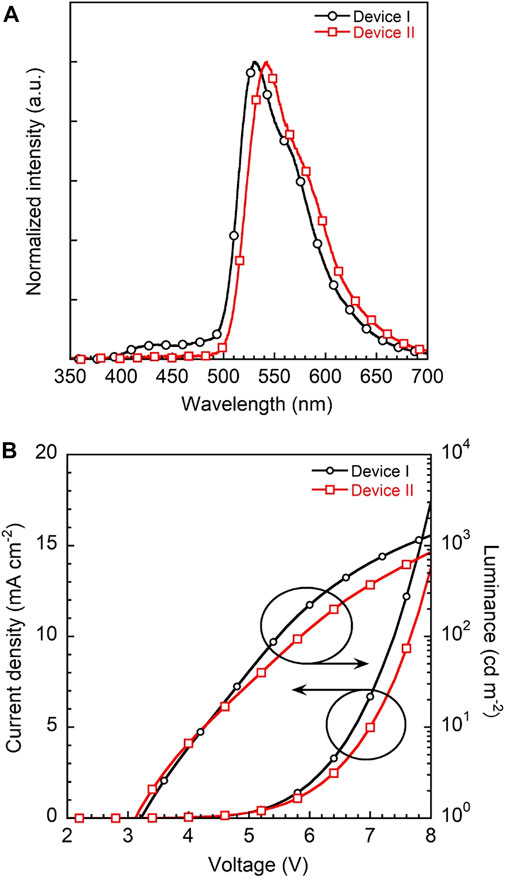
FIGURE 8. (A) EL spectra of devices I and II with 1 and 5 wt% 3a-doped CBP as an emissive layer, respectively and (B) current density-voltage-luminance (J-V-L) plots of two devices.
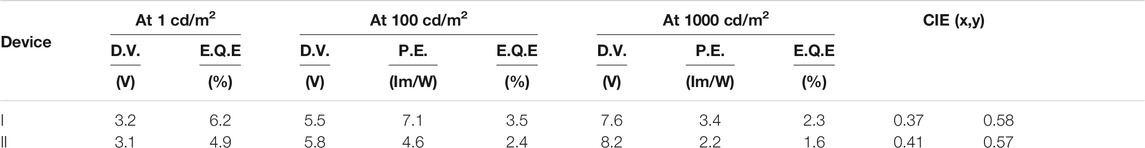
TABLE 1. Characteristics of OLED devices I (1 wt%) and II (5 wt%).
The EL spectra of devices I and II with a λmax at 530 nm are quite similar to the corresponding PL spectra of the emission layers on their own (Supplementary Figure 10). Both devices show a yellowish-green emission with CIEx,y coordinates of (0.37, 0.58) and (0.41, 0.57) and low turn-on voltages of 3.2 and 3.1 V, respectively. However, device I features a higher maximum brightness of 1290 cd m−2 at 8 V than device II with 844 cd m−2.
Notably, the EQEs in both devices (Supplementary Figure 9) are higher than the theoretical limit. In particular, device I exhibits an EQE of 6.2%, which is 35% higher than the theoretical maximum of about 4.6% as calculated according to the equation EQE(ηext) = γ×ηr×ηPL×ηout, γ: charge balance (∼100%), ηr: exciton formation ratio (25% for fluorescence, 100% for phosphorescence), ηPL: PLQE (61%) and ηout: outcoupling efficiency (∼30%). Kim et al. demonstrated that energy transfer from an exciplex formed between the host material CBP and the electron transporting layer bis-4,6-(3,5-di-3-pyridylphenyl)-2-methylpyrimidine (B3PYMPM) to the dopants enhanced the efficiency significantly (Park et al., 2011). Recently, they could make use of energy transfer from the host exciplex to the phosphorescent dopant, finally achieving high performance OLEDs (Park et al., 2013). The same holds for our present case. The EL spectrum of device I shows two emission peaks at 530 nm from 3a and a weaker one at 430 nm from a CBP/B4PyMPM exciplex (Figure 8). In contrast, the corresponding EL spectrum of device II displays no emission around 430 nm, probably due to complete energy transfer from the exciplex to the EML with a higher concentration of dopant.
