 Lingqi Qiu†
Lingqi Qiu† Nicolás M. Morato
Nicolás M. Morato Kai-Hung Huang
Kai-Hung Huang R. Graham Cooks
R. Graham Cooks- Department of Chemistry and Center for Analytical Instrumentation Development, Purdue University, West Lafayette, IN, United States
Spontaneous oxidation of compounds containing diverse X=Y moieties (e.g., sulfonamides, ketones, esters, sulfones) occurs readily in organic-solvent microdroplets. This surprising phenomenon is proposed to be driven by the generation of an intermediate species [M+H2O]+·: a covalent adduct of water radical cation (H2O+·) with the reactant molecule (M). The adduct is observed in the positive ion mass spectrum while its formation in the interfacial region of the microdroplet (i.e., at the air-droplet interface) is indicated by the strong dependence of the oxidation product formation on the spray distance (which reflects the droplet size and consequently the surface-to-volume ratio) and the solvent composition. Importantly, based on the screening of a ca. 21,000-compound library and the detailed consideration of six functional groups, the formation of a molecular adduct with the water radical cation is a significant route to ionization in positive ion mode electrospray, where it is favored in those compounds with X=Y moieties which lack basic groups. A set of model monofunctional systems was studied and in one case, benzyl benzoate, evidence was found for oxidation driven by hydroxyl radical adduct formation followed by protonation in addition to the dominant water radical cation addition process. Significant implications of molecular ionization by water radical cations for oxidation processes in atmospheric aerosols, analytical mass spectrometry and small-scale synthesis are noted.
1 Introduction
Microdroplet chemistry is an emerging field in chemistry due to the remarkable reaction acceleration (Girod et al., 2011; Müller et al., 2012; Yan et al., 2016, 2017; Lee et al., 2017; Nam et al., 2017; Cooks and Yan, 2018; Sahota et al., 2019; Zhang et al., 2019, Zhang et al., 2021 Y.; Wei et al., 2020, 2022; Cao et al., 2020; Kuai et al., 2021; Vannoy et al., 2021; Zheng et al., 2021), and the unique properties of the reaction environment (Li et al., 2014; Lee et al., 2019a, 2019b; Gao et al., 2019; Huang et al., 2021; Wang et al., 2021; Gong et al., 2022; Qiu et al., 2022). These features give rise to numerous applications (Wei et al., 2017; Yan et al., 2018; Zhu et al., 2019; Nie et al., 2020; Li Z. et al., 2021; Gunawardena et al., 2021; Yan, 2021; Zhang, 2021). Reaction acceleration is firmly attributed to the reactions at the solution/air interface (Yan et al., 2016; Lee et al., 2019b; Wei et al., 2020). Partial solvation of the reactants at the droplet surface has been proposed to increase the reaction rate constant at the interface compared to the bulk phase (Yan et al., 2016; Wei et al., 2020), a suggestion supported by multiple experimental results (Bain et al., 2015; Yan et al., 2016; Wei et al., 2020; Li et al., 2021b; Qiu et al., 2021) and limited high-level quantum mechanical modeling (Narendra et al., 2020). A detailed study of reaction rate constants in microdroplets showed that the acceleration factor (i.e., ratio of rate constant in confined volume vs. rate constant in bulk) is between 104 to 106 for all the bimolecular reactions investigated (Qiu et al., 2021). The partial solvation hypothesis explains reaction acceleration on the following basis: 1) the solvation energy of reactants is greater than that of the (more charge-dispersed) transition state, and 2) partial solvation (at the air/droplet interface) reduces solvation energies relative to complete solvation (in bulk). These solvation energy effects decrease the activation energy for interfacial reactions relative to bulk so producing accelerated reactions. Note that the first assumption holds for bimolecular reactions but not for unimolecular transformations, a fact consistent with the experimental observation that only bimolecular reactions show large rate acceleration effects (Qiu et al., 2021). One factor that can affect reaction rates without changing the rate constant is solvent evaporation, which increases concentrations (Rovelli et al., 2020). Other interfacial effects have been considered in explaining observed reaction acceleration (Fallah-Araghi et al., 2014; Yan et al., 2016; Lee et al., 2017, 2019a, 2019b, 2020; Zhou et al., 2018; Wei et al., 2020; Xiong et al., 2020; Huang et al., 2021), including the extreme pH at the droplet surface (Basuri et al., 2020; Huang et al., 2021), preferential orientation of reactants (Zhou et al., 2018; Narendra et al., 2020), and strong electric fields (on the order of MV/cm) at or near the microdroplet interface (Lee et al., 2019b, 2020; Xiong et al., 2020). The existence of the strong fields is supported by spectroscopic (Xiong et al., 2020) and computational (Leung, 2010; Kathmann et al., 2011; Yesibolati et al., 2020; Hao et al., 2022) data for aqueous microdroplets. The electric field lowers the energy barrier for reaction by stabilizing the transition state or by activating the reactant. Both effects accelerate reactions (Kreuzer, 2004; Aragonès et al., 2016; Fried and Boxer, 2017; Welborn and Head-Gordon, 2019; Ashton et al., 2020; Shaik et al., 2020). Moreover, reactive species can reasonably be proposed to be generated by strong electric fields causing unique reactions (Lee et al., 2019a, 2019b, 2020; Gao et al., 2019; Gong et al., 2022; Qiu et al., 2022).
Spontaneous oxidation and reduction without the addition of reducing/oxidizing agents has been reported in microdroplets (Lee et al., 2019a; Gao et al., 2019; Gong et al., 2022; Qiu et al., 2022), although the species responsible for these redox reactions is still under debate. Data from Zare and coworkers led them to conclude that the reactive species are the hydroxyl radical and hydrogen peroxide (Gao et al., 2019; Lee et al., 2019b, 2019a). These authors hypothesize that the high electric field at aqueous microdroplet interfaces can oxidize hydroxide to hydroxyl radicals, which can in turn generate hydrogen peroxide. They proposed that these reactive species can induce both reduction, e.g., acetophenone to 1-phenylethanol (Lee et al., 2019a), and oxidation, e.g., aromatic aldehydes to phenols (Gao et al., 2019). On the other hand, Zhang and collaborators postulated that electrons at the microdroplet air-water interface account for the reduction of doubly-charged ethyl viologen to the singly-charged radical cation and subsequent degradation reactions (Gong et al., 2022). Recently, our group suggested that water radical cations can be generated in organic-solvent microdroplets containing trace amounts of water and that they play a key role in the oxidation of aromatic sulfones to sulfonic acids in microdroplets (Qiu et al., 2022). Water radical cations are reported to be naturally abundant in pure bulk water (Ben-Amotz, 2011; Lee and Rick, 2011) and are generated by electric discharge in water vapor under otherwise mild conditions (Wang et al., 2021). Notably, in a study preceding this one, water radical cations produced in charged microdroplets were shown to trigger spontaneous oxidation (Qiu et al., 2022). The argument that water radical cation is a key reactive species is supported by the fact that the water radical cation has a higher oxidative power than either the hydroxyl radical or hydrogen peroxide in aqueous solution (Ma et al., 2014, 2018). The generation of water radical cations could occur by 1) the ionization of water due to the strong electric field at the microdroplet interface or 2) by hydrogen bond-induced electron transfer to form a water radical cation/anion pair (H2O∙∙∙OH2
To study this spontaneous oxidation, we utilized both nano-electrospray ionization (nESI), a common technique utilized in microdroplet reaction acceleration studies (Bain et al., 2015; Yan et al., 2016; Sahota et al., 2019; Wei et al., 2020) and desorption electrospray ionization (DESI) to generate charged microdroplets. DESI is an ambient ionization technique (Takáts et al., 2004; Cooks et al., 2006) that has found a wide application (Ifa et al., 2008; Morelato et al., 2013; Ifa and Eberlin, 2016; Zheng and Chen, 2016; Pirro et al., 2017). The micron-sized secondary droplets of DESI (Costa and Graham Cooks, 2008), facilitate reaction acceleration and its applications to on-line derivatization and small-scale organic synthesis (Girod et al., 2011; Morato et al., 2021). The compatibility of DESI-MS with automated high-throughput screening enables chemical analysis at throughputs better than one sample per second using high-density arrays (50 nL samples, up to 6,144 samples per array) (Morato et al., 2021). Applications include screening of organic reactions (Wleklinski et al., 2018; Jaman et al., 2019, 2020; Loren et al., 2019; Biyani et al., 2020; Ewan et al., 2020; Sobreira et al., 2020; Li et al., 2021a; Morato et al., 2021), label-free enzymatic assays (Morato et al., 2020; Kulathunga et al., 2022), and the generation of spectral libraries (Le et al., 2021). Here, we take advantage of high-throughput DESI to investigate adduct formation by water radical cations in a large compound set. We also performed a detailed study, using a small set of monofunctional model compounds on the mechanism of adduct formation and the subsequent oxidation.
2 Results and Discussion
2.1 High-Throughput Systematic Exploration of Molecular Ionization by Water Radical Cations
Given that sulfones have been shown to be ionized as [M + H2O]+· (Qiu et al., 2022), the question of how general is this behavior must arise. Automated DESI-MS analysis allows rapid (<1 s per sample) generation of spectral libraries, as recently demonstrated utilizing a set of 20,798 non-proprietary compounds (Le et al., 2021). That study focused on the fragmentation of protonated ([M+H]+) compounds, but here the same MS spectral library was utilized to evaluate the nature of the molecular ion. Protonation and alkali ion attachment are the expected ionization reactions but the presence of the [M+H2O]+˙ ion is a previously unrecognized complication in its use for molecular weight determination by MS. This information was obtained by retrieving the intensities of ions with m/z values corresponding to [M+18]+˙ in the large set of polyfunctional library compounds with vast structural diversity. Note that a small contribution by artifactual peaks at this m/z value are to be expected. A schematic of the automated DESI-MS experiment is shown in Supplementary Figure S1.
