 Aftab Hussain1*
Aftab Hussain1* Ahmad Irfan2
Ahmad Irfan2 Farah Kanwal1*Muhammad Afzal1Aijaz Rasool Chaudhry3Mohamed Hussien2
Farah Kanwal1*Muhammad Afzal1Aijaz Rasool Chaudhry3Mohamed Hussien2 Muhammad Arif Ali4
Muhammad Arif Ali4- 1School of Chemistry, University of the Punjab, Lahore, Pakistan
- 2Department of Chemistry, College of Science, King Khalid University, Abha, Saudi Arabia
- 3Department of Physics, College of Science, University of Bisha, Bisha, Saudi Arabia
- 4Institute of Chemistry, Baghdad Campus, The Islamia University of Bahawalpur, Bahawalpur, Pakistan
The violet-to-blue thermally activated delayed fluorescence (TADF) emitters were created employing several substituents based on 5,5-dimethyl-5,10-dihydropyrido [2,3-b][1,8] naphthyridine-diphenylsulphone (DMDHPN-DPS) called 1a via “CH/N” and “H/CN” substitutions at the diphenylsulphone acceptor (DPS) moiety. The parent compound 1a was selected from our former work after extensive research employing “CH/N” substitution on Dimethyl-acridine (DMAC) donor moiety. There is a little overlap amid the highest occupied molecular orbitals (HOMOs) and lowest un-occupied molecular orbitals (LUMOs) due to the distribution of HOMOs and LUMOs primarily on the DMDHPN donor and the DPS acceptor moieties, respectively. It resulted in a narrower energy gap (∆EST) between the lowest singlet (S1) and triplet (T1) excited state. In nearly all derivatives, the steric hindrance results in a larger torsional angle (85°–98°) between the plane of the DMDHPN and the DPS moieties. The predicted ΔEST values of the compounds with “H/CN” substitution were lower than those of the comparable “CH/N” substituents, demonstrating the superiority of the reversible inter-system crossing (RISC) from the T1 → S1 state. All derivatives have emission wavelengths (λem) in the range of 357–449 nm. The LUMO → HOMO transition energies in the S1 states are lowered by the presence of –CN groups or –N = atoms at the ortho or meta sites of a DPS acceptor unit, causing the λem values to red-shift. Furthermore, the λem showed a greater red-shift as there were more–CN groups or –N = atoms. Three of the derivatives named 1b, 1g, and 1h, emit violet (394 nm, 399 nm, and 398 nm, respectively), while two others, 1f and 1i, emit blue shade (449 nm each) with reasonable emission intensity peak demonstrating that these derivatives are effective violet-to-blue TADF nominees. The lower ΔEST value for derivative 1i (0.01 eV) with λem values of 449 nm make this molecule the finest choice for blue TADF emitter amongst all the studied derivatives. We believe our research might lead to the development of more proficient blue TADF-OLEDs in the future.
1 Introduction
Owing to their potential use as solid-state lightening sources and high-resolution flat panel display, organic light-emitting diodes (OLEDs) having multi-layer structures got a lot of interest since Tang and his colleagues initially described them (Tang and VanSlyke, 1987; Adachi et al., 2001; Zhang et al., 2014a; Chan et al., 2018; Liu et al., 2018; Wada et al., 2020). Because traditional fluorescent material can only harvest singlet excitons, OLEDs can only attain an internal quantum efficiency (IQE) of 25% (Shuai et al., 2000; Mehes et al., 2012; Sun et al., 2015). In contrast, noble heavy-metal-based phosphorescent materials may harvest both singlets as well as triplet excitons and hence can achieve an IQE of ≈ 100% through effective spin-orbit coupling interaction (SOC) (Adachi et al., 2001; Deaton et al., 2010; Zhang et al., 2012a; Xu et al., 2021). Noble metal-containing phosphorescent materials are, however, rather expensive and unreliable for blue emission. Therefore, a novel approach for obtaining high photoluminescence is required (Finkenzeller, 2008; Chi and Chou, 2010; Zhang et al., 2014a; Yu et al., 2022).
Adachi’s group, recently, has purported a novel triplet-harvesting mechanism named thermally activated delayed fluorescence (TADF) for highly proficient OLEDs as a workable alternative method with greater singlet yield (Endo et al., 2009; Mehes et al., 2012; Uoyama et al., 2012; Youn Lee et al., 2012; Li et al., 2020). For proficient TADF emitters, a narrower energy gap (∆EST) amid the lowest singlet excited state (S1) and lowest triplet excited state (T1) is essential for reversible inter-system crossing (RISC) from the T1 → S1 state (Berberan-Santos and Garcia, 1996; Wada et al., 2020). The ∆EST is associated with exchange energy (j) between the HOMOs and LUMOs in a molecule (Lu et al., 2015a). A tiny spatial overlap (ρ) amid HOMO-LUMO is believed to be an essential component (Lu et al., 2015b) for obtaining a low ∆EST value, and this is realized by joining the donor (D) and the acceptor (A) fragments in structures via steric hindrance, for instance, bulk, spirojunction, or homoconjugation (Kawasumi et al., 2015; Lu et al., 2016; Liu et al., 2020; Zhang et al., 2022).
Diphenylsulphone (DPS) unit is a versatile fragment with favorable features for TADF materials owing to a twist angle in the center and a high electron-accepting capacity (Huang et al., 2014; Tao et al., 2014; Bryden and Zysman-Colman, 2021). The sulfonyl group has an electron-withdrawing characteristic because of significant electronegativity of the oxygen atom in the group. It can also prevent compounds from undergoing π-conjugation because of its tetrahedral structure. High-performance TADF-OLEDs have been recognized by several sulfone-based compounds. As a result, in recent years, DPS has emerged as the most popular electron-accepting moiety for TADF emitters (Ye et al., 2013; Wu et al., 2014; Li et al., 2015; Liu et al., 2015). Several DPS-containing compounds with TADF properties have previously been described by Adachi’s group (Zhang et al., 2012b; Ye et al., 2013; Wu et al., 2014; Li et al., 2015; Liu et al., 2015; Cui et al., 2020). Dimethylacridine-Diphenylsulphone (DMAC-DPS), among all the previously described DPS-based emitters, has been revealed to be a powerful blue TADF emitter with emission wavelength (λem) of 460 nm in toluene and ∆EST (CT) value of 0.02 eV (Zhang et al., 2014a; Luo et al., 2016). In order to adjust the emission color, our group has published a chain of derivatives using H/R substitution on the D and A units (R = CH3 and CN) and “CH/N” substitution on the D-fragment (Wang et al., 2017a). It was discovered that modifying the D and A fragments with push-pull substituents is a useful technique for adjusting the λem, reducing the ∆EST, and enhancing the optical characteristics of designed molecules (Fan et al., 2016a; Lin et al., 2017a; Wang et al., 2017b; Yang et al., 2020; Jiao et al., 2021; Zhang et al., 2022). The “CH/N” as well as “H/CN” substitution on the acceptor unit, which results in bathochromically-shifted λem values and decreases ∆EST values, is a successful strategy, according to our analysis (Sun et al., 2008a; Sun et al., 2008b; Li et al., 2012).