To verify this speculative emission peaking around 430 nm from the exciplex, the PL spectrum of a co-evaporated CBP/B4PyMPM blend with a weight ratio of 1:1 was recorded. As expected, an exciplex emission around 440 nm (Figure 9), which is red-shifted compared to either CBP or B4PYMPM emission band, matches well with our observations in the EL spectrum of device I. The energy level of the exciplexes is 2.8 eV, which is similar to the difference between the HOMO level of the CBP and the LUMO level of the B4PYMPM, which underlines that the CBP and B4PYMPM molecules form an exciplex upon excitation. Recently, Zhao and co-workers reported that oligofluorene-based emitters in CBP host films fabricated by spin-coating method exhibit moderate horizontal orientation (orientation parameter, S ∼ −0.3) for those exhibiting a large molecular shape anisotropy (Zhao et al., 2016). Therefore, another possible mechanism for this enhancement in EQE is the contribution from the horizontal molecular orientation because 3a has a relatively large molecular shape anisotropy similar to oligofluorene-based emitters, and we also used CBP as a host material in solution-processed OLEDs. However, at this stage, we are not able to quantify the contributions from the energy transfer from exciplex and/or the horizontal molecular orientation of the emitter molecule.
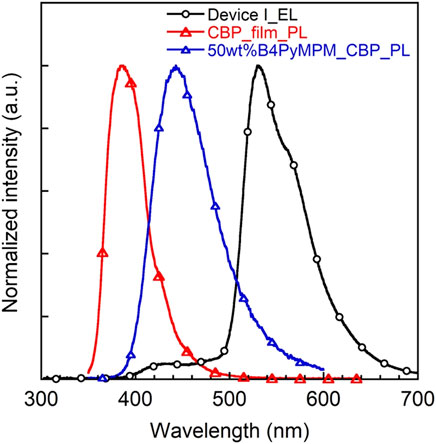
FIGURE 9. EL spectrum of device I and PL of CBP neat film and CBP/B4PyMPM blend.
Conclusion
We have developed facile synthetic routes to triphenylamine-substituted benzodifuran triads 1–3. All of them have been fully characterized, and their electronic absorption and PL properties as well as redox behaviour have been investigated in detail. Compared to non-planar 2, π-extended conjugation via vinyl linkers leads to the coplanarity between the relevant redox TPA and BDF moieties in 1 and 3, with comparatively strong electronic interactions. Due to intriguing electronic properties and unique structural features, the OLED device with 1 wt% 3a-doped CBP as an emissive layer achieves a high external quantum efficiency (EQE) of 6.2%. Given such promising results, 3a can serve as an attractive dopant emitter in OLEDs, and our present study should assist in future research in the development of efficient fluorescent chromophores by emphasizing the full use of energy transfer from an exciplex to an emissive layer.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: CCDC 2062823 contains the supplementary crystallographic data for 2. These data can be obtained free of charge viawww.ccdc.cam.ac.uk/data/cif, or by emailing ZGF0YV9yZXF1ZXN0QGNjZGMuY2FtLmFjLnVr, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, United Kingdom; fax: +44 1223 336033.
Author Contributions
AH, SD, JK, and S-XL conceived the idea and designed the experiments. HL, BO, and SC synthesized 1–3 and characterized them by NMR, MS and cyclic voltammetry. JiH conducted the absorption, emission and spectroelectrochemical measurements and analyzed the experimental data. JüH solved the single crystal structure of 2 by X-ray crystallography. LD carried out the DFT calculations on 1. RK performed the OLED device fabrication and analyzed the experimental data. RK, AH, and S-XL prepared the manuscript. HS, SD, AH, JK, and S-XL supervised the project. HL, HS, and SD co-wrote the manuscript.
