 Saghi Sepehri
Saghi Sepehri Mina Saeedi
Mina Saeedi Bagher Larijani
Bagher Larijani Mohammad Mahdavi
Mohammad Mahdavi- 1Department of Medicinal Chemistry, School of Pharmacy, Ardabil University of Medical Sciences, Ardabil, Iran
- 2Pharmaceutical Sciences Research Center, Ardabil University of Medical Sciences, Ardabil, Iran
- 3Medicinal Plants Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
- 4Persian Medicine and Pharmacy Research Center, Tehran University of Medical Sciences, Tehran, Iran
- 5Endocrinology and Metabolism Research Center, Endocrinology and Metabolism Research Institute, Tehran University of Medical Sciences, Tehran, Iran
Background: Alzheimer’s disease (AD) is an advanced and irreversible degenerative disease of the brain, recognized as the key reason for dementia among elderly people. The disease is related to the reduced level of acetylcholine (ACh) in the brain that interferes with memory, learning, emotional, and behavior responses. Deficits in cholinergic neurotransmission are responsible for the creation and progression of numerous neurochemical and neurological illnesses such as AD.
Aim: Herein, focusing on the fact that benzylpyridinium salts mimic the structure of donepezil hydrochlorideas a FDA-approved drug in the treatment of AD, their synthetic approaches and inhibitory activity against cholinesterases (ChEs) were discussed. Also, molecular docking results and structure–activity relationship (SAR) as the most significant concept in drug design and development were considered to introduce potential lead compounds. Key scientific concepts: AChE plays a chief role in the end of nerve impulse transmission at the cholinergic synapses. In this respect, the inhibition of AChE has been recognized as a key factor in the treatment of AD, Parkinson’s disease, senile dementia, myasthenia gravis, and ataxia. A few drugs such as donepezil hydrochloride are prescribed for the improvement of cognitive dysfunction and memory loss caused by AD. Donepezil hydrochloride is a piperidine-containing compound, identified as a well-known member of the second generation of AChE inhibitors. It was established to treat AD when it was assumed that the disease is associated with a central cholinergic loss in the early 1980s. In this review, synthesis and anti-ChE activity of a library of benzylpyridinium salts were reported and discussed based on SAR studies looking for the most potent substituents and moieties, which are responsible for inducing the desired activity even more potent than donepezil. It was found that linking heterocyclic moieties to the benzylpyridinium salts leads to the potent ChE inhibitors. In this respect, this review focused on the recent reports on benzylpyridinium salts and addressed the structural features and SARs to get an in-depth understanding of the potential of this biologically improved scaffold in the drug discovery of AD.
Introduction
More than 100 years ago, Alzheimer’s disease (AD) was exposed by Alois Alzheimer in 1907. Despite a lot of effort in the treatment of the disease, it has emerged as a crucial public health issue in the 21st century due to the lack of effective and no clinically accepted therapeutic approach, as well as due to a huge economic burden on the society (Prince et al., 2013). AD is known as an irreversible chronic neurodegenerative disturbance of the CNS start-up on a continuing loss of cognitive skill (Goyal et al., 2017). It generally occurs in the elderly population; however, it appears as an autosomal dominant trait in families in 1%–2% of cases (Bekris et al., 2010). AD is usually characterized by the loss of short-term memory, disorientation, impairment of judgment and reasoning and decision making, language, and learning. Patients misplace their capability to connect, fail to identify their near and dear ones, and become bedridden at the last steps of the disease (Sharma, 2019).
The pathogenesis of AD has not yet been definitely clarified. It is extensively recognized that a mixture of environmental activities and genetic vulnerability features is responsible for outspread late-onset AD. Comprehending the mechanism of creation of the disease has been remained as a key factor for developing effective anti-AD drugs. AD is a multifactorial disease in which the formation of toxic amyloid beta (Aβ) (Matsuzaki, 2011), tau protein hyperphosphorylation (Miao et al., 2019), neuroinflammation (Calsolaro and Edison, 2016), oxidative stress (Tonnies and Trushina, 2017), and biometals (Lavado et al., 2019) play vital aspects in the creation and progression of illness. Also, it has been extensively assumed that low levels of acetylcholine (ACh) play a crucial role in the creation of AD (McGleenon et al., 1999; Lan et al., 2017a).
Now, there are a few U.S. FDA-approved drugs, which are prescribed to the patients with AD that only relieve the symptoms of AD. Among them, donepezil, galantamine, and rivastigmine are cholinesterase (ChE) inhibitors. They avoid the hydrolysis of acetylcholine (ACh), which is the main neurotransmitter of the parasympathetic nervous system in the brain and responsible for learning and memory (Picciotto et al., 2012). It should be noted that tacrine was also the approved drug in the same category, which was removed from the market due to hepatotoxicity (Girek and Szymanski, 2019). Another drug is memantine belonging to different classes of anti-AD agents, NMDAR antagonists (Mangialasche et al., 2010). However, the combination of donepezil and memantine is currently used to improve physical and mental health in patients with AD (Wake et al., 2018). As mentioned above, all these drugs do not treat the disease definitely and relieve its signs to increase the quality of life of patients and caregivers. In this regard, the design and development of anti-AD drugs is in urgent demand (Lee and Kim, 2017).
The cholinergic hypothesis is the most general description of the mechanism of AD progression, which directly contributes to the cognitive decline (Bartus, 2000). Amyloid protein plaques can be created using both ChEs, AChE and butyrylcholinesterase (BuChE), in which their inhibitors can reduce them (Yu L. et al., 2010). BuChE is generally of glial origin, while AChE is typically of neuronal origin (Mesulam et al., 2002). Under usual conditions, ACh is frequently disintegrated using AChE instead of BuChE (Mack and Robitzki, 2000).
As the decline of ACh levels in the hippocampus and cortex leads to reasoning and memory shortfalls, refining cholinergic function has been completely measured in the treatment of AD (Stanciu et al., 2020). Several studies have also indicated that AChE seems to be implicated in the pathogenesis of AD by promoting the formation of both Aβ fibrils. Fortunately, AChEIs possibly affect the metabolic processing of the amyloid precursor protein (APP) and thus may influence the generation of Aβ (Garcia-Ayllon et al., 2011). Using BuChE, acetylcholine can be hydrolyzed and the levels of AChE can be compensated when they are decreased. In the brain affected by AD with variations becoming more pronounced during the disease course, there is a reduction in AChE levels, whereas BuChE levels are obviously unchanged or increased. Moreover, the BuChE genotype can affect AD risk and the rate of illness development (Nordberg et al., 2013).
It is worth emphasizing that although many hypotheses and strategies are currently being proposed for the treatment of AD, ChEIs still remain to be a supreme clinical success in the treatment of AD, which fully demonstrates the value of this target (Nadri et al., 2010; Jiang et al., 2018).
ChEs have very similar structures. Both of them comprise a deep gorge and a peripheral anionic site (PAS) and a catalytic active site (CAS). Amino acid sequences in the AChE and BuChE are almost 65% homologic (Nicolet et al., 2003). Catalytic triads of human AChE (hAChE) and human BuChE (hBuChE) contain preserved residues: His438, Glu325, and Ser198 in hBuChE, and Glu334, His447, and Ser203 in hAChE (Shafferman et al., 1992). Yet, the existence and number of amino acids inside the gorge are diverse, particularly exhibited using the acyl-binding pocket, which includes an acyl moiety to catalyze the substrate (Brus et al., 2014). There are Phe295 and Phe297 residues in the hAChE pocket, whereas Leu286 and Val288 are in the hBuChE pocket. Two aromatic amino acids of hAChE bulging into the gorge somewhat occupy the space, while the presence of smaller amino acids in hBuChE prepares an extensive space and permits bigger substrates to bind to be hydrolyzed (Li et al., 2017). Diverse structural characteristics of the both enzymes contribute to their substrate particularity: Small molecules such as ACh have a higher affinity toward AChE, whereas numerous peptides have more selectivity for BuChE (Taylor and Radic, 1994). Based on studies, the benzylic moiety of donepezil interacts with the CAS and dimethoxyindanone moiety interacts with the PAS of the AChE (Mohsin and Ahmad, 2020).
Donepezil (donepezil hydrochloride, under the brand name Aricept) is a medication used for the symptomatic treatment of mild-to-moderate AD, which was introduced in 1996 (Figure 1). It is a non-competitive and reversible inhibitor of AChE, thus inhibiting ACh hydrolysis. Donepezil appears to compensate for the loss of functioning cholinergic neurons through maintaining high ACh levels (Knowles, 2006). It has demonstrated a 1000-fold selectivity for the inhibition of AChE over BuChE in in vitro experiments (Mushtaq et al., 2014). Other mechanisms have been also postulated for the anti-AD activity of donepezil. It has an impact on nicotinic receptors (Cacabelos, 2007) and reduces the decrease in expression in cerebral cortex and avoids the diminution in nicotinic binding that is related to illness harshness (Wu et al., 2010). It declines glutamate neurotoxicity and inhibits excitotoxic damage to keep neuroprotective actions (Shen et al., 2010). As for another important characteristics of AD pathology, oxidative stress has stimulated a great attention. Donepezil has been potentially established to fight against free radicals and improve the effects of oxidative stress in a streptozotocin-induced model of AD mice (Li et al., 2018). The brain AChE is inhibited by the oral administration of this drug in a dose-dependent manner. It has revealed impartially good permeability to the brain. The concentration of donepezil in the brain is about sixfold to sevenfold more than that in plasma. It is a group of ChE inhibitor bearing indanone and N-benzylpiperidine moieties that displays longer and more selective action.
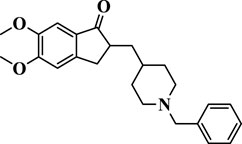
FIGURE 1. Structure of donepezil.
Donepezil or donepezil hydrochloride is being synthesized from arylidene-2-indanone or alkylidene provided using Aldol condensation as crucial intermediates followed by the catalytic reduction (Dubey et al., 2010; Rawat et al., 2013; Costanzo et al., 2016) (Scheme 1).

SCHEME 1. Synthesis procedure of donepezil.
Based on molecular docking studies, it was found that the dimethoxyindanone moiety is responsible for binding to the peripheral anionic site (PAS) of the AChE via aromatic π–π stacking interactions with Trp279, Arg289, Ser286, Phe331, and Tyr121, while the piperidine ring interacts with amino acids Tyr337 and Tyr334 located in the anionic part of catalytic active site (Figure 1). The benzyl moiety of donepezil is in nearby Trp86, Gly118, and Trp84; and Phe330, His447, Glu199, His440, and Ser203 amino acids, both parts of the catalytic triad (Figure 1). Previously, it has been revealed that the modification of the benzyl ring can lead to a strong AChE inhibition (Kryger et al., 1999; Makarian et al., 2022).
As anti-AD activity of donepezil has been proven from various mechanistic points of view, it has been considered as the main scaffold in the design and synthesis of a varied range of compounds against AD (Sugimoto et al., 2002; Agatonovic-Kustrin et al., 2018; Piemonyese et al., 2018; Mohsin and Ahmad, 2020). Herein, we focused on the benzylpyridinium salts, which have shown very good ChE inhibitory activity mimicking the structure of donepezil.
Coumarin hybrids
Alipour et al. (2012) reported the synthesis of coumarin derivatives bearing N-benzylpyridinium moiety (Supplementary Scheme S1). Among the synthesized compounds, 5a (IC50 = 0.11 nM) exhibited higher activity than donepezil (IC50 = 14 nM). The compound 5b exhibited the high selectivity for AChE. Based on results, the introduction of substituents into 2-, 3-, and 4-positions of the benzyl moiety influenced the activity and selectivity (5a (IC50 = 0.11 nM) vs. 5c (IC50 = 0.46 nM)). The existence of the group at 2- and 4-positions showed the highest and lowest anti-AChE activity, respectively, while the nature of the group (electron-withdrawing and electron-donating) was not important for the inhibitory activity. Moreover, the presence of two substituents on the benzyl moiety drastically reduced the activity (5m, IC50 = 330 nM; 5n, IC50 = 440 nM). In addition, the size of the substituent at 2- and 4-positions [5d (IC50 = 1,600 nM) and 5e (IC50 = 1,470 nM); (5h, IC50 = 0.16 nM) vs. (5i, IC50 = 26 nM)] of compounds is important for the activity. Moreover, according to the data, the presence of more electron-withdrawing substituent at the 3-position of the benzyl moiety of derivatives revealed higher activity in the order of 5b (IC50 = 0.47 nM) > 5j (IC50 = 76 nM) > 5k (IC50 = 86 nM) > 5l (IC50 = 800 nM) (Figure 2).
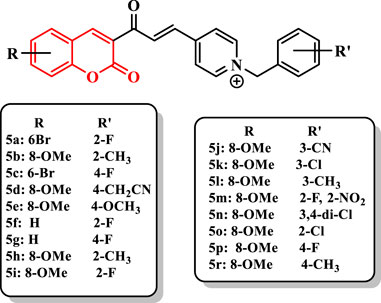
FIGURE 2. Coumarin derivatives bearing N-benzylpyridinium moiety.
The same research group (Alipour et al., 2013) synthesized a series of derivatives with coumarin and 3-coumaranone scaffolds connecting to phenacylpyridinium moiety as ChE inhibitors (Supplementary Schemes S2, S3).
All screened compounds showed weaker activity than donepezil as a standard drug (IC50 = 0.014 and 5.38 µM against ChEs). Among the synthesized compounds, 16a showed the most potent activity (IC50 = 1.3 µM) against AChE, while it had lower activity than donepezil. According to results, 3-coumaranone derivatives were generally more potent and selective than coumarin derivatives against AChE. By contrast, derivatives unsubstituted at the benzyl moiety displayed the same activities (compounds 9a and 16b with IC50 values of 10 and 7.4 µM, respectively). Thus, the substituent on the benzyl moiety plays a key role in activity (Supplementary Figure S1).
Khoobi et al. (2013) reported a series of coumarins connected to N-benzylpyridinium moiety as ACh inhibitors (Supplementary Scheme S4).