In this contribution, we use the parent molecule DMDHPN-DPS (1a) as a starting point from our aforementioned report (Wang et al., 2017a). Then, by “CH/N” as well as “H/CN” substitution at meta and ortho sites of the DPS A-fragment, we were able to change the emission color to fashion blue TADF emitters. Figure 1 illustrates the structures of the parent molecule and all the symmetric substituted molecules. By computing ∆EST and λem values with the optimal Hartree-Fock percentage (OHF%) method in the exchange-correlation of the time-dependent density functional (TD-DFT) theory, we were able to analyse the TADF of these designed molecules.
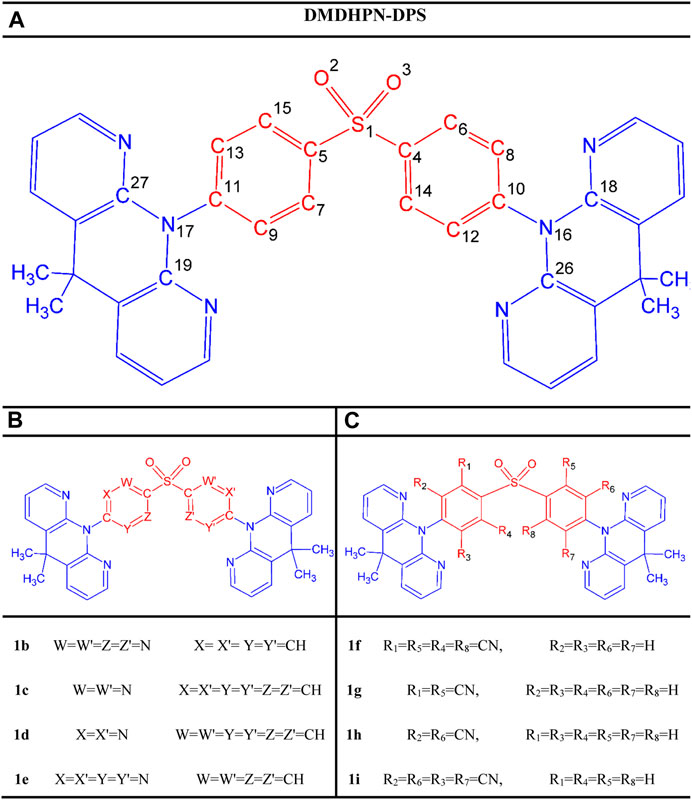
FIGURE 1. Scheme of the study showing the molecular structures of the parent molecule (A) and designed molecules (B,C) representing the CH/N and H/CN substituted derivatives, respectively.
2 Computational methodology
At first, the Gaussian-09 program was utilized to perform the ground state geometry optimization (S0) for all of the derivatives employing B3LYP/6-31G(d) method. In addition, vibrational frequency analysis was accomplished to validate the local minima, and they turned up no imaginary frequencies (Irfan et al., 2014; Gao et al., 2017a; Gao et al., 2017b; Liu et al., 2022). Following that, the charge-transfer index (q), which is a measure of electron density re-distribution within a molecule, was determined from D to A using the HOMO and LUMO distribution. Subsequently, to examine the orbital composition using the Multiwfn tool, the optimal HF% (OHF%) was calculated using the relationship OHF = 42q. Based on S0 geometry, the vertical absorption energies for singlet EVA (S1) as well as triplet EVA (T1) were computed using different functionals including M06-HF, M06-2X, BMK, MPW1B95, PBE0, and B3LYP having different HF% of 100%, 54%, 42%, 31%, 25%, and 20%, respectively, with 6-31G(d) basis set. The HF percentage (HF%) of various functionals is given in Supplementary Figure S1. Afterward, the best-fit straight line of the double log plots of EVA (S1, T1) against HF% was used to calculate the vertical excitation energy EVA (S1, OHF) as seen in Figure 2 and S1. Eventually, the ∆EST and zero-zero transition energies (E0-0) were predicted using the following proven formulae of Adachi et al. (Huang et al., 2013; Zhang et al., 2014b; Tian et al., 2016; Cui et al., 2020).
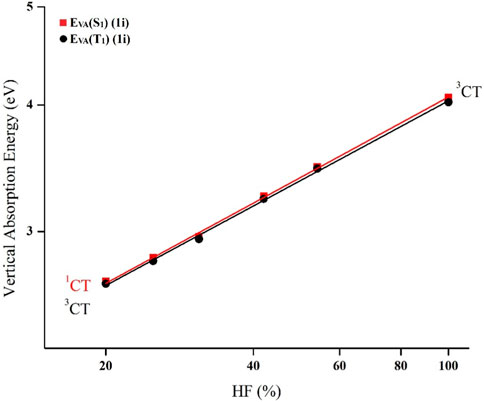
FIGURE 2. TD-DFT dependence of EVA (S1) as well as EVA (T1) on HF% in plotted on double log scale for the substituent 1i.
Here, ∆Estokes (energy loss during Stokes-shift) is about 0.09 eV, and ∆EV (difference in vibrational energy levels between the zero-zero transitions and the vertical transitions) is around 0.15 eV for the conjugated compounds. EVA (T1, B3LYP) depicts the vertical excitation energy for the triplet (T1) state calculated via B3LYP/6-31G(d) method. The C is the correction factor whose value for BMK, M06-2X, and, M06-HF functionals is about 1.10, 1.18, and 1.30, respectively. The S1 state geometries were subsequently optimized using functionals with an HF% near the OHF (%). The MPW1B95 functional was selected for S1 state optimization for all the studied derivatives as the OHF was determined to be closest to 31% as shown in Table 1. Then, using the TD-MPW1B95/6-31G(d) method with a polarizable continuum model (PCM) in the toluene medium, the absorption (λab) and emission (λem) wavelengths were determined based on optimized S0 and S1 state geometries respectively. The Gaussian-09 software is used for all calculations (Fan et al., 2016b; Cai et al., 2016; Liu et al., 2020). Software such as Multiwfn, PyMOlyze, Origin, Gaussview, and Gausssum were used for postprocessing the findings.