Funding
This work was supported by the Center of Innovation (COI) Program funded by the Japan Science and Technology Agency, JST and also by JSPS KAKENHI (17H03131) from JSPS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate the help of Eric Vauthey and Oleksandr Yushchenko for luminescence decay measurements.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.721272/full#supplementary-material
References
Aeschi, Y., Li, H., Cao, Z., Chen, S., Amacher, A., Bieri, N., et al. (2013). Directed Metalation Cascade to Access Highly Functionalized Thieno[2,3-F]benzofuran and Exploration as Building Blocks for Organic Electronics. Org. Lett. 15 (21), 5586–5589. doi:10.1021/ol402787d
Aprà, E., Bylaska, E. J., de Jong, W. A., Govind, N., Kowalski, K., Straatsma, T. P., et al. (2020). NWChem: Past, Present, and Future. J. Chem. Phys. 152 (18), 184102. doi:10.1063/5.0004997
Baghernejad, M., Yang, Y., Al‐Owaedi, O. A., Aeschi, Y., Zeng, B. F., Abd Dawood, Z. M., et al. (2020). Constructive Quantum Interference in Single‐Molecule Benzodichalcogenophene Junctions. Chem. Eur. J. 26 (23), 5264–5269. doi:10.1002/chem.201905878
Baldo, M. A., Thompson, M. E., and Forrest, S. R. (2000). High-efficiency Fluorescent Organic Light-Emitting Devices Using a Phosphorescent Sensitizer. Nature 403 (6771), 750–753. doi:10.1038/35001541
Becke, A. D. (1993). Density‐functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 98 (7), 5648–5652. doi:10.1063/1.464913
Burn, P. L., Lo, S.-C., and Samuel, I. D. W. (2007). The Development of Light-Emitting Dendrimers for Displays. Adv. Mater. 19 (13), 1675–1688. doi:10.1002/adma.200601592
Casida, M. E. (1995). “Time-Dependent Density Functional Response Theory for Molecules,” in Recent Advances in Density Functional Methods. Editor D. P. Chong (Singapore: World Scientific), 155–192. doi:10.1142/9789812830586_0005
Cheng, G., Zhang, Y., Zhao, Y., Liu, S., Xie, Z., Xia, H., et al. (2005). Tunable Electroluminescent Color for 2, 5-diphenyl -1, 4-distyrylbenzene with Two Trans-double Bonds. Appl. Phys. Lett. 87 (1), 013506. doi:10.1063/1.1994950
Oxford Diffraction (2010). CrysAlisPro (Version 1.171.34.36). Oxford Diffraction Ltd., Yarnton, Oxfordshire, UK.
de Sousa, J. A., Bejarano, F., Gutiérrez, D., Leroux, Y. R., Nowik-Boltyk, E. M., Junghoefer, T., et al. (2020). Exploiting the Versatile Alkyne-Based Chemistry for Expanding the Applications of a Stable Triphenylmethyl Organic Radical on Surfaces. Chem. Sci. 11 (2), 516–524. doi:10.1039/c9sc04499j
Fan, C., Wang, X., Ding, P., Wang, J., Liang, Z., and Tao, X. (2012). Synthesis, Photophysical and Iron-Sensing Properties of Terpyridyl-Based Triphenylamine Derivatives. Dyes Pigm. 95 (3), 757–767. doi:10.1016/j.dyepig.2012.05.004
Faurie, A., Gohier, F., and Frère, P. (2018). Facile Synthesis and Optical Properties of Extended TPA-Benzodifuran Derivatives Connected by Cyano-Vinylene Junctions. Dyes Pigm. 154, 38–43. doi:10.1016/j.dyepig.2018.02.025
Gao, Y., Shen, Z., Tan, F., Yue, G., Liu, R., Wang, Z., et al. (2020). Novel Benzo[1,2-B:4,5-B']difuran-Based Copolymer Enables Efficient Polymer Solar Cells with Small Energy Loss and High VOC. Nano Energy 76, 104964. doi:10.1016/j.nanoen.2020.104964
Godbout, N., Salahub, D. R., Andzelm, J., and Wimmer, E. (1992). Optimization of Gaussian-type Basis Sets for Local Spin Density Functional Calculations. Part I. Boron through Neon, Optimization Technique and Validation. Can. J. Chem. 70 (2), 560–571. doi:10.1139/v92-079