To identify the presence of a peak, the intensity of the [M+18]+˙ species was compared to a blank and signal-to-noise ratio, SNR >3, was used to recognize the peak. Comparisons with the intensities of the [M+H]+, [M+Na]+, and [M+17]+ intensities were made as described in the Supplementary Material. We identified 3,683 hits with a substantial [M+18]+˙ peak compared to 16,662 and 14,121 compounds showing [M+H]+ and [M+Na]+, respectively. Some representative spectra (of compounds 1–4) corresponding to high and low [M+18]+˙ intensities are shown in Figure 1A. Note that the low intensity [M+18]+˙ peaks were in many cases broad, indicating that in those instances the interaction between the molecule and the water radical cation is weak, and the resultant ion is consequently fragile. Such a relationship between weakly-bound ion clusters and the peak broadening observed during mass analysis in quadrupole ion traps is well known (McClellan et al., 2002). Of course, the low intensity of the peak by itself is already an indication of the low stability of the [M+18]+˙ species. It is worth highlighting, however, that more than 65% of the hits identified showed intense (>30% relative intensity) [M+18]+˙ peaks, and that these all displayed unit resolution (characteristic resolution of the instrument utilized) indicating strong bonding between the molecule and the water radical cation, as previously demonstrated in the case of sulfones (Qiu et al., 2022).
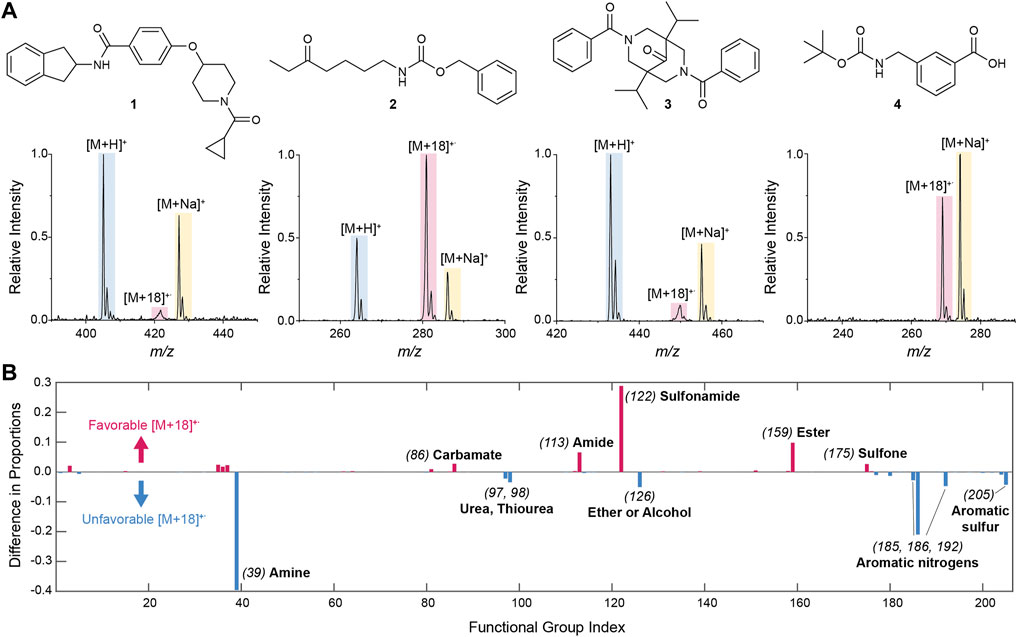
FIGURE 1. (A) Representative examples of mass spectra from the 20,798-compound library showing cases of high and low [M+18]+˙ intensity. Note the peak broadening in the low intensity cases. The structures of the molecules corresponding to the example spectra are also shown. (B) Effect of functional groups present in the molecule on the generation of [M+18]+˙ as estimated from the difference in the proportion of compounds with a functional group within the set of [M+18]+˙ hits and the same proportion in the complete library (Supplementary Equation S3). A difference of zero indicates no effect of the functional group. Significant positive differences indicate that the formation of the [M+18]+˙ species is favored by the presence of the functional group, whereas significant negative differences indicate a disfavoring effect. For the sake of simplicity, functional groups are indicated by indices specified in Supplementary Table S1.
To gain further insight into the molecular interactions leading to the generation of [M+18]+˙ species, we investigated the effect of the functional groups present in the molecules that gave hits. To achieve this we first extracted all the functional groups present in the complete library of compounds utilizing their SMILES strings as input and Ertl’s algorithm as previously described (Ertl, 2017; Le et al., 2021). Note that this algorithm designates functional groups as groups of special atoms (i.e., heteroatoms as well as sp2, sp, and acetal carbons). Following this approach, a total of 205 functional groups were identified in the whole library (Supplementary Table S1). The proportion of any particular functional group within the library was then calculated by counting all compounds that have said functional group and dividing it by the total number of compounds in the library. In a similar manner, one can find the proportion of every functional group within the set of species that provided [M+18]+˙ hits (Supplementary Equations S1, S2). If there is no effect of a functional group in the generation of [M+18]+˙ ions, the proportion of compounds with that functional group in the overall library and in the [M+18]+ hits set is expected to be similar. By contrast, if those proportions differ significantly, this indicates that the presence or absence of the functional group influences the generation of a [M+18]+˙ species.
Clear trends were observed showing the influence of functional groups on the formation of water radical cation adducts (Figure 1B, Supplementary Table S2). In particular, the presence of sulfonamides, amides, esters and carbamates was found to significantly favor the generation of [M+18]+˙ species; this is in accordance with the expectation that H2O+· will react with X=Y groups, and in agreement with the proposed mechanism of adduct formation (Wang et al., 2021; Qiu et al., 2022). The absence of basic nitrogen atoms in these functional groups reinforces this result. On the other hand, water radical cation adduct formation is relatively unfavorable for compounds containing basic nitrogen atoms (i.e., amines, ureas, and nitrogen-containing heterocycles). Both results indicate a competition between protonation and formation of the water radical cation adduct in which protonation is favored in the presence of basic groups and covalent adducts being formed with X=Y functionalities.
Note that the ionization of library compounds to give the radical cation form of the molecular ion (M+˙) was also investigated using similar searching and filtering strategies as for the [M+18]+˙ species. Overall, this radical cation was far less abundant than the water radical cation adducts. In fact, of all the compounds showing hits for the [M+18]+˙ species, only 132 (ca. 3% of the hits) showed a M+˙ peak, indicating that the observed adduct is likely a water radical cation derived species (M + H2O+˙), as proposed, and not the water adduct of the molecular radical cation (M+˙ + H2O).
2.2 Processes Leading to Water Radical Cation Adducts
It is known that the water radical cation forms as an O-O weakly bonded dimer (i.e., [H2O∙∙∙OH2]+˙) and that this species can react with various unsaturated compounds to form water radical cation adducts. Examples include alkenes (Zhang et al., 2020), benzene (Mi et al., 2022), acetone (Zhang X. et al., 2021), ethyl acetate (Wang et al., 2021) and various sulfones (Qiu et al., 2022). The variety of substrates that form adducts with the water radical cation and the spontaneous oxidation driven as a result of adduct formation, triggered our interest in systematically exploring the properties of water radical cation adducts generated in microdroplets. As mentioned before, these ions occur in the positive ion mass spectra as [M+18]+˙ species. Note that MS/MS data (Section 2.5 below) show the adduct to be strongly bound; several structures can be attributed to the general adducts, including a three-membered ring and a geminal diol structure (when Y is oxygen) (Figure 2A). The adduct can undergo 1,2-migration and radical induced bond cleavage (Figure 2B) or direct bond cleavage (when the bond is fragile, so generating a geminal diol cation, Figure 2C).
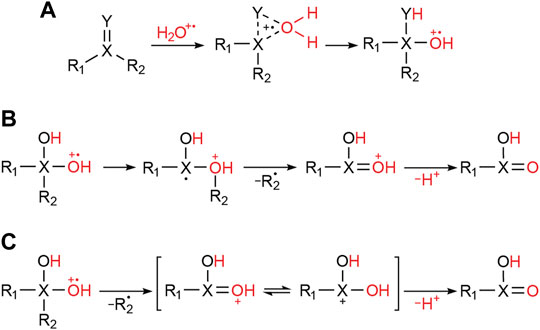
FIGURE 2. Possible processes for water radical cation adduct formation with a double bond-containing compound. (A) Direct attachment of the water radical cation (monomer or dimer) to the reactant. (B) Oxidation of the substrate by 1,2-migration and sigma bond cleavage. (C) Oxidation of the substrate by geminal diol cation formation after direct sigma bond cleavage.
2.3 Mechanistic Study of Water Radical Cation Adduct Formation
To further explore the effect of functional groups on the competitive generation of water radical cation adducts ([M+18]+˙) and protonated molecules ([M+H]+), we chose six monofunctional compounds (5–10) bearing different functional groups that were found to favor the generation of water radical cation adducts during the large compound library analysis (sulfone, sulfonamide, ketone, ester, amide, and carbamate). Methanolic solutions of these compounds were analyzed using nESI-MS and DESI-MS (commercial methanol was used as solvent). As expected from the compound library analysis, [M+18]+˙ species were observed in all cases (Figure 3 and Supplementary Figure S3), albeit with higher intensities (i.e., [M+18]+˙/[M+H]+ ratios) in nESI than in DESI, probably due to the smaller sizes and higher effective concentrations of the nESI-generated droplets.