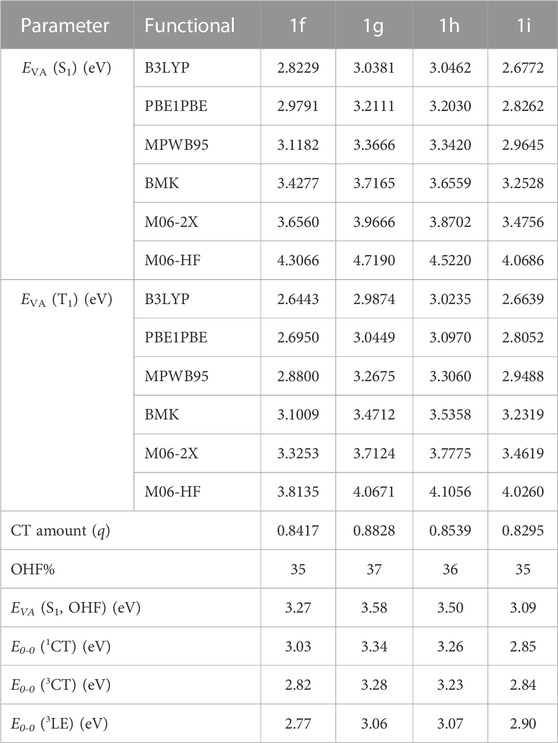
TABLE 1. Computed EVA (S1), EVA (T1), CT amount (q), OHF%, E0-0(1CT), E0-0(3CT), and E0-0 (3LE) employing several functionals and 6-31G(d) basis set based on B3LYP/6-31G(d) optimized geometries of the designed substituents 1f–1i. (1a–1e are shown in Supplementary Table S6).
3 Results and discussion
3.1 Optimized geometries at S0 and S1 states
Generally speaking, the photophysical characteristics of molecules with conjugated systems depend greatly on the dihedral angle and bond length. Stronger absorption, more effective emission, and improved fluorescence characteristics are frequently the results of optimal conjugation and planarity. Supplementary Table S1 shows the optimized geometrical parameters at S0 and S1 state of the parent and designed derivatives at B3LYP/6-31G(d) coupled with TD-MPW1B95/6-31G(d) level, employing DFT and TD-DFT, respectively. It is observed that the “CH/N” derivatives possess smaller C–N = bond lengths than the original C-C bond lengths at the pyrimidine/pyridine ring of the A-fragment because of the greater electronegativity of the nitrogen atom which draws the electron density towards itself and shortens the C–N = bond. In contrast, the C–C bond lengths in “H/CN” derivatives are longer than the prior C–C bond lengths because the –CN group will increase the electron-withdrawing strength of the A-moiety and elongates the C–C bond length inside the ring.
Bond lengths mostly alter on the substituent atom and nearby atom when compared to the original molecule 1a. For instance, C4–N14 and C4–N6 bond lengths of 1b, where C14 and C6 have been replaced by –N = atom, have decreased by 0.064 and 0.069 Å, respectively, in the S0 state and 0.071 and 0.070 Å in the S1 optimized state. Also, the lengths of the neighboring bonds C12–N14 and N6–C8 have lessened by 0.054 Å and 0.051 Å, respectively, in the S0 state and 0.072 Å in the S1 state. Conversely, for 1i, where –CN group has been used to replace the H-atom at C8 and C12, and the C8–C6, C10–C8, C12–C10, and C14–C12 bond lengths have risen by 0.065, 0.011, 0.010 and, 0.066 Å in S0 state and 0.076, 0.004, 0.005, and 0.076 Å in S1 state, respectively. The bond length values for C12–C14 and C6–C8 are decreased by 0.019 Å in S0 to S1 transition while increased by 0.003 Å for C8–C10 and C10–C12. The geometrical parameters compared in the S0 state and S1 states for the parent and designed molecules showed a bond lengths alteration of up to 0.038 Å in C–C and C–N = bonds and up to 0.067 Å in C–S bonds. For the C–S bonds, the bond lengths alteration in the S0 state and S1 state are more pronounced. All of the proposed compounds have shorter C–S bonds in the S1 (0.045–0.067 Å) than they do in the S0. But their neighboring N–C and C–C bond lengths increase in the range of (0.002–0.023 Å) in S1 than those in the S0 state with only a few exceptions. Hence, it is obvious that the “CH/N” derivatives possess smaller C–N = bond lengths while “H/CN” derivatives possess larger C–C bond lengths. In both types of derivatives, “CH/N” and “H/CN”, the conjugation is increased compared with parent molecule 1a, hence there is a red-shift in wavelength.
We know that the greater the value of the torsional/dihedral angle (β) between D and A fragments, the smaller will be the HOMO-LUMO overlap and the lower will be the value of △EST. It can be seen from Supplementary Table S1 that all the derivatives have large dihedral angle values (∼85°–98°) between the D and A fragments in the S0 state except 1b (70.2°) due to the greater steric hindrance and 1f (115.8°) due to the smaller steric hindrance. The larger β values for almost all the derivatives are optimistic for blocking the electrical interaction between D and A units (Zhang et al., 2014b; Lin et al., 2017b).
3.2 Frontier molecular orbitals
It is acknowledged that the frontier molecular orbitals (FMOs) offer crucial details on the nature of the TADF. The HOMO-LUMO electron density graphs at the S0 state are displayed in Figure 3. Using the below relation in the Multiwfn program, the overlap between HOMO and LUMO (ρ) was calculated using.
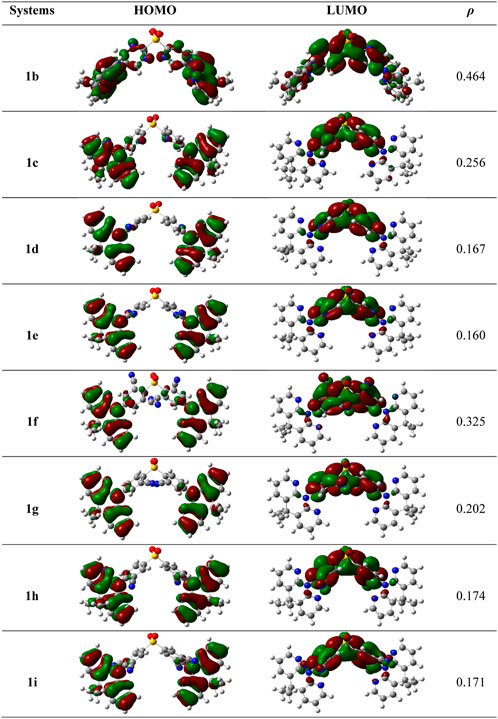
FIGURE 3. The electron density diagrams of HOMOs and LUMOs and the overlap between them in whole space for these studies compounds in S0 geometry. The isosurface value for FMOs is 0.02.