Gong, S., He, X., Chen, Y., Jiang, Z., Zhong, C., Ma, D., et al. (2012). Simple CBP Isomers with High Triplet Energies for Highly Efficient Blue Electrophosphorescence. J. Mater. Chem. 22 (7), 2894–2899. doi:10.1039/c1jm14903b
Hirata, S., and Head-Gordon, M. (1999). Time-dependent Density Functional Theory within the Tamm-Dancoff Approximation. Chem. Phys. Lett. 314 (3), 291–299. doi:10.1016/s0009-2614(99)01149-5
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous Electron Gas. Phys. Rev. 136 (3B), B864–B871. doi:10.1103/physrev.136.b864
Huang, C., Chen, S., Baruël Ørnsø, K., Reber, D., Baghernejad, M., Fu, Y., et al. (2015). Controlling Electrical Conductance through a π-Conjugated Cruciform Molecule by Selective Anchoring to Gold Electrodes. Angew. Chem. Int. Ed. 54 (48), 14304–14307. doi:10.1002/anie.201506026
Huang, W., Yang, B., Sun, J., Liu, B., Yang, J., Zou, Y., et al. (2014). Organic Field-Effect Transistor and its Photoresponse Using a Benzo[1,2-B:4,5-B′]difuran-Based Donor-Acceptor Conjugated Polymer. Org. Electronics 15 (5), 1050–1055. doi:10.1016/j.orgel.2014.02.020
Huo, L., Huang, Y., Fan, B., Guo, X., Jing, Y., Zhang, M., et al. (2012a). Synthesis of a 4,8-Dialkoxy-Benzo[1,2-B:4,5-B′]difuran Unit and its Application in Photovoltaic Polymer. Chem. Commun. 48 (27), 3318–3320. doi:10.1039/c2cc17708k
Huo, L., Ye, L., Wu, Y., Li, Z., Guo, X., Zhang, M., et al. (2012b). Conjugated and Nonconjugated Substitution Effect on Photovoltaic Properties of Benzodifuran-Based Photovoltaic Polymers. Macromolecules 45 (17), 6923–6929. doi:10.1021/ma301254x
Kalinowski, J., Cocchi, M., Virgili, D., Fattori, V., and Williams, J. A. G. (2007). Mixing of Excimer and Exciplex Emission: A New Way to Improve White Light Emitting Organic Electrophosphorescent Diodes. Adv. Mater. 19 (22), 4000–4005. doi:10.1002/adma.200700655
Keller, S., Yi, C., Li, C., Liu, S.-X., Blum, C., Frei, G., et al. (2011). Synthesis, Structures, Redox and Photophysical Properties of Benzodifuran-Functionalised Pyrene and Anthracene Fluorophores. Org. Biomol. Chem. 9 (18), 6410–6416. doi:10.1039/c1ob05778b
Kim, H.-B., and Kim, J.-J. (2019). Recent Progress on Exciplex-Emitting OLEDs. J. Inf. Display 20 (3), 105–121. doi:10.1080/15980316.2019.1650838
Ko, S.-B., Cho, A.-N., Kim, M.-J., Lee, C.-R., and Park, N.-G. (2012). Alkyloxy Substituted Organic Dyes for High Voltage Dye-Sensitized Solar Cell: Effect of Alkyloxy Chain Length on Open-Circuit Voltage. Dyes Pigm. 94 (1), 88–98. doi:10.1016/j.dyepig.2011.10.014
Kohn, W., and Sham, L. J. (1965). Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 140 (4A), A1133–A1138. doi:10.1103/physrev.140.a1133
Kularatne, R. S., Magurudeniya, H. D., Sista, P., Biewer, M. C., and Stefan, M. C. (2013). Donor-acceptor Semiconducting Polymers for Organic Solar Cells. J. Polym. Sci. A. Polym. Chem. 51 (4), 743–768. doi:10.1002/pola.26425
Lambert, C., Nöll, G., and Schelter, J. (2002). Bridge-mediated Hopping or Superexchange Electron-Transfer Processes in Bis(triarylamine) Systems. Nat. Mater. 1 (1), 69–73. doi:10.1038/nmat706
Li, H., Jiang, P., Yi, C., Li, C., Liu, S.-X., Tan, S., et al. (2010). Benzodifuran-Based π-Conjugated Copolymers for Bulk Heterojunction Solar Cells. Macromolecules 43 (19), 8058–8062. doi:10.1021/ma101693d
Li, H., Schubert, C., Dral, P. O., Costa, R. D., La Rosa, A., Thüring, J., et al. (2013a). Probing Charge Transfer in Benzodifuran-C60Dumbbell-type Electron Donor-Acceptor Conjugates: Ground- and Excited-State Assays. ChemPhysChem 14 (13), 2910–2919. doi:10.1002/cphc.201300378