The evaluation of 4-pyridinium derivatives against AChE demonstrated that derivative 21a (IC50 = 0.038 µM) was the most potent compound, while 2- or 3-flouro derivatives (21b (IC50 = 2.9 µM) and 21c (IC50 = 2.8 µM)) showed lower activity in analogs bearing halides. Furthermore, the presence of 3-chloro, 2,4-dichloro or 3,4-dichloro in the N-benzyl group of the 4-pyridinium series enhanced the anti-AChE activity as detected in compounds 21d (IC50 = 0.48 µM), 21e (IC50 = 0.044 µM), and 21f (IC50 = 1.8 µM). Comparing the IC50 values of 2- or 3-chloro compounds 21g (IC50 = 1.5 µM) and 21d (IC50 = 0.48 µM) with their 2- or 3-fluoro analogs 21b and 21c revealed that the chlorine substituent is more effective than fluorine at 2- and 3-positions.
In the 3-pyridinium series, the fluorine-substituted analogs 21h (IC50 = 1.8 µM), 21i (IC50 = 2.0 µM), and 21j (IC50 = 2.17 µM) showed approximately similar inhibitory activity on AChE. It was found that the fluorine substituent on the N-benzyl group of the 3-pyridinum series had no noteworthy effect on inhibitory activity. Unlike, the chlorine substituent on the benzyl group of derivative 21k (IC50 = 1.0 µM) was significantly more active than 3-chlorobenzyl analog 21l (IC50 = 0.79 µM). All compounds showed less activity than donepezil (IC50 = 0.014 µM) (Supplementary Figure S2).
Based on results, the position and halogen element on the benzyl ring can control anti-AChE activity and AChE/BuChE selectivity. Among compounds, the derivative 21a depicted the highest anti-AChE activity (IC50 value = 0.038 µM) and the most AChE/BuChE selectivity (SI > 48).
Arab et al. (2015) designed and synthesized a series of chroman-4-one derivatives containing the N-benzylpyridinium moiety (29a-l) and evaluated them against AChE (Supplementary Scheme S5).
The compound 29a showed the most potent anti-AChE activity (IC50 = 0.048 µM). All compounds showed weaker activity than donepezil as a standard drug (IC50 = 0.022 µM).
Based on results, the alkoxy group at the 7-position as well as the type and position of group on the benzyl moiety are important in the anti-AChE activity. Derivatives having the ethoxy group at the 7-position of chroman-4-one showed higher activity than those having the methoxy group at the same position. Thus, an increase in chain length at the 7-position of chroman-4-one was favorable for the desired inhibitory activity.
Introduction of two bromine substituents into 2- and 3-positions of the benzyl ring (compound 29a) led to more potent activity than that of one substituent. In addition, the presence of bromine at the 4-position was inappropriate for the activity. Among halides at the 4-position, chlorine showed the highest activity [compound 29b (IC50 = 0.091 µM)], and also, the electron-withdrawing group (NO2) was favorable compared with the electron-donating group (CH3) [compound 29c (IC50 = 2.713 µM) vs. compound 29d (IC50 = 7.240 µM)] (Figure 3).
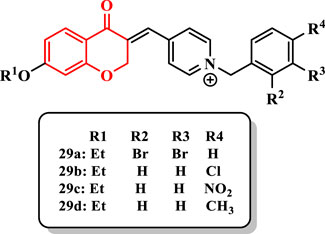
FIGURE 3. Chroman-4-one derivatives.
Khunnawutmanotham et al. (2016) synthesized some derivatives of scopoletin linked to the pyridinium moiety and investigated them against AChE (Supplementary Scheme S6).
Derivative 33a showed the highest activity (IC50 value of 0.215 µM), which was lower than donepezil (IC50 = 0.060 µM). The introduction of chlorine into the 2- and 3-positions of the benzyl group (33c and 33d) showed similar IC50 values (0.360 and 0.397 µM, respectively), which were about 15-fold more potent than that of chlorine substituent at the 4-position. Introducing the fluorine into the 2-position dramatically decreased the activity of compounds 33a and 33b. In addition, anti-AChE activities of difluorine substituents (33f and 33g) were not remarkably diverse from those of 33a. Fascinatingly, the IC50 value of 33a was very nearby to that of tacrine; while galantamine exhibited higher activity than the compound 33a (about 10-fold) (Supplementary Figure S3).
Wang et al. (2015) described a series of 4-isochromanone hybrids as selective AChE inhibitors (Supplementary Scheme S7).
The compound 40a showed an IC50 = 8.9 nM against AChE, in which its inhibitory activity was sixfold greater than that of donepezil (IC50 = 59.9 nM). Furthermore, the unsubstituted compound 40b and halogen-substituted analogs such as 40c (IC50 = 21.1 nM) and 40d (IC50 = 15.9 nM) all revealed good activities against AChE, more potent than donepezil. Particularly, as to the intermediate 39, missing the benzylpyridinium fragment, a dramatic decrease was observed in the anti-AChE activity (IC50 > 100 µM). Thus, it was recommended that this moiety was essential for higher activity.
The introduction of the chlorine and the fluorine groups into 2 (40c, IC50 = 21.1 nM)-, 3 (40e, IC50 = 26.3 nM)-, and 4 (40a, IC50 = 8.93 nM)-positions retains or improves the activity of the corresponding compounds, whereas the replacement of the halide on the benzyl moiety with the nitro or methoxy group dramatically reduced the activity of compounds such as 40f (IC50 = 640 nM) and 40g (IC50 = 503 nM) (Figure 6). Remarkably, the groups at the 4-position had the most influence on the AChE inhibitory activity. Based on results, the presence of the group except for the fluorine at the 4-position of the benzyl moiety decreased the inhibitory activity of the compounds such as 40h (IC50 = 1,193 nM), 40i (IC50 = 646 nM), and 40j (IC50 = 3,548 nM) strangely (Supplementary Figure S4). It was recognized that the small size of fluorine was as similar as hydrogen.
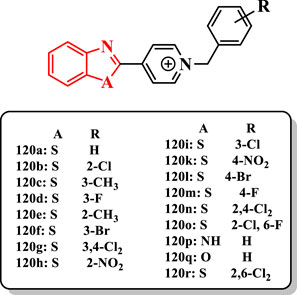
FIGURE 6. Benzimidazole, benzoxazole, and benzothiazole derivatives.
Lan et al. (2017b) reported a series of donepezil derivatives as ChE inhibitors (Supplementary Scheme S8).
The compound 45a (IC50 = 1.9 nM) displayed the highest AChE inhibitory activity. This compound was about 21-fold more potent than donepezil (IC50 = 40.2 nM). Fluorine and hydrogen substituents compared with diverse substituents such as NO2, Br, and CH3 were in attention of maintaining and increasing the anti-AChE activity. For instance, compounds 45b (IC50 = 2.9 nM) and 45c (IC50 = 4.6 nM) with fluorine and hydrogen substituents were more potent than compounds 45d (IC50 = 25.6 nM), 45e (IC50 = 77.3 nM), and 45f (IC50 = 74.6 nM) (Supplementary Figure S4). Also, the presence of substituents at the 2- or 3-position showed higher activity than at the 4-position (compounds 45g (IC50 = 2.9 nM) and 45e) (Supplementary Figure S5). All tested compounds presented more potent inhibitors for hAChE than for eeAChE. The compound 45a (IC50 = 0.8 nM) showed the most potent inhibition about 47-fold higher than donepezil (IC50 = 37.6 nM). The compound 45a (SI > 5263.1) was the highest selective AChE/BuChE inhibitor.
The same research group (Lan et al., 2017c) designed and synthesized several coumarin derivatives and investigated them as ChEs (Supplementary Scheme S9).
Compounds 49a (IC50 = 24.9 nM) and 49b (IC50 = 25.9 nM) exhibited the highest activity against AChE. They were 1.9-fold more than donepezil (IC50 = 47.4 nM) (Supplementary Figure S6).
The presence of methyl, nitro, fluorine, and bromine groups on the benzyl moiety improved the AChE inhibitory activity compared with that of unsubstituted derivatives (49c (IC50 = 29.5 nM), 49b (IC50 = 24.9 nM), 49d (IC50 = 36.7 nM), 49e (IC50 = 38.3 nM) vs. 49f (IC50 = 380 nM)) (Supplementary Figure S6). Replacing the fluorine on the benzyl moiety with the nitro or bromine group somewhat diminished the inhibitory activity. Additionally, compounds having a methyl group showed a slight increase in the AChE inhibition [e.g., 49g (IC50 = 37.3 nM) vs. 49h (IC50 = 63.2 nM) and 49e] (Supplementary Figure S6). Based on results, the presence of electron-donating groups on the benzyl moiety might be useful for favorite AChE inhibitory activity.
The substituents at the 4-position had the most activity against AChE. The existence of numerous groups except for the fluorine at the 4-position of the benzyl moiety significantly decreased the inhibitory activity [49i (IC50 = 29.1 nM), 49j (IC50 = 119 nM), and 49k (IC50 = 1,187 nM)]) (Supplementary Figure S6).
The compound 49b (IC50 = 35.4 nM) displayed the highest activity against hAChE, which was as similar as donepezil (IC50 = 31.8 nM).
Khunnawutmanotham et al. (2018) synthesized and evaluated some coumarins conjugated with the benzylpyridinium moiety via an amide spacer against AChE (Supplementary Scheme S10).
3-Aminocoumarins 52a and 52b showed no inhibitory activity against AChE (12% and 5%, respectively). Moreover, N-acyl-3-aminocoumarin derivatives such as 53a, 53b, 53c, and 53d did not increase activity (0%, 8%, 9%, and 9%, respectively). The change of 53a–54a increased AChE inhibitory activity (IC50 = inactive to 71.88 nM). Also, the compound 54b showed poor activity (10% inhibition). According to IC50 results of 54a and 54b, the existence of a methylene spacer between the pyridine ring and the carbonyl group diminished the inhibitory activity. The removal of the methylene group from the compound 54c made an IC50 = 12.48 nM, whereas the compound 54d showed an IC50 = 1,087.7 nM. Furthermore, the placement of methoxy groups at 6- and 7-positions on coumarin ring strangely improved the anti-AChE activity of compounds 54c (IC50 = 12.48 nM) and 54a (IC50 = 71.88 nM, and of compounds 54d (IC50 = 1,087.7 nM) and 54b (10%) (Figure 4). Compared with 54c, the presence of chlorine at the 2-position [54e (IC50 = 6.03 nM)] led to enhanced activity while that at 3- and 4-positions [54f (IC50 = 11.47 nM) and 54g (IC50 = 293.17 nM), respectively] gave similar and diminished inhibitory activities, respectively (Figure 4). Similarity, the presence of fluorine at the 2-position [54h (IC50 = 3.05 nM)] augmented the inhibitory activity, while the presence of fluorine at 3- and 4-positions diminished the activity of compounds 54i (IC50 = 5.04 nM) and 54j (IC50 = 5.31 nM). Difluorinated compounds were more active than monofluorinated compounds [54k (IC50 = 1.53 nM) vs. 54l (IC50 = 2.43 nM)]. In addition, the compound 54k was the most active compound in this study that the IC50 value was 35-fold less than donepezil (IC50 = 53.51 nM) and 124-fold lower than tacrine (IC50 = 190.37 nM) (Figure 4).
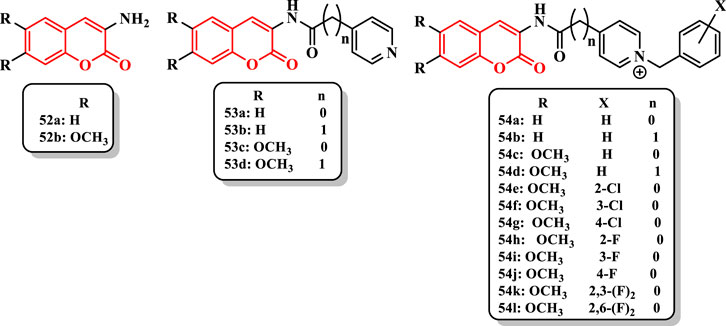
FIGURE 4. Amino-dialkoxy coumarin derivatives.
Vafadarnejad et al. (2018) synthesized different coumarin–pyridinium hybrids and investigated them as the inhibitors of ChEs (Supplementary Scheme S11).
The compound 59a (IC50 = 10.14 µM compared with rivastigmine (IC50 = 11.07 µM)) from the 4-pyridinium series exhibited the best anti-AChE activity. The compound 59h represented lower activity (IC50 = 36.82 µM). Yet, compounds 59h, 59i (IC50 = 26.25 µM), and 59j (IC50 = 33.02 µM) having fluorine at 2- and 4-positions on the benzyl moiety from the 3-pyridinium series exhibited somewhat better activity. In continuance, the compound 59b was established to exhibit the weakest anti-AChE activity (IC50 = 95.81 µM). The compound 59c indicated the second potent anti-AChE activity from the 4-pyridinium series with IC50 = 12.15 µM, and the compound 59k was established to be weaker (IC50 = 32.38 µM) (Supplementary Figure S7).
Opposite results were detected in compounds having 2-chloro and 2,3-dichlorobenzyl groups from the 3-pyridinium series (59l and 59m), which displayed better inhibitory activity toward AChE (IC50 = 17.70 and 24.93 µM, respectively) than their analogs (59d and 59e, IC50 = 28.66 and 54.64 µM, respectively). The compound 59f from the 4-pyridinium series having the 2-methylbenzyl group revealed inhibitory activity with IC50 = 16.29 µM. The compound 59g, in the same class, bearing 4-methylbenzyl moiety did not reveal any activity (IC50 > 100). Compounds 59n and 59o showed moderate activity (IC50 = 47.73 and 51.69 µM, respectively) and were weaker than their analog, the compound 59f (IC50 = 16.29 µM) (Supplementary Figure S7).
Hosseini et al. (2019) reported synthesis and evaluation of a series of the 4‐oxobenzo[d]1,2,3‐triazin‐pyridiniums against ChEs (Supplementary Scheme S12).