As is obvious from Figure 3, the HOMOs are particularly localized on the DMDHPN donor fragment while the LUMOs are largely distributed on the DPS acceptor moiety. Additionally, we found that the ρ values are ranging from 0.160 to 0.256, showing an ease of charge transfer between HOMO and LUMO, and a low electron exchange energy (j), which makes the △EST very modest except 1f (0.325) and 1b (0.464). Because the LUMO is mainly found on the A-unit of substituted derivatives, more substantial changes can be found in LUMO energy levels than HOMO energy levels, as seen in Figure 4. The energetic gaps (ΔEH-L) between HOMO and LUMO are in the range of 3.84–4.69 eV in the S1 state and 3.40–4.45 eV in the S0 state.
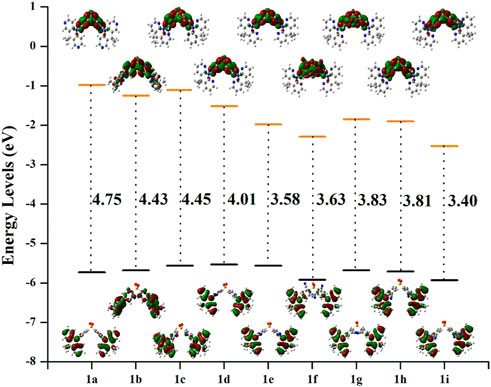
FIGURE 4. The electron density diagrams and HOMO-LUMO energetic gap of the studies molecules in the S0 state. The isosurface value for FMOs is 0.02.
When –N = atom or –CN groups are added to A-fragments, the values of ΔEH-L are lower than they would be for the parent molecule because the enhanced electron-accepting capacity of the DPS acceptors lowers the LUMO energy level, as shown in Supplementary Table S2. However, the LUMO energy levels are reduced more effectively by the –CN group than by the –N = atom, which reduces the ΔEH-L.
Additionally, the shift in ∆EH–L values is more apparent as the greater number of –CN groups or –N = atoms there are. Given that the λab and λem values are connected to the ∆EH–L values, (Li et al., 2012), the import of –CN groups or of –N = atoms to the A-fragments may alter the absorption and emission spectra, as illustrated in Supplementary Tables S4, S5. As a result, it is expected that the newly created derivatives would exhibit red-shifted λem when compared to the parent molecule 1a.
3.3 Singlet-triplet energy gap
Using the Multiwfn program, the E0-0 (S1), E0-0 (3CT), as well as E0-0 (3LE) are attained based on the computed CT amount (q) (Table 1). At first, double log plots of EVA (S1, T1) versus HF% for the compounds under investigation were plotted (Figure 2). Then the E0-0 (S1), E0-0 (3CT), as well as E0-0 (3LE) of these designed derivatives were computed using Eqs 1–3 as shown in Table 3. The calculated findings for the substituted molecules show that the addition of –CN groups or of –N = atoms on the A-moiety improves its capacity to pull electrons, which eventually leads to a decrease in the ∆EST value. Additionally, it is clear that as there are more –CN groups or of –N = atoms, the ∆EST values decrease, particularly at meta-position. It is also obvious that the “CH/N” derivatives have lower ΔEST values than the original molecule but are larger than the “H/CN” derivatives. When the lowest T1 state is taken into account, the computed E0-0 (3CT), as well as E0-0 (3LE), appear to have the lowest energy levels. The 1CT–3CT splitting is denoted by ∆EST (CT), while the energy difference amid the lowest T1 and the S1 state is denoted by ΔES1-T1 (Huang et al., 2013). All the newly designed molecules are symmetric substitution derivatives and have lower ΔEST values as compared with a parent molecule. According to Table 2, for 1i, the T1 state is 3CT in nature, which is ideal for an effectual RISC, whereas, for others, T1 states are 3LE in nature. The calculated ∆EST (CT) value using the OHF method for 1i is 0.01 eV resulting from the small ρ value. The 1i has the smallest ∆EST value compared with all the other substituted derivatives because 1) it is a “H/CN” substituted derivative which is more effective than “CH/N” substituted derivatives 2) two H-atoms have been replaced with two CN-groups which will further enhance the electron-withdrawing strength of the A-fragment 3) it is meta-substituted and is at a greater distance from sulphonyl group. Even though some other designed molecules, including the selected violet-blue to blue derivates like 1b, 1f, 1g, and 1h, possess a lower 3LE state, the effective TADF is made feasible via reversible internal conversion from 3LE to 3CT state followed by a subsequent RISC from 3CT to 1CT state. So, by enhancing the electron-withdrawing capacity of A, it is beneficial to raise the 3LE state and lower the ∆EST (Hussain et al., 2019; Hassan et al., 2022; Hussain et al., 2023).
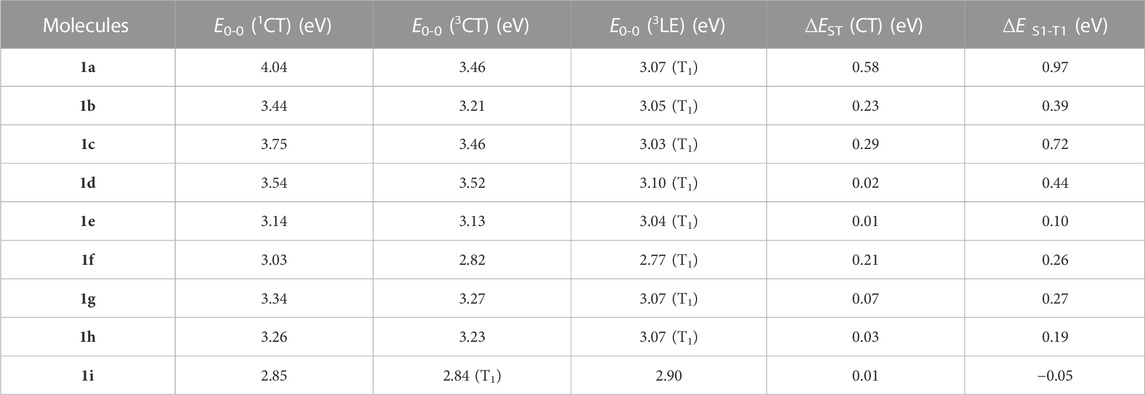
TABLE 2. Computed ∆EST (CT) and ΔES1-T1 for the studies molecules.