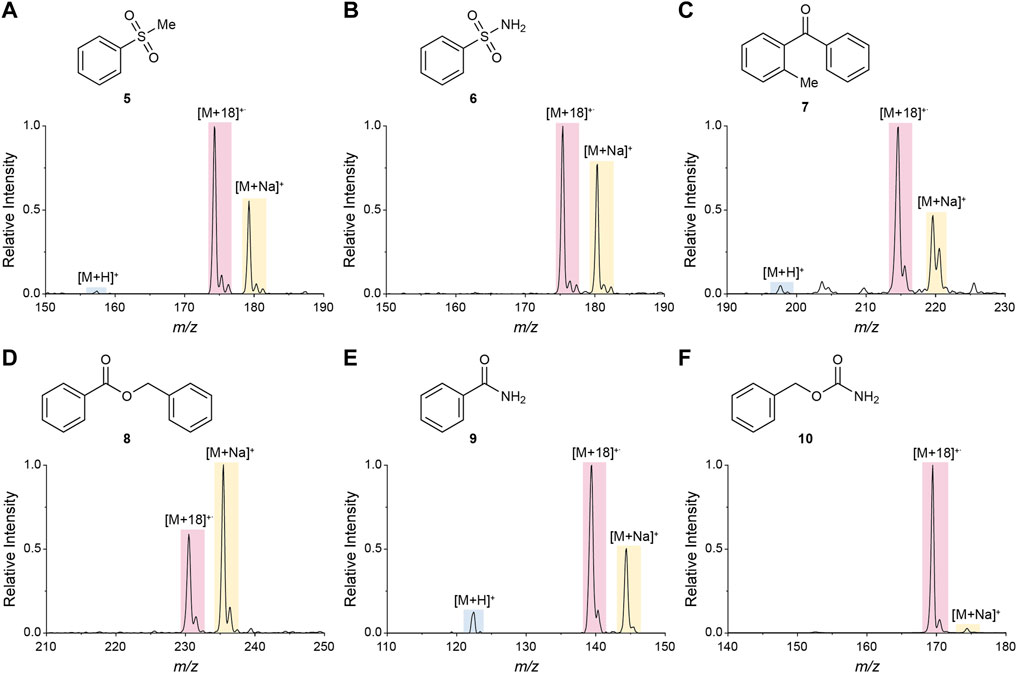
FIGURE 3. Mass spectra of six monofunctional compounds in nESI-generated microdroplets. Sulfone (A), sulfonamide (B), ketone (C), ester (D), amide (E), and carbamate (F) functionalities were explored.
The DESI-MS analysis was repeated using methanol with 0.1% formic acid as DESI solvent (same conditions of analysis as the library compounds). Under these conditions, the [M+18]+˙ species were still observed (Supplementary Figure S4), however with lower intensities than in the absence of formic acid. This indicates that the formation of [M+18]+˙ is unfavorable in microdroplets containing this acid and is consistent with the decrease in product conversion observed for the benzenesulfonamide oxidation after formic acid addition (Section 2.4 below), as is expected according to the proposed reaction mechanism. This effect likely comes from the properties of formic acid as a reducing agent (Garron and Epron, 2005; Choi et al., 2013; Al-Naji et al., 2020), allowing it to react directly with the oxidizing water radical cation, or to react as a proton donor to enhance the competitive direct protonation of the substrate (vs. intermediate adduct formation). Note that this effect of formic acid suggests that the number of compounds shown to be capable of generating substantial water radical cation adducts in the library analyzed is likely an underestimation.
The spray distance (i.e., microdroplet flight distance from the nESI emitter to the mass spectrometer inlet) was varied from 5 to 40 mm, while monitoring the ratio of [M+18]+˙/[M+H]+ as a proxy for the stability of the [M+18]+˙ species in the microdroplets (Figure 4). The strongest tendency to form [M+18]+˙ over [M+H]+ was found in the sulfonamide case which provided [M+18]+˙/[M+H]+ ratios as high as 103. The sulfone, ester and carbamate also showed high ratios. All these results are in complete agreement with those obtained from the high throughput library exploration. In contrast, the model amide analyzed exhibited a low [M+18]+˙/[M+H]+ ratio (ca. 10), despite the favorability estimated with the compound library analysis. This minor discrepancy is likely the result of an increased basicity of the -CONH2 present in the model compound when compared to the -CO-NH-R (R ≠ H) moiety, which is the common amide structure within the library. Across all functional groups, increasing the spray distance (i.e., decreasing droplet size and increasing surface-to-volume ratios), resulted in lower [M+18]+˙/[M+H]+ ratios. This result contrasts with the negative ion data shown in Figure 6E below (Section 2.4). Several factors are at play here: 1) fragmentation of [M+18]+˙ to [M+H]+ by loss of a hydroxyl radical increases with distance and inverts the expected ratio, 2) conversion of [M+18]+˙ to oxidation products, and 3) competitive direct protonation of the molecule (M) during flight to provide [M+H]+.
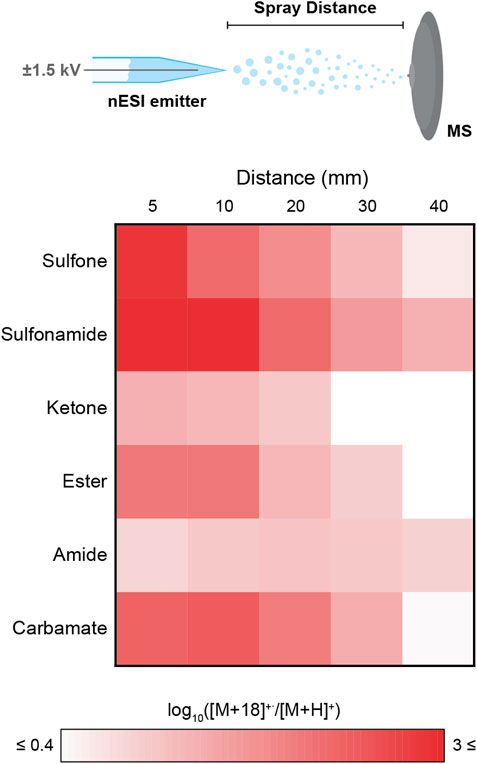
FIGURE 4. Effect of the functional group and spray distance (i.e., distance between the nESI emitter and the mass spectrometer inlet) on the generation of [M+18]+˙, monitored as the ratio [M+18]+˙/[M+H]+. Note the color scale is logarithmic to facilitate visual comparison. Results are for six representative monofunctional compounds identified by their functional groups: sulfone (benzyl methyl sulfone, 5), sulfonamide (benzenesulfonamide, 6), ketone (2-methylbenzophenone, 7), ester (benzyl benzoate, 8), amide (benzamide, 9), and carbamate (benzyl carbamate, 10).
2.4 Reactions Driven by Water Radical Cation Adducts
To investigate the oxidation reactions driven by the water radical cation adduct, we selected three of the six monofunctional model compounds 6–8 containing X=Y bonds namely 2-methylbenzophenone (7, ketone), benzyl benzoate (8, ester) and benzenesulfonamide (6, sulfonamide). Using nESI-MS, methanolic solutions of these compounds (20 mM) were analyzed in the negative ion mode. In all cases the corresponding products (11–13), which are benzoic or sulfonic acids, were detected (Figure 5) and their structures were confirmed by MS/MS (Supplementary Figure S5). Formation of these products is proposed to follow the processes shown in Figure 2 and the specific mechanism for each case is illustrated in Supplementary Figure S6. Note the transition from the positive to the negative ion mode. Also note the strong product preference in the ketone oxidation (i.e., benzoic acid vs. 2-methylbenozic acid; Figure 5A) is presumably derived from the steric hindrance of the methyl group at the ortho position which modifies the normal almost equal phenyl/tolyl migratory aptitude (Supplementary Figure S7) (Aureliano Antunes et al., 2005). This effect is further amplified by the reaction acceleration phenomenon in microdroplets, thus providing a remarkably high 10-fold regioselectivity.
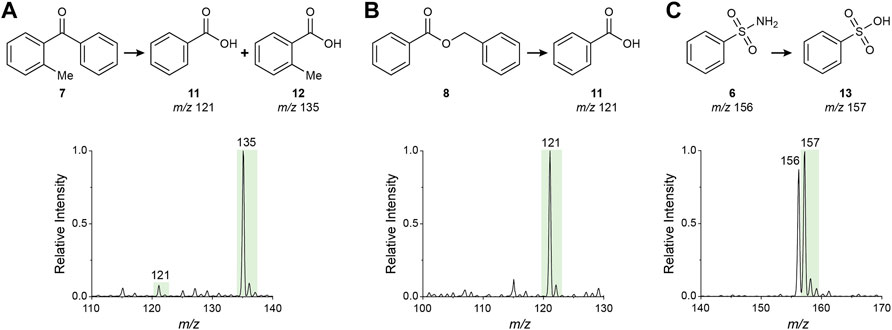
FIGURE 5. Negative ion mode nESI-MS spectra of methanolic solutions of (A) 2-methylbenzophenone (7), (B) benzyl benzoate (8) and (C) benzenesulfonamide (6) showing the product(s) of oxidation (for the ketone and ester) and/or hydrolysis (for the ester and sulfonamide) driven by the water radical cation in microdroplets.