Among the synthesized compounds, the compound 66a (IC50 = 0.10 μM) was found as the most potent compound against AChE (Supplementary Figure S8). In the 3-pyridinium series, the compound 66b (IC50 = 0.17 μM) without the substituted benzyl group revealed good anti-AChE activity. The placement of methyl or fluorine substituent at the 2-position of the benzyl group of 66b, as in compounds 66c (IC50 = 0.20 μM) and 66d (IC50 = 0.29 μM), respectively, reduced the anti-AChE inhibitory activity. Furthermore, the presence of fluorine at 3- and 4-positions, bromine at the 2-position, or nitro at the 4-position on the benzyl group of the compound 66b had good effect on the inhibitory activity of compounds 66e (IC50 = 0.17 μM), 66f (IC50 = 0.15 μM), 66g (IC50 = 0.15 μM), and 66h (IC50 = 0.16 μM) (Supplementary Figure S8). By the way, the inhibitory activities of the fluorinated analogs 66d, 66e, and 66f were in the order of 4 > 3 > 2, respectively, while the inhibitory activities of the brominated analogs 66g, 66i (IC50 = 0.30 μM), and 66j (IC50 = 0.84 μM) were in the order of 2 > 3 > 4, respectively (Supplementary Figure S8).
In the 4-pyridinium series, the compound 66k showed the highest activity (IC50 = 1.18 μM). In this series, the derivatives 66l, 66m, and 66n with hydrogen, chlorine, and nitro at the 2-position, respectively, onto the benzyl ring exhibited approximately the same anti-AChE inhibitory activity (IC50 = 1.34–1.37 μM). Fluorinated derivatives 66o (IC50 = 1.68 μM) and 66p (IC50 = 1.64 μM) were the least active derivatives in this series (Supplementary Figure S8). According to the anti-AChE activity of the 4-pyridinium series with 3-pyridinium, it was discovered that the switching of nitrogen in pyridinium ring from the 3- to the 4-position led to a significant reduction in the activity of, e.g., the compound 66k vs. the compound 66j.
Mollazadeh et al. (2019) described the synthesis of 2,4-dioxochroman derivatives linked to benzylpyridinium and investigated them as ChEs (Supplementary Scheme S13).
The most potent compounds had chlorine, bromine, and nitro at the 2-position on the benzyl moiety [compounds 73a (IC50 = 0.89 μM), 73b (IC50 = 1.10 μM), and 73c (IC50 = 1.41 μM), respectively] (Supplementary Figure S9), which were less active than donepezil (IC50 = 0.028 μM). Furthermore, compounds 73d (IC50 = 3.00 μM), 73e (IC50 = 2.34 μM), 73f (IC50 = 2.29 μM), and 73g (IC50 = 2.54 μM) exhibited good anti-AChE activity (Supplementary Figure S9). The placement of a methyl group at the 2-position of the benzyl ring [compound 73h (IC50 = 9.48 μM)] exhibited a dramatic reduction in the inhibitory activity against AChE. Shifting the methyl group from the 2- to the 4-position created 73i (IC50 = 4.75 μM) that improved the inhibitory activity. The presence of fluorine at the 3-position of the benzyl ring enhanced the anti-AChE activity of the compound 73j (IC50 = 2.34 μM)]. The compound 73j having the fluorine group and the compound 73i having the methyl group at the 4-position of the benzyl ring revealed approximately a similar anti-AChE activity (Supplementary Figure S9).
The compound 73a, having the chlorine at the 2-position of the benzyl group revealed the highest anti-AChE activity (IC50 = 0.89 μM). Moving the chlorine from the 2- to the 3-position and/or inserting the second chlorine into the 3-position remarkably diminished the inhibitory activity (compound 73a vs. compounds 73f and 73g). Furthermore, the presence of chlorine at the 4-position of the benzyl group [compound 73k (IC50 > 100 μM)] removed the anti-AChE activity. The compound 73b (IC50 = 1.10 μM) was the second most potent compound that had the bromine at the 2-position; moving the bromine from the 2- to the 3-position [compound 73l (IC50 = 8.23 μM)] decreased the inhibitory activity, while attachment of the substituent at the 4-position (the compound 73l) led to the removal of inhibitory activity (Supplementary Figure S9). According to results, the existence of an electron-withdrawing substituent with a suitable size at the 2-position of the benzyl ring can aid in making a better interaction with the AChE. The compound 73n (IC50 = 12.48 μM) having CN at the 4-position on the benzyl group displayed a moderate anti-AChE activity (Supplementary Figure S9).
Shuai et al. (2019) synthesized some isothio- and isoselenochromanone derivatives having N-benzylpyridinium moiety (Supplementary Scheme S14).
When pyridine moiety was exchanged by piperidine, the anti-AChE activity of compounds 82a (IC50 = 7,470 nM), 82b (IC50 = 8,020 nM), 82c (IC50 = 16,400 nM), and 82d (IC50 = 15,600 nM) (Supplementary Figure S10) was dramatically reduced. The unsubstituted benzyl group compound 82e having an isothiochromanone moiety showed the highest anti-AChE activity (IC50 value of 2.7 nM), which was 4.7-fold more potent than donepezil (IC50 = 12.7 nM). In the presence of fluorine at the 4-position on the benzyl group, the compound 82f showed a potent inhibitory activity (IC50 = 5.8 nM), which was 2.2-fold more potent than donepezil.
The unsubstituted benzylpyridinium compounds 82e and 82g having an isothiochromanone moiety showed the highest anti-AChE activity (IC50 = 440 and 564 nM, respectively), which was approximately 1.5-fold more potent than donepezil (IC50 = 737 nM) (Supplementary Figure S10).
Indole hybrids
Rook et al. (2010) synthesized a series of N2 and N9-bivalent β-carboline derivatives and introduced them as potent inhibitors of ChEs.
Compounds 84c (IC50 = 278 nM), 84d (IC50 > 10,000 nM), and 84e (IC50 = 4,261 nM) with a spacer less than six carbons showed moderate activity, whereas compounds 84f (IC50 = 81 nM) and 84g (IC50 = 63 nM) with a spacer more than six carbons displayed stronger activities for both ChEs (Supplementary Figure S11). Substituents on the aromatic moiety (compounds 84d and 84e), the replacement of the tricyclic aromatic moiety with pyridinium [compounds 86 (IC50 = 564 nM) and 87 (IC50 > 10,000 nM)], and the variation of the spacer [compounds 84h (IC50 = 3,141 nM) and 84i (IC50 = 147 nM)] reduced the activities of the compounds (Supplementary Figure S11).
By contrast, methylation at the 2-position and introducing a permanent positive charge into the structure strongly increased the activity of the resulting compounds with spacers longer than 5 carbons [compounds 90a (IC50 = 0.5 nM) and 90b (IC50 = 1.2 nM)] (Supplementary Figure S11).
The N9-bivalent β-carbolines without a permanent positive charge (89a-c) generally showed very low ChEs inhibitory activities. Interestingly, partial reduction of the compound 90a (IC50 = 0.5 nM for AChE and 5.7 nM for BuChE) to 91 (IC50 = 27 nM for AChE and 38 nM for BuChE) resulted in a moderate decrease in the ChE inhibitory activity (Supplementary Figure S11).
Yu Q. et al. (2010) synthesized the N-monophenylcarbamate analogs of neostigmine methyl sulfate (92a), pyridostigmine bromide (92b), and N (1)-methylammonium analogs of (-)-phenserine (92d), (-)-tolserine (92f), (-)-cymserine (92h), and (-)-phenethylcymserine (92j) to produce long-acting peripheral inhibitors of AChE and BuChE.
The presence of a phenylcarbamoyl in either neostigmine (92k) or pyridostigmine (92l) (for AChE and BuChE, IC50 = 360 nM and 900 nM, respectively), to afford analogs of 92a and 92b (for AChE and BuChE, IC50 > 30,000 nM and 550 nM, respectively), resulted in a loss of anti-ChE activity. The IC50 value of neostigmine (92k) decreased by 100- and 300-fold for AChE and BuChE (from 18.8 to 1875 nM, and from 60 to 18,000 nM), for 92a, respectively. This similar modification in pyridostigmine (92l) exhibited a loss of AChE activity for 92b but unlike 92a, retained anti-BuChE activity (Supplementary Figure S12).
By contrast, the quaternization of (-)-physostigmine (92m) (for AChE and BuChE, IC50 = 27.9 nM and 16.0 nM, respectively) and related phenylcarbamates to provide 92n (for AChE and BuChE, IC50 = 26.1 nM and 130 nM, respectively), 92d (for AChE and BuChE, IC50 = 25.4 nM and 210 nM, respectively), 92f (for AChE and BuChE, IC50 = 14.6 nM and 140 nM, respectively), 92h (for AChE and BuChE, IC50 = 145 nM and 43 nM, respectively), and 92j (for AChE and BuChE, IC50 = 300 nM and 51 nM, respectively), with charge characteristics akin to neostigmine (92k) and pyridostigmine (92l), retained or enhanced the AChE inhibitory activity. For N (1)-methylammonium bromides (92n (for AChE and BuChE, IC50 = 26.1 nM and 130 nM), 92d, and 92f) of (-)-physostigmine (92m), (-)-phenserine (92c) (for AChE and BuChE, IC50 = 24.0 nM and 1,560 nM, respectively), and (-)-tolserine (92e) (for AChE and BuChE, IC50 = 10.3 nM and 1950 nM, respectively), the high AChE activity of the parent compounds was kept, but the differential selectivity of 92m and 92c for AChE was missing resulting in the enhancement of BuChE inhibition. In the case of the BuChE-selective inhibitors, (-)-cymserine (92g) (for AChE and BuChE, IC50 = 760 nM and 51 nM, respectively), and (-)-phenethylcymserine (92i) (for AChE and BuChE, IC50 > 30,000 nM and 6.0 nM, respectively), quaternization caused a remarkable increase in the AChE inhibitory activity for 92h and 92j that, together with a less 10-fold against BuChE activity for 92j, resulted in a decrease in the BuChE selectivity of these quaternary compounds (Supplementary Figure S12). Yet, the resulting AChE IC50 values of these quaternary (-)-physostigmine phenylcarbamates (92n, 92d, 92f, 92h, and 92j) were compared approvingly with those of neostigmine (92k) and were more potent than those of pyridostigmine (92l). Furthermore, the AChE and BuChE IC50 values were compared favorably with those of prior synthesized quaternary (-)-physostigmine phenylcarbamate iodide salts.
Khorana et al. (2012) evaluated various indoles, β-carbolines, and quinolines against AChE (Supplementary Figure S13).
For indoles with the electron-donating group such as compounds 93a (4.26% inhibition) and 93b (11.41% inhibition), % inhibition lower than 20% was reported, and for indoles containing electron-withdrawing groups such as compounds 93c (36.82% inhibition) and 93d (19.04% inhibition), % inhibition lower than 40% was obtained. It seems that the small compounds cannot be suitable for the AChE active site occupancy. Moving the substituent to the 2-position of the pyrrole ring also led to low activity, e.g., 2-methylindole 93e with %inhibition of 17.97%. The introduction of a longer substituent such as ethylamine into the pyrrole moiety and 5-methoxy into the benzene ring (93f, 17.19% inhibition) did not improve AChE inhibition compared with that of 93a. However, serotonin (93g, 62.59% inhibition) having the hydroxyl group at the 5-position exhibited important enhancement in %inhibition compared with 93a-f. Therefore, the more rigid structures of β-carboline derivatives were evaluated with an increase in the anti-AChE activity. Like 93h (83.19% inhibition), they showed a good %inhibition on AChE.
The substitution in 93h at the 7-position using methoxy (93i, 74.10% inhibition) did not show the anti-AChE activity, while replacement of methoxy by the hydroxyl group (93j, 87.07% inhibition) increased the activity. Reduction of one of the double bonds in pyridine ring (93k) improved the activity of about fivefold (85.52% inhibition) (Supplementary Figure S13). It showed that the flexibility of the compound in the suitable direction was essential for binding to the active site. Unlike the compound 93l (88.61% inhibition), the reduced form of 93j did not display the inhibitory activity. The tetrahydro-β-carboline analog (93m, 60.56% inhibition) exhibited the better inhibitory activity than the other less flexible β-carboline.
1-Carboxylic and 6-methoxy substituents on the tetrahydro-β-carboline ring are not suitable for desired activity as in 93n (43.07% inhibition) and 93o (14.29% inhibition). The 6-methoxyquinoline (93p) showed no inhibitory activity similar to its bioisostere 93a. 6-Methoxy-1-methylquinolinium iodide (93q (87.17% inhibition) and 1-benzyl-6-methoxyquinolinium iodide (93r (99.68% inhibition) significantly enhanced the anti-AChE activity higher than the 6-methoxyquinoline (93p) with IC50 = 7.67 µM and 2.46 µM, respectively (Supplementary Figure S13).
In a research, Peng et al. (2012) reported various bisindole derivatives. Among them, 100b showed enhanced activity with a Ki = 1,437.00 nM against AChE (Supplementary Figure S14.
The indole ring of 100a was switched with a positively charged quaternary nitrogen (compounds 100g and 100h). These compounds did display remarkably increased activity (Ki = 197.78 and 123.73 nM, respectively), approximately 70- and 100-fold higher than 100a (Supplementary Figure S14).
The indole ring was exchanged with acetophenone moiety (105a) that would play as hydrogen bond acceptors of the compound with the goal of making new interactions in the CAS additionally to the π–π stacking interactions. The activity of the compound 105a was increased in a promising manner (Ki = 53.34 nM) (Supplementary Figure S15).
Compounds 105b-g possessing linkers of diverse lengths, were synthesized. Compounds 105b (Ki = 45.81 nM) and 105c (Ki = 44.66 nM) with isoquinoline- and pyridine-substituted indole ring displayed somewhat enhanced activity, while the compound 105d (linker contained six methylene units) was established to reveal a much higher activity (Ki = 9.41 nM). The compound 105e having a linker one carbon longer than in the compound 105a exhibited the maximum inhibitory activity against hAChE (Ki value of 6.47 nM), approximately with sevenfold enhanced activity than 105a (Ki = 53.34 nM). Yet, a higher increase in the linker showed a reduction in the activity. For instance, compounds 105f (Ki = 81.37 nM) and 105g (Ki = 47.25 nM) having five and six methylene units revealed less activity than the compound 105e. The compound 105e exhibited higher activity than the compound 100a against AChE (Supplementary Figure S15).