Li, H., Tang, P., Zhao, Y., Liu, S. X., Aeschi, Y., Deng, L., et al. (2012). Benzodifuran‐containing Well‐defined π‐conjugated Polymers for Photovoltaic Cells. J. Polym. Sci. A. Polym. Chem. 50 (14), 2935–2943. doi:10.1002/pola.26075
Li, H., Yi, C., Moussi, S., Liu, S.-X., Daul, C., Grätzel, M., et al. (2013b). Benzo[1,2-b:4,5-b′]difuran-based Sensitizers for Dye-Sensitized Solar Cells. RSC Adv. 3 (43), 19798–19801. doi:10.1039/c3ra43669a
Li, Z., Li, H., Chen, S., Froehlich, T., Yi, C., Schönenberger, C., et al. (2014). Regulating a Benzodifuran Single Molecule Redox Switch via Electrochemical Gating and Optimization of Molecule/Electrode Coupling. J. Am. Chem. Soc. 136 (25), 8867–8870. doi:10.1021/ja5034606
Mitsui, C., Tsuji, H., Sato, Y., and Nakamura, E. (2012). Carbazolyl Benzo[1,2-B:4,5-B′]difuran: An Ambipolar Host Material for Full-Color Organic Light-Emitting Diodes. Chem. Asian J. 7 (6), 1443–1450. doi:10.1002/asia.201200062
Palatinus, L., and Chapuis, G. (2007). SUPERFLIP- a Computer Program for the Solution of crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Cryst. 40 (4), 786–790. doi:10.1107/s0021889807029238
Park, Y.-S., Jeong, W.-I., and Kim, J.-J. (2011). Energy Transfer from Exciplexes to Dopants and its Effect on Efficiency of Organic Light-Emitting Diodes. J. Appl. Phys. 110 (12), 124519. doi:10.1063/1.3672836
Park, Y.-S., Lee, S., Kim, K.-H., Kim, S.-Y., Lee, J.-H., and Kim, J.-J. (2013). Exciplex-Forming Co-host for Organic Light-Emitting Diodes with Ultimate Efficiency. Adv. Funct. Mater. 23 (39), 4914–4920. doi:10.1002/adfm.201300547
Poriel, C., and Rault‐Berthelot, J. (2020). Blue Single‐Layer Organic Light‐Emitting Diodes Using Fluorescent Materials: A Molecular Design View Point. Adv. Funct. Mater. 30 (17), 1910040. doi:10.1002/adfm.201910040
Pu, Y.-J., Nakata, G., Satoh, F., Sasabe, H., Yokoyama, D., and Kido, J. (2012). Optimizing the Charge Balance of Fluorescent Organic Light-Emitting Devices to Achieve High External Quantum Efficiency beyond the Conventional Upper Limit. Adv. Mater. 24 (13), 1765–1770. doi:10.1002/adma.201104403
Sheldrick, G. M. (2008). A Short History of SHELX. Acta Cryst. Sect A. 64 (1), 112–122. doi:10.1107/s0108767307043930
Shukla, R., Wadumethrige, S. H., Lindeman, S. V., and Rathore, R. (2008). Synthesis, Electronic Properties, and X-ray Structural Characterization of Tetrarylbenzo[1,2-B:4,5-B′]difuran Cation Radicals. Org. Lett. 10 (16), 3587–3590. doi:10.1021/ol801356e
Teng, J.-M., Wang, Y.-F., and Chen, C.-F. (2020). Recent Progress of Narrowband TADF Emitters and Their Applications in OLEDs. J. Mater. Chem. C 8 (33), 11340–11353. doi:10.1039/d0tc02682d
Thejo Kalyani, N., and Dhoble, S. J. (2012). Organic Light Emitting Diodes: Energy Saving Lighting Technology-A Review. Renew. Sustainable Energy Rev. 16 (5), 2696–2723. doi:10.1016/j.rser.2012.02.021
Tong, Q.-X., Lai, S.-L., Chan, M.-Y., Tang, J.-X., Kwong, H.-L., Lee, C.-S., et al. (2007). High-efficiency Nondoped white Organic Light-Emitting Devices. Appl. Phys. Lett. 91 (2), 023503. doi:10.1063/1.2756137
Tsuji, H., Mitsui, C., Ilies, L., Sato, Y., and Nakamura, E. (2007). Synthesis and Properties of 2,3,6,7-Tetraarylbenzo[1,2-B:4,5-B']difurans as Hole-Transporting Material. J. Am. Chem. Soc. 129 (39), 11902–11903. doi:10.1021/ja074365w