The simultaneous detection of the sulfonamide (reactant) and the corresponding sulfonic acid (product) in the negative ion mode spectrum (Figure 5C) allows exploration of this transformation by monitoring the reaction conversion under different conditions. One factor explored was the solvent composition. Compared to commercial methanol, the addition of a desiccant (MgSO4) completely eliminated oxidation (Figure 6B), demonstrating the importance of water in the reaction. However, the addition of water (50% v/v) also shut off the reaction (Figure 6C), possibly by converting the water radical cation to the hydroxyl radical (Ma et al., 2014, 2018). Thus, trace amounts of water enhance the water radical cation mediated oxidation reaction. This outcome supports the suggestion that the sulfonamide hydrolysis is driven by the water radical cation rather than being due to direct hydrolysis by hydroxide. This conclusion is also supported by the experimental results that 1) the water dimer radical cation is observed in the spray of sulfonamide solution (Supplementary Figure S2) and 2) introduction of water dimer radical cation into the sulfonamide solution boosted the transformation to sulfonic acid (Supplementary Figure S8). It was observed also that adding formic acid traces (0.1%) to the methanolic solution reduced the reaction conversion (Figure 6D), in direct agreement with the effect of this acid in the abundance of the water radical cation adduct as previously discussed. The effect of the spray distance was also evaluated. Over the range explored (5–50 mm) larger distances provided higher conversion (Figure 6E; cf. Figure 6F at 50 mm and Figure 6A at 5 mm) with the maximum distance leaving virtually no unconverted reactant. This phenomenon has been reported for several microdroplet reactions and has been linked to the smaller sizes (and consequently larger surface-to-volume ratios) of the droplets with increased evaporation at longer flight times (Bain et al., 2015; Marsh et al., 2019).
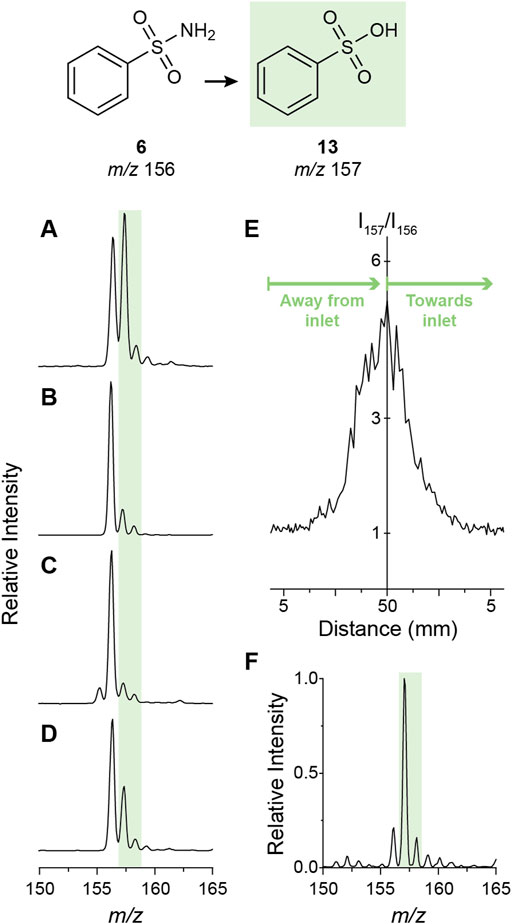
FIGURE 6. Mechanistic study of the microdroplet oxidation of sulfonamide 6. (A) through (D) show the effect of solvent composition on the amount of oxidation product observed using nESI-MS in the negative ion mode. Solvents explored were methanol (A), methanol dried over MgSO4 (B), 1:1 methanol-water (C), and methanol with 0.1% formic acid (D). The effect of the spray distance (i.e. distance from the nESI emitter to the mass spectrometer inlet) is shown in (E). Note that from left to right the graph indicates moving away from the inlet (5–50 mm) and then back (50–5 mm). The mass spectrum corresponding to the maximum spray distance of 50 mm shows maximum conversion (F).
The observed sulfonic acid product was characterized online via MS/MS (Supplementary Figure S5), and off-line by IR after microdroplet collection. To obtain a substantial amount of product, a multiplexed nESI sprayer containing 16 nESI emitters was built (Supplementary Figure S9; Supplementary Material for experimental details). From 2 mg of starting sulfonamide material, 1.7 mg of solid was collected when spray distance was 30 mm to optimize both collection efficiency and yield. The IR spectrum of the collected sample showed the characteristic O-H stretch of benzenesulfonic acid at 2,700–3,000 cm−1, a feature which is absent from the spectrum of the starting material (Supplementary Figure S10). The product yield in the collected material was estimated as 6.5% by MS analysis after correction for the ionization efficiency differences between reactant and product (Supplementary Figure S11; Supplementary Material for details). Note that microdroplet reaction yields can be increased using a solvent recycling system (Nie et al., 2020).
2.5 MS/MS Characterization of Water Radical Cation Adducts
The structures of the [M+18]+˙ species were probed using tandem mass spectrometry (MS/MS). Several collision energies as well as three different DESI solvents (methanol with 0.1% formic acid, methanol, and methanol-d4), were utilized. The major product ions and corresponding neutral losses are summarized in Table 1 (for spectra Supplementary Figure S12). Overall, the neutral loss of hydroxyl radical (−17) in MS2 is a general fragmentation channel of the [M+H2O]+˙ species, indicating again that these ions are not weakly-bound water adducts of radical cations. This is also reinforced by the fact that in many cases the adducts fragment to other product ions generated by cleavage of covalent bonds within the original compound structures (e.g., benzyl carbamate water radical cation fragments yielding a tropylium ion at m/z 91). Furthermore, upon ramping the collision energy (0–30 arbitrary units), it was observed that the six adducts formed are not weakly bound (as it would be in the case of the water adduct of a molecular radical cation) because they do not show significant fragmentation until high energies (≥20), and even then, a significant precursor ion population remains unfragmented (Supplementary Figure S13). Using methanol-d4, several adduct species were obtained, in particular [M+H2O]+˙, [M+HDO]+˙, and [M+D2O]+˙, detected at m/z values of [M+18]+˙, [M+19]+˙ and [M+20]+˙, respectively (Supplementary Figure S15). Analogously to the cases using non-deuterated solvent, the main fragmentation pathway of the [M+D2O]+˙ ions was the loss of DO˙, while neutral loss of HDO and D2O was also observed, especially in the sulfone and sulfonamide (Table 1). The presence of these multiple fragments agrees with the proposed existence of multiple water radical cation adduct structures (Figure 2).
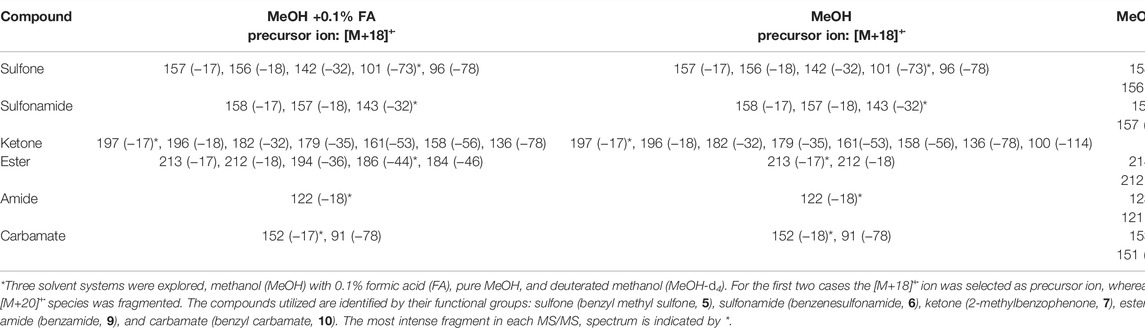
TABLE 1. Main product ions and neutral losses (in parenthesis) of the [M+18]+˙ ions as determined by DESI-MS/MS analysis.*
2.6 Isomeric Forms of Water Radical Cation Adducts
Interestingly, during the MS/MS analysis of one of the model compounds, benzoyl benzoate (8), significant differences were observed between the spectra obtained when using methanol with 0.1% formic acid and neat methanol (Table 1), with more product ions being observed in the presence of the acid (note that the new fragments were absent from the corresponding experimental blanks under identical conditions). These differences held true over a large range of collision energies, up to the energy where no precursor ion population remained, eliminating any effect of energy differences as an explanation of this phenomenon. These observations strongly suggest that the addition of the acid is inducing the formation of an isomeric [M+18]+˙ species (Supplementary Figure S15). A potential isomer would result from protonation of the compound, favored by formic acid, followed by hydroxy radical addition. Such a process agrees with reported redox phenomena involving the generation of hydroxyl radicals at the microdroplet surface thanks to the action of high energy fields on hydroxide ions (Gao et al., 2019; Lee et al., 2019b, 2019a, 2020). The direct conversion of H2O+˙ to hydroxyl radicals may contribute, too. The formation of [M+18]+˙ species has been proposed to be favored by the promiscuity of electrons at the air-water interface (Ben-Amotz, 2011) and it correlates with molecular dynamics results suggesting an enhanced surface concentration of hydroxyl radicals compared to the one in aqueous bulk phase (Roeselová et al., 2004). Thus, the presence of this particular isomeric [M+18]+˙ ion might represent the coexistence of two mechanisms leading to accelerated redox chemistry in microdroplets. One, as described here, involves the water radical cation, a short-lived but abundant species spontaneously generated in water, and adduct formation with X=Y moieties as an intermediate step in an overall oxidation process; the second, involves hydroxyl radicals generated under highly energetic conditions at the droplet interface.