3.4 Photophysical properties
Figure 5 displays the emission and absorption spectra of the parent molecule and all derivatives. Table 3 lists the estimated emission and absorption wavelengths (λem and λab) in the toluene medium at the TD-MPW1B95/6-31G(d) theory level. All the investigation molecules have λem and λab values corresponding to the values of ΔEH-L. The designed derivatives show that the λem and λab values range from 357 to 449 nm and 321–418 nm, respectively. The λem bands instigate from S1 → S0 transitions which is mainly the transference of the electrons from LUMO → HOMO, and the electronic shifts are π* → π type. The addition of –CN group or –N = atom to DPS acceptor moiety has resulted in the bathochromically-shifted values for λem and λab. Furthermore, by increasing the numbers of –CN groups or –N = atoms, the λem, and λab values display a more significant red-shift. Particularly, the derivatives 1f and 1i, display a more noticeable red-shift in λem of about 97 nm each. Additionally, the meta-positioned derivatives show more red-shift in λem and λab values than ortho-positions with the same number of –CN groups or of –N = atoms. Actually, at meta-position, the –CN group or –N = atom is at a larger distance from the sulphonyl group and experiences less steric hindrance compared to the ortho position which experiences more steric repulsion. It will increase the stability and charge transfer characters of the substituted derivatives which results in lowering the ΔEST and increasing the λem value. It is evident from the calculated results that the three molecules named 1b, 1g, and 1h are showing violet-blue emission (394, 399, and 398 nm) and two of them named 1f and 1i are showing blue emission of 449 nm with reasonable emission intensity peak demonstrating that the studied compounds are effective violet-blue to blue TADF materials. We can see that both 1f and 1i have the same value of emission wavelength (λem) of 449 nm lying in the pure blue region but 1i is regarded as a comparatively better TADF candidate as it has a lower ΔEST 0.01 eV value as compared to 1f (0.26 eV). The constructed molecules demonstrated that the sequence of λab, as well as λem, is coherent with the propensity of the electron-withdrawing strength of A-fragment.
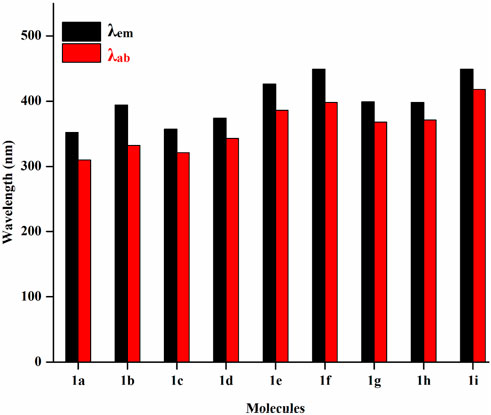
FIGURE 5. Absorption and emission spectrum of the studied molecules.
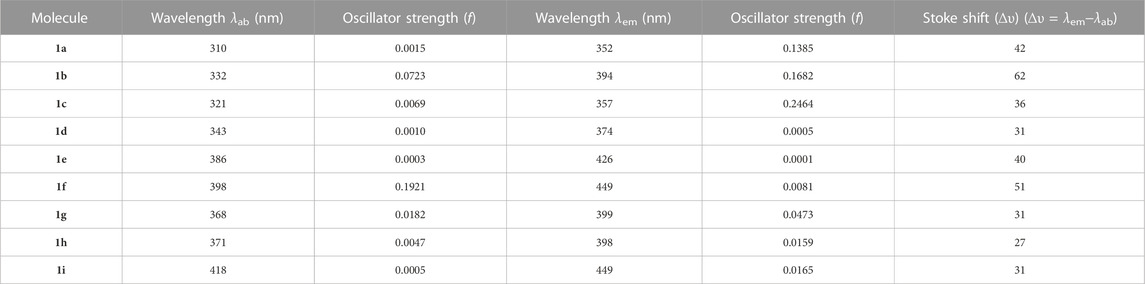
TABLE 3. Calculated λab and λem values in toluene for the studied molecules employing TD-MPW1B95/6-31G(d) method based on S0 and S1 state optimized geometries, respectively, along with Stoke Shift (∆υ) values.
The energy (or wavelength) differential between a molecule’s absorption (λab) and emission (λem) maxima is called as the Stokes-shift (∆υ). It represents the energy loss occurring during the S1 → S0 transition. In general, a smaller Stokes-shift is advantageous since it decreases the spectral overlap between excitation and emission signals. Since self-absorption is reduced by a narrow Stokes-shift, more of the absorbed energy is transformed into light output, enhancing OLEDs’ total energy efficiency. Every constructed molecule, except 1b and 1f, exhibits a smaller stokes-shift than the original molecule as shown in Table 3. It is also obvious that the “H/CN” substituents exhibit a relatively smaller value compared with “CH/N” derivatives hence more effective in reducing the energy loss during the S1 → S0 transition.
4 Conclusion
In conclusion, we have projected the photophysical as well as electronic characteristics for a variety of freshly created substituents to improve the efficacies for blue TADF emitters based on the parent system DMDHPN-DPS. The HOMOs are primarily concentrated on the DMDHPN-donor moiety, whereas the LUMOs are located at the DPS-acceptor unit. The λem of all the designed derivatives ranges from 357 to 449 nm. The import of –CN groups or of –N = atoms on DPS moiety lowers the transition energy from LUMO to HOMO, causing a red-shift in λem. Additionally, the red-shift in λem values becomes more significant by increasing the quantity of –CN groups or of –N = atoms. The derivatives 1f and 1i display a more evident red-shift of 97 nm. Additionally, meta-position substituents exhibit greater red-shifted λem values than ortho-position substituents with the same number of –CN groups or of –N = atoms. Actually, at meta-position, the –CN group or –N = atom is at a larger distance from the sulphonyl group and experiences less steric hindrance compared to the ortho position which experiences more steric repulsion. It will increase the stability and charge transfer characters of the substituted derivatives which result in lowering the ΔEST and increasing the λem value. The small ρ values for the designed molecules due to the greater β values of 85°–98° contribute to the realization of the charge transfer state and small ΔEST. The estimated findings showed that, out of all the analyzed molecules, three violet-blue emitters (394–399 nm) and two blue emitters (449 nm) with reasonable emission intensity and smaller ΔEST are favorable to be effective violet-blue to blue TADF emitters with 1i being the best TADF candidate as it has smallest ΔEST values of 0.01 eV. We believe that in the future, understanding and building effective violet-blue to blue TADF-based OLEDs will be made easier with the aid of our theoretical designs.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
AH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing–original draft, Writing–review and editing. AI: Formal Analysis, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing–original draft. FK: Conceptualization, Formal Analysis, Investigation, Methodology, Supervision, Writing–original draft. MAf: Investigation, Methodology, Writing–review and editing. AC: Investigation, Methodology, Software, Writing–review and editing. MH: Formal analysis, Investigation, Methodology, Writing–review and editing. MAl: Formal Analysis, Investigation, Writing–review and editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
AI extends his appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through Large Groups Project under grant number (RGP2/276/44). AC is thankful to the Deanship of Scientific Research at the University of Bisha, for supporting this work through the Fast-Track Research Support Program. We are also acknowledging the school of Chemistry, University of the Punjab, Lahore, Pakistan for providing research facilities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MQ declared a past co-authorship with the author AI to the handling editor.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1279355/full#supplementary-material
References
Adachi, C., Baldo, M. A., Thompson, M. E., and Forrest, S. R. (2001). Nearly 100% internal phosphorescence efficiency in an organic light-emitting device. J. Appl. Phys. 90 (10), 5048–5051. doi:10.1063/1.1409582
Berberan-Santos, M. N., and Garcia, J. M. M. (1996). Unusually strong delayed fluorescence of C70. J. Am. Chem. Soc. 118 (39), 9391–9394. doi:10.1021/ja961782s
Bryden, M. A., and Zysman-Colman, E. (2021). Organic thermally activated delayed fluorescence (TADF) compounds used in photocatalysis. Chem. Soc. Rev. 50 (13), 7587–7680. doi:10.1039/d1cs00198a
Cai, X., Li, X., Xie, G., He, Z., Gao, K., Liu, K., et al. (2016). Rate-limited effect of reverse intersystem crossing process: the key for tuning thermally activated delayed fluorescence lifetime and efficiency roll-off of organic light emitting diodes. Chem. Sci. 7 (7), 4264–4275. doi:10.1039/c6sc00542j
Chan, C.-Y., Cui, L.-S., Kim, J. U., Nakanotani, H., and Adachi, C. (2018). Rational molecular design for deep-blue thermally activated delayed fluorescence emitters. Adv. Funct. Mater. 28 (11), 1706023. doi:10.1002/adfm.201706023
Chi, Y., and Chou, P. T. (2010). Transition-metal phosphors with cyclometalating ligands: fundamentals and applications. Chem. Soc. Rev. 39 (2), 638–655. doi:10.1039/b916237b
Cui, L.-S., Gillett, A. J., Zhang, S.-F., Ye, H., Liu, Y., Chen, X.-K., et al. (2020). Fast spin-flip enables efficient and stable organic electroluminescence from charge-transfer states. Nat. Photonics 14 (10), 636–642. doi:10.1038/s41566-020-0668-z
Deaton, J. C., Switalski, S. C., Kondakov, D. Y., Young, R. H., Pawlik, T. D., Giesen, D. J., et al. (2010). E-type delayed fluorescence of a phosphine-supported Cu2(μ-NAr2)2 diamond core: harvesting singlet and triplet excitons in OLEDs. J. Am. Chem. Soc. 132 (27), 9499–9508. doi:10.1021/ja1004575
Endo, A., Ogasawara, M., Takahashi, A., Yokoyama, D., Kato, Y., and Adachi, C. (2009). Thermally activated delayed fluorescence from Sn(4+)-porphyrin complexes and their application to organic light emitting diodes--a novel mechanism for electroluminescence. Adv. Mater. 21 (47), 4802–4806. doi:10.1002/adma.200900983
Fan, J., Lin, L., and Wang, C. (2016a). Modulating excited state properties of thermally activated delayed fluorescence molecules by tuning the connecting pattern. Theor. Chem. Accounts 135 (7), 169. doi:10.1007/s00214-016-1928-3
Fan, J.-z., Lin, L.-l., and Wang, C.-k. (2016b). Decreasing the singlet–triplet gap for thermally activated delayed fluorescence molecules by structural modification on the donor fragment: first-principles study. Chem. Phys. Lett. 652, 16–21. doi:10.1016/j.cplett.2016.04.027
Finkenzeller, H. Y. A. W. J. (2008). Highly efficient OLEDs with phosphorescent materials. Weinheim: Wiley VCH: KGaA, 1–97.
Gao, Y., Geng, Y., Wu, Y., Zhang, M., and Su, Z.-M. (2017a). Investigation on the effect of connected bridge on thermally activated delayed fluorescence property for DCBPy emitter. Dyes Pigments 145, 277–284. doi:10.1016/j.dyepig.2017.04.001
Gao, Y., Su, T., Zhao, L., Geng, Y., Wu, Y., Zhang, M., et al. (2017b). How does a little difference in structure determine whether molecules have thermally activated delayed fluorescence characteristic or not? Org. Electron. 50, 70–76. doi:10.1016/j.orgel.2017.07.024
Hassan, M., Baig, M. M., Shah, K. H., Hussain, A., Hassan, S. A., and Ali, A. (2022). MOF-based bimetallic diselenide nanospheres as a bifunctional efficient electrocatalysts for overall water splitting. J. Phys. Chem. Solids 167, 110780. doi:10.1016/j.jpcs.2022.110780
Huang, B., Qi, Q., Jiang, W., Tang, J., Liu, Y., Fan, W., et al. (2014). Thermally activated delayed fluorescence materials based on 3,6-di-tert-butyl-9-((phenylsulfonyl)phenyl)-9H-carbazoles. Dyes Pigments 111, 135–144. doi:10.1016/j.dyepig.2014.06.008
Huang, S., Zhang, Q., Shiota, Y., Nakagawa, T., Kuwabara, K., Yoshizawa, K., et al. (2013). Computational prediction for singlet- and triplet-transition energies of charge-transfer compounds. J. Chem. Theory Comput. 9 (9), 3872–3877. doi:10.1021/ct400415r
Hussain, A., Kanwal, F., Irfan, A., Hassan, M., and Zhang, J. (2023). Exploring the influence of engineering the linker between the donor and acceptor fragments on thermally activated delayed fluorescence characteristics. ACS Omega 8, 15638–15649. doi:10.1021/acsomega.3c01098
Hussain, A., Yuan, H., Li, W., and Zhang, J. (2019). Theoretical investigations of the realization of sky-blue to blue TADF materials via CH/N and H/CN substitution at the diphenylsulphone acceptor. J. Saudi Chem. Soc. 7 (22), 6685–6691. doi:10.1039/c9tc01449g
Irfan, A., Aftab, H., and Al-Sehemi, A. G. (2014). Push–pull effect on the geometries, electronic and optical properties of thiophene based dye-sensitized solar cell materials. J. Saudi Chem. Soc. 18 (6), 914–919. doi:10.1016/j.jscs.2011.11.013
Jiao, Y., Đorđević, L., Mao, H., Young, R. M., Jaynes, T., Chen, H., et al. (2021). A donor–acceptor [2] catenane for visible light photocatalysis. J. Am. Chem. Soc. 143 (21), 8000–8010. doi:10.1021/jacs.1c01493
Kawasumi, K., Wu, T., Zhu, T., Chae, H. S., Van Voorhis, T., Baldo, M. A., et al. (2015). Thermally activated delayed fluorescence materials based on homoconjugation effect of donor-acceptor triptycenes. J. Am. Chem. Soc. 137 (37), 11908–11911. doi:10.1021/jacs.5b07932
Li, W., Li, Z., Si, C., Wong, M. Y., Jinnai, K., Gupta, A. K., et al. (2020). Organic long-persistent luminescence from a thermally activated delayed fluorescence compound. Adv. Mater. 32 (45), 2003911. doi:10.1002/adma.202003911
Li, Y., Wang, Z., Li, X., Xie, G., Chen, D., Wang, Y.-F., et al. (2015). Highly efficient spiro[fluorene-9,9′-thioxanthene] core derived blue emitters and fluorescent/phosphorescent hybrid white organic light-emitting diodes. Chem. Mater. 27 (3), 1100–1109. doi:10.1021/cm504441v
Li, Y., Zou, L.-Y., Ren, A.-M., and Feng, J.-K. (2012). Theoretical study on the electronic structures and photophysical properties of a series of dithienylbenzothiazole derivatives. Comput. Theor. Chem. 981, 14–24. doi:10.1016/j.comptc.2011.11.021
Lin, L., Fan, J., and Wang, C.-K. (2017b). Theoretical perspective for internal quantum efficiency of thermally activated delayed fluorescence emitter in solid phase: a QM/MM study. Org. Electron. 51, 349–356. doi:10.1016/j.orgel.2017.09.021
Lin, L., Wang, Z., Fan, J., and Wang, C. (2017a). Theoretical insights on the electroluminescent mechanism of thermally activated delayed fluorescence emitters. Org. Electron. 41, 17–25. doi:10.1016/j.orgel.2016.11.035
Liu, M., Seino, Y., Chen, D., Inomata, S., Su, S. J., Sasabe, H., et al. (2015). Blue thermally activated delayed fluorescence materials based on bis(phenylsulfonyl)benzene derivatives. Chem. Commun. 51 (91), 16353–16356. doi:10.1039/c5cc05435d
Liu, T., Deng, C., Duan, K., Tsuboi, T., Niu, S., Wang, D., et al. (2022). Zero–zero energy-dominated degradation in blue organic light-emitting diodes employing thermally activated delayed fluorescence. ACS Appl. Material Interfaces 14 (19), 22332–22340. doi:10.1021/acsami.2c02623
Liu, Y., Wei, X., Li, Z., Liu, J., Wang, R., Hu, X., et al. (2018). Highly efficient, solution-processed organic light-emitting diodes based on thermally activated delayed-fluorescence emitter with a mixed polymer interlayer. ACS Appl. Energy Mater. 1 (2), 543–551. doi:10.1021/acsaem.7b00131
Liu, Y., Yuan, Y., Tian, X., and Sun, J. (2020). Theoretical investigation on reverse intersystem crossing from upper triplet to lowest singlet: a “hot exciton” path for blue fluorescent OLEDs. Int. J. Quantum Chem. 120 (23), e26399. doi:10.1002/qua.26399
Lu, J., Zheng, Y., and Zhang, J. (2015a). Tuning the color of thermally activated delayed fluorescent properties for spiro-acridine derivatives by structural modification of the acceptor fragment: a DFT study. RSC Adv. 5 (24), 18588–18592. doi:10.1039/c4ra15155k
Lu, J., Zheng, Y., and Zhang, J. (2015b). Rational design of phenoxazine-based donor-acceptor-donor thermally activated delayed fluorescent molecules with high performance. Phys. Chem. Chem. Phys. 17 (30), 20014–20020. doi:10.1039/c5cp02810h
Lu, J., Zheng, Y., and Zhang, J. (2016). Computational design of benzo [1,2-b:4,5-b′] dithiophene based thermally activated delayed fluorescent materials. Dyes Pigments 127, 189–196. doi:10.1016/j.dyepig.2015.12.030
Luo, J., Gong, S., Gu, Y., Chen, T., Li, Y., Zhong, C., et al. (2016). Multi-carbazole encapsulation as a simple strategy for the construction of solution-processed, non-doped thermally activated delayed fluorescence emitters. J. Mater. Chem. C 4 (13), 2442–2446. doi:10.1039/c6tc00418k
Mehes, G., Nomura, H., Zhang, Q., Nakagawa, T., and Adachi, C. (2012). Enhanced electroluminescence efficiency in a spiro-acridine derivative through thermally activated delayed fluorescence. Angew. Chem. Int. Ed. Engl. 51 (45), 11311–11315. doi:10.1002/anie.201206289
Shuai, Z., Beljonne, D., Silbey, R. J., and Brédas, J. L. (2000). Singlet and triplet exciton formation rates in conjugated polymer light-emitting diodes. Phys. Rev. Lett. 84 (1), 131–134. doi:10.1103/physrevlett.84.131
Sun, H., Zhong, C., and Bredas, J. L. (2015). Reliable prediction with tuned range-separated functionals of the singlet-triplet gap in organic emitters for thermally activated delayed fluorescence. J. Chem. Theory Comput. 11 (8), 3851–3858. doi:10.1021/acs.jctc.5b00431
Sun, M., Niu, B., and Zhang, J. (2008a). Computational study on optical and electronic properties of the “CH”/N substituted emitting materials based on spirosilabifluorene derivatives. J. Mol. Struct. THEOCHEM 862 (1-3), 85–91. doi:10.1016/j.theochem.2008.04.038
Sun, M., Niu, B., and Zhang, J. (2008b). Theoretical design of blue emitting materials based on symmetric and asymmetric spirosilabifluorene derivatives. Theor. Chem. Accounts 119 (5-6), 489–500. doi:10.1007/s00214-008-0410-2
Tang, C. W., and VanSlyke, S. A. (1987). Organic electroluminescent diodes. Appl. Phys. Lett. 51 (12), 913–915. doi:10.1063/1.98799
Tao, Y., Yuan, K., Chen, T., Xu, P., Li, H., Chen, R., et al. (2014). Thermally activated delayed fluorescence materials towards the breakthrough of organoelectronics. Adv. Mater. 26 (47), 7931–7958. doi:10.1002/adma.201402532
Tian, X., Sun, H., Zhang, Q., and Adachi, C. (2016). Theoretical predication for transition energies of thermally activated delayed fluorescence molecules. Chin. Chem. Lett. 27 (8), 1445–1452. doi:10.1016/j.cclet.2016.07.017
Uoyama, H., Goushi, K., Shizu, K., Nomura, H., and Adachi, C. (2012). Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492 (7428), 234–238. doi:10.1038/nature11687
Wada, Y., Nakagawa, H., Matsumoto, S., Wakisaka, Y., and Kaji, H. (2020). Organic light emitters exhibiting very fast reverse intersystem crossing. Nat. Photonics 14 (10), 643–649. doi:10.1038/s41566-020-0667-0
Wang, J., Lu, J., and Zhang, J. (2017a). Tuning the electronic and optical properties of diphenylsulphone based thermally activated delayed fluorescent materials via structural modification: a theoretical study. Dyes Pigments 143, 42–47. doi:10.1016/j.dyepig.2017.03.064
Wang, L., Li, T., Feng, P., and Song, Y. (2017b). Theoretical tuning of the singlet-triplet energy gap to achieve efficient long-wavelength thermally activated delayed fluorescence emitters: the impact of substituents. Phys. Chem. Chem. Phys. 19 (32), 21639–21647. doi:10.1039/c7cp02615c
Wu, S., Aonuma, M., Zhang, Q., Huang, S., Nakagawa, T., Kuwabara, K., et al. (2014). High-efficiency deep-blue organic light-emitting diodes based on a thermally activated delayed fluorescence emitter. J. Mater. Chem. C 2 (3), 421–424. doi:10.1039/c3tc31936a
Xu, Y., Wang, Q., Cai, X., Li, C., and Wang, Y. (2021). Highly efficient electroluminescence from narrowband green circularly polarized multiple resonance thermally activated delayed fluorescence enantiomers. Adv. Mater. 33 (21), 2100652. doi:10.1002/adma.202100652
Yang, S.-Y., Wang, Y.-K., Peng, C.-C., Wu, Z.-G., Yuan, S., Yu, Y.-J., et al. (2020). Circularly polarized thermally activated delayed fluorescence emitters in through-space charge transfer on asymmetric spiro skeletons. J. Am. Chem. Soc. 142 (41), 17756–17765. doi:10.1021/jacs.0c08980
Ye, J., Chen, Z., Fung, M.-K., Zheng, C., Ou, X., Zhang, X., et al. (2013). Carbazole/sulfone hybrid D-π-A-structured bipolar fluorophores for high-efficiency blue-violet electroluminescence. Chem. Mater. 25 (13), 2630–2637. doi:10.1021/cm400945h
Youn Lee, S., Yasuda, T., Nomura, H., and Adachi, C. (2012). High-efficiency organic light-emitting diodes utilizing thermally activated delayed fluorescence from triazine-based donor–acceptor hybrid molecules. Appl. Phys. Lett. 101 (9), 093306. doi:10.1063/1.4749285
Yu, R., Song, Y., Zhang, K., Pang, X., Tian, M., and He, L. (2022). Intrinsically ionic, thermally activated delayed fluorescent materials for efficient, bright, and stable light-emitting electrochemical cells. Adv. Funct. Mater. 32 (13), 2110623. doi:10.1002/adfm.202110623
Zhang, Q., Komino, T., Huang, S., Matsunami, S., Goushi, K., and Adachi, C. (2012a). Triplet exciton confinement in green organic light-emitting diodes containing luminescent charge-transfer Cu(I) complexes. Adv. Funct. Mater. 22 (11), 2327–2336. doi:10.1002/adfm.201101907
Zhang, Q., Kuwabara, H., Potscavage, W. J., Huang, S., Hatae, Y., Shibata, T., et al. (2014b). Anthraquinone-based intramolecular charge-transfer compounds: computational molecular design, thermally activated delayed fluorescence, and highly efficient red electroluminescence. J. Am. Chem. Soc. 136 (52), 18070–18081. doi:10.1021/ja510144h
Zhang, Q., Li, B., Huang, S., Nomura, H., Tanaka, H., and Adachi, C. (2014a). Efficient blue organic light-emitting diodes employing thermally activated delayed fluorescence. Nat. Photonics 8 (4), 326–332. doi:10.1038/nphoton.2014.12
Zhang, Q., Li, J., Shizu, K., Huang, S., Hirata, S., Miyazaki, H., et al. (2012b). Design of efficient thermally activated delayed fluorescence materials for pure blue organic light emitting diodes. J. Am. Chem. Soc. 134 (36), 14706–14709. doi:10.1021/ja306538w
Keywords: diphenylsulphone (DPS), thermally activated delayed fluorescence (TADF), singlet-triplet energy gap (ΔEST), organic light emitting diode (OLED), reverse intersystem crossing (RISC), DFT, TD-DFT, HOMO-LUMO
Citation: Hussain A, Irfan A, Kanwal F, Afzal M, Chaudhry AR, Hussien M and Ali MA (2023) Exploration of violet-to-blue thermally activated delayed fluorescence emitters based on “CH/N” and “H/CN” substitutions at diphenylsulphone acceptor. A DFT study. Front. Chem. 11:1279355. doi: 10.3389/fchem.2023.1279355
Received: 17 August 2023; Accepted: 20 October 2023;
Published: 09 November 2023.
Edited by:
Shamsa Bibi, University of Agriculture, Faisalabad, PakistanReviewed by:
Anex Jose, Stanford University, United StatesMuhammad Abdul Qayyum, University of Education Lahore, Pakistan
Copyright © 2023 Hussain, Irfan, Kanwal, Afzal, Chaudhry, Hussien and Ali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aftab Hussain, YWZ0YWIuY2hlbUBwdS5lZHUucGs=; Farah Kanwal, ZmFyYWhrY2hlbUB5YWhvby5jb20=