 Alkhair Adam Khalil Mohamed
Alkhair Adam Khalil Mohamed Isaac Asiamah
Isaac Asiamah Ghazi Elamin3
Ghazi Elamin3 James Darkwa
James Darkwa Christian K. Adokoh
Christian K. Adokoh- 1Department of Biomedical Sciences, School of Allied Health Sciences, College of Health and Allied Sciences, University of Cape Coast, Cape Coast, Ghana
- 2Department of Chemistry, School of Physical Sciences, College of Agriculture and Natural Sciences, University of Cape Coast, Cape Coast, Ghana
- 3Department of Pharmaceutical Chemistry, College of Pharmacy, Karary University, Khartoum, Sudan
- 4Department of Chemical Sciences, Faculty of Science, University of Johannesburg, Johannesburg, South Africa
- 5Department of Forensic Science, School of Biological Sciences, College of Agriculture and Natural Sciences University of Cape Coast, Cape Coast, Ghana
Introduction:: This study employed in silico methods to investigate the anticancer potential and mechanisms of twenty novel phosphinogold(I) thiocarbohydrate complexes.
Methods:: Molecular docking and Prime MM-GBSA screening of seventeen cancer-related protein targets, including Human Double Minute 2 protein (HDM2), DNA methyltransferase-1 (DNMT1), Protein Kinase B (AKT2), and Poly (ADP-ribose) polymerase 1 (PARP-1), were conducted. Molecular dynamics simulations were performed for complex 9.
Results:: Virtual screening revealed strong binding affinities for several complexes, often surpassing native ligands. All the complexes except 16, 18, and 19 exhibited strong binding affinity with one or two cancer protein targets compared to native ligands. Complex 9 emerged as the best candidate, demonstrating promising binding affinity particularly against AKT2 (–82.40 kcal/mol) and PARP-1 (–75.7 kcal/mol). Molecular dynamics simulations of complex 9 with PARP-1 and AKT2 revealed distinct binding profiles, with a more stable interaction with PARP-1, suggesting its potential for disrupting DNA repair mechanisms. Binuclear complexes generally exhibited higher affinities than mononuclear counterparts, particularly for DNMT1 and HDM2. Complex 13 demonstrated high in vitro activity against prostate, colon, and breast cancer cell lines (IC50 = 0.03, 0.25, and 0.07 μM respectively), collaborating with a significant interaction with Human Epidermal Growth Factor Receptor 2 (HER2) (–71.15 kcal/mol binding affinity) in silico. While acetylation decreased binding affinity; it enhanced cellular activity as reported in in vitro studies indicative of the need to balance lipophilicity and binding strength in future ligand design.
Discussion:: These findings provide valuable insights into multi-target anticancer mechanisms, with a particular emphasis on complex 9 as a potential PARP-1 inhibitor, and guide future optimization and experimental validation of these novel gold-based complexes. The stable interaction of complex 9 with PARP-1 highlights PARP-1 as a particularly promising therapeutic target. Binuclear complexes' superior affinities for DNMT1 and HDM2 suggest structural advantages for multi-target inhibition.
Conclusion:: The paradoxical effect of acetylation underscores the importance of balancing lipophilicity and binding strength in ligand design.
Introduction
The exploration of gold-based compounds for therapeutic applications has a rich history, dating back to ancient civilizations where gold was utilized for treating various ailments. Recent advancements have reignited interest in these compounds, particularly due to their potential anticancer properties. However, the clinical application of gold complexes is often limited by the toxicity of their ligands and their biocompatibility issues (Biebuyck et al., 1994).
In a previous study, we synthesized and characterized novel phosphinogold(I) thiocarbohydrate complexes, which were designed to overcome the limitations associated with traditional gold complexes (Adokoh et al., 2017). The choice of targeting these phosphinogold(I) thiocarbohydrate complexes is as result of their promising cancer treatment due to their potent cytotoxicity, ability to induce apoptosis through mitochondrial pathways, the tunable nature of their phosphine ligands to enhance anticancer activity and selective targeting characteristics of thiocarbohydrate ligands. By combining gold(I) with phosphine and thiocarbohydrate ligands, the complex can leverage multiple mechanisms: enzyme inhibition, oxidative stress induction, and selective targeting of cancer cells, thus, a potential for synergistic effects is expected (Zarewa et al., 2023; Keter et al., 2014). These new compounds were synthesized through the reaction of n-gluconamidoalkyl thiols with various gold precursors, resulting in a series of complexes that exhibit promising anticancer activities against different cancer cell lines, including breast and prostate cancer (Adokoh et al., 2017). The anticancer evaluation of these complexes revealed that certain dinuclear complexes demonstrate significantly higher tumor selectivity and activity than their mononuclear counterparts. One such complex exhibited remarkable tumor selectivity (TS) value of approximately 24, indicating its potential as a targeted therapeutic agent (Adokoh et al., 2017). Furthermore, in vitro, studies highlight the crucial role of the length of the alkyl chains in anticancer efficacy, with longer chains generally correlating with improved selectivity and activity (Keter et al., 2014).
Despite these promising results, the precise mechanism of action of these phosphinogold(I) complexes remains to be elucidated. Therefore, the present in silico investigation is expected to provide insights into how these complexes interact at the molecular level with cancer cells. This computational approach is designed to help identify potential targets and pathways involved in the anticancer activities reported for these complexes to pave the way for future experimental validation and optimization of these novel compounds. The findings from this investigation could significantly contribute to the development of safer and more effective gold-based anticancer therapies.
The in silico analysis of phosphinogold(I) thiocarbohydrate complexes can provide insights into their potential mechanisms of action as anticancer agents. We hope to verify several hypotheses based on computational modeling and molecular docking studies. Firstly, we hypothesized that the complexes may bind effectively to specific target proteins involved in cancer cell signaling pathways. In silico docking studies could reveal high-affinity interactions with proteins such as kinases or transcription factors that regulate cell proliferation and survival, indicating a potential mechanism for inhibiting tumor growth (Steven, 2003; Yip and Papa, 2021). Secondly, the computational analysis may suggest that the phosphinogold(I) complexes exhibit higher binding affinities for cancer cell-specific targets compared to normal cell targets. This selectivity could be attributed to the unique structural features of the complexes, such as the presence of thiocarbohydrate ligands, which may enhance their interaction with tumor-specific receptors or enzymes (Sankarganesh et al., 2019). Also, it is possible that the binding of these complexes to target proteins induces conformational changes that disrupt normal protein function. In silico simulations could help to visualize these changes, providing evidence that the complexes interfere with the activity of critical proteins involved in cancer cell survival and proliferation (Bajracharya et al., 2022). Additionally, the complexes may be hypothesized to generate reactive oxygen species through their interactions with cellular components. In silico studies could model the redox potential of the complexes, suggesting that they may promote oxidative stress in cancer cells, leading to cell death (Arojojoye et al., 2022; Bhattacharjee et al., 2022; Nath et al., 2023; Ndagi et al., 2017; Selivanov et al., 2011; Yu et al., 2022). Again, computational studies might indicate that these complexes can bind to DNA repair proteins, inhibiting their function. This could lead to an accumulation of DNA damage in cancer cells, ultimately resulting in cell cycle arrest and apoptosis. In silico analysis could identify potential binding sites on these proteins (Kim et al., 2021; Kim et al., 2019).
Successfully verifying these hypotheses can guide future experimental studies to validate the proposed mechanisms of action for phosphinogold(I) thiocarbohydrate complexes as anticancer agents. By leveraging in silico approaches, we hope to gain a deeper understanding of the molecular interactions and pathways involved in the anticancer activity of these complexes.
Methods
3D modeling and preparation of the Phosphinogold(I) thiocarbohydrate complexes
The complexes were first sketched using the 2D sketcher of Maestro (Schrödinger), by drawing the triphenylphosphine, bisdiphenylphosphines, and seven ligands as building blocks for the final assembly of the mono and binuclear Phosphinogold(I) thiocarbohydrate complexes as illustrated in Figure 1. The sketched molecules in Figure 1 were then assembled as the 20 mono and binuclear complexes described by (Adokoh et al., 2017) utilizing the single complex builder within Maestro (Schrödinger, 2023a). This was done by selecting gold(I) as the central atom(s), and linear geometry for the final complex around the central atom (Au). The complex builder was run to join the gold atom(s) to the phosphine’s phosphorus atom(s) at one end, and to the sulfur atom(s) of the thiocarbohydrate ligand(s) at the opposite end. The final assembled complexes are illustrated in Figure 1. The LigPrep tool in (Schrödinger, 2023b) was used to convert the 2D structures into 3D structures. Subsequently, each complex’s geometry was optimized using OPLS4 force field minimization by running the complex cleanup tool in Maestro. The final library of prepared complexes was then used to perform the subsequent molecular docking and MM-GBSA free energy calculation studies.

Figure 1. Two-dimensional structures of 20 Phosphinogold(I) Thiocarbohydrate Complexes.