3 Conclusion
Water radical cation adduct formation was found to be a general phenomenon for X=Y functionalized molecules through the analysis of a large (>20,000) set of complex polyfunctional compounds, which represents 18% occurrence in all ca. 21,000 molecules and more than 65% in the set of X=Y compounds. Tandem mass spectrometry revealed that the adducts are covalently bound, and a closer examination of a small subset of model monofunctional molecules suggested that gem diol structures are involved. Upon exploration of a model ester (8), we identified structural differences in the generated [M+18]+˙ ion upon addition of formic acid traces to the microdroplet solvent. This observation suggests that there are two processes that compete in the generation of this particular [M+18]+˙ species, the major one being direct H2O+˙ addition, while the minor process involves OH˙ addition followed by protonation. In other words, there is evidence for two reactive oxidizing agents (H2O+˙ and OH˙). Note that OH˙ has been previously suggested to have this role (Gao et al., 2019; Lee et al., 2019b, 2019a) while H2O+˙ has only recently been identified as a strong oxidizing agent in microdroplets (Qiu et al., 2022). The competition between these two species is still to be fully explored. Similarly, some experimental factors, especially the emitter-inlet distance effect on the adduct generation, are incompletely elucidated.
The possibility of using this chemistry to generate small amounts of oxidized products without any external oxidants is clear. Note that these reactions occur under very mild conditions and are accelerated by such large factors that the corresponding reactions are not even observable in bulk solution. Extensive research has been devoted over the past decades to find green oxidation approaches for diverse functionalities (Goti and Cardona, 2008; Jiao and Stahl, 2019). In particular, the use of green solvent alternatives, the absence of catalysts, and reaction under mild conditions are relevant factors for efficient and environmentally-friendly oxidation (Yu et al., 2012; Amaniampong et al., 2017; Khan et al., 2022). All these requirements are fulfilled by the microdroplet oxidation process described in this work, which adds advantages such as the need for no external oxidizing agents, the wide applicability to various chemical functionalities, and the enhanced reaction rates as well as increased regioselectivities (Qiu et al., 2022). Furthermore, leveraging the properties of automated DESI-MS (Morato et al., 2021) or spray solvent recycling systems (Nie et al., 2020), the microdroplet-generated oxidation products could be easily screened and collected (nanogram scale) in a high-throughput fashion, or scaled up to obtain yields in milligram-gram amounts, respectively.
Similarly, the spontaneous oxidation induced by the water radical cation has several implications for atmospheric and aerosol chemistry, topics that involve sub-micrometer size droplets (e.g., spray and organic aerosols) (Gantt and Meskhidze, 2013; Ault and Axson, 2017; Kirpes et al., 2018). Although an enhanced rate of oxidation has been reported in aerosols via other mechanisms and other reactive species (e.g., oxygen, ozone, hydroxyl radicals or hydrogen peroxide) (Glasius et al., 2000; Richards-Henderson et al., 2016; Liu et al., 2020; Angle et al., 2021), we suggest that the possible role of the water radical cation chemistry is worthy of attention. The effects of this radical species might be significant considering the generality here demonstrated for X=Y containing compounds (common moieties in VOCs). Thus, the characterization and quantification of molecular composition in aerosols (Jeong et al., 2011) may be affected by the adduct formation with water radical cations and subsequent oxidation processes, especially in the case of organosulfate (Farmer et al., 2010; Tao et al., 2014; Wang et al., 2017) or carbonyl group-containing molecules (Kautzman et al., 2010; Takahama et al., 2013).
Finally, the facile water radical cation adduct formation seen in this study has direct implications for chemical analysis using mass spectrometry, particularly with regards to the automated chemical annotation of compounds. Currently, the identification of molecules (in the positive ion mode) is almost exclusively based in seeking the protonated form of the molecules with due consideration of sodium ([M+Na]+) and potassium adducts ([M+K]+) (Jaeger et al., 2017; Domingo-Almenara et al., 2018; Stricker et al., 2020). Adducts with water radical cation are not part of the identification of molecules in samples examined by MS. Remarkably, as shown in this work, these adducts represent significant peaks, in a substantial fraction of cases with greater intensities than the commonly searched protonated and sodiated forms of the molecules. Note that the leveraging of the spontaneous water radical cation adduct formation to obtain increased sensitivity for compounds with low ionization efficiencies, such as acetone (Zhang X. et al., 2021), may represent a novel derivatization-free strategy for mass spectrometry analysis.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
Conceptualization, RC and LQ; methodology, LQ, NM, and RC; formal analysis, LQ, NM, and RC; investigation, LQ and NM; data curation and visualization, NM and LQ; writing—original draft, LQ, NM and KH; Writing—review and editing, LQ, NM, KH, and RC; supervision, project administration and funding acquisition, RC. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the Multi-University Research Initiative (MURI) of the Air Force Office of Scientific Research (FA9550-21-1-0170) via Stanford University (sub-award 62741613-204669), the National Science Foundation (grant CHE-1905087), and Waters Corporation (grant 40002775). NM acknowledges support from the American Chemical Society Division of Analytical Chemistry Graduate Fellowship sponsored by Agilent Technologies.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge My Phuong T. Le for her contribution to the implementation of the functional group identification algorithm. Authors also thank Andreas Kaerner and Eli Lilly and Company for the gift of the compound library utilized to generate the high throughput data used in this study and Christopher Welch for his interest in this topic.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.903774/full#supplementary-material
References
Al-Naji, M., Popova, M., Chen, Z., Wilde, N., and Gläser, R. (2020). Aqueous-Phase Hydrogenation of Levulinic Acid Using Formic Acid as a Sustainable Reducing Agent over Pt Catalysts Supported on Mesoporous Zirconia. ACS Sustain. Chem. Eng. 8, 393–402. doi:10.1021/acssuschemeng.9b05546
Amaniampong, P. N., Karam, A., Trinh, Q. T., Xu, K., Hirao, H., Jérôme, F., et al. (2017). Selective and Catalyst-free Oxidation of D-Glucose to D-Glucuronic Acid Induced by High-Frequency Ultrasound. Sci. Rep. 7, 40650. doi:10.1038/srep40650
Angle, K. J., Neal, E. E., and Grassian, V. H. (2021). Enhanced Rates of Transition-Metal-Ion-Catalyzed Oxidation of S(IV) in Aqueous Aerosols: Insights into Sulfate Aerosol Formation in the Atmosphere. Environ. Sci. Technol. 55, 10291–10299. doi:10.1021/acs.est.1c01932
Aragonès, A. C., Haworth, N. L., Darwish, N., Ciampi, S., Bloomfield, N. J., Wallace, G. G., et al. (2016). Electrostatic Catalysis of a Diels-Alder Reaction. Nature 531, 88–91. doi:10.1038/nature16989
Ashton, M., Mishra, A., Neugebauer, J., and Freysoldt, C. (2020). Ab Initio Description of Bond Breaking in Large Electric Fields. Phys. Rev. Lett. 124, 176801. doi:10.1103/PhysRevLett.124.176801
Ault, A. P., and Axson, J. L. (2017). Atmospheric Aerosol Chemistry: Spectroscopic and Microscopic Advances. Anal. Chem. 89, 430–452. doi:10.1021/acs.analchem.6b04670