Mainly, the compound 105d reduced approximately 38-fold compared with tacrine, and it displayed around 10-fold higher anti-AChE activity than anti-BuChE activity. The compound 105f exhibited also almost the same inhibitory activity against both AChE and BuChE (Supplementary Figure S15).
Some indolinone derivatives having benzylpyridinium moiety were evaluated as dual-binding inhibitors of AChE by Akrami et al. (2014). According to IC50 values, compounds 108a-d, 108l-o, and 108r (IC50 = 0.44–12.8 nM) were more potent than donepezil (IC50 = 14 nM). The derivative 108a showed the highest anti-AChE activity with an IC50 value of 0.44 nM (Figure 5). Furthermore, fluorine and bromine at 2-position (compounds 108b and 108c, respectively) with IC50 values 1.25 and 1.46 nM exhibited more activity than AChE. The unsubstituted compound 108d (IC50 = 47.10 nM) with 2- or 3-substituted analogs established that the presence of methyl (e.g., 108e (IC50 = 5.2 nM)), halide (e.g., 108a (IC50 = 0.44 nM)), and methoxy (e.g., 108f (IC50 = 6.6 nM)) groups at the 2- or 3-position of N-benzyl moiety remarkably increased the anti-AChE activity. Among them, the chlorine at the 2-position had the most effect on the AChE. Unlike, the presence of diverse substituents at the 4-position of the benzyl group reduced the AChE inhibitory activity of, e.g., compounds 108g (IC50 = 590 nM), 108h (IC50 = 677 nM), and 108i (IC50 = 744 nM). The compound 108i bearing the nitro group at the 4-position more dramatically diminished the activity. The attachment of second chlorine at 2- and 3-positions of the 4-chlorobenzyl derivative 108g showed a more potent activity in compounds 108j (IC50 = 29.4 nM) and 108k (IC50 = 257 nM). Nevertheless, compounds 108a or 108l (IC50 = 4.9 nM) bearing chlorine at 2- or 3-position on the benzyl group, respectively, showed that the presence of second halogen declined the anti-AChE activity as detected with compounds 108m (IC50 = 4.1 nM), 108n (IC50 = 17 nM), 108j (IC50 = 29.4 nM), and 108k (IC50 = 257 nM). The position of the halogen group on the benzyl moiety significantly affects the anti-AChE activity. The order of activity was as follows: 2 > 3 > 4 for, e.g., 108c (IC50 = 1.46 nM), 108o (IC50 = 10.3 nM), and 108p (IC50 = 653.4 nM) (Figure 5).
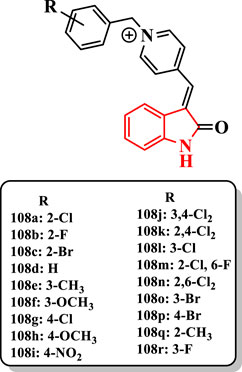
FIGURE 5. Indolinone-based compounds.
The compound 108a with the highest activity against AChE exhibited higher selectivity for this enzyme (SI = 3,113).
Luo et al. (2015) synthesized a series of melatonin-derived benzylpyridinium bromides (Supplementary Scheme S22).
Among all compounds, the compound 116b (IC50 = 0.11 µM) exhibited the highest activity against AChE, and its potency was 10-fold less than donepezil (IC50 = 0.014 µM). However, 112k revealed the most potent activity toward BuChE (IC50 value of 0.08 µM), which was 70-fold more than donepezil (IC50 = 5.6 µM). It seemed that the inhibitory activities of 4-pyridinium derivatives against hAChE [112c (IC50 = 4.2 µM) and 112d (IC50 = 3.2 µM)] were more potent than those of the 3-pyridinium series [112a (IC50 = 30.5 µM) and 112b (IC50 = 28.6 µM)], while the opposite trend was established on the inhibitory activity against BuChE [112c (IC50 = 2.8 µM) and 112d (IC50 = 4.1 µM)) vs. 112a (IC50 = 0.28 µM) and 112b (IC50 = 0.34 µM)] (Supplementary Figure S11). The presence of cyanide group [112g (IC50 = 22.9 µM for AChE) (IC50 > 100 µM for BuChE)] at the 4-position of the benzyl group decreased strangely the inhibitory activity against ChEs (Supplementary Figure S15).
Derivatives with a methoxy group at the 5-position of the indole ring were synthesized. Compounds 112h-j (IC50s = 3.3, 3.8, and 5.1 µM, respectively, for AChE and IC50s = 6.8, 7.9, and 7.5 µM, respectively for BuChE) exhibited the same activity of the corresponding unsubstituted analogs (112d-f) (IC50s = 3.2, 3.9, and 3.4 µM, respectively, for AChE and IC50s = 4.1, 5.1, and = 4.8 µM, respectively, for BuChE) (Supplementary Figure S15). Those results indicated that the presence of methoxy at the 5-position of the indole ring exhibited a low effect on ChEs. Furthermore, the derivative 112k (IC50 = 1.3 µM for AChE and IC50 = 0.08 µM for BuChE) having carbamate at the 4-position of the benzyl ring exhibited enhanced anti-ChE activity; particularly, the inhibitory activity of 112k was closely 94-fold higher than that of the compound 112j (IC50 = 5.1 µM for AChE and IC50 = 7.5 µM for BuChE) against BuChE (Supplementary Figure S11). Moreover, to study the possible activities, e.g., the effect of length of the spacer between benzylpyridinium and tryptamine on the ChE inhibitory activity, compounds 116a-f were synthesized. It was established that both AChE and BuChE inhibitory activities of compounds 116a-c (IC50s = 0.26, 0.11, and 0.21 µM, respectively, for AChE and IC50s = 2.3, 1.1, and 0.71 µM, respectively, for BuChE) with an additional double bond on the spacer, were improved significantly with respect to the corresponding shorter analogs (112h-j) (Supplementary Figure S15). Reduction of the additional double bond of compounds 116d-f (IC50s = 0.53, 0.44, and 0.58 µM, respectively, for AChE and IC50s = 0.86, 0.72, and 0.65 µM, respectively, for BuChE) exhibited a minor reduction in anti-AChE activity and enhanced anti-BuChE activities (Supplementary Figure S15).
Salehi et al. (2019) reported a range of benzoheterocycles linked to benzylpyridinium (benzimidazole, benzoxazole, or benzothiazole) as ChE inhibitors (Supplementary Scheme S23).
Benzothiazole derivatives 120a, 120b, 120c, and 120d (Figure 6) showed a similar or more potent anti-AChE activity than donepezil (IC50 = 14–23 nM). The compound 120a (IC50 value of 14 nM) exhibited the more potent activity against AChE. The unsubstituted compound 120a with substituted benzyl derivatives demonstrated that the presence of diverse substituents decreased the anti-AChE activity of the compounds [e.g., compound 120e (IC50 = 30 nM), compound 120f (IC50 = 53 nM), and compound 120g (IC50 > 300 nM)]. Based on results, the hydrophobic substituent such as chlorine at the 2-position was more suitable than others [e.g., compound 120b (IC50 = 22 nM) vs. compound 120h (IC50 = 159 nM)]. The shift of the methyl group from the 2- to the 3-position increased the activity of compounds 120c (IC50 = 21 nM) vs. 120e (IC50 = 30 nM). Compounds having halide at the 3-position of benzyl displayed that fluorine is more effective than chlorine and bromine substituents against ChE [e.g., compound 120d (IC50 = 23 nM) vs. compound 120i (IC50 = 61 nM) and compound 120f (IC50 = 53 nM)]. In the 4-substituted congeners, the compounds 120k (IC50 = 1900 nM) and 120l (IC50 > 300 nM) were less potent against AChE than 120m (IC50 = 36 nM). The nitro group dramatically reduced the activity of compounds 120h and 120k, whereas the existence of electron-withdrawing groups with small size such as fluorine on the benzyl ring showed good inhibitory activity against AChE (120d and 120m). The introduction of the second chlorine into the 2- or 3-position (120r (IC50 = 78 nM) and 120n (IC50 > 300 nM)] of 2-chlorobenzyl derivative 120b showed a reduction in the anti-AChE activity, while the introduction of fluorine into the 2-position [120o (IC50 = 27 nM)] exhibited the same activity. Particularly, the comparison of the compound 120a with the compound 120p (IC50 = 147 nM) or 120q (IC50 = 100 nM) indicated that the switching of sulfur with oxygen or NH did not have suitable effect on the anti-AChE activity.
Baussanne et al. (2021) reported the synthesis of N-alkylpyridinium-indolizine hybrids and evaluated them against ChEs (Supplementary Scheme S24).
The uncharged pyridine–indolizines (125a-c) were inactive against both enzymes. Compounds 126a (IC50 = 5.4 µM for eeAChE and IC50 = 55.4 µM for eqBuChE) and its analog 126b (IC50 = 7.9 µM for eeAChE and IC50 = 72.1 µM for eqBuChE) displayed the highest selectivity (being 10 times more active against eeAChE than against eqBuChE) (Figure 7). Donepezil as the standard drug showed IC50 = 2.0 µM for eeAChE and IC50 = 8.8 µM for eqBuChE. The Ind-PyC3 (3 carbons between the two rings) molecules comprising the 3-p-methoxybenzoyl group seemed to be more active against the two enzymes than their Ind-PyC2 (2 carbons between the two rings) analogs (126c (IC50 = 2.6 µM for eeAChE and IC50 = 4.8 µM for eqBuChE) and 126d (IC50 > 10 µM for eeAChE and IC50 = 4.8 µM for eqBuChE) vs. 126e (IC50 = 4.4 µM for eeAChE and IC50 = 17.5 µM for eqBuChE) and 126f (inactive for both them), respectively) (Figure 7). This effect may be related to the higher flexibility of the propyl linker compared with the shorter ethyl one. The compound 126g, analog of 126d, exhibited a good activity against both eeAChE (IC50 = 2.7 μM) and eqBuChE (IC50 = 7.3 μM) (Figure 7). The compound 126h was not active (IC50 > 100 μM) confirming its previously observed selectivity for AChE vs. BuChE.
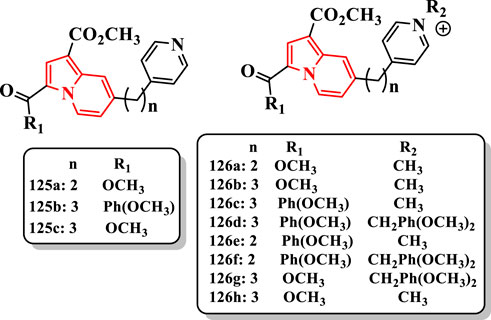
FIGURE 7. N-alkylpyridinium–indolizine hybrids.
Phthalimide hybrids
Saeedi et al. (2016) synthesized phthalimide derivatives liked to the benzylpyridinium moiety and evaluated them against ChEs (Supplementary Scheme S25).
All compounds showed less anti-AChE activity than donepezil (IC50 = 0.023 µM). The compound 132a having fluorine at the 2-position (IC50 = 0.77 µM) showed the highest anti-AChE activity. Its potency was fivefold more than that of unsubstituted benzyl derivative 132b (IC50 = 4.29 µM). Similarly, the 2-bromobenzyl analog 132c (IC50 = 2.16 µM) showed higher activity than the compound 132b. Therefore, the presence of fluorine (132a) or bromine (132c) at the 2-position of the benzyl moiety enhanced the anti-AChE activity. However, the substitution of halogen at 3- and 4-positions had negative effect on the inhibitory activity against AChE (e.g., compounds 132d (IC50 = 6.14 µM) and 132e (IC50 = 6.90 µM)). Among the dichlorine derivatives, analog 132f with dichlorine at 2- and 6-positions (IC50 = 5.81 µM) showed better activity toward AChE (Figure 8). Reduction of activity of compounds bearing a 3- or 4-substituent may be related to the steric effects.
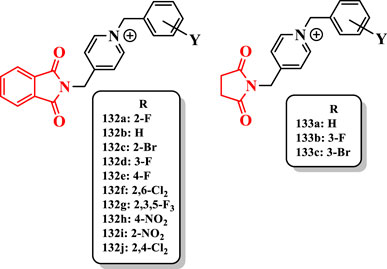
FIGURE 8. Phthalimide-based derivatives.
Derivatives bearing fluorine at the 4- or 3-position and fluorine at 2-, 3-, and 5-positions [compounds 132e, 132d, and 132g (IC50 = 6.77 µM)] showed a similar AChE inhibitory activity. The replacement of fluorine on the benzyl group with bromine showed a decreased activity (e.g., compound 132a vs. compound 132c). Moreover, the presence of the nitro group at the 2- or 4-position did not increase the activity of compounds 132h (IC50 = 11.23 µM) and 132i (IC50 = 9.28 µM) (Figure 8).
The achieved results from the limited series of succinimide derivatives 133a (IC50 = 18.3 µM), 133b (IC50 = 7.19 µM), and 133c (IC50 = 22.12 µM) showed that the replacement of phthalimide with succinimide decreased the anti-AChE activity. The presence of fluorine at the 3-position of the benzyl group in the succinimide series increased the anti-AChE activity (compound 132d vs. compound 133b) (Figure 8).
Benzothiophene hybrids
Palin et al. (2002) synthesized some pyridinium and piperidinium salts containing 5,6-dimethoxybenzothiophene as AChE inhibitors.
The compound 134a (IC50 = 0.19 µM) exhibited about a fourfold reduction in the AChE inhibition compared with 134b (IC50 = 0.043 µM). The hydroxyl-containing compounds 134b and 134a showed fivefold to 20-fold weaker activity in the AChE inhibition than 134c (IC50 = 0.008 µM) (Supplementary Figure S16).
The compound 134d (IC50 = 0.09 µM) exhibited a twofold decline toward AChE 134b. Both exo and endo compounds [134e (IC50 = 0.52 µM) and 134f (IC50 = 0.75 µM)] had weak AChE inhibitory activity (0.52 and 0.75 mM, respectively) (Supplementary Figure S16). Decreasing the spacer significantly diminished the activity against AChE. All these compounds showed weaker activity than 134c and 134b, suggesting the propanone is the optimum linker.