Tsuji, H., Mitsui, C., Sato, Y., and Nakamura, E. (2009). Bis(carbazolyl)benzodifuran: A High-Mobility Ambipolar Material for Homojunction Organic Light-Emitting Diode Devices. Adv. Mater. 21 (37), 3776–3779. doi:10.1002/adma.200900634
Wang, F., Dai, Y., Wang, W., Lu, H., Qiu, L., Ding, Y., et al. (2018). Incorporation of Heteroatoms in Conjugated Polymers Backbone toward Air-Stable, High-Performance N-Channel Unencapsulated Polymer Transistors. Chem. Mater. 30 (15), 5451–5459. doi:10.1021/acs.chemmater.8b02359
Wang, X., Choi, H. H., Zhang, G., Ding, Y., Lu, H., Cho, K., et al. (2016). Bis(2-oxoindolin-3-ylidene)-benzodifuran-dione and Bithiophene-Based Conjugated Polymers for High Performance Ambipolar Organic Thin-Film Transistors: the Impact of Substitution Positions on Bithiophene Units. J. Mater. Chem. C 4 (26), 6391–6400. doi:10.1039/c6tc01617k
Wei, Q., Fei, N., Islam, A., Lei, T., Hong, L., Peng, R., et al. (2018). Small-Molecule Emitters with High Quantum Efficiency: Mechanisms, Structures, and Applications in OLED Devices. Adv. Opt. Mater. 6 (20), 1800512. doi:10.1002/adom.201800512
Wu, C.-C., Lin, Y.-T., Wong, K.-T., Chen, R.-T., and Chien, Y.-Y. (2004). Efficient Organic Blue-Light-Emitting Devices with Double Confinement on Terfluorenes with Ambipolar Carrier Transport Properties. Adv. Mater. 16 (1), 61–65. doi:10.1002/adma.200305619
Xiang, A., Li, H., Chen, S., Liu, S.-X., Decurtins, S., Bai, M., et al. (2015). Electronic Transport in Benzodifuran Single-Molecule Transistors. Nanoscale 7 (17), 7665–7673. doi:10.1039/c5nr00402k
Xu, Y., Xu, P., Hu, D., and Ma, Y. (2021). Recent Progress in Hot Exciton Materials for Organic Light-Emitting Diodes. Chem. Soc. Rev. 50 (2), 1030–1069. doi:10.1039/d0cs00391c
Yi, C., Blum, C., Lehmann, M., Keller, S., Liu, S.-X., Frei, G., et al. (2010). Versatile Strategy to Access Fully Functionalized Benzodifurans: Redox-Active Chromophores for the Construction of Extended π-Conjugated Materials. J. Org. Chem. 75 (10), 3350–3357. doi:10.1021/jo100323s
Zhang, G., Dai, Y., Song, K., Lee, H., Ge, F., Qiu, L., et al. (2017). Bis(2-oxo-7-azaindolin-3-ylidene)benzodifuran-dione-based Donor-Acceptor Polymers for High-Performance N-type Field-Effect Transistors. Polym. Chem. 8 (15), 2381–2389. doi:10.1039/c7py00295e
Zhao, L., Komino, T., Kim, D. H., Sazzad, M. H., Pitrat, D., Mulatier, J.-C., et al. (2016). Horizontal Molecular Orientation of Light-Emitting Oligofluorenes in Spin-Coated Glassy Organic Thin Films. J. Mater. Chem. C 4 (48), 11557–11565. doi:10.1039/c6tc02861f
Keywords: benzodifuran, triphenylamine, spectroelectrochemistry, optical spectroscopy, organic light-emitting diode
Citation: Li H, Komatsu R, Hankache J, Sasabe H, Lawson Daku LM, Özen B, Chen S, Hauser J, Hauser A, Decurtins S, Kido J and Liu S-X (2021) Bis(Triphenylamine)Benzodifuran Chromophores: Synthesis, Electronic Properties and Application in Organic Light-Emitting Diodes. Front. Chem. 9:721272. doi: 10.3389/fchem.2021.721272
Received: 06 June 2021; Accepted: 08 July 2021;
Published: 21 July 2021.
Edited by:
Zhendong Jin, The University of Iowa, United StatesReviewed by:
Xianfeng Qiao, South China University of Technology, ChinaHaibo Qiu, Zhejiang Hisun Pharmaceutical, China
Copyright © 2021 Li, Komatsu, Hankache, Sasabe, Lawson Daku, Özen, Chen, Hauser, Hauser, Decurtins, Kido and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junji Kido, a2lkQHl6LnlhbWFnYXRhLXUuYWMuanA=; Shi-Xia Liu, bGl1QGRjYi51bmliZS5jaA==