Aureliano Antunes, C. S., Bietti, M., Ercolani, G., Lanzalunga, O., and Salamone, M. (2005). The Effect of Ring Substitution on the O-Neophyl Rearrangement of 1,1-diarylalkoxyl Radicals. A Product and Time-Resolved Kinetic Study. J. Org. Chem. 70, 3884–3891. doi:10.1021/jo0502448
Bain, R. M., Pulliam, C. J., and Cooks, R. G. (2015). Accelerated Hantzsch Electrospray Synthesis with Temporal Control of Reaction Intermediates. Chem. Sci. 6, 397–401. doi:10.1039/c4sc02436b
Basuri, P., Gonzalez, L. E., Morato, N. M., Pradeep, T., and Cooks, R. G. (2020). Accelerated Microdroplet Synthesis of Benzimidazoles by Nucleophilic Addition to Protonated Carboxylic Acids. Chem. Sci. 11, 12686–12694. doi:10.1039/d0sc02467h
Ben-Amotz, D. (2011). Unveiling Electron Promiscuity. J. Phys. Chem. Lett. 2, 1216–1222. doi:10.1021/jz2002875
Biyani, S. A., Qi, Q., Wu, J., Moriuchi, Y., Larocque, E. A., Sintim, H. O., et al. (2020). Use of High-Throughput Tools for Telescoped Continuous Flow Synthesis of an Alkynylnaphthyridine Anticancer Agent, HSN608. Org. Process. Res. Dev. 24, 2240–2251. doi:10.1021/acs.oprd.0c00289
Cao, J., Wang, Q., An, S., Lu, S., and Jia, Q. (2020). Facile and Efficient Preparation of Organoimido Derivatives of [Mo6O19]2− Using Accelerated Reactions in Leidenfrost Droplets. Analyst 145, 4844–4851. doi:10.1039/d0an00578a
Choi, E.-k., Park, K.-h., Lee, H.-b., Cho, M., and Ahn, S. (2013). Formic Acid as an Alternative Reducing Agent for the Catalytic Nitrate Reduction in Aqueous media. J. Environ. Sci. 25, 1696–1702. doi:10.1016/S1001-0742(12)60226-5
Cooks, R. G., Ouyang, Z., Takats, Z., and Wiseman, J. M. (2006). Ambient Mass Spectrometry. Science 311, 1566–1570. doi:10.1126/science.1119426
Cooks, R. G., and Yan, X. (2018). Mass Spectrometry for Synthesis and Analysis. Annu. Rev. Anal. Chem. 11, 1–28. doi:10.1146/annurev-anchem-061417-125820
Costa, A. B., and Graham Cooks, R. (2008). Simulated Splashes: Elucidating the Mechanism of Desorption Electrospray Ionization Mass Spectrometry. Chem. Phys. Lett. 464, 1–8. doi:10.1016/j.cplett.2008.08.020
Domingo-Almenara, X., Montenegro-Burke, J. R., Benton, H. P., and Siuzdak, G. (2018). Annotation: A Computational Solution for Streamlining Metabolomics Analysis. Anal. Chem. 90, 480–489. doi:10.1021/acs.analchem.7b03929
Ertl, P. (2017). An Algorithm to Identify Functional Groups in Organic Molecules. J. Cheminform. 9, 36. doi:10.1186/s13321-017-0225-z
Ewan, H. S., Biyani, S. A., Didomenico, J., Logsdon, D., Sobreira, T. J. P., Avramova, L., et al. (2020). Aldol Reactions of Biorenewable Triacetic Acid Lactone Precursor Evaluated Using Desorption Electrospray Ionization Mass Spectrometry High-Throughput Experimentation and Validated by Continuous Flow Synthesis. ACS Comb. Sci. 22, 796–803. doi:10.1021/acscombsci.0c00119
Fallah-Araghi, A., Meguellati, K., Baret, J.-C., Harrak, A. E., Mangeat, T., Karplus, M., et al. (2014). Enhanced Chemical Synthesis at Soft Interfaces: A Universal Reaction-Adsorption Mechanism in Microcompartments. Phys. Rev. Lett. 112, 028301. doi:10.1103/PhysRevLett.112.028301
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., et al. (2010). Response of an Aerosol Mass Spectrometer to Organonitrates and Organosulfates and Implications for Atmospheric Chemistry. Proc. Natl. Acad. Sci. U.S.A. 107, 6670–6675. doi:10.1073/pnas.0912340107
Fried, S. D., and Boxer, S. G. (2017). Electric fields and Enzyme Catalysis. Annu. Rev. Biochem. 86, 387–415. doi:10.1146/annurev-biochem-061516-044432
Gantt, B., and Meskhidze, N. (2013). The Physical and Chemical Characteristics of marine Primary Organic Aerosol: A Review. Atmos. Chem. Phys. 13, 3979–3996. doi:10.5194/acp-13-3979-2013
Gao, D., Jin, F., Lee, J. K., and Zare, R. N. (2019). Aqueous Microdroplets Containing Only Ketones or Aldehydes Undergo Dakin and Baeyer-Villiger Reactions. Chem. Sci. 10, 10974–10978. doi:10.1039/c9sc05112k
Garron, A., and Epron, F. (2005). Use of Formic Acid as Reducing Agent for Application in Catalytic Reduction of Nitrate in Water. Water Res. 39, 3073–3081. doi:10.1016/j.watres.2005.05.012
Girod, M., Moyano, E., Campbell, D. I., and Cooks, R. G. (2011). Accelerated Bimolecular Reactions in Microdroplets Studied by Desorption Electrospray Ionization Mass Spectrometry. Chem. Sci. 2, 501–510. doi:10.1039/c0sc00416b
Glasius, M., Lahaniati, M., Calogirou, A., Di Bella, D., Jensen, N. R., Hjorth, J., et al. (2000). Carboxylic Acids in Secondary Aerosols from Oxidation of Cyclic Monoterpenes by Ozone. Environ. Sci. Technol. 34, 1001–1010. doi:10.1021/es990445r
Gong, C., Li, D., Li, X., Zhang, D., Xing, D., Zhao, L., et al. (2022). Spontaneous Reduction-Induced Degradation of Viologen Compounds in Water Microdroplets and its Inhibition by Host-Guest Complexation. J. Am. Chem. Soc. 144, 3510–3516. doi:10.1021/jacs.1c12028
Goti, A., and Cardona, F. (2008). “Hydrogen Peroxide in Green Oxidation Reactions: Recent Catalytic Processes,” in Green Chemical Reactions (Dordrecht: Springer Netherlands), 191–212. doi:10.1007/978-1-4020-8457-7_9
Hao, H., Leven, I., and Head-Gordon, T. (2022). Can Electric fields Drive Chemistry for an Aqueous Microdroplet? Nat. Commun. 13, 280. doi:10.1038/s41467-021-27941-x
Huang, K.-H., Wei, Z., and Cooks, R. G. (2021). Accelerated Reactions of Amines with Carbon Dioxide Driven by Superacid at the Microdroplet Interface. Chem. Sci. 12, 2242–2250. doi:10.1039/d0sc05625a
Ifa, D. R., and Eberlin, L. S. (2016). Ambient Ionization Mass Spectrometry for Cancer Diagnosis and Surgical Margin Evaluation. Clin. Chem. 62, 111–123. doi:10.1373/clinchem.2014.237172
Ifa, D. R., Manicke, N. E., Dill, A. L., and Cooks, R. G. (2008). Latent Fingerprint Chemical Imaging by Mass Spectrometry. Science 321, 805. doi:10.1126/science.1157199
Jaeger, C., Méret, M., Schmitt, C. A., and Lisec, J. (2017). Compound Annotation in Liquid Chromatography/high-Resolution Mass Spectrometry Based Metabolomics: Robust Adduct Ion Determination as a Prerequisite to Structure Prediction in Electrospray Ionization Mass Spectra. Rapid Commun. Mass. Spectrom. 31, 1261–1266. doi:10.1002/rcm.7905
Jaman, Z., Logsdon, D. L., Szilágyi, B., Sobreira, T. J. P., Aremu, D., Avramova, L., et al. (2020). High-Throughput Experimentation and Continuous Flow Evaluation of Nucleophilic Aromatic Substitution Reactions. ACS Comb. Sci. 22, 184–196. doi:10.1021/acscombsci.9b00212
Jaman, Z., Sobreira, T. J. P., Mufti, A., Ferreira, C. R., Cooks, R. G., and Thompson, D. H. (2019). Rapid On-Demand Synthesis of Lomustine under Continuous Flow Conditions. Org. Process. Res. Dev. 23, 334–341. doi:10.1021/acs.oprd.8b00387
Jeong, C.-H., McGuire, M. L., Godri, K. J., Slowik, J. G., Rehbein, P. J. G., and Evans, G. J. (2011). Quantification of Aerosol Chemical Composition Using Continuous Single Particle Measurements. Atmos. Chem. Phys. 11, 7027–7044. doi:10.5194/acp-11-7027-2011
Jiao, N., and Stahl, S. S. (2019). “Green Oxidative Synthesis of Epoxides,” in Green Oxidation in Organic Synthesis. Editors N. Jiao,, and S. S. Stahl (Hoboken, NJ, USA: Wiley). doi:10.1002/9781119304197
Kathmann, S. M., Kuo, I.-F. W., Mundy, C. J., and Schenter, G. K. (2011). Understanding the Surface Potential of Water. J. Phys. Chem. B 115, 4369–4377. doi:10.1021/jp1116036
Kautzman, K. E., Surratt, J. D., Chan, M. N., Chan, A. W. H., Hersey, S. P., Chhabra, P. S., et al. (2010). Chemical Composition of Gas- and Aerosol-phase Products from the Photooxidation of Naphthalene. J. Phys. Chem. A. 114, 913–934. doi:10.1021/jp908530s
Khan, S. R., Saini, S., Naresh, K., Kumari, A., Aniya, V., Khatri, P. K., et al. (2022). CO2 as Oxidant: an Unusual Light-Assisted Catalyst Free Oxidation of Aldehydes to Acids under Mild Conditions. Chem. Commun. 58, 2208–2211. doi:10.1039/d1cc06057k