Protein targets selection and preparation
The 20 complexes have previously shown remarkable anticancer activities in 3 cell lines of the breast (MCF7), prostate (PC3), and colon (HCT116) cancers (Adokoh et al., 2017). However, the mechanism of action of these complexes is not yet known. To investigate the possible mechanisms for the observed anticancer activities, a thorough literature review was initiated to look for the available known targets involved in the three types of cancer cells that have significant effects on their life cycle. The review discovered 17 targets that could be investigated through an in silico approach to evaluate their binding interactions and to estimate binding affinities toward the Phosphinogold(I) Thiocarbohydrate Complexes, which will make it possible to hypothesize possible mechanisms for the observed anticancer activities. Table 1 summarizes the selected targets and their involvement in various types of cancer. The crystallized structures of the targets were downloaded as pdb files from the protein data bank, accessible at https://www.rcsb.org/, then they were loaded into the protein preparation workflow in Maestro (Schrödinger, 2023c) at the default settings. Protein preparation for molecular docking simulations was conducted through a series of sequential steps, involving filling in missing side chains, assigning bond orders, placing hydrogens, generating het states at pH 7.4 ± 2, deleting bulk waters, optimizing hydrogen-bond assignments at pH 7.4, and lastly, energy minimization of the protein using the OPLS4 force field. The co-crystallized ligands or inhibitors (Table 1) from the protein targets were extracted, prepared in LigPrep (Schrödinger, 2023d), and subsequently used to validate docking protocols and they were used as reference to compare binding affinities across the 20 phosphinogold(I) thiocarbohydrate complexes using Glide (Schrödinger). It is important to mention that, the target protein β-catenin with PDB ID: 1JDH had no cocrystallized ligand or inhibitor, therefore, the binding site was determined utilizing the SiteMap function within Maestro software, and for subsequent docking studies, PRI-724 (also known as Foscenvivint); a known potent inhibitor for β-catenin signaling pathway (Schmidtova et al., 2021), was selected as the standard ligand for 1JDH to compare binding affinity between ligands to this target.
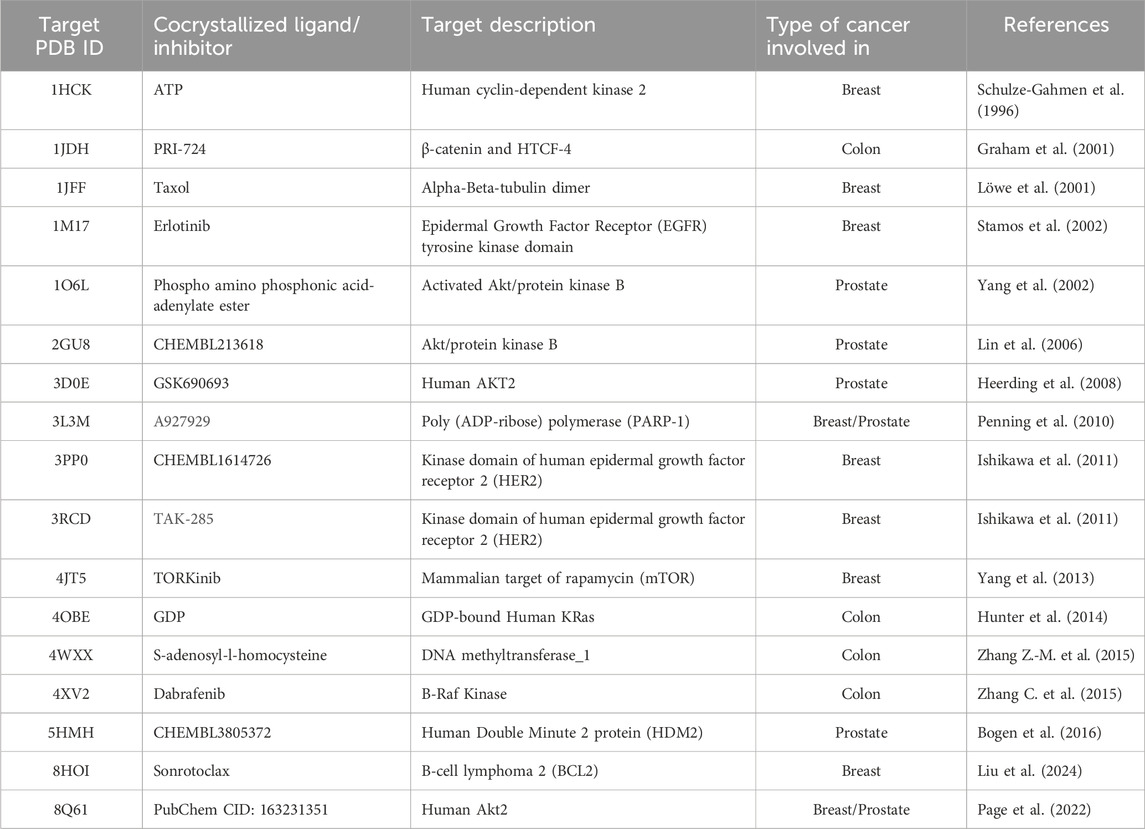
Table 1. Protein targets used for virtual screening of the gold(I) complexes.
Molecular docking
Molecular docking was performed using Glide (Schrödinger, 2023a) software. The procedure was initiated by selecting each prepared target protein as the macromolecule and the 20 prepared gold(I) complexes as the ligands. A receptor grid box using glide was previously generated, centered on the position of the cocrystallized ligand, and a midpoint box of 10 Å diameter in all three coordinates. Flexible ligand sampling with extra precision (XP) was selected to run the molecular docking simulations. The output settings of the docking results were kept at their default values to report the best pose with the highest docking score for each ligand.
Validation of docking protocol
The reliance on redocking in validating docking protocols (Table 2) and assessing accuracy will help assess benchmarking performance. When there is a high level of repetition between the experimentally determined and docked poses, it gives confidence that the docking results reflect, to a high degree, what is happening. In the present study, the native ligands were docked using the same grid box generated for ligand docking, and the poses of the co-crystallized and docked ligands were compared by calculating root mean square deviation (RMSD) (Figure 2). Following this outcome, if the RMSD is small (preferably less than 2.0), then the docking protocol is considered valid (Hevener et al., 2009; Jain, 2008).
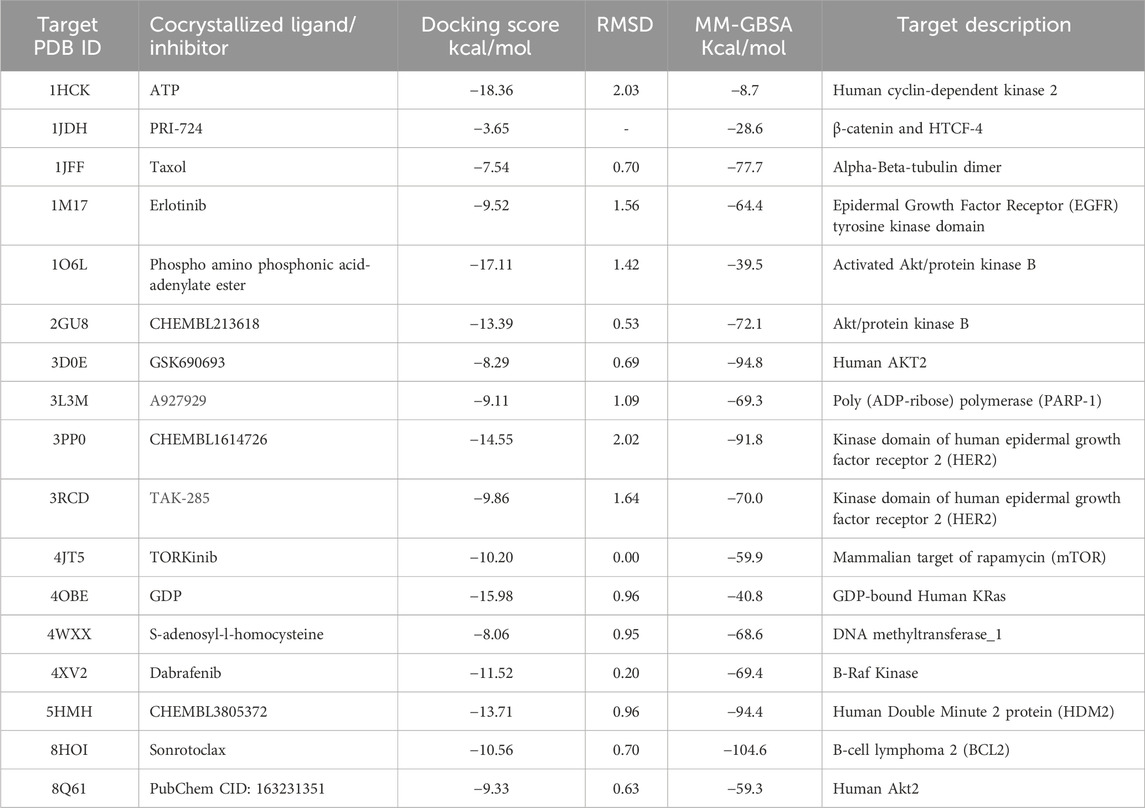
Table 2. Docking scores and binding free energy of target’s cocrystallized ligands or inhibitors.
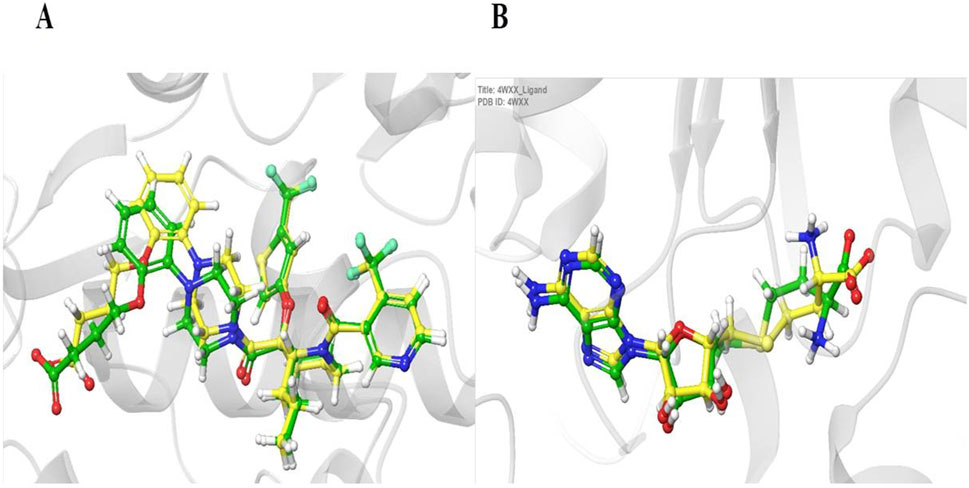
Figure 2. Validation of the docking protocol using redocking. Panels (A, B) show the superimposition of the experimentally determined (cocrystallized) ligand (yellow) and the computationally redocked ligand (green) for two target proteins: HDM2 (PDB ID: 5HMH, A) and DNAMT1 (PDB ID: 4WXX, B). The low root mean square deviation (RMSD) values of 0.96 Å (A) and 0.95 Å (B) demonstrate the reliability of the docking methodology in accurately reproducing known ligand binding poses.