The N-methoxyethyl and N-carboxymethyl derivatives (134g and 134h) showed the same anti-AChE activity (IC50 = 0.054 and 0.053 µM, respectively). The presence of nitrofuran at the 2-position of 134i and tetrahydropyran at that of 134j derivatives provided the highest anti-AChE activity (IC50 = 0.032 and 0.02 µM, respectively). Phenoxyethyl derivative 134k (IC50 = 0.39 µM) and cyanomethyl derivative 134l (IC50 > 1.0 µM) showed a decrease in the inhibition of AChE (Supplementary Figure S16).
The presence of simple alkyl groups such as methyl 134m (IC50 = 0.9 µM), ethyl 134n (IC50 = 0.28 µM), and allyl 134o (IC50 = 0.54 µM) reduced AChE inhibitory activity.
The presence of a choline (e.g., 134p and 134r) increased the anti-AChE activity (IC50 = 0.03 µM and 0.007 µM, respectively). The in vitro reversal potency of 134p was also higher than the alkyl derivatives [e.g., compound 134q (IC50 = 2.57 µM) and 134s (IC50 = 0.11 µM)].
N-Benzylpyridinium 134t exhibited high activity against AChE (IC50 = 0.0046 µM). The presence of fluorine at the 4-position on the benzyl ring 134u (IC50 = 0.0026 µM) enhanced the AChE inhibition. However, 4-carboxyl substituent in compound 134v (IC50 = 1.0 µM) diminished anti-AChE activity. Heteroaromatic derivatives 134w and 134x possessing thiophene and 2-nitrofuran, respectively, gave excellent AChE inhibition (IC50 = 0.006 and 0.007 µM, respectively) (Supplementary Figure S16).
Benzofuranone hybrids
Nadri et al. (2013) synthesized benzofuranone-ylidene-methyl benzylpyridinium derivatives as AChE inhibitors (Supplementary Scheme S26).
The compound 140a having the unsubstituted benzyl ring showed a significant anti-AChE activity (IC50 = 41 nM) compared with donepezil (IC50 = 28 nM). The presence of a fluorine at the 2-position [140b (IC50 = 10 nM)] or 4-position [140c (IC50 = 22 nM)] of the benzyl moiety resulted in an increase in the anti-AChE activity. The compound 140b bearing fluorine at the 2-position of the benzyl group revealed the highest activity (IC50 = 10 nM) and was more potent than donepezil. Shifting the position of fluorine from the 2- to the 3-position showed a stronger decline of activity in the compound 140d (60 nM), while the fluorine at the 4-position of the compound 140c revealed a rather minor reduction compared with compound 140b. The presence of the methyl group on the benzyl ring showed less activity, except the compound 140e, which displayed moderate anti-AChE activity (IC50 = 68 nM) (Figure 9).
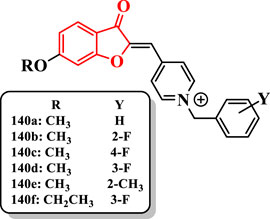
FIGURE 9. Benzofuranone-based benzylpyridinium derivatives.
Compounds 140b and 140c having the methoxy group showed a higher activity than donepezil. However, increasing of the alkoxy group length revealed an unfavorable effect on AChE inhibitory activity. Moreover, it was detected that replacing the methoxy group by ethoxy and propoxy groups led to a decrease in the activity except the compound 140f (IC50 = 48 nM).
The same research group (Nadri et al., 2010) reported benzofuranone derivatives linked to the pyridinium moiety. The compounds were evaluated against AChE (Supplementary Scheme S27).
The methyl on the benzyl moiety [145a (IC50 = 262 nM), 145b (IC50 = 208 nM), and 145c (IC50 = 514 nM)] showed weaker activity than the unsubstituted analogs [145d (IC50 = 86 nM)] (Calsolaro and Edison, 2016).
The compound 145e (IC50 = 52 nM) having fluorine at the 2-position showed the most potent activity against AChE. Nevertheless, compounds having methoxy (IC50 = 10 nM), ethoxy (IC50 = 32 nM), and propoxy (IC50 = 50 nM) at the 6-position showed less activity than its analogs (Supplementary Figure S17).
Likewise, the fluorine at the 3- and 4-position of the benzyl moiety displayed less activity than the alkoxy at the 6-position. Moving the fluorine from the 2- to either the 3- or the 4-position reduced the activity of compounds 145f (IC50 = 115 nM) and 145g (IC50 = 74 nM). Therefore, methyl group showed the order of activity as follows: 145b > 145a > 145c. The attachment of the methyl group to any position of benzyl moiety decreased the activity compared with unsubstituted analog (145d).
Baharloo et al. (2015) reported benzofuran scaffold linked to benzylpyridinium derivatives as AChE inhibitors (Supplementary Scheme S28).
The derivative 150b with IC50 = 4.1 nM showed the highest activity, in which its activity was sevenfold higher than donepezil (IC50 = 31 nM).
The studies on the benzofuran ring showed that the presence of bromine at the 5-position reduced the activity [compounds 150a and 150c (IC50 = 16 nM) vs. 150d (IC50 = 8.1 nM) and 150e (IC50 = 5.8 nM)]. Furthermore, the attachment of the methoxy group to the 7-position reduced the anti-AChE activity of compounds 150f (IC50 = 29.5 nM) and 150g (IC50 = 10.6 nM) compared with those corresponding analogs 150c and 150e. Compounds 150h (IC50 =9.6 nM), 150i (IC50 =8.7 nM), and 150j (IC50 =18.9 nM) having the fluorobenzyl group showed higher activity than compounds 150k (IC50 =13.5 nM) and 150l (IC50 =26.5 nM) having benzyl part, respectively. However, bromine at the 4-position of benzyl derivatives 150a and 150f exhibited lower activity compared with their corresponding benzyl analogs 150k and 150l. Furthermore, the presence of nitro at the 4-position of the benzyl moiety of compounds 150k and 150l resulted in higher active compounds 150d and 150g (Supplementary Figure S18).
Mostofi et al. (2015) described different benzofuran-based chalconoids as potential AChE inhibitors (Supplementary Scheme S29).
Compounds were synthesized in two classes: 3-pyridinium and 4-pyridinium derivatives. The compound 156c was potent AChE inhibitor (IC50 = 0.027 µM). The introduction of chlorine into the 2- and 3-positions of the pyridinium derivatives enhanced the activity of compounds156d (IC50 = 2.85 µM) and 156e (IC50 = 0.985 µM) compared with that of 156f (IC50 = 5.41 µM) and 156g (IC50 = 3.89 µM)]; in the 4-pyridinium series, this substituent reduced the activity. The bromine at the 2-position of benzyl derivatives 156h (IC50 = 0.035 µM), 156i (IC50 = 2.15 µM), 156c (IC50 = 0.027 µM), and 156n (IC50 = 0.041 µM) showed higher activity than their benzyl analogs 156j (IC50 = 0.058 µM), 156f (IC50 = 5.41 µM), 156g (IC50 = 3.89 µM), and 156r (IC50 = 0.064 µM). Changing the position of bromine from the 2- to the 4-position of the benzyl moiety significantly diminished the activity of compound 156i (IC50 = 2.15 µM) compared with that of the compound 156k (IC50 = 31 µM) (Figure 10).
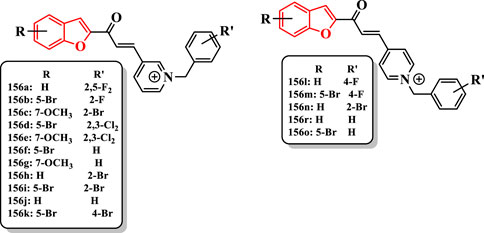
FIGURE 10. Benzofuran-based chalconoid derivatives.
The compound 156c having bromine at the 2-position of the benzyl moiety and methoxy at the 7-position of benzofuran ring revealed the highest activity (IC50 = 0.027 µM), which was comparable to donepezil (IC50 = 0.023 µM) (Figure 10).
Abedinifar et al. (2018) synthesized benzofuran-2-carboxamide-N-benzylpyridinium halide derivatives as ChE inhibitors (Supplementary Scheme S30).
All compounds exhibited lower AChE inhibition than donepezil (IC50 = 0.031 µM for AChE and 5.4 µM for BuChE), but all of them except 162a (IC50 = 9.6 µM) were better BuChE inhibitors. The compound 162b (IC50 = 2.1 µM) exhibited the best inhibitory activity against AChE (Supplementary Figure S19).
The introduction of the substituent into 2- and 3-positions led to higher inhibition against AChE than that of the derivative having substituent at the derivative having 4-position [e.g., compound 162b (IC50 = 2.1 µM) and compound 162c (IC50 = 13.8 µM) vs. compound 162d (IC50 = 19.8 µM)]. The presence of strong electron-withdrawing groups at the 4-position in 162e (IC50 = 40.0 µM) decreased the inhibitory activity against AChE in the 3-pyridinium series (Supplementary Figure S19).
Bispyridinium hybrids
Musilek et al. (2011) evaluated symmetrical bispyridinium on human erythrocyte ChEs (Supplementary Scheme S31).
Among compounds, 163l, 163m, and 163v (0.7–0.2 µM) showed the highest activity against hAChE. Moreover, the compound 163v showed the same activity on hAChE like the compound 163b (neostigmine) (IC50 = 0.1 µM). Moreover, the inhibitory ability of compounds 163k-m and 163v exceeded the frequently used commercial compound 163a (pyridostigmine) (IC50 = 40 µM) (Supplementary Figure S20).
The length of the spacer was the important factor for all compounds. Among compounds 163h-l, those having methylene units (163k-m) (IC50 = 2, 0.4, and 0.7 µM, respectively) showed the highest activity against hAChE. Compounds having shorter (163f-h) (IC50 = 505, 1,270, and 63 µM, respectively, for AChE and 9,800, 120, and 130 µM, respectively, for BuChE) or longer (163n) (no activity) methylene spacers were inactive against both enzymes (Supplementary Figure S20).
The length of the spacer in these compounds varied from 4 to 6 methylene units that were inadequate to interact like the compounds 163j-m (IC50 = 31, 2, 0.4, and 0.7 µM, respectively, for AChE and 29, 6, 5, and 7 µM, respectively, for BuChE). The compound 163v with a naphtylene spacer revealed the highest inhibitory activity for both enzymes (IC50 = 0.2 µM for AChE and 0.8 µM for BuChE) (Supplementary Figure S20). These compounds did not show selectivity for AChE over BuChE.
The same research group (Musilek et al., 2011) prepared bis-isoquinolinium ChEIs to compare their in vitro ability with that of standard myasthenia gravis (MG) drugs (Supplementary Scheme S32).
The compound 164h (IC50 = 0.005 µM) having an aliphatic spacer showed the most potent activity against AChE, but the selectivity of the compound was poor. The most potent compounds 164h-j (IC50 = 0.005, 0.04, and 0.05 µM, respectively, for AChE and 0.4, 0.6, and 1.6 µM, respectively, for BuChE) displayed only poor selectivity of AChE over BuChE (Supplementary Figure S21).
Among compounds 164d-i, those having methylene units [164f-k (IC50 = 0.3, 0.5, 0.005, 0.04, 0.05, and 0.1 µM, respectively)] revealed the most potent inhibitory activity against hAChE. Compounds having shorter spacers [164a-d (IC50 = 654, 0, 446, and 36 µM, respectively, for AChE and 1,400, 0, 2,600, and 40 µM, respectively, for BuChE)] were found to be ineffective toward both enzymes.
The compound 164t (IC50 = 0.3 µM for AChE and 4 µM for BuChE) having a naphthalenyl spacer showed an improvement in the inhibitory activity against both enzymes. The linker length of the compound 164t was the same as compound 164g (7 C-C bonds) and therefore exhibited the same binding to the AChE or BuChE. These compounds depicted no selectivity for AChE over BuChE.
This group (Musilek et al., 2011) also reported a series of SAD-128 analogs as ChEIs (Supplementary Scheme S33).
The commercial oximes [pralidoxime (IC50 = 878 µM) and obidoxime (IC50 = 577 µM)] showed a weak inhibition toward hAChE, while the selective standards (BW284c51 (IC50 = 0.03 µM) and ethopropazine (IC50 = 1,020 µM)) were favorite inhibitors of hAChE (Supplementary Figure S22).
Some synthesized compounds [165g (IC50 = 0.016 µM), 165h (IC50 = 0.005 µM), 165j (IC50 = 0.012 µM), 165k (IC50 = 0.026 µM), 165l (IC50 = 0.007 µM), and 165t (IC50 = 0.024 µM)] exhibited good inhibitory activity against hAChE. Compounds 165h, 165j, and 165l having an aliphatic spacer revealed the highest activity against AChE, and the compound 165t showed the highest activity with diverse spacers.
According to results, compounds with short spacer C1-C5 (165a-e), that are aliphatic with heteroatom (165m and 165n), and with double bonded linkers (165o and 165p) or linkers bearing xylene moiety (165q-s) were found to be effective as hAChE inhibitors. By contrast, compounds bearing longer aliphatic C6-C12 (165f-l) and naphtylene linkers (165t) showed the highest activity against eeAChE. The even spacers C8-C10-C12 (165h, 165j, and 165l) displayed higher activity than odd spacers (165g, 165i, and 165k). Moreover, the naphthalenyl-connected compound 165t revealed slightly lower activity than aliphatic-connected compounds 165g, d 165h, 165j, and 165l.
Komloova et al. (2013) investigated the isoquinolinium–pyridinium and quinolinium–pyridinium bisquaternary compounds 168a-k in vitro against ChEs (Supplementary Scheme S34).
There are two classes of the bisquaternary derivatives: Quinolinium-pyridinium and isoquinolinium-pyridinium groups (Supplementary Figure S23). Commonly, isoquinolinium-pyridinium compounds exhibited slightly less activity toward AChE than quinolinium-pyridinium compounds. Each compound with odd number of methylene groups in the spacer had a slightly lower activity than those compartments having the even number.
Compounds with 10 methylene groups (168c, IC50 = 0.005 µM) and 12 methylene groups (168d, IC50 = 0.0047 µM) in the spacer demonstrated the most potent activity, edrophonium (IC50 = 5.17 µM) and BW284C51 (IC50 = 0.03 µM) (Supplementary Figure S23). Furthermore, both compounds revealed the high selectivity for hAChE.
Pyridinium hybrids
Parlar et al. (2016) synthesized some hydrazone derivatives possessing pyridinium moiety and investigated their activity against ChEs (Supplementary Scheme S35).
Compounds 170d and 170e having benzofuran ring showed the highest activity against eeAChE. Furthermore, 170d and 170e (IC50 = 0.32 and 0.23 μM, respectively) revealed higher activity than galantamine (IC50 = 0.43 μM) (Supplementary Figure S24).
Considering results from hAChE inhibitory activity all compounds made the same inhibitory activity against eeAChE. Moreover, compounds 170d (IC50 = 0.62 μM) and 170e (IC50 = 0.24 μM) showed the highest activity toward hAChE. The AChE inhibitory activity improved when the length of linker between pyridinium nitrogen and the benzyl ring was increased from two to three methylene units (Supplementary Figure S24). Replacing the methylene group by ether in propyl chain resulted in a strange reduction of AChE inhibitory activity.
Compounds 170a (IC50 = 0.84 µM), 170b (IC50 = 0.74 µM), 170d (IC50 = 0.32 µM), and 170e (IC50 = 0.23 µM) showed the most potent activity against AChE and exhibited the most selectivity over BuChE.
Shi et al. (2013) synthesized various aloe emodin compounds and investigated them against AChE. Most of the compounds displayed significant AChE inhibitory activities toward AChE. The results showed that the phenolic hydroxyl groups of the aloe-emodin played a vital role in the AChE inhibitory activity. The aloe-emodin derivatives containing quaternary ammonium fragment 171a-f (IC50 = 0.09, 3.76, 25.38, 0, 0.54, and 13.56 µM, respectively) showed potent AChE inhibitory activity. The compound 171a possessed the best AChE inhibitory activity with IC50 value of 0.09 µM, which was higher than the positive control tacrine (IC50 = 0.26 µM) (Figure 11).
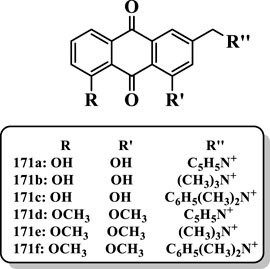
FIGURE 11. Aloe emodin derivatives.
Lan et al. (2017a) reported a series of cinnamic acid derivatives to be multifunctional cholinesterase inhibitors against AD by linking to the N-benzylpyridinium part and diverse substituted cinnamic acids (Supplementary Scheme S36).
The compound 176a (IC50 = 12.1 nM) exhibited the highest activity against AChE, and it was 3.3-fold more potent than donepezil (IC50 = 40.2 nM). Furthermore, the compound 176a revealed the high selectivity for AChE over BuChE. The compound 176h showed the most potent activity against BuChE (IC50 = 1.9 µM), which was 2.3-fold higher than donepezil (IC50 = 4.5 µM). Yet, cinnamic acid displayed significantly lower anti-AChE activity (IC50 > 100 µM), so it seems that the N-benzylpyridinium part is inescapably essential for higher activity (Figure 12).
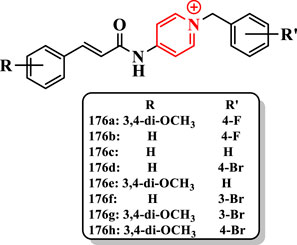
FIGURE 12. Cinnamic acid derivatives.
Based on the IC50 results, compounds 176a-n having methoxy at 3- and 4-positions of the benzylpyridinium part showed excellent anti-ChE activity. For instance, compound 176a (IC50 = 12.1 nM for AChE and 2.6 µM for BuChE) was more potent than compound 176b (IC50 = 90.8 nM for AChE and 10.0 µM for BuChE) against ChEs. The presence of fluorine, methyl, and bromine at 3- and 4-positions of the benzyl moiety declined the anti-AChE activity of compound 176c (IC50 = 54.1 nM for AChE), which was higher than that of the compound 176d (IC50 = 1,450.5 nM for AChE), while compounds with diverse groups on the benzyl moiety depicted an improved anti-AChE activity (compound 176a (IC50 = 12.1 nM for AChE) having fluorine at the 4-position of the benzyl moiety displayed higher AChE inhibition (11-fold) than compound 176e). Also, compounds 176f (IC50 = 2.5 µM), 176d (IC50 = 2.1 µM), 176a (IC50 = 2.5 µM) and 176g (IC50 = 1.9 µM) bearing bromine substituent revealed potent inhibitory activity.
The compound 176a (IC50 = 8.6 nM for hAChE) showed the most potent activity, which was 4-fold higher than donepezil (IC50 = 33.5 nM) (Figure 12).
Ghotbi et al. (2020) synthesized compounds containing thiazole and pyridinium moieties (Supplementary Scheme S37).
The derivative 183a (IC50 = 0.40 μM) having fluorine at the 2-position showed the highest anti-AChE activity, and compound 183b possessing bromine at the 2-position (IC50 = 0.69 μM) was the second potent inhibitor. In both compounds, fluorine and bromine were at the 2-position of the benzyl ring. Changing these substituents in 183a and 183b to 3 (183c and 183d)- and 4 [183e (IC50 = 6.48 μM and 183f (IC50 = 30.49 μM)]-positions revealed a slight reduction of inhibitory activity. According to results, the 2- and 3-substituted derivatives provided stronger inhibition against AChE than the 4-substituted derivatives (the order of substitution position is 2 > 3 > 4). For derivatives 183g (IC50 = 40.80 μM) as well as 183e, 183h (IC50 = 21.49 μM), and 183f (IC50 = 30.49 μM), containing nitro or halide group at the 4-position, the electron-withdrawing group decreased the activity (Supplementary Figure S25).
The presence of nitro at the 4-position of the compound showed lower activity (IC50 = 54.58 μM) than the halogenated derivatives, and among the diverse halogen groups, fluorine revealed the most potent anti-AChE activity to the compound 183l (IC50 = 1.95 μM), while the compound 183m unsubstituted in the 3-pyridinium series exhibited the most potent anti-AChE activity (IC50 = 1.64 μM) (Supplementary Figure S25). Commonly, in the 3-pyridinium series, similar to 4-substituted compounds in 4-pyridinium derivatives, the presence of a substituent led to the decreased activity.
Abdullaha et al. (2020) described the synthesis of pyridinium benzamides and screened for the inhibition of ChEs (Supplementary Schemes S37, 38).
Donepezil showed IC50 = 0.049 µM for the inhibition of AChE and IC50 = 5.52 µM for the inhibition of BuChE.
As a general trend, the 2-substituted derivatives (e.g., 187x (88.59% for AChE), 187z (91.67% for AChE), 191ac (90.68% for AChE), and 191af (49.27% for AChE)) were superior to other substitutions. The comparison of this series of compounds with the parent lead 187w indicated that only derivatives bearing substituent at the 2-position of benzyl ring displayed a similar AChE inhibitory activity to that of 187w (Supplementary Figure S26). The ethylene linker [191ah (78.93% for AChE and 53.25% for BuChE)] was significantly superior to methylene linker [191 ag (48.33% for AChE and 2.41% for BuChE)]. However, several compounds [191ah, 191aj (78.69% for AChE and 54.17% for BuChE), and 191ao (46.58% for AChE and 47.75% for BuChE)] displayed superior inhibition of BuChE to benzamide 187w (Supplementary Figure S26). This series provided critical information that placing a small -CH2CH2- linker between naphthylamide and pyridine ring imparts a significant positive impact on the potency. This additional spacer might be helping the compound to occupy the active site gorge perfectly, and helping it to interact with all key residues of both the active sites.
Next, when the pyridinium moiety of the first stage lead compound 187w was replaced with piperidine (compound 193a), the ChE inhibitory activity was completely lost (7.70% for AChE and 1.77% for BuChE). However, the introduction of an ethylene linker between piperidine ring and naphthylamide moiety [compound 193b (80.02% for AChE and 60.98% for BuChE)] resulted in the gain of activity against both ChEs (Supplementary Figure S26). In fact, the compound 193b, though it does not bear quaternary nitrogen, still displayed the same level of AChE inhibition, and superior BuChE inhibition compared with 187w. The replacement of naphthyl (compound 187w) with phenoxy-phenyl, biphenyl, benzoyloxy-phenyl, and phenoxy-benzyl (191aq-at) resulted in a loss of activity, indicating that naphthalene ring is essential for dual cholinesterase inhibition.
Zarei et al. (2021) reported the synthesis of benzyl-oxoquinazolin-pyridinium derivatives assessed as ChEs inhibitors (Supplementary Scheme S40).
203a-l could be categorized into two group: 1) having methoxy group substituted on oxoquinazoline ring and 2) without methoxy group on oxoquinazoline ring. In the first group of tested compounds 203a-e, 203a having bromine at the 3-position of benzyl group showed the strongest AChE inhibitory effect (IC50 = 5.90 μM) (Figure 13). Donepezil showed IC50 = 0.079 µM for the inhibition of AChE and IC50 = 5.19 µM for the inhibition of BuChE.
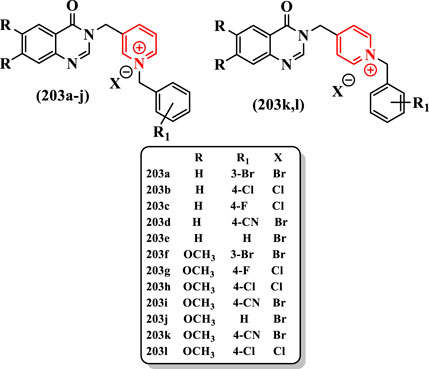
FIGURE 13. Moieties of compounds oxoquinazoline and pyridinium.
Supplanting the chlorine by the fluorine in 203c (IC50 > 100 µM) or CN in 203d (IC50 > 100 µM) resulted in derivatives with very low activities. Although the removal of this group from the benzyl ring led to an increase in the AChE inhibition for the compound 203e (IC50 = 41.21 μM), the compound 203a remained the most potent AChE inhibitor in this group of compounds.
In the second group containing methoxy substitution on oxoquinazoline ring, the best compound was 203h with a chlorine group at the 4-position, which showed a promising potency as an AChE inhibitor (IC50 = 1.11 μM). However, 203h had no inhibitory activity against BuChE, which indicated that this compound is a good selective AChE inhibitor. The replacement of chlorine with hydrogen, fluorine, and CN at the 4-position caused a depletion of inhibitory activity for AChE in compounds 203j, 203g, and 203i with IC50 = 10.08 μM, IC50 = 21.92 μM, and IC50 > 100 μM, respectively. The 203f with bromine at the 3-position of the benzyl ring revealed an intermediate potency among other compounds in this group (IC50 = 6.77 μM).
Two compounds with 4-(methyl)pyridine moiety (203k and 203l) showed lower inhibitory activities both for AChE and for BuChE than other compounds containing the 3-(methyl)pyridine group (IC50 > 100 µM for both enzyme). According to the observed results, on the one hand, the first group had a higher activity for BuChE inhibition. On the other hand, the second group with the substituted methoxy groups had better results for AChE inhibition. It is worthwhile to note that compounds 203s in the series of 3-methylpyridines showed superior activity to 4-methylpyridine derivatives and the electron-withdrawing CN group decreased the inhibitory activity. Finally, the compound 203a with IC50 = 5.90 μM for AChE and IC50 = 6.76 μM for BuChE was the most potent dual inhibitor and the compound 203h with IC50 = 1.11 μM for AChE was the strongest derivative among tested compounds against AChE (Figure 13).
Hassanzadeh et al. (2021) reported synthesis of isoindoline-1,3-dione-N-benzylpyridinium hybrids and evaluated them against AChE (Supplementary Scheme S41).
The best anti-AChE activity was obtained by compounds 210a and 210f (IC50 = 2.1 μM possessing fluorine at the 4-position of benzylpyridinium moiety, compared with rivastigmine (IC50 = 11.07 μM). The shift of the fluorine group from the 4- to the 3-position in 210c and 210h led to a reduction in the inhibitory activity (IC50s = 2.7 and 2.9 μM, respectively). The switching from the fluorine to the chlorine group in compounds 210d (IC50 = 7.4 μM) and 210i (IC50 = 6.7 μM) led to a more than twofold reduction in the inhibitory activity than in 210c and 210h so that chlorine-substituted derivatives showed the weakest inhibitory activity among compounds. The 3-methyl-substituted compounds 210b (IC50 = 5.4 μM) and 210g (IC50 = 4.8 μM) showed improved AChE inhibitory activities compared with chlorine-substituted compounds 210d and 210i (Figure 14).
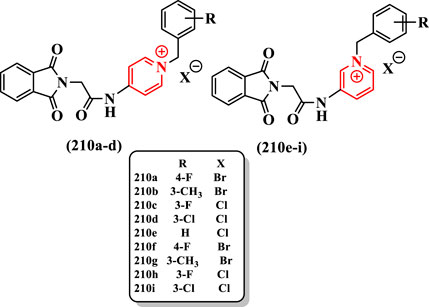
FIGURE 14. Isoindoline-1,3-dione–N-benzylpyridinium hybrid compounds.
Molecular docking results, kinetic analysis, and BuChE activity of reported compounds are given in the Supplementary Data section.
Conclusion
According to the reported results in the literature, both anti-AChE and anti-BuChE activities showed almost similar SAR. AChE inhibitory activity of reported compounds was sensitive to the electronic and steric properties of substituents at different positions of the benzyl moiety of pyridinium salts. Thus, it could be concluded that irrespective of the electronic properties and the size of substituents on the benzyl group are important in inducing desired inhibitory activity. According to results, all compounds exhibited high selectivity for AChE.
1 Benzylpiperidine moiety of donepezil seems to be important for optimal AChE inhibitory activity since various studies have shown that donepezil binds to the CAS and PAS of the enzyme.
2 Dual site binding donepezil-based scaffolds have been found to elicit other vital pharmacological activities such as prevention of amyloid aggregation, which is crucial for the management of AD. Furthermore, various hybrids have also displayed MAO, BuChE inhibitory activity, metal chelating, and ROS scavenging ability.
3 Based on results, the inhibitory activity of compounds toward ChEs was sensitive to the length of the spacers between different moieties.
4 The type of functional group in the spacer, linking benzylpyridine group to other moieties, is an imperative parameter in the inhibitory activity. Generally, the presence of the amide group has depicted a positive result.
5 It was found that the type and position of substituents on the benzyl group is very important in AChE inhibitory activity. The presence of substituents at 2- or 4- or 2- and 3- positions of the aryl ring affected the activity. Substitution at 2- or 4- or 2- and 3-positions by halides was found to increase the selectivity and inhibitory activity toward AChE. The introduction of NO2 into these positions decreased activity. In addition, substitution at the 3-position of aryl ring seems to be unfavorable for activity. Also, the presence of substitution on benzylpyridine moiety increased anti-ChE activity in comparison with nonsubstituted compounds. The position and type of substituents on the benzyl part could change the steric and electronic properties of the aryl ring. Then, the affinity of the compounds could be improved by altering the substituent on the benzyl moiety.
6 Most derivatives showed stronger inhibitory activity than donepezil. It seems that the presence of substituents on the aryl ring and spacers in the corresponding compounds, which are not present in the donepezil structure, play important roles in the anti-ChE activity.
7 Replacing phthalimide by succinimide reduced the anti-AChE activity. It was perceived that the aromatic ring linked to the succinimide moiety played a vital role in the inhibitory activity of compounds.
8 Generally, 4-pyridinium salts series exhibited the highest anti-AChE activity and the high selectivity for AChE over BuChE compared with 3-pyrimidinum series.
9 The presence of a positive charge on the pyridinium ring is essential for inducing the inhibitory activity. Based on the results, uncharged structures showed lower ChE inhibitory activity.
10 Docking and kinetic studies exhibited that all derivatives attached to the CAS and PAS of the enzyme active site.
11 The geometric orientation of the benzylpyridinium moiety is an imperative parameter in the inhibitory activity.
It is concluded that benzylpyridinium moiety that linked to other moieties and functional groups through a spacer with appropriate length demonstrated potent AChE inhibitory activity as donepezil analogs.
And based on studies, the benzylpyridinium moiety of inhibitors interacts with the CAS and other moieties interact with the PAS of the AChE.
Author contributions
SS searched the database and wrote the manuscript. MS edited the manuscript. MM and BL supervised all processes.
Funding
The financial support from Ardabil University of Medical Sciences under Grant IR. ARUMS.REC.1399.511 and Tehran University of Medical Sciences is acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2022.936240/full#supplementary-material
References
Abdullaha, M., Nuthakki, V. K., and Bharate, S. B. (2020). Discovery of methoxy-naphthyl linked N-(1-benzylpiperidine) benzamide as a blood-brain permeable dual inhibitor of acetylcholinesterase and butyrylcholinesterase. Eur. J. Med. Chem. 207, 112761. doi:10.1016/j.ejmech.2020.112761
Abedinifar, F., Farnia, S. M. F., Mahdavi, M., Nadri, H., Moradi, A., Ghasemi, J. B., et al. (2018). Synthesis and cholinesterase inhibitory activity of new 2-benzofuran carboxamide-benzylpyridinum salts. Bioorg. Chem. 80, 180–188. doi:10.1016/j.bioorg.2018.06.006
Agatonovic-Kustrin, S., Kettle, C., and Morton, D. W. (2018). A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 106, 553–565. doi:10.1016/j.biopha.2018.06.147
Akrami, H., Mirjalili, B. F., Khoobi, M., Nadri, H., Moradi, A., Sakhteman, A., et al. (2014). Indolinone-based acetylcholinesterase inhibitors: Synthesis, biologicalactivity and molecular modeling. Eur. J. Med. Chem. 84, 375–381. doi:10.1016/j.ejmech.2014.01.017
Alipour, M., Khoobi, M., Foroumadi, A., Nadri, H., Moradi, A., Sakhteman, A., et al. (2012). Novel coumarin derivatives bearing N-benzyl pyridinium moiety: potent and dual binding site acetylcholinesterase inhibitors. Bioorg. Med. Chem. 20, 7214–7222. doi:10.1016/j.bmc.2012.08.052
Alipour, M., Khoobi, M., Nadri, H., Sakhteman, A., Moradi, A., Ghandi, M., et al. (2013). Synthesis of some new 3-coumaranone and coumarin derivatives as dual inhibitors of acetyl- and butyrylcholinesterase. Arch. Pharm. Weinh. 346, 577–587. doi:10.1002/ardp.201300080
Arab, S., Sadat-Ebrahimi, S. E., Mohammadi-Khanaposhtani, M., Moradi, A., Nadri, H., Mahdavi, M., et al. (2015). Synthesis and evaluation of chroman-4-one linked to N-benzyl pyridinium derivatives as new acetylcholinesterase inhibitors. Arch. Pharm. Weinh. 348, 643–649. doi:10.1002/ardp.201500149
Baharloo, F., Moslemin, M. H., Nadri, H., Asadipour, A., Mahdavi, M., Emami, S., et al. (2015). Benzofuran-derived benzylpyridinium bromides as potentacetylcholinesterase inhibitors. Eur. J. Med. Chem. 93, 196–201. doi:10.1016/j.ejmech.2015.02.009
Bartus, R. T. (2000). On neurodegenerative diseases, models, and treatment strategies: Lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 163, 495–529. doi:10.1006/exnr.2000.7397
Baussanne, I., Firstova, O., Dediu, A. B., Larosa, C., Furdui, B., Ghinea, I. O., et al. (2021). Interest of novel N-alkylpyridinium-indolizine hybrids in the field of Alzheimer’s disease: Synthesis, characterization and evaluation of antioxidant activity, cholinesterase inhibition, and amyloid fibrillation interference. Bioorg. Chem. 116, 105390. doi:10.1016/j.bioorg.2021.105390
Bekris, L. M., Yu, C. E., Bird, T. D., and Tsuang, D. W. (2010). Review article: genetics of alzheimer disease. J. Geriatr. Psychiatry Neurol. 23, 213–227. doi:10.1177/0891988710383571
Brus, B., Kosak, U., Turk, S., Pislar, A., Coquelle, N., Kos, J., et al. (2014). Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J. Med. Chem. 57, 8167–8179. doi:10.1021/jm501195e
Cacabelos, R. (2007). Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr. Dis. Treat. 3, 303–333.
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer's disease: Current evidence and future directions. Alzheimer's. Dement. 12, 719–732. doi:10.1016/j.jalz.2016.02.010
Costanzo, P., Cariati, L., Desiderio, D., Sgammato, R., Lamberti, A., Arcone, R., et al. (2016). Design, synthesis, and evaluation of donepezil-like compounds as AChE and BACE-1 inhibitors. ACS Med. Chem. Lett. 7, 470–475. doi:10.1021/acsmedchemlett.5b00483
Dubey, S. K., Kharbanda, M., Dubey, S. K., and Mathela, C. S. (2010). A new commercially viable synthetic route for donepezil hydrochloride: anti-Alzheimer’s drug. Chem. Pharm. Bull. 58 (9), 1157–1160. doi:10.1248/cpb.58.1157
Garcia-Ayllon, M. S., Small, D. H., Avila, J., and Saez-Valero, J. (2011). Revisiting the role of acetylcholinesterase in Alzheimer’s disease: Cross-talk with P-tau and β-amyloid. Front. Mol. Neurosci. 4, 22. doi:10.3389/fnmol.2011.00022
Ghotbi, G., Mahdavi, M., Najaf, Z., Moghadam, F. H., Hamzeh-Mivehroud, M., Davaran, S., et al. (2020). Design, synthesis, biological evaluation, and docking study of novel dualacting thiazole-pyridiniums inhibiting acetylcholinesterase and β-amyloid aggregation for Alzheimer’s disease. Bioorg. Chem. 103, 104186. doi:10.1016/j.bioorg.2020.104186
Girek, M., and Szymanski, P. (2019). Tacrine hybrids as multi-target-directed ligands in Alzheimer’s disease: Influence of chemical structures on biological activities. Chem. Pap. 73, 269–289. doi:10.1007/s11696-018-0590-8
Goyal, D. D., Shuaib, S., Mann, S., and Goyal, B. (2017). Rationally designed peptides and peptidomimetics as inhibitors of amyloid-β (Aβ) aggregation: potential therapeutics of Alzheimer’s disease. ACS Comb. Sci. 19, 55–80. doi:10.1021/acscombsci.6b00116
Hassanzadeh, M., Hassanzadeh, F., khodarahmi, G. A., Rostami, M., Azimi, F., Nadri, H., et al. (2021). Design, synthesis, and bio-evaluation of new isoindoline-1, 3-dione derivatives as possible inhibitors of acetylcholinesterase. Res. Pharm. Sci. 16, 482–492. doi:10.4103/1735-5362.323915
Hosseini, F., Ramazani, A., Mohammadi-Khanaposhtani, M., Tehrani, M. B., Nadri, H., Larijani, B., et al. (2019). Design, synthesis, and biological evaluation of novel 4-oxobenzo[d]1, 2, 3-triazin-benzylpyridinum derivatives as potent anti-Alzheimer agents. Bioorg. Med. Chem. 27, 2914–2922. doi:10.1016/j.bmc.2019.05.023
Jiang, X. Y., Chen, T. K., Zhou, J. T., He, S. Y., Yang, H. Y., Chen, Y., et al. (2018). Dual GSK-3β/AChE inhibitors as a new strategy for multitargeting anti-Alzheimer’s disease drug discovery. ACS Med. Chem. Lett. 9, 171–176. doi:10.1021/acsmedchemlett.7b00463
Khoobi, M., Alipour, M., Sakhteman, A., Nadri, H., Moradi, A., Ghandi, M., et al. (2013). Design, synthesis, biological evaluation and docking study of 5-oxo-4, 5-dihydropyrano[3, 2-c]chromene derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors. Eur. J. Med. Chem. 68, 260–269. doi:10.1016/j.ejmech.2013.07.038
Khorana, N., Changwichit, K., Ingkaninan, K., and Utsintong, M. (2012). Prospective acetylcholinesterase inhibitory activity of indole and its analogs. Bioorg. Med. Chem. Lett. 22, 2885–2888. doi:10.1016/j.bmcl.2012.02.057
Khunnawutmanotham, N., Chimnoi, N., Saparpakorn, P., and Techasakul, S. (2016). Synthesis and anti-acetylcholinesterase activity of scopoletin derivatives. Bioorg. Chem. 65, 137–145. doi:10.1016/j.bioorg.2015.12.002
Khunnawutmanotham, N., Laongthipparos, C., Saparpakorn, P., Chimnoi, N., and Techasakul, S. (2018). Synthesis of 3-aminocoumarin-N-benzylpyridinium conjugates with nanomolar inhibitory activity against acetylcholinesterase. Beilstein J. Org. Chem. 14, 2545–2552. doi:10.3762/bjoc.14.231
Knowles, J. (2006). Donepezil in Alzheimer’s disease: An evidence-based review of its impact on clinical and economic outcomes. Core Evid. 1, 195–219.
Komloova, M., Horova, A., Hrabinova, M., Jun, D., Dolezal, M., Vinsova, J., et al. (2013). Preparation, in vitro evaluation and molecular modelling of pyridinium-quinolinium/isoquinolinium non-symmetrical bisquaternary cholinesterase inhibitors. Bioorg. Med. Chem. Lett. 23, 6663–6666. doi:10.1016/j.bmcl.2013.10.043
Kryger, G., Silman, I., and Sussman, J. L. (1999). Structure of acetylcholinesterase complexed with E2020 (Aricept): Implications for the design of new anti-alzheimer drugs. Structure 15, 297–307. doi:10.1016/s0969-2126(99)80040-9
Lan, J. S., Hou, J. W., Liu, Y., Ding, Y., Zhang, Y., Lia, L., et al. (2017). Design, synthesis and evaluation of novel cinnamic acid derivatives bearing N-benzyl pyridinium moiety as multifunctional cholinesterase inhibitors for Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 32, 776–788. doi:10.1080/14756366.2016.1256883
Lan, J. S., Zhang, T., Liu, Y., Yang, J., Xie, S. S., Liu, J., et al. (2017). Design, synthesis and biological activity of novel donepezil derivatives bearing N-benzyl pyridinium moiety as potent and dual binding site acetylcholinesterase inhibitors. Eur. J. Med. Chem. 133, 184–196. doi:10.1016/j.ejmech.2017.02.045
Lan, J. S., Ding, Y., Liu, Y., Kang, P., Hou, J. W., Zhang, X. Y., et al. (2017). Design, synthesis and biological evaluation of novel coumarin-Nbenzyl pyridinium hybrids as multi-target agents for the treatment of Alzheimer's disease. Eur. J. Med. Chem. 139, 48–59. doi:10.1016/j.ejmech.2017.07.055
Lavado, L., Zhang, M. H., Patel, K., Khan, S., and Patel, U. K. (2019). Biometals as potential predictors of the neurodegenerative decline in Alzheimer’s disease. Cureus 11, e5573. doi:10.7759/cureus.5573
Lee, J. K., and Kim, N. J. (2017). Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 22, 1287. doi:10.3390/molecules22081287
Li, Q., Yang, H., Chen, Y., and Sun, H. (2017). Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer's disease. Eur. J. Med. Chem. 132, 294–309. doi:10.1016/j.ejmech.2017.03.062
Li, Q., He, S., Chen, Y., Feng, F., Qu, W., and Sun, H. (2018). Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer's disease. Eur. J. Med. Chem. 158, 463–477. doi:10.1016/j.ejmech.2018.09.031
Luo, X. T., Wang, C. M., Liu, Y., and Huang, Z. G. (2015). New multifunctional melatonin-derived benzylpyridinium bromides with potent cholinergic, antioxidant, and neuroprotective properties as innovative drugs for Alzheimer's disease. Eur. J. Med. Chem. 103, 302–311. doi:10.1016/j.ejmech.2015.08.052
Mack, A., and Robitzki, A. (2000). The key role of butyrylcholinesterase during neurogenesis and neural disorders: An antisense-5'butyrylcholinesterase-DNA study. Prog. Neurobiol. 60, 607–628. doi:10.1016/s0301-0082(99)00047-7
Makarian, M., Gonzalez, M., Salvador, S. M., Lorzadeh, S., Hudson, P. K., and Pecic, S. (2022). Synthesis, kinetic evaluation and molecular docking studies of donepezil-based acetylcholinesterase inhibitors. J. Mol. Struct. 1247, 131425. doi:10.1016/j.molstruc.2021.131425
Mangialasche, F., Solomon, A., Winblad, B., Mecocci, P., and Kivipelto, M. (2010). Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 9, 702–716. doi:10.1016/s1474-4422(10)70119-8
Matsuzaki, K. (2011). formation of toxic amyloid fibrils by amyloid β-protein on ganglioside clusters. Int. J. Alzheimer's. Dis. 2011, 1–7. doi:10.4061/2011/956104
McGleenon, B. M., Dynan, K. B., and Passmore, A. P. (1999). Acetylcholinesterase inhibitors in Alzheimer’s disease. Br. J. Clin. Pharmacol. 48, 471–480. doi:10.1046/j.1365-2125.1999.00026.x
Mesulam, M. M., Guillozet, A., Shaw, P., Levey, A., Duysen, E. G., and Lockridge, O. (2002). Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 110, 627–639. doi:10.1016/s0306-4522(01)00613-3
Miao, J., Shi, R., Li, L., Chen, F., Zhou, Y., Tung, Y. C., et al. (2019). Pathological tau from Alzheimer’s brain induces site-specific hyperphosphorylation and SDS- and reducing agent-resistant aggregation of tau in vivo. Front. Aging Neurosci. 11, 34. doi:10.3389/fnagi.2019.00034
Mohsin, N. A., and Ahmad, M. (2020). Donepezil: A review of the recent structural modifications and their impact on anti-alzheimer activity. Braz. J. Pharm. Sci. 56, e18325. doi:10.1590/s2175-97902019000418325
Mollazadeh, M., Mohammadi-Khanaposhtani, M., Zonouzi, A., Nadri, H., Najafi, Z., Larijani, B., et al. (2019). New benzyl pyridinium derivatives bearing 2, 4-dioxochroman moiety as potent agents for treatment of Alzheimer’s disease: Design, synthesis, biological evaluation, and docking study. Bioorg. Chem. 87, 506–515. doi:10.1016/j.bioorg.2019.03.012
Mostofi, M., Mohammadi Ziarani, G., Mahdavi, M., Moradi, A., Nadri, H., Emami, S., et al. (2015). Synthesis and structure-activity relationship study of benzofuranbased chalconoids bearing benzylpyridinium moiety as potent acetylcholinesterase inhibitors. Eur. J. Med. Chem. 103, 361–369. doi:10.1016/j.ejmech.2015.08.061
Mushtaq, G., Greig, N. H., Khan, J. A., and Kamal, M. A. (2014). Status of acetylcholinesterase and butyrylcholinesterase in Alzheimer’s disease and type 2 diabetes mellitus. CNS Neurol. Disord. Drug Targets. 13, 1432–1439. doi:10.2174/1871527313666141023141545
Musilek, K., Komloova, M., Zavadova, V., Holas, O., Hrabinova, M., Pohanka, M., et al. (2010). Preparation and in vitro screening of symmetrical bispyridinium cholinesterase inhibitors bearing different connecting linkage-initial study for Myasthenia gravis implications. Bioorg. Med. Chem. Lett. 20, 1763–1766. doi:10.1016/j.bmcl.2010.01.034
Musilek, K., Komloova, M., Holas, O., Hrabinova, M., Pohanka, M., Dohnal, V., et al. (2011). Preparation and in vitro screening of symmetrical bis-isoquinoliniumcholinesterase inhibitors bearing various connecting linkage-implicationsfor early myasthenia gravis treatment. Eur. J. Med. Chem. 46, 811–818. doi:10.1016/j.ejmech.2010.12.011
Nadri, H., Pirali-Hamedani, M., Shekarchi, M., Abdollahi, M., Sheibani, V., Amanlou, M., et al. (2010). Design, synthesis and anticholinesterase activity of a novel seriesof 1-benzyl-4-((6-alkoxy-3-oxobenzofuran-2(3H)-ylidene) methyl) pyridinium derivatives. Bioorg. Med. Chem. 18, 6360–6366. doi:10.1016/j.bmc.2010.07.012
Nadri, H., Pirali-Hamedani, M., Moradi, A., Sakhteman, A., Vahidi, A., Sheibani, V., et al. (2013). 5, 6-Dimethoxybenzofuran-3-one derivatives: A novel series of dual acetylcholinesterase/butyrylcholinesterase inhibitors bearing benzylpyridinium moiety. DARU J. Pharm. Sci. 21, 15. doi:10.1186/2008-2231-21-15
Nicolet, Y., Lockridge, O., Masson, P., Fontecilla-Camps, J. C., and Nachon, F. (2003). Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 278, 41141–41147. doi:10.1074/jbc.m210241200
Nordberg, A., Ballard, C., Bullock, R., Darreh-Shori, T., and Somogyi, M. (2013). A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord. 15, PCC.12r01412. doi:10.4088/pcc.12r01412
Palin, R., Clark, J. K., Cowley, P., Muir, A. W., Pow, E., Prosser, A. B., et al. (2002). Novel piperidinium and pyridinium agents as water-soluble acetylcholinesterase inhibitors for the reversal of neuromuscular blockade. Bioorg. Med. Chem. Lett. 12, 2569–2572. doi:10.1016/s0960-894x(02)00483-3
Parlar, S., Bayraktar, G., Tarikogullari, A. H., Alptüzün, V., and Erciyas, E. (2016). Synthesis, biological evaluation and molecular docking study of hydrazone-containing pyridinium salts as cholinesterase inhibitors. Chem. Pharm. Bull. 64, 1281–1287. doi:10.1248/cpb.c16-00221
Peng, D. Y., Sun, Q., Zhu, X. L., Lin, H. Y., Chen, Q., Yu, N. X., et al. (2012). Design, synthesis, and bioevaluation of benzamides: novel acetylcholinesterase inhibitors with multi-functions on butylcholinesterase, Aβ aggregation, and β-secretase. Bioorg. Med. Chem. 20, 6739–6750. doi:10.1016/j.bmc.2012.09.016
Picciotto, M. R., Higley, M. J., and Mineur, Y. S. (2012). Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76, 116–129. doi:10.1016/j.neuron.2012.08.036
Piemonyese, L., Tomas, D., Hiremathad, A., Capriati, V., Candeias, E., Cardoso, S. M., et al. (2018). Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J. Enzyme Inhib. Med. Chem. 33, 1212–1224. doi:10.1080/14756366.2018.1491564
Prince, M., Bryce, R., Albanese, E., Wimo, A., Ribeiro, W., and Ferri, C. P. (2013). The global prevalence of dementia: a systematic review and metaanalysis. Alzheimer's. &. Dement. 9, 63–75. doi:10.1016/j.jalz.2012.11.007
Rawat, A. S., Pande, S., Bhatt, N., Kharatkar, R., Belwal, C., and Vardhan, A. (2013). Synthesis of donepezil hydrochloride via chemoselective hydrogenation. Org. Process Res. Dev. 17, 1617. doi:10.1021/op400007p
Rook, Y., Schmidtke, K. U., Gaube, F., Schepmann, D., Wunsch, B., Heilmann, J., et al. (2010). Bivalent β-carbolines as potential multitarget anti-alzheimer agents. J. Med. Chem. 53, 3611–3617. doi:10.1021/jm1000024
Saeedi, M., Golipoor, M., Mahdavi, M., Moradi, A., Nadri, H., Emami, S., et al. (2016). Phthalimide-derived N-benzylpyridinium halides targeting cholinesterases: synthesis and bioactivity of new potential anti-alzheimer’s disease agents. Arch. Pharm. Weinh. 349, 293–301. doi:10.1002/ardp.201500425
Salehi, N., Mirjalili, B. F., Nadri, H., Abdolahi, Z., Forootanfar, H., Samzadeh-Kermani, A., et al. (2019). Synthesis and biological evaluation of new N-benzylpyridinium-basedbenzoheterocycles as potential anti-Alzheimer’s agents. Bioorg. Chem. 83, 559–568. doi:10.1016/j.bioorg.2018.11.010
Shafferman, A., Kronman, C., Flashner, Y., Leitner, M., Grosfeld, H., Ordentlich, A., et al. (1992). Mutagenesis of human acetylcholinesterase. Identification of residues involved in catalytic activity and in polypeptide folding. J. Biol. Chem. 267, 17640–17648. doi:10.1016/s0021-9258(19)37091-7
Sharma, K. (2019). Cholinesterase inhibitors as Alzheimer's therapeutics (Review). Mol. Med. Rep. 20, 1479–1487.
Shen, H., Kihara, T., Hongo, H., Wu, X., Kem, W. R., Shimohama, S., et al. (2010). Neuroprotection by donepezil against glutamate excitotoxicity involves stimulation of α7 nicotinic receptors and internalization of NMDA receptors. Br. J. Pharmacol. 161, 127–139. doi:10.1111/j.1476-5381.2010.00894.x
Shi, D. H., Huang, W., Li, C., Wang, L. T., and Wang, S. F. (2013). Synthesis, biological evaluation and molecular modeling of aloe-emodin derivatives as new acetylcholinesterase inhibitors. Bioorg. Med. Chem. 21, 1064–1073. doi:10.1016/j.bmc.2013.01.015
Shuai, W., Li, W., Yin, Y., Yang, L., Xu, F., Xu, S., et al. (2019). Design, synthesis and molecular modeling of isothiochromanone derivatives as acetylcholinesterase inhibitors. Future Med. Chem. 11, 2687–2699. doi:10.4155/fmc-2019-0125
Stanciu, G. D., Luca, A., Rusu, R. N., Bild, V., Chiriac, S. I. B., Solcan, C., et al. (2020). Alzheimer’s disease pharmacotherapy in relation to cholinergic system involvement. Biomolecules 10, 40. doi:10.3390/biom10010040
Sugimoto, H., Ogura, H., Arai, Y., Iimura, Y., and Yamanishi, Y. (2002). Research and development of donepezil hydrochloride, a new type of acetylcholinesterase inhibitor. Jpn. J. Pharmacol. 89, 7–20. doi:10.1254/jjp.89.7
Taylor, P., and Radic, Z. (1994). The cholinesterases: from genes to proteins. Annu. Rev. Pharmacol. Toxicol. 34, 281–320. doi:10.1146/annurev.pa.34.040194.001433
Tonnies, E., and Trushina, E. (2017). Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 57, 1105–1121. doi:10.3233/jad-161088
Vafadarnejad, F., Mahdavi, M., Karimpour-Razkenari, E., Edraki, N., Sameem, B., Khanavi, M., et al. (2018). Design and synthesis of novel coumarin-pyridinium hybrids: In vitro cholinesterase inhibitory activity. Bioorg. Chem. 77, 311–319. doi:10.1016/j.bioorg.2018.01.013
Wake, R., Araki, T., Miyaoka, T., Nagahama, M., Furuya, M., Hayashida, M., et al. (2018). Long-term effects of combined treatment with memantine and donepezil on Alzheimer’s disease patients: 72-Week study. Neuropsychiatry (London) 8, 951–960. doi:10.4172/neuropsychiatry.1000421
Wang, C., Wua, Z., Cai, H., Xu, S., Liu, J., Jiang, J., et al. (2015). Design, synthesis, biological evaluation and docking study of 4-isochromanone hybrids bearing N-benzyl pyridinium moiety as dual binding site acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 25, 5212–5216. doi:10.1016/j.bmcl.2015.09.063
Wu, J., Ishikawa, M., Zhang, J., and Hashimoto, K. (2010). Brain imaging of nicotinic receptors in alzheimer's disease. Int. J. Alzheimer's. Dis. 2010, 1–11. doi:10.4061/2010/548913
Yu, L., Cao, R., Yi, W., Yan, Q., Chen, Z., Ma, L., et al. (2010). Synthesis and binding ability of 1, 2, 3-triazole-based triterpenoid receptors for recognition of Hg(2+) ion. Bioorg. Med. Chem. Lett. 20, 3254–3258. doi:10.1016/j.bmcl.2010.04.059
Yu, Q., Holloway, H. W., Luo, W., Lahiri, D. K., Brossi, A., and Greig, N. H. (2010). Long-acting anticholinesterases for myasthenia gravis: synthesis and activities of quaternary phenylcarbamates of neostigmine, pyridostigmine and physostigmine. Bioorg. Med. Chem. 18, 4687–4693. doi:10.1016/j.bmc.2010.05.022
Zarei, S., Shafiei, M., Firouzi, M., Firoozpour, L., Divsalar, K., Asadipour, A., et al. (2021). Design, synthesis and biological assessment of new 1 -benzyl-4-((4-oxoquinazolin-3(4H)-yl)methyl) pyridin-1-ium derivatives (BOPs) as potential dual inhibitors of acetylcholinesterase and butyrylcholinesterase. Heliyon 7, e06683. doi:10.1016/j.heliyon.2021.e06683
Keywords: Alzheimer’s disease, acetylcholinesterase, butyrylcholine esterase, donepezil, cholinesterase, benzylpyridinium salts
Citation: Sepehri S, Saeedi M, Larijani B and Mahdavi M (2022) Recent developments in the design and synthesis of benzylpyridinium salts: Mimicking donepezil hydrochloride in the treatment of Alzheimer’s disease. Front. Chem. 10:936240. doi: 10.3389/fchem.2022.936240
Received: 05 May 2022; Accepted: 06 July 2022;
Published: 26 September 2022.
Edited by:
Shah Alam Khan, National University of Science and Technology (Muscat), OmanReviewed by:
Asif Husain, Jamia Hamdard University, IndiaMohd Imran, Northern Border University, Saudi Arabia
Da-Hua Shi, Jiangsu Ocean University, China
Copyright © 2022 Sepehri, Saeedi, Larijani and Mahdavi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saghi Sepehri, c2VwZWhyaS5zYWdoaUB5YWhvby5jb20=; Mohammad Mahdavi, bW9tYWhkYXZpQHNpbmEudHVtcy5hYy5pcg==