Kirpes, R. M., Bondy, A. L., Bonanno, D., Moffet, R. C., Wang, B., Laskin, A., et al. (2018). Secondary Sulfate Is Internally Mixed with Sea spray Aerosol and Organic Aerosol in the winter Arctic. Atmos. Chem. Phys. 18, 3937–3949. doi:10.5194/acp-18-3937-2018
Kreuzer, H. J. (2004). Physics and Chemistry in High Electric fields. Surf. Interf. Anal. 36, 372–379. doi:10.1002/sia.1895
Kuai, D., Cheng, H., Kuan, K.-Y., and Yan, X. (2021). Accelerated Five-Component spiro-pyrrolidine Construction at the Air-Liquid Interface. Chem. Commun. 57, 3757–3760. doi:10.1039/d1cc00574j
Kulathunga, S. C., Morato, N. M., Zhou, Q., Cooks, R. G., and Mesecar, A. D. (2022). Desorption Electrospray Ionization Mass Spectrometry Assay for Label‐Free Characterization of SULT2B1b Enzyme Kinetics. ChemMedChem 2022, e202200043. doi:10.1002/cmdc.202200043
Le, M. T., Morato, N. M., Kaerner, A., Welch, C. J., and Cooks, R. G. (2021). Fragmentation of Polyfunctional Compounds Recorded Using Automated High-Throughput Desorption Electrospray Ionization. J. Am. Soc. Mass. Spectrom. 32, 2261–2273. doi:10.1021/jasms.1c00176
Lee, A. J., and Rick, S. W. (2011). The Effects of Charge Transfer on the Properties of Liquid Water. J. Chem. Phys. 134, 184507. doi:10.1063/1.3589419
Lee, J. K., Han, H. S., Chaikasetsin, S., Marron, D. P., Waymouth, R. M., Prinz, F. B., et al. (2020). Condensing Water Vapor to Droplets Generates Hydrogen Peroxide. Proc. Natl. Acad. Sci. U.S.A. 117, 30934–30941. doi:10.1073/pnas.2020158117
Lee, J. K., Nam, H. G., and Zare, R. N. (2017). Microdroplet Fusion Mass Spectrometry: Accelerated Kinetics of Acid-Induced Chlorophyll Demetallation. Quart. Rev. Biophys. 50, e2. doi:10.1017/S0033583517000014
Lee, J. K., Samanta, D., Nam, H. G., and Zare, R. N. (2019a). Micrometer-Sized Water Droplets Induce Spontaneous Reduction. J. Am. Chem. Soc. 141, 10585–10589. doi:10.1021/jacs.9b03227
Lee, J. K., Walker, K. L., Han, H. S., Kang, J., Prinz, F. B., Waymouth, R. M., et al. (2019b). Spontaneous Generation of Hydrogen Peroxide from Aqueous Microdroplets. Proc. Natl. Acad. Sci. U.S.A. 116, 19294–19298. doi:10.1073/pnas.1911883116
Leung, K. (2010). Surface Potential at the Air−Water Interface Computed Using Density Functional Theory. J. Phys. Chem. Lett. 1, 496–499. doi:10.1021/jz900268s
Li, A., Luo, Q., Park, S.-J., and Cooks, R. G. (2014). Synthesis and Catalytic Reactions of Nanoparticles Formed by Electrospray Ionization of Coinage Metals. Angew. Chem. Int. Ed. 53, 3147–3150. doi:10.1002/anie.201309193
Li, Y., Huang, K.-H., Morato, N. M., and Cooks, R. G. (2021a). Glass Surface as strong Base, 'green' Heterogeneous Catalyst and Degradation Reagent. Chem. Sci. 12, 9816–9822. doi:10.1039/d1sc02708e
Li, Y., Mehari, T. F., Wei, Z., Liu, Y., and Cooks, R. G. (2021b). Reaction Acceleration at Air‐solution Interfaces: Anisotropic Rate Constants for Katritzky Transamination. J. Mass. Spectrom. 56, e4585. doi:10.1002/jms.4585
Li, Z., Li, Y., Zhan, L., Meng, L., Huang, X., Wang, T., et al. (2021c). Point-of-Care Test Paper for Exhaled Breath Aldehyde Analysis via Mass Spectrometry. Anal. Chem. 93, 9158–9165. doi:10.1021/acs.analchem.1c01011
Liu, T., Clegg, S. L., and Abbatt, J. P. D. (2020). Fast Oxidation of Sulfur Dioxide by Hydrogen Peroxide in Deliquesced Aerosol Particles. Proc. Natl. Acad. Sci. U.S.A. 117, 1354–1359. doi:10.1073/pnas.1916401117
Loren, B. P., Ewan, H. S., Avramova, L., Ferreira, C. R., Sobreira, T. J. P., Yammine, K., et al. (2019). High Throughput Experimentation Using DESI-MS to Guide Continuous-Flow Synthesis. Sci. Rep. 9, 1–8. doi:10.1038/s41598-019-50638-7
Ma, J., Schmidhammer, U., Pernot, P., and Mostafavi, M. (2014). Reactivity of the Strongest Oxidizing Species in Aqueous Solutions: The Short-Lived Radical Cation H2O+. J. Phys. Chem. Lett. 5, 258–261. doi:10.1021/jz402411x
Ma, J., Wang, F., and Mostafavi, M. (2018). Ultrafast Chemistry of Water Radical Cation, H2O+, in Aqueous Solutions. Molecules 23, 244. doi:10.3390/molecules23020244
Marsh, B. M., Iyer, K., and Cooks, R. G. (2019). Reaction Acceleration in Electrospray Droplets: Size, Distance, and Surfactant Effects. J. Am. Soc. Mass. Spectrom. 30, 2022–2030. doi:10.1007/s13361-019-02264-w
McClellan, J. E., Murphy, J. P., Mulholland, J. J., and Yost, R. A. (2002). Effects of Fragile Ions on Mass Resolution and on Isolation for Tandem Mass Spectrometry in the Quadrupole Ion Trap Mass Spectrometer. Anal. Chem. 74, 402–412. doi:10.1021/ac015610b
Mi, D., Mao, Y., Wei, B., Li, Y.-C., Dong, X., and Chingin, K. (2022). Generation of Phenol and Molecular Hydrogen through Catalyst-free C-H Activation of Benzene by Water Radical Cations. J. Am. Soc. Mass. Spectrom. 33, 68–73. doi:10.1021/jasms.1c00268
Morato, N. M., Holden, D. T., and Cooks, R. G. (2020). High‐Throughput Label‐Free Enzymatic Assays Using Desorption Electrospray‐Ionization Mass Spectrometry. Angew. Chem. Int. Ed. 59, 20459–20464. doi:10.1002/anie.202009598
Morato, N. M., Le, M. T., Holden, D. T., and Graham Cooks, R. (2021). Automated High-Throughput System Combining Small-Scale Synthesis with Bioassays and Reaction Screening. SLAS Technol. 26, 555–571. doi:10.1177/24726303211047839
Morelato, M., Beavis, A., Kirkbride, P., and Roux, C. (2013). Forensic Applications of Desorption Electrospray Ionisation Mass Spectrometry (DESI-MS). Forensic Sci. Int. 226, 10–21. doi:10.1016/j.forsciint.2013.01.011
Müller, T., Badu-Tawiah, A., and Cooks, R. G. (2012). Accelerated CarbonCarbon Bond-Forming Reactions in Preparative Electrospray. Angew. Chem. Int. Ed. 51, 11832–11835. doi:10.1002/anie.201206632
Nam, I., Lee, J. K., Nam, H. G., and Zare, R. N. (2017). Abiotic Production of Sugar Phosphates and Uridine Ribonucleoside in Aqueous Microdroplets. Proc. Natl. Acad. Sci. U.S.A. 114, 12396–12400. doi:10.1073/pnas.1714896114
Narendra, N., Chen, X., Wang, J., Charles, J., Cooks, R. G., and Kubis, T. (2020). Quantum Mechanical Modeling of Reaction Rate Acceleration in Microdroplets. J. Phys. Chem. A. 124, 4984–4989. doi:10.1021/acs.jpca.0c03225
Nie, H., Wei, Z., Qiu, L., Chen, X., Holden, D. T., and Cooks, R. G. (2020). High-yield Gram-Scale Organic Synthesis Using Accelerated Microdroplet/thin Film Reactions with Solvent Recycling. Chem. Sci. 11, 2356–2361. doi:10.1039/c9sc06265c
Pirro, V., Alfaro, C. M., Jarmusch, A. K., Hattab, E. M., Cohen-Gadol, A. A., and Cooks, R. G. (2017). Intraoperative Assessment of Tumor Margins during Glioma Resection by Desorption Electrospray Ionization-Mass Spectrometry. Proc. Natl. Acad. Sci. U.S.A. 114, 6700–6705. doi:10.1073/pnas.1706459114
Qiu, L., Psimos, M. D., and Cooks, R. G. (2022). Spontaneous Oxidation of Aromatic Sulfones to Sulfonic Acids in Microdroplets. J. Am. Soc. Mass. Spectrom. 2022, 2c00029. doi:10.1021/jasms.2c00029
Qiu, L., Wei, Z., Nie, H., and Cooks, R. G. (2021). Reaction Acceleration Promoted by Partial Solvation at the Gas/Solution Interface. Chempluschem 86, 1362–1365. doi:10.1002/cplu.202100373
Richards-Henderson, N. K., Goldstein, A. H., and Wilson, K. R. (2016). Sulfur Dioxide Accelerates the Heterogeneous Oxidation Rate of Organic Aerosol by Hydroxyl Radicals. Environ. Sci. Technol. 50, 3554–3561. doi:10.1021/acs.est.5b05369
Roeselová, M., Vieceli, J., Dang, L. X., Garrett, B. C., and Tobias, D. J. (2004). Hydroxyl Radical at the Air−Water Interface. J. Am. Chem. Soc. 126, 16308–16309. doi:10.1021/ja045552m
Rovelli, G., Jacobs, M. I., Willis, M. D., Rapf, R. J., Prophet, A. M., and Wilson, K. R. (2020). A Critical Analysis of Electrospray Techniques for the Determination of Accelerated Rates and Mechanisms of Chemical Reactions in Droplets. Chem. Sci. 11, 13026–13043. doi:10.1039/D0SC04611F
Sahota, N., Abusalim, D. I., Wang, M. L., Brown, C. J., Zhang, Z., El-Baba, T. J., et al. (2019). A Microdroplet-Accelerated Biginelli Reaction: Mechanisms and Separation of Isomers Using IMS-MS. Chem. Sci. 10, 4822–4827. doi:10.1039/c9sc00704k
Shaik, S., Danovich, D., Joy, J., Wang, Z., and Stuyver, T. (2020). Electric-Field Mediated Chemistry: Uncovering and Exploiting the Potential of (Oriented) Electric Fields to Exert Chemical Catalysis and Reaction Control. J. Am. Chem. Soc. 142, 12551–12562. doi:10.1021/jacs.0c05128
Sobreira, T. J. P., Avramova, L., Szilagyi, B., Logsdon, D. L., Loren, B. P., Jaman, Z., et al. (2020). High-throughput Screening of Organic Reactions in Microdroplets Using Desorption Electrospray Ionization Mass Spectrometry (DESI-MS): Hardware and Software Implementation. Anal. Methods 12, 3654–3669. doi:10.1039/d0ay00072h
Stricker, T., Bonner, R., Lisacek, F., and Hopfgartner, G. (2020). Adduct Annotation in Liquid Chromatography/high-Resolution Mass Spectrometry to Enhance Compound Identification. Anal. Bioanal. Chem. 413, 503–517. doi:10.1007/s00216-020-03019-3
Takahama, S., Johnson, A., Russell, L. M., Takahama, S., and Johnson, A. (2013). Quantification of Carboxylic and Carbonyl Functional Groups in Organic Aerosol Infrared Absorbance Spectra. Aerosol Sci. Technol. 47, 310–325. doi:10.1080/02786826.2012.752065
Takáts, Z., Wiseman, J. M., Gologan, B., and Cooks, R. G. (2004). Mass Spectrometry Sampling under Ambient Conditions with Desorption Electrospray Ionization. Science 306, 471–473. doi:10.1126/science.1104404
Tao, S., Lu, X., Levac, N., Bateman, A. P., Nguyen, T. B., Bones, D. L., et al. (2014). Molecular Characterization of Organosulfates in Organic Aerosols from Shanghai and Los Angeles Urban Areas by Nanospray-Desorption Electrospray Ionization High-Resolution Mass Spectrometry. Environ. Sci. Technol. 48, 10993–11001. doi:10.1021/es5024674
Vannoy, K. J., Lee, I., Sode, K., and Dick, J. E. (2021). Electrochemical Quantification of Accelerated FADGDH Rates in Aqueous Nanodroplets. Proc. Natl. Acad. Sci. U.S.A. 118, e2025726118. doi:10.1073/pnas.2025726118
Wang, M., Gao, X.-F., Su, R., He, P., Cheng, Y.-Y., Li, K., et al. (2022). Abundant Production of Reactive Water Radical Cations under Ambient Conditions. CCS Chem. 4, 1224–1231. doi:10.31635/ccschem.021.202101427
Wang, Y., Ren, J., Huang, X. H. H., Tong, R., and Yu, J. Z. (2017). Synthesis of Four Monoterpene-Derived Organosulfates and Their Quantification in Atmospheric Aerosol Samples. Environ. Sci. Technol. 51, 6791–6801. doi:10.1021/acs.est.7b01179
Wei, J., Sun, J., Yin, Y., Zeng, N., Ouyang, J., and Na, N. (2022). Accelerated Photocatalytic [3+2] Cycloaddition of Phenols and Olefins in nESI-MS. Int. J. Mass Spectrom. 471, 116754. doi:10.1016/j.ijms.2021.116754
Wei, Z., Li, Y., Cooks, R. G., and Yan, X. (2020). Accelerated Reaction Kinetics in Microdroplets: Overview and Recent Developments. Annu. Rev. Phys. Chem. 71, 31–51. doi:10.1146/annurev-physchem-121319-110654
Wei, Z., Wleklinski, M., Ferreira, C., and Cooks, R. G. (2017). Reaction Acceleration in Thin Films with Continuous Product Deposition for Organic Synthesis. Angew. Chem. Int. Ed. 56, 9386–9390. doi:10.1002/anie.201704520
Welborn, V. V., and Head-Gordon, T. (2019). Fluctuations of Electric Fields in the Active Site of the Enzyme Ketosteroid Isomerase. J. Am. Chem. Soc. 141, 12487–12492. doi:10.1021/jacs.9b05323
Wleklinski, M., Loren, B. P., Ferreira, C. R., Jaman, Z., Avramova, L., Sobreira, T. J. P., et al. (2018). High Throughput Reaction Screening Using Desorption Electrospray Ionization Mass Spectrometry. Chem. Sci. 9, 1647–1653. doi:10.1039/c7sc04606e
Xiong, H., Lee, J. K., Zare, R. N., and Min, W. (2020). Strong Concentration Enhancement of Molecules at the Interface of Aqueous Microdroplets. J. Phys. Chem. B 124, 9938–9944. doi:10.1021/acs.jpcb.0c07718
Yan, X., Bain, R. M., and Cooks, R. G. (2016). Organic Reactions in Microdroplets: Reaction Acceleration Revealed by Mass Spectrometry. Angew. Chem. Int. Ed. 55, 12960–12972. doi:10.1002/anie.201602270
Yan, X., Cheng, H., and Zare, R. N. (2017). Two‐Phase Reactions in Microdroplets without the Use of Phase‐Transfer Catalysts. Angew. Chem. Int. Ed. 56, 3562–3565. doi:10.1002/anie.201612308
Yan, X. (2021). Emerging Microdroplet Chemistry for Synthesis and Analysis. Int. J. Mass Spectrom. 468, 116639. doi:10.1016/j.ijms.2021.116639
Yan, X., Lai, Y.-H., and Zare, R. N. (2018). Preparative Microdroplet Synthesis of Carboxylic Acids from Aerobic Oxidation of Aldehydes. Chem. Sci. 9, 5207–5211. doi:10.1039/c8sc01580e
Yesibolati, M. N., Laganà, S., Sun, H., Beleggia, M., Kathmann, S. M., Kasama, T., et al. (2020). Mean Inner Potential of Liquid Water. Phys. Rev. Lett. 124, 065502. doi:10.1103/PhysRevLett.124.065502
Yu, B., Liu, A.-H., He, L.-N., Li, B., Diao, Z.-F., and Li, Y.-N. (2012). Catalyst-free Approach for Solvent-dependent Selective Oxidation of Organic Sulfides with Oxone. Green. Chem. 14, 957–962. doi:10.1039/c2gc00027j
Zhang, W., Zheng, B., Jin, X., Cheng, H., and Liu, J. (2019). Rapid Epoxidation of α,β-Unsaturated Olefin in Microdroplets without Any Catalysts. ACS Sustain. Chem. Eng. 7, 14389–14393. doi:10.1021/acssuschemeng.9b04059
Zhang, X. (2021). Mass Spectrometry at the Air-Water Interface. Int. J. Mass Spectrom. 462, 116527. doi:10.1016/j.ijms.2021.116527
Zhang, X., Ren, X., Chingin, K., Xu, J., Yan, X., and Chen, H. (2020). Mass Spectrometry Distinguishing C=C Location and Cis/trans Isomers: A Strategy Initiated by Water Radical Cations. Analytica Chim. Acta 1139, 146–154. doi:10.1016/j.aca.2020.09.027
Zhang, X., Ren, X., Zhong, Y., Chingin, K., and Chen, H. (2021a). Rapid and Sensitive Detection of Acetone in Exhaled Breath through the Ambient Reaction with Water Radical Cations. Analyst 146, 5037–5044. doi:10.1039/d1an00402f
Zhang, Y., Apsokardu, M. J., Kerecman, D. E., Achtenhagen, M., and Johnston, M. V. (2021b). Reaction Kinetics of Organic Aerosol Studied by Droplet Assisted Ionization: Enhanced Reactivity in Droplets Relative to Bulk Solution. J. Am. Soc. Mass. Spectrom. 32, 46–54. doi:10.1021/jasms.0c00057
Zhao, P., Gunawardena, H. P., Zhong, X., Zare, R. N., and Chen, H. (2021). Microdroplet Ultrafast Reactions Speed Antibody Characterization. Anal. Chem. 93, 3997–4005. doi:10.1021/acs.analchem.0c04974
Zheng, B., Jin, X., Liu, J., and Cheng, H. (2021). Accelerated Metal-free Hydration of Alkynes within Milliseconds in Microdroplets. ACS Sustain. Chem. Eng. 9, 4383–4390. doi:10.1021/acssuschemeng.1c00887
Zheng, Q., and Chen, H. (2016). Development and Applications of Liquid Sample Desorption Electrospray Ionization Mass Spectrometry. Annu. Rev. Anal. Chem. 9, 411–448. doi:10.1146/annurev-anchem-071015-041620
Zhou, Z., Yan, X., Lai, Y.-H., and Zare, R. N. (2018). Fluorescence Polarization Anisotropy in Microdroplets. J. Phys. Chem. Lett. 9, 2928–2932. doi:10.1021/acs.jpclett.8b01129
Keywords: mass spectrometry, reaction acceleration, interfacial reaction, ionization, solvation, ion thermochemistry, high-throughput screening, fragmentation mechanism
Citation: Qiu L, Morato NM, Huang K-H and Cooks RG (2022) Spontaneous Water Radical Cation Oxidation at Double Bonds in Microdroplets. Front. Chem. 10:903774. doi: 10.3389/fchem.2022.903774
Received: 24 March 2022; Accepted: 11 April 2022;
Published: 26 April 2022.
Edited by:
Xin Yan, Texas A&M University, United StatesReviewed by:
Heyong Cheng, Hangzhou Normal University, ChinaHao Chen, New Jersey Institute of Technology, United States
Copyright © 2022 Qiu, Morato, Huang and Cooks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. Graham Cooks, Y29va3NAcHVyZHVlLmVkdQ==
†These authors have contributed equally to this work and share first authorship