Prime MM-GBSA calculations
The MM-GBSA method combines molecular mechanics force fields (MM) with implicit solvation models (Generalized Born and Surface Area, (GBSA)) to estimate the binding free energy (binding affinity). MM-GBSA is computationally efficient and often used as a first-pass estimate of binding affinity. Prime MM-GBSA can be performed without running a full-fledged MD simulation, utilizing the docked poses as a starting point.
Molecular docking relies on scoring functions to assess the quality of the predicted binding poses generated during conformational search. These functions utilize approximations to streamline the calculations, enabling high-throughput screening of potential drug candidates. Due to the use of approximate scoring functions, molecular docking often falls short of accurately predicting binding energies when compared to experimental measurements. Although, numerous docking programs effectively identify potential ligand binding conformations, a universal scoring function that accurately predicts binding energies for all molecules and protein families remains elusive. Consequently, rescoring steps after molecular docking are often essential to refine the initial predictions (Zhang et al., 2017). On the other hand, MM-GBSA utilizes molecular mechanics and free energy calculations to consider entropic contributions and thus provide more accurate binding affinity predictions (Sgobba et al., 2012). MM-GBSA is computationally more expensive but offers greater accuracy and insights into binding mechanisms, making it ideal for ranking and refining promising ligands (Genheden and Ryde, 2015; Sgobba et al., 2012). Instead of simulating the system’s molecular dynamics over time, the Prime MM-GBSA module in the Schrodinger suite, performs a series of calculations on a single optimized pose from Glide’s docking output, using a Generalized Born solvation model (VSGB 2.0) (Li et al., 2011) to estimate the binding free energy. These calculations involve minimization to relax any strained interactions, energy calculation of the complex and its components using the MM-GBSA method, and finally, determining the binding affinity by calculating the difference in energies (E) of the protein-ligand’s complex and its components (Muddagoni et al., 2021):
This approach offers significant time efficiency compared to standard MM-GBSA, which relies on lengthy MD simulations. However, it is important to note that this method is limited by the single, optimized docked pose, potentially missing the full range of conformations the ligand can adopt in the binding site and potentially neglecting entropic effects. Overall, Prime MM-GBSA without MD can be a useful tool for quick binding affinity estimation. However, for more accurate and complete understanding of the binding process, a full MD simulation is recommended. Therefore, in our study, the poses obtained with the highest docking score for each ligand across all 17 targets were re-scored using the Prime (Schrödinger, 2023a) MM-GBSA to calculate binding free energy (ΔG) in kcal/mol. Then, MD simulations were performed for the best scoring complex.
Molecular dynamics (MD) simulations
Building upon the promising MM-GBSA and docking results, Complex 9, which emerged as the top contender from a group of twenty Phosphinogold(I) Thiocarbohydrate Complexes, was subjected to detailed molecular dynamics (MD) simulations. The 100 ns simulations, performed using Desmond software (Schrödinger, 2018), aimed to explore the dynamic interactions between Complex 9 and its target proteins: Poly (ADP-ribose) polymerase (PARP-1, PDB ID 3L3M) and Protein Kinase B (AKT2, PDB ID 8Q61). To prepare the simulations, the protein-ligand complexes were placed in minimized, solvated orthorhombic boxes, ensuring a 10 Å buffer of TIP3P water molecules. The simulation environments were brought to physiological ionic strength by adding counter ions for neutralization and 0.15 M NaCl. The MD simulations were performed under NPT conditions (constant particle number, pressure, and temperature) at 300 K and 1.01325 bar, with the OPLS3e force field. Prior to the 100 ns production run, all systems were relaxed using Desmond’s default 160 picosecond protocol. Following the simulation, Maestro (Schrödinger, 2023b) was employed for comprehensive analysis of simulation data. Key parameters assessed included root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), the dynamic protein-ligand contact profiles, and finally, the calculation of post-simulation MM-GBSA binding free energy (ΔGbind in kcal/mol).
Results
Docking validation and evaluation of complex interactions
The reliability of the molecular docking protocol was established through redocking of native ligands into their respective protein structures, confirming accurate reproduction of experimentally observed binding poses (Figure 2; Table 2). Root mean square deviation (RMSD) was calculated to quantify the difference in position between co-crystallized and re-docked native ligands. An RMSD value equal to or less than 2.0 Å was used as a criterion to validate the docking protocol (Hevener et al., 2009; Jain, 2008). Following validation, molecular docking was performed using Glide XP, and subsequently, the more rigorous Prime MM-GBSA method was employed to refine binding affinity estimations. A detailed breakdown of observations for each complex can be found in the supplementary materials (Supplementary Tables S1–20; Supplementary Figurea S1–14).
Molecular docking and prime MM-GBSA analysis
The analysis identified several key protein targets, the most significant are Human Double Minute 2 protein (HDM2), DNA methyltransferase-1 (DNMT1), Human AKT2, and Poly (ADP-ribose) polymerase (PARP-1), which frequently exhibited strong interactions with multiple complexes. Number of complexes exhibited strong binding affinities, with several of them surpassing the binding strength of native ligands (Tables 3, 4). This suggests potential multi-targeting mechanisms and broad-spectrum activity. A general trend observed was that binuclear complex (those numbered 8 and above) consistently exhibited higher binding affinities than mononuclear complexes, as illustrated in Figures 3, 4. This suggests that the presence of two gold centers within the complex may be associated with enhanced binding. This relationship was confirmed by a statistically significant (α = 0.05) negative correlation between complex type (mononuclear or binuclear) and docking scores with two targets; DNMT-1 (Spearman’s correlation coefficient r = −0.837, p = 0.0061) and HDM2 (r = −0.717, p = 0.0242). The negative correlation indicates that binuclear complexes, which correspond to lower docking scores, bind more tightly to the proteins (Supplementary Tables S21, 22). The noted increase in binding affinity with binuclear complexes is entirely consistent with the results of Adokoh et al., who found that dinuclear gold(I) complexes (8-20) displayed significantly enhanced growth inhibition of cancer cells when compared to their mononuclear counterparts (1-7). Acetylation has been found to have a significant effect on binding affinity, with acetylated complexes (complexes 5-7 and 12-20) consistently exhibiting higher docking scores (lower binding affinity) (as detailed in Supplementary Tables S1, 20) than their non-acetylated analogs (complexes 1-4 and 8-11). This observation is supported by statistically significant positive correlations between acetylation and docking scores for DNMT-1 (Spearman’s r = 0.8452, p = 0.0016), HDM2 (r = 0.8563, p = 0.0001), PARP-1 (r = 0.8452, p = 0.0016), and AKT2 (r = 0.7681, p = 0.0081) (Supplementary Tables S22, 23). These positive correlations indicate that acetylation is associated with higher (less negative) docking scores, suggesting a weaker binding affinity. While this reduction in binding affinity can be explained by reduced hydrogen bond forming capability, Adokoh et al. observed that acetylation paradoxically enhanced the activity of gold(I) complexes (specifically complexes 5-7 in their study). This difference between the in silico finding and the experimental in vitro activity underscores that acetylation has a complex and multifaceted effect, where its impact on binding affinity does not necessarily correlate directly with its impact on overall cellular activity due to factors such as cell penetration, which Adokoh et al. attributed to increased lipophilicity as a result of reduced hydrogen bonding potential. The following illustrative examples underscore the significant interactions observed in this study. Table 4 provides a complete listing of the strongest interactions.

Table 3. Compounds with the highest binding free energy across the 17 cancer targets.
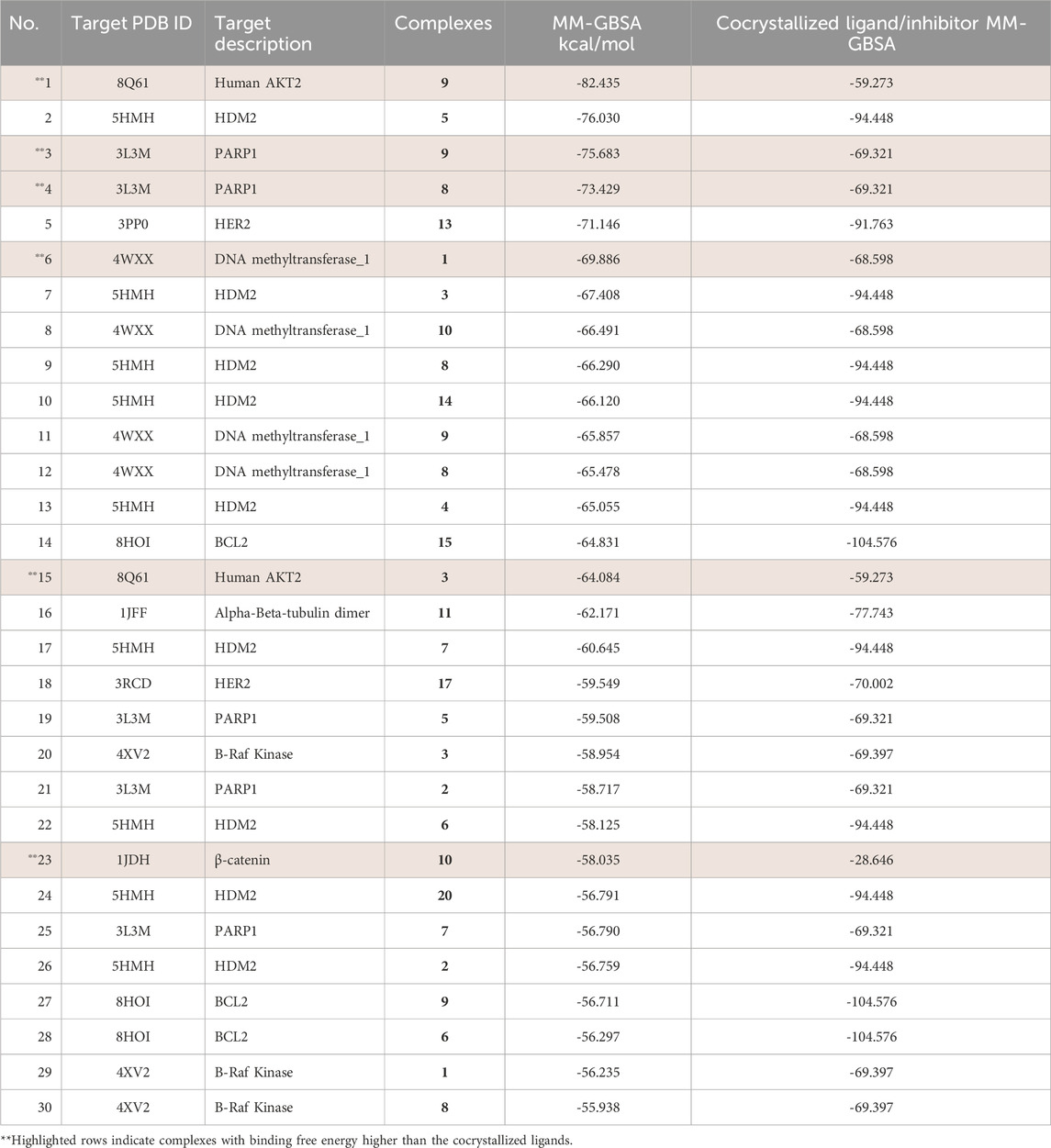
Table 4. Free binding energies of the top 30 interactions compared to native ligands.
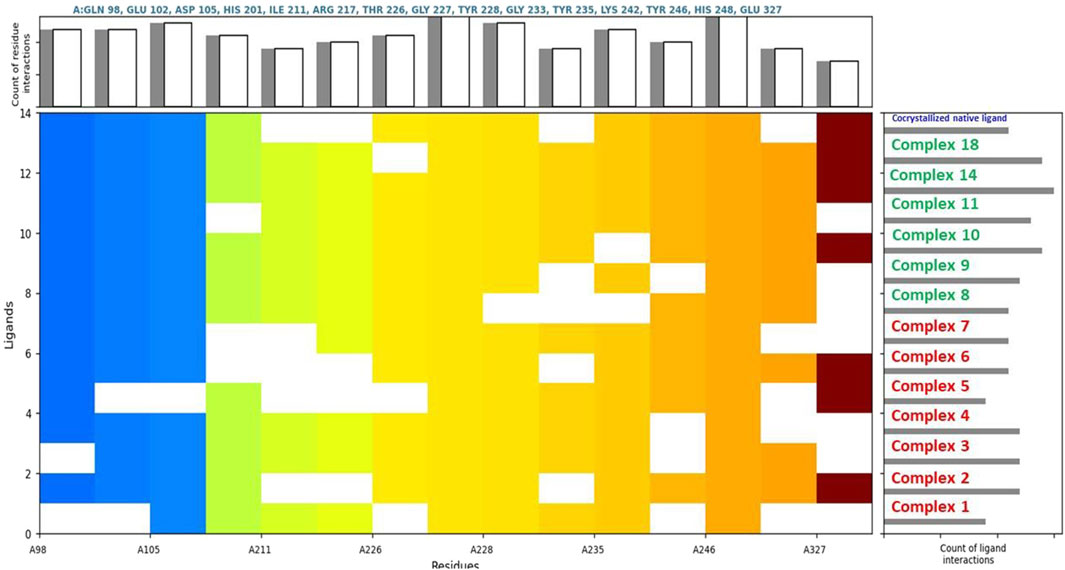
Figure 3. Comparative ligand-residue interaction profiles for thirteen Phosphinogold(I) Thiocarbohydrate complexes and the native ligand (indicated at the top right corner in blue) within the Poly (ADP-ribose) polymerase 1 (PARP1) binding site (PDB ID: 3L3M). The figure displays a matrix where the presence of an interaction between each ligand and a specific residue (A:GLN 98 – A:GLU 327) in PARP1 is indicated by a colored square. Colors are assigned arbitrarily and do not represent interaction strength or frequency. The total number of interactions per residue and per ligand are shown in the top and right panels, respectively, allowing for a comparative analysis of the interaction patterns. This figure clearly shows that binuclear complexes 8–20 have generally more interactions than mononuclear complexes 1 – 7.
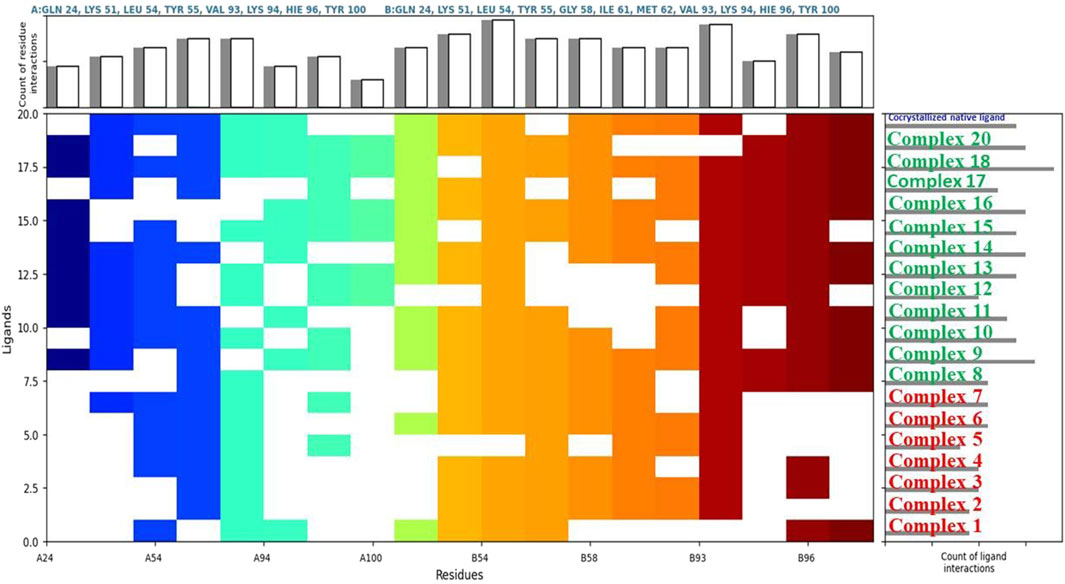
Figure 4. Comparative ligand-residue interaction profiles for nineteen Phosphinogold(I) Thiocarbohydrate complexes and the native ligand (indicated at the top right corner in blue) within the Human Double Minute 2 protein (HDM2) binding site (PDB ID: 5HMH). The figure displays a matrix where the presence of an interaction between each ligand and a specific residue (A:GLN 24 – A: TYR 100, and B:GLN 24 – B:TYR 100) in HDM2 is indicated by a colored square. Colors are assigned arbitrarily and do not represent interaction strength or frequency. The total number of interactions per residue and per ligand are shown in the top and right panels, respectively, allowing for a comparative analysis of the interaction patterns. This figure clearly shows that binuclear complexes 8–20 have generally more interactions than mononuclear complexes 1 – 7.
Complex 9: This complex stood out as a top performer against Human AKT2 isoform 8Q61, displaying an MM-GBSA binding score of −82.4 kcal/mol (Supplementary Table S9; Figure 5C), which exceeded even the native ligand’s score of −59.3 kcal/mol. This strong interaction aligns well with the strong anti-proliferative effects that have been seen with complexes 8-11 in vitro, and suggests that disruption of AKT2 may be a key factor in its success. Complex 9 also demonstrated strong affinity for PARP1 with MM-GBSA score of −75.7 kcal/mol and was therefore, considered for further MD study. Unfortunately, complex 9 was selected for in vitro studies at the time and however, it is imperative to further investigate complex 9. Complex 5 showed the highest binding affinity towards the HDM2 with a binding free energy of −76.0 kcal/mol. Several other complexes also showed promising interactions with HDM2, this suggests that inhibiting 5HMH/p53 interaction plays a crucial role in the mechanism of action of these complexes. Complex 10, demonstrated multi-target binding interactions, in particular HDM2, DNMT-1, and a particular affinity towards β-catenin (1JDH), were exceeding the MM-GBSA score of the reference inhibitor by a notable margin (Table 4). The tight binding of complex 10 with beta-catenin is crucial in the Wnt signaling pathway (Zhao et al., 2022) which supports its potent activity in colon cancer (HCT116 cell line) in vitro (IC50 = 0.90 µM). Similarly, complex 11, also displayed a diverse array of binding targets, notably with DNMT-1, PARP-1, and the strongest binding affinity observed for the Alpha-Beta-tubulin dimer (1JFF) which could also account for strong in vitro anti-proliferative effects against colon and prostate cancer with IC50S of 0.63 µM and 0.22 µM respectively (Adokoh et al., 2017). Complex 8 being the shorter chain of 9 and 11 demonstrated a significant interaction with both DNMT-1 and HDM2, surpassing the respective native ligand of PARP1 in line with in vitro data, which was the most potent growth inhibitor of prostate (PC3) cell line (IC50 = 0.003 μM) (Adokoh et al., 2017).
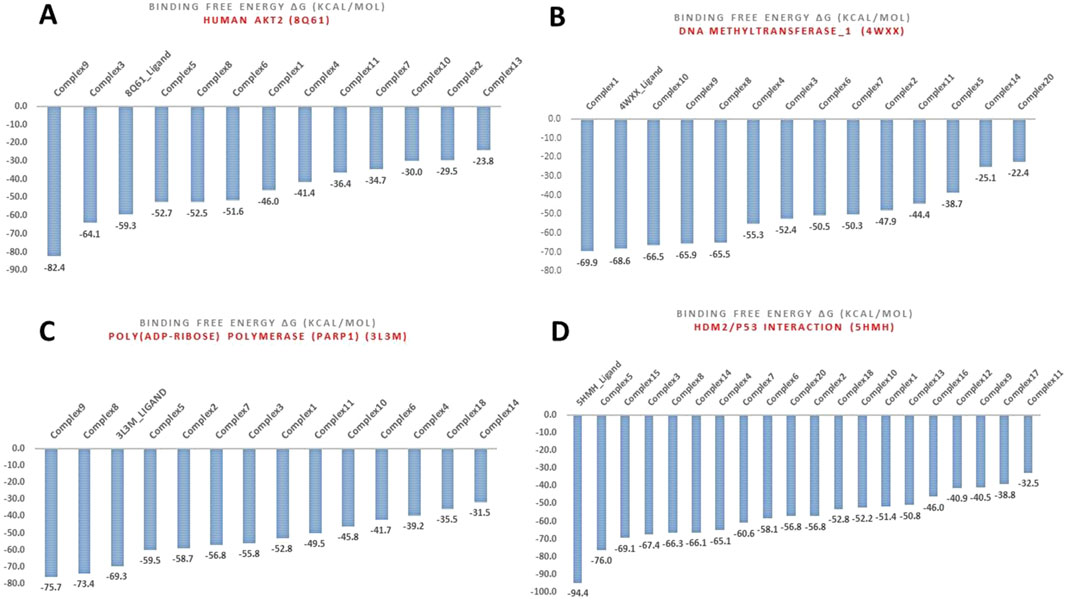
Figure 5. MM-GBSA binding free energies (Kcal/mol) for the various complexes with targets: (A) Human AKT2 (8Q61), (B) DNA methyltransferase-1 (DNMT1) (4WXX), (C) Poly (ADP-ribose) polymerase (PARP1) (3L3M), and (D) Human Double Minute 2 protein (HDM2) (5HMH).
MD simulations
Complex 9’s binding stability with PARP-1 and AKT2 was investigated via 100 ns MD simulations. As shown in Figure 6A and Table 5, the PARP-1-Complex 9 system achieved equilibrium after approximately 10 ns, exhibiting a stable protein Cα RMSD averaging 2.0 Å (range: 1.8–2.4 Å). This suggests minimal overall protein structural fluctuation. While the ligand RMSD relative to the protein (ligand fitted on protein) showed some fluctuations, reaching up to ∼6 Å in few instances, it was generally stabilized between 2.0 and 3.0 Å, averaging 2.72 Å. This suggests sustained binding despite some ligand mobility within the pocket, likely due to conformational adjustments. The ligand’s internal structure remained rigid, as evidenced by the low average RMSD of 2.21 Å (range: 1.6–2.5 Å) when fitted on itself. In contrast, the AKT2-Complex 9 simulation (Figure 6B) revealed a less stable protein Cα RMSD profile, although equilibrium was reached around 20 ns with an average of 3.03 Å (Table 5). Ligand mobility was more pronounced in this system, with an average ligand RMSD (relative to the protein) of 4.13 Å (Table 5). A period of increased fluctuation between 46 and 76 ns suggests substantial ligand conformational changes during this timeframe, potentially impacting the binding site and correlating with the observed jump in protein RMSD (Figure 6B). Despite this increased mobility relative to the protein, the ligand’s internal structure remained relatively rigid, averaging 2.74 Å RMSD when fitted on itself. This indicates the ligand’s conformational changes involve shifts in position or orientation within the binding pocket rather than significant internal rearrangements.

Figure 6. Dynamic Stability of complex 9 with (A) PARP-1 (3L3M) and (B) AKT2 (8Q61) during MD Simulation. This figure visualizes the root mean square deviation (RMSD) of the proteins and Complex 9, over the 100 ns MD simulation. The plot shows three RMSD traces: dark blue for the protein’s C-alpha atoms, red for Complex 9 fitted to the protein (highlighting the ligand’s movement within the binding pocket), and pink for Complex 9 fitted to itself (demonstrating its conformational flexibility).
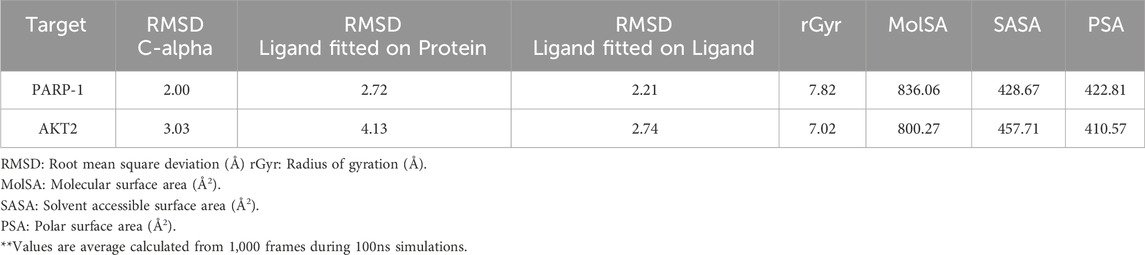
Table 5. **Average structural and dynamic analysis of complex 9 during MD Simulations.
Figure 7 summarizes the properties of complex 9 bound to (A) PARP-1 and (B) AKT2 during MD simulations, including radius of gyration (rGyr), molecular surface area (MolSA), solvent accessible surface area (SASA), and polar surface area (PSA). Complex 9 displayed a more compact average structure and reduced solvent exposure with AKT2 (average rGyr ≈7.0 Å, range 6.2–8.6 Å; average SASA ≈458 Å2, range 330–783 Å2) compared to PARP-1 (average rGyr ≈7.8 Å, range 6.8–8.9 Å; average SASA ≈428 Å2, range 300–590 Å2), although the PARP-1 system showed greater rGyr fluctuation. Intramolecular hydrogen bonding was similarly low and variable in both simulations (average <2, range 0–4 H-bonds), indicating it is unlikely to be a key stabilizing factor. Polar surface area values were comparable (≈411 Å2 for AKT2 and ≈423 Å2 for PARP-1). These results indicate Complex 9 binds more dynamically to AKT2 than to PARP-1. Table 5 also summarizes the average values for these properties.
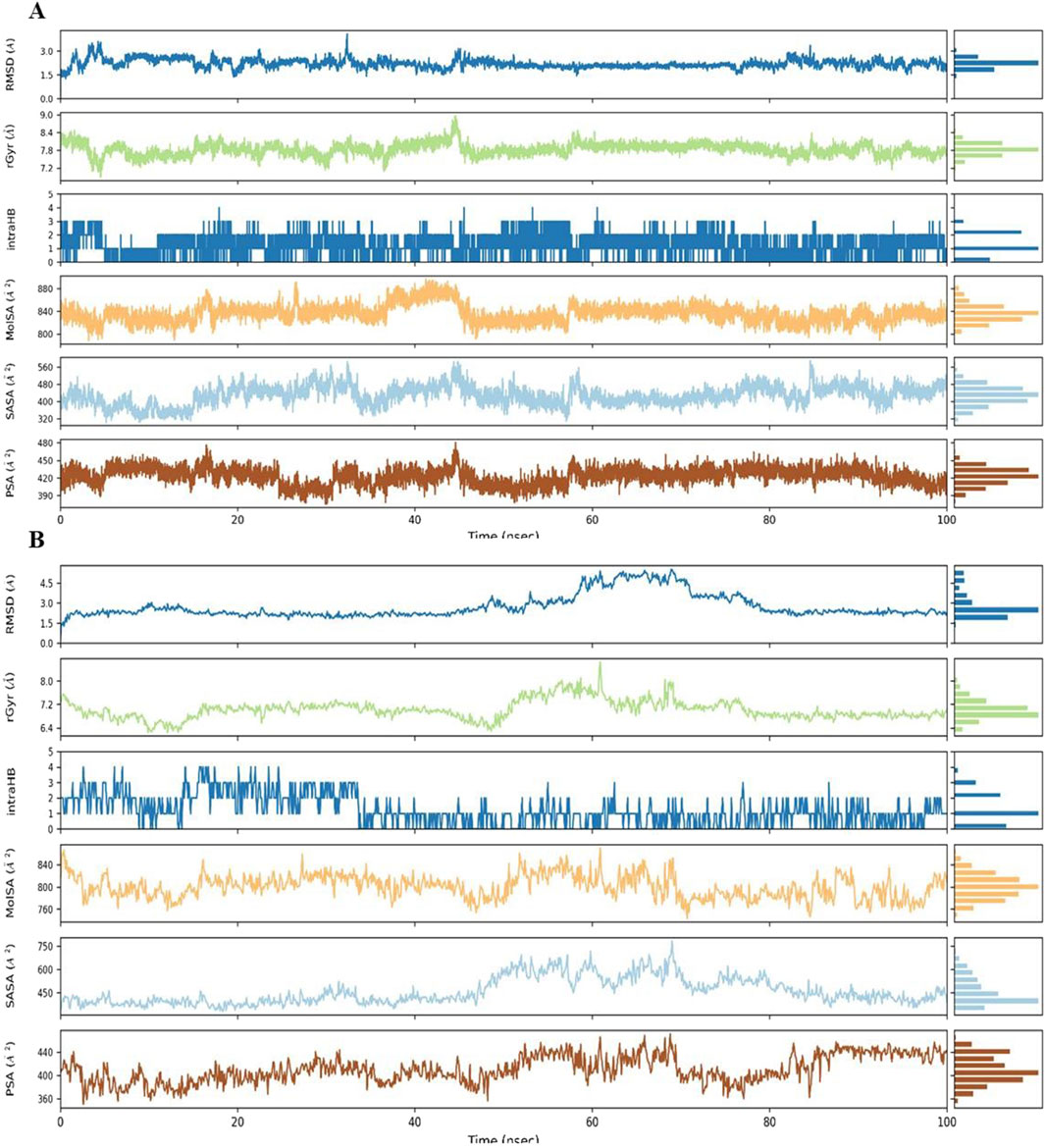
Figure 7. The figure displays the dynamic behavior of Complex 9 within the binding site of (A) PARP-1 and (B) AKT2, tracking changes in Root Mean Square Deviation (RMSD), radius of gyration (rGyr), intramolecular hydrogen bonds (intraHB), Molecular Surface Area (MolSA), Solvent Accessible Surface Area (SASA), and Polar Surface Area (PSA). Each parameter (with units of Å or Å2) is shown as a trace across the simulation time with a histogram of the parameter’s distribution.
Analysis of Cα RMSF for PARP-1 bound to complex 9 (Figure 8A) suggests that ligand binding modulates protein flexibility. While the N- and C-termini appear generally rigid, ligand contacts at various locations, including residues 27-28, 55-60, 92-109, 200-275, and 322, may contribute to localized changes in flexibility. For example, increased RMSF values around residues 57-64 could indicate ligand-induced conformational changes. It is important to acknowledge that these observations are based on a single MD simulation and require further validation, including comparison to apo PARP-1 dynamics and experimental studies, to definitively establish the effects of complex 9 binding. On the other hand, complex 9 binding differentially affects the flexibility of AKT2. PARP-1’s N-terminus becomes rigid upon binding, whereas AKT2’s N-terminus shows a substantial increase in flexibility (i.e., higher RMSF) (Figure 8B). Overall, complex 9 has a more dramatic effect on AKT2’s flexibility, particularly at the N- and C-termini. In contrast, complex 9’s impact on PARP-1 is more localized. AKT2 also displays a greater number of contact points with complex 9. This broader interaction interface may suggest a more extensive allosteric effect of complex 9 on AKT2 compared to PARP-1. Further studies are needed to validate how these distinct dynamic responses relate to the functional regulation of each protein. Figures 9, 10 illustrate the interaction profiles of complex 9 with PARP-1 and AKT2 binding site residues, respectively. The prominent mode of interaction in both cases is through H-bonding and to a lesser extent, water bridges, which is a special form of H-bonds mediated by surrounding water molecules. This explain the higher affinities of non-acetylated series of complexes such as complex 9, compared to those of the per-acetylated complexes such as 12-20 as we have discussed earlier.
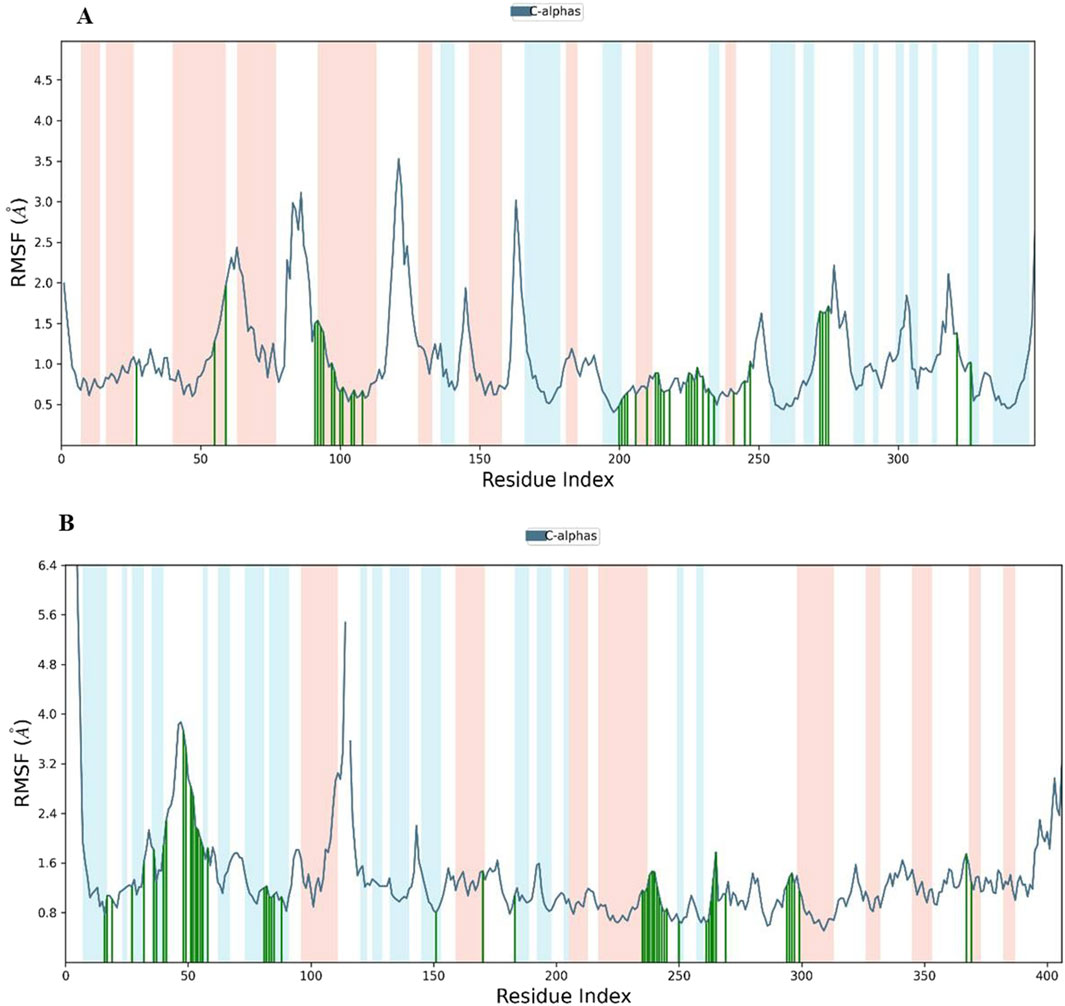
Figure 8. Root Mean Square Fluctuation (RMSF) of (A) PARP-1 and (B) AKT2 in complex with complex 9. The blue line shows the RMSF of the protein’s C-alpha atoms. Green vertical lines indicate residues interacting with Complex 9, and the shaded background highlights the protein’s secondary structure elements (red for alpha helices and light blue for beta strands).
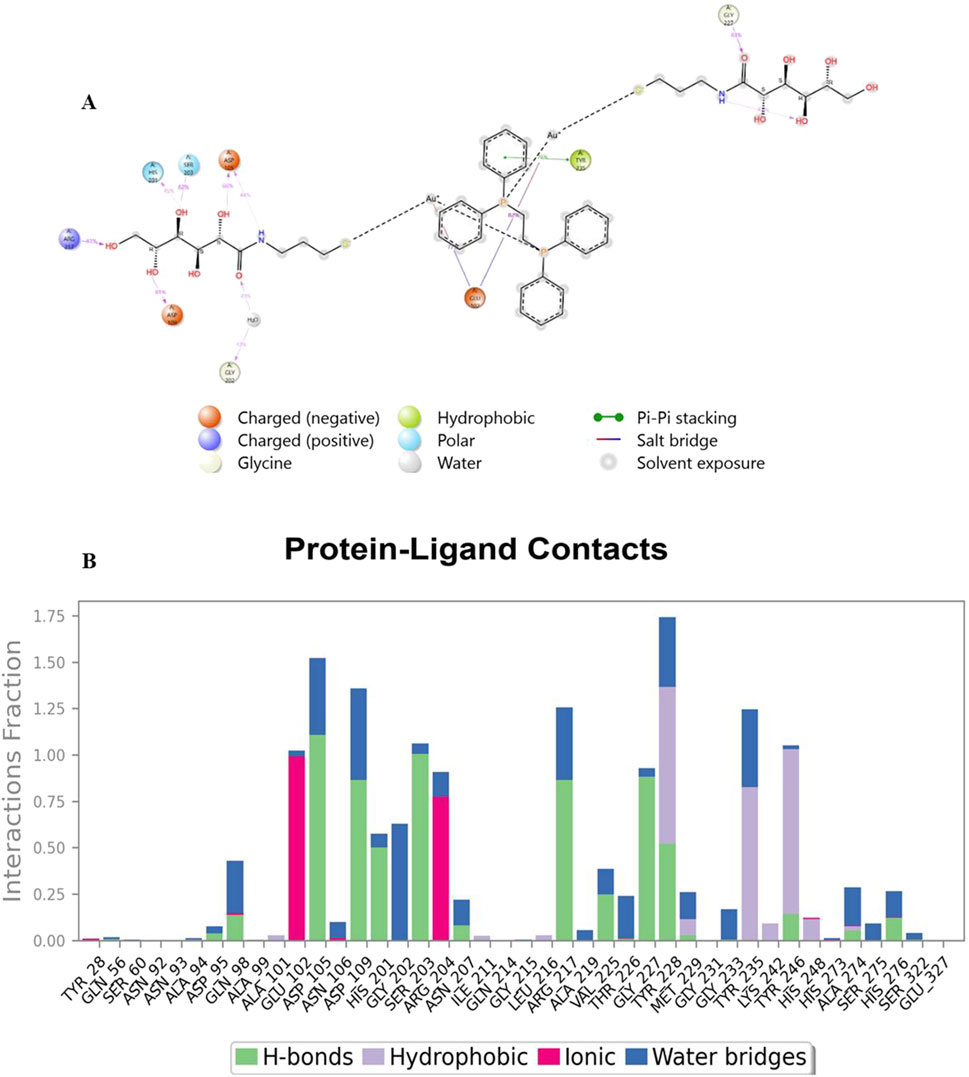
Figure 9. Interaction profile of complex 9 with PARP-1 binding site residues. Panel (A) displays a 2D interaction diagram, highlighting the specific contacts (ionic interactions, hydrogen bonds, hydrophobic interactions, and water bridges) between Complex 9 and key PARP-1 residues. Panel (B) shows an interaction fraction plot, quantifying the frequency with which each residue interacts with Complex 9 across the molecular dynamics simulation.
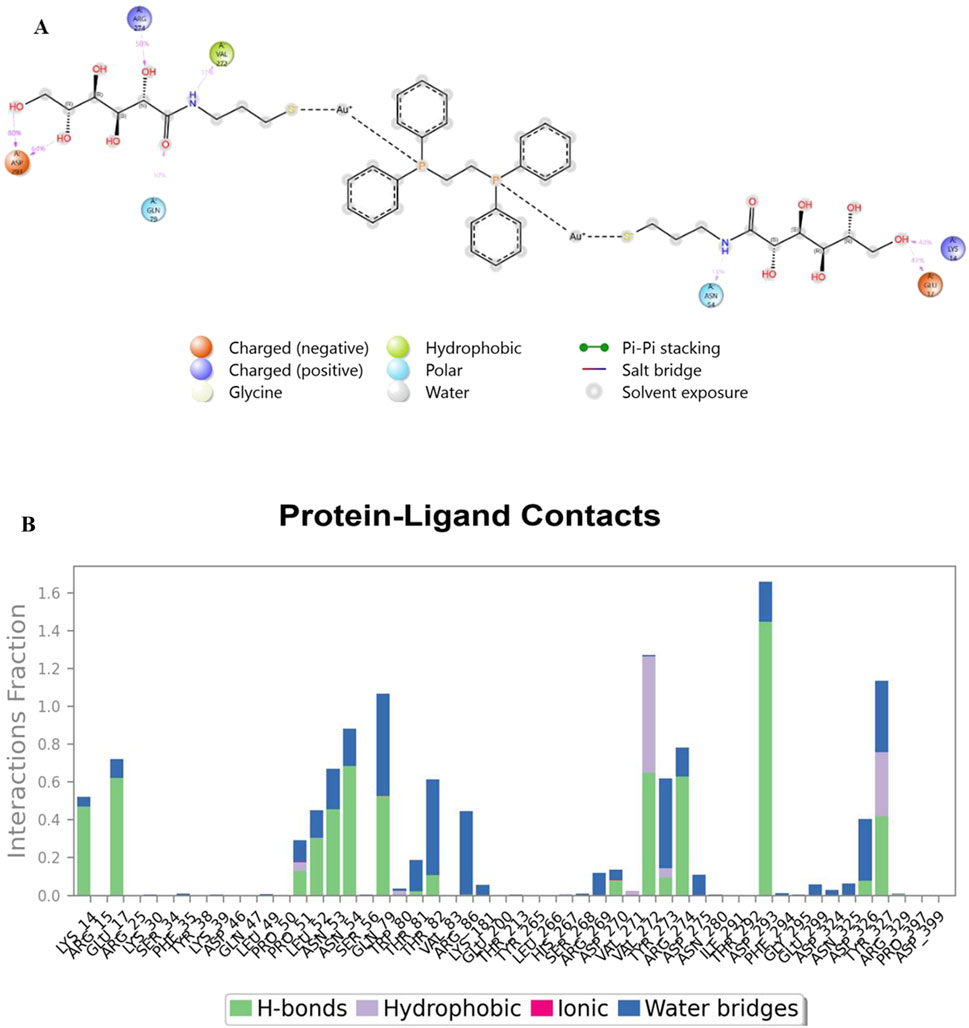
Figure 10. Interaction profile of complex 9 with AKT2 binding site residues. Panel (A) displays a 2D interaction diagram, highlighting the specific contacts (ionic interactions, hydrogen bonds, hydrophobic interactions, and water bridges) between Complex 9 and key AKT2 residues. Panel (B) shows an interaction fraction plot, quantifying the frequency with which each residue interacts with Complex 9 across the molecular dynamics simulation.
Prime MM-GBSA calculations, using 31 frames extracted at ∼3.3 ns intervals from 100 ns MD trajectories (Figure 11), provide a more comprehensive understanding of the binding profile of complex 9 with PARP-1 and AKT2 compared to previous calculations based on the single docked poses in the molecular docking study. These post-MD calculations, incorporating protein and ligand flexibility by sampling representative snapshots across the simulation, offer a more realistic representation of the binding dynamics. Complex 9 displays a more favorable and stable binding profile with PARP-1, exhibiting an average binding free energy of −71.82 kcal/mol and fluctuations mostly within a −50 to −91 kcal/mol range. In contrast, complex 9 binding to AKT2 is characterized by a lower average binding free energy (−63.31 kcal/mol) and wider fluctuations (−38 to −91 kcal/mol), suggesting a weaker or more dynamic interaction.
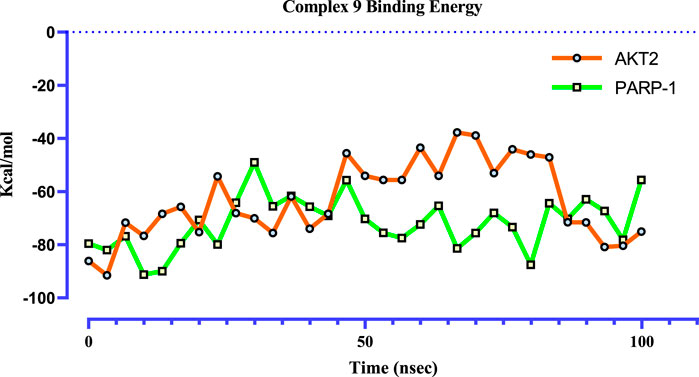
Figure 11. Time-dependent binding free energy profiles of complex 9 with PARP1 (green) and AKT2 (orange), computed via the MM-GBSA method from 100 ns molecular dynamics trajectories (n = 31). The calculated average binding free energies were −71.82 ± 3.65 kcal/mol for PARP1 and -63.31 ± 5.33 kcal/mol for AKT2. These values suggest a significantly more thermodynamically favorable binding between complex 9 and PARP1, implying a stronger and more stable interaction relative to AKT2.
Discussion
The integration of in silico and in vitro data underscores the significance of computational approaches in identifying promising lead compounds and elucidating their mechanisms of action. In this study, in silico analyses revealed substantial interactions between phosphinogold(I) thiocarbohydrate complexes and key proteins implicated in cancer progression. These interactions suggest mechanisms targeting cell survival, proliferation, DNA repair, and apoptosis. Notably, complexes 5, 8, 9, 10, and 11 exhibited strong binding to HDM2, DNMT1, AKT2, and PARP-1, indicating a multi-target strategy that could enhance therapeutic efficacy by simultaneously modulating several signaling pathways.
Among these interactions, the binding to HDM2 is particularly significant, given its role in regulating the tumor suppressor protein p53 (Wang et al., 2023). Overexpression of HDM2 in cancer inhibits p53 function, leading to uncontrolled cell growth (Nag et al., 2014; Wang et al., 2023; Yang et al., 2002). The in silico findings suggest that these complexes, particularly complex 5, may disrupt the HDM2/p53 interaction, thereby restoring p53 activity and promoting tumor suppression. Similarly, complexes 1, 8, 9, and 10 demonstrated strong interactions with DNMT1 (Figure 5B), an enzyme responsible for maintaining DNA methylation patterns (Chen et al., 2010; Massie et al., 2017; Mohd Kamal et al., 2024). Aberrant methylation can silence tumor suppressor genes, and inhibition of DNMT1 by these complexes may restore their expression, thereby suppressing cancer cell proliferation (Mohd Kamal et al., 2024).
AKT2, a key player in the PI3K/AKT/mTOR signaling pathway, is frequently dysregulated in cancers (Attoub et al., 2022), promoting tumor growth and therapeutic resistance (Chau and Ashcroft, 2004; Riggio et al., 2017; Rychahou et al., 2008; Su et al., 2021) The strong binding of complex 9 to AKT2 suggests its potential to disrupt this pathway, thereby inhibiting tumor proliferation. Furthermore, complex 9 exhibited a significant affinity for PARP-1, an enzyme involved in DNA damage repair (Bondar and Karpichev, 2024; Deshmukh and Qiu, 2015; Puentes-Pardo et al., 2023). Given the established efficacy of PARP inhibitors in targeting cancers with defective DNA repair mechanisms, complex 9 may potentiate anti-cancer effects by disrupting PARP-1 activity (Bondar and Karpichev, 2024).
To further characterize complex 9, molecular dynamics simulations were performed with PARP-1 and AKT2, revealing distinct binding dynamics. Complex 9 demonstrated a stable interaction with PARP-1, as indicated by lower RMSD values and a narrower range of MM-GBSA binding free energies, suggesting a more favorable binding profile. Conversely, its interaction with AKT2 was more dynamic, characterized by higher RMSD and RMSF values, particularly at the termini, and a broader range of MM-GBSA energies. These findings suggest that complex 9 preferentially stabilizes PARP-1 binding, warranting further experimental validation to assess its therapeutic potential.
The in silico findings were correlated with in vitro observations from Adokoh et al., where complexes with high binding affinities to key targets exhibited potent activity in cell-based assays (Table 6). A critical consideration in ligand design is the balance between lipophilicity, modulated by acetylation, and direct binding affinity. While acetylation enhances cellular uptake by increasing lipophilicity, our findings suggest that it may simultaneously reduce direct binding affinity by limiting hydrogen bonding interactions. Docking and MM-GBSA analyses revealed that non-acetylated complexes (8, 9, 10, and 11) exhibited the strongest interactions, emphasizing the importance of free hydroxyl groups in target binding. However, the acetylated complex 5 and 13, for example, demonstrated strong binding affinity, suggesting a nuanced balance between hydrogen bonding and lipophilicity. Complex 13, for example, typically showed high binding affinity (−71.1 kcal/mol) to HER2, collaborating the high in vitro activity against prostate, colon and breast cancer cell lines (IC50 = 0.03, 0.25, and 0.07 µM respectively) reported by our group. Strangely, no statistically significant correlation was found for complex 13 in this work and the in vitro work by Adokoh et al. (2017). These results indicate that a uniform acetylation strategy may not be optimal for all target proteins.
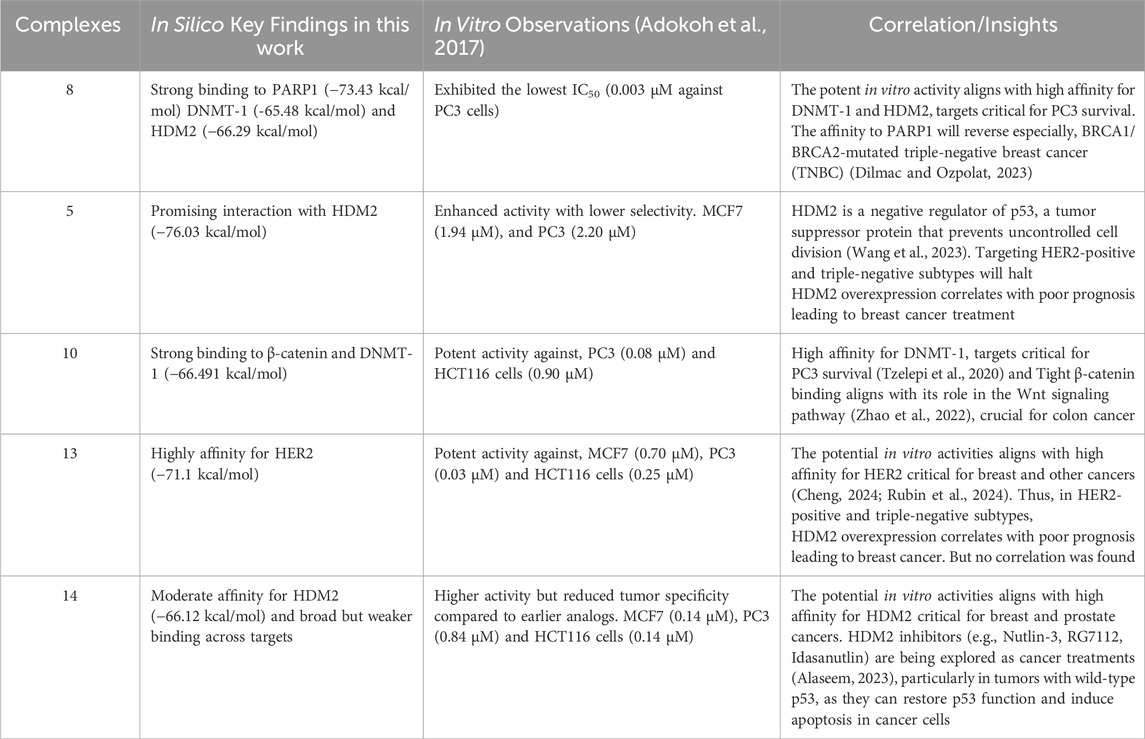
Table 6. Summarizing and linking some in silico findings to the in vitro data from the studies of Adokoh et al.
Future research should focus on systematically optimizing acetylation patterns to enhance both binding affinity and cellular permeability. Strategies may include: (1) exploring partial acetylation to modulate lipophilicity while preserving hydrogen bonding, (2) incorporating alternative modifications to improve permeability without compromising target binding, (3) screening libraries of analogs with varying acetylation degrees to assess their impact on binding affinity and cellular activity, and (4) developing computational models to predict ligand permeability and interaction strength. Moreover, in vitro and in vivo validation, particularly of complex 9, is crucial to confirm its therapeutic potential. These investigations could identify an optimal balance between acetylation and binding efficacy, thereby guiding the rational design of future anticancer agents.
Conclusion
This in silico investigation provides compelling evidence for the anticancer potential of phosphinogold(I) thiocarbohydrate complexes, particularly complex 9. The study highlights a multi-target mechanism, with strong interactions observed against key protein targets in cancer pathways, including HDM2, DNMT1, AKT2, and PARP-1. The correlation between in silico binding affinities and previously reported in vitro activity strengthens the validity of our computational approach. Molecular dynamics simulations further differentiated the binding dynamics of complex 9 with PARP-1 and AKT2, revealing a more stable interaction with PARP-1. This emphasizes PARP-1 as a particularly promising target for complex 9 and warrants further investigation and experimental validation. Furthermore, our findings underscore the importance of balancing lipophilicity, influenced by acetylation, with target binding affinity in future ligand design efforts. This research lays the groundwork for the development of more effective and selective gold-based anticancer therapies.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
AM: Data curation, Formal Analysis, Methodology, Software, Writing – original draft. IA: Formal Analysis, Supervision, Validation, Writing – review and editing. GE: Formal Analysis, Methodology, Software, Writing – review and editing. JD: Conceptualization, Funding acquisition, Supervision, Writing – review and editing. CKA: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1533026/full#supplementary-material
References
Adokoh, C. K., Darkwa, J., and Kinfe, H. H. (2017). Synthesis, characterization and anticancer evaluation of phosphinogold(I) thiocarbohydrate complexes. Polyhedron 138, 57–67. doi:10.1016/j.poly.2017.09.010
Alaseem, A. M. (2023). Advancements in MDM2 inhibition: clinical and pre-clinical investigations of combination therapeutic regimens. Saudi Pharm. J. 31 (10), 101790. doi:10.1016/j.jsps.2023.101790
Arojojoye, A. S. K., Olelewe, J. H., Parkin, C., and Awuah, S. (2022). Chiral gold complexes: speciation, in vitro, and in vivo anticancer profile. Chem. Commun. 58, 10237–10240. doi:10.1039/D2CC03081K
Attoub, S., Arafat, K, Hammadi, N.K., Mester, J., and Gaben, A.-M. (2022). Author correction: Akt2 knock-down reveals its contribution to human lung cancer cell proliferation, growth, motility, invasion and endothelial cell tube formation. Sci. Reports 12 (1), 10431. doi:10.1038/s41598-022-14136-7
Bajracharya, R., Song, J. G., Patil, B. R., Lee, S. H., Noh, H. M., Kim, D. H., et al. (2022). Functional ligands for improving anticancer drug therapy: current status and applications to drug delivery systems. Drug Deliv. 29 (1), 1959–1970. doi:10.1080/10717544.2022.2089296
Bhattacharjee, T. A., Bhattacharjee, S., Debnath, S., Das, S., Daniliuc, A. G., Thirumoorthy, C., et al. (2022). Exploring dithiolate-amine binary ligand systems for the supramolecular assemblies of Ni(II) coordination compounds: crystal structures, theoretical studies, cytotoxicity studies, and molecular docking studies. Inorg. Chim. Acta 543, 121157. doi:10.1016/j.ica.2022.121157
Biebuyck, H. A. B., D, C., and Whitesides, G. M. (1994). Comparison of organic monolayers on polycrystalline gold spontaneously assembled from solutions containing dialkyl disulfides or alkanethiols. Langmuir 10, 1825–1831. doi:10.1021/la00018a034
Bogen, S. L., Pan, W., Gibeau, C. R., Lahue, B. R., Ma, Y., Nair, L. G., et al. (2016). Discovery of novel 3,3-disubstituted piperidines as orally bioavailable, potent, and efficacious HDM2-p53 inhibitors. ACS Med. Chem. Lett. 7 (3), 324–329. doi:10.1021/acsmedchemlett.5b00472
Bondar, D., and Karpichev, Y. (2024). Poly(ADP-Ribose) polymerase (PARP) inhibitors for cancer therapy: advances, challenges, and future directions. Biomolecules 14 (10), 1269. doi:10.3390/biom14101269
Chau, N. M., and Ashcroft, M. (2004). Akt2: a role in breast cancer metastasis. Breast Cancer Res. 6 (1), 55–57. doi:10.1186/bcr739
Chen, M.-F., Chen, W.-C., Chang, Y.-J., Wu, C.-F., and Wu, C.-T. (2010). Role of DNA methyltransferase 1 in hormone-resistant prostate cancer. J. Mol. Med. 88 (9), 953–962. doi:10.1007/s00109-010-0640-3
Cheng, X. (2024). A comprehensive review of HER2 in cancer biology and therapeutics. Genes (Basel) 15 (7), 903. doi:10.3390/genes15070903
Deshmukh, D., and Qiu, Y. (2015). Role of PARP-1 in prostate cancer. Am. J. Clin. Exp. Urol 3 (1), 1–12.
Dilmac, S., and Ozpolat, B. (2023). Mechanisms of PARP-inhibitor-resistance in BRCA-mutated breast cancer and new therapeutic approaches. Cancers (Basel) 15 (14), 3642. doi:10.3390/cancers15143642
Genheden, S., and Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10 (5), 449–461. doi:10.1517/17460441.2015.1032936
Graham, T. A., Ferkey, D. M., Mao, F., Kimelman, D., and Xu, W. (2001). Tcf4 can specifically recognize β-catenin using alternative conformations. Nat. Struct. Biol. 8 (12), 1048–1052. doi:10.1038/nsb718
Heerding, D. A., Rhodes, N., Leber, J. D., Clark, T. J., Keenan, R. M., Lafrance, L. V., et al. (2008). Identification of 4-(2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-{[(3S)-3-piperidinylmethyl]oxy}-1H-imidazo[4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a novel inhibitor of AKT kinase. J. Med. Chem. 51 (18), 5663–5679. doi:10.1021/jm8004527
Hevener, K. E., Zhao, W., Ball, D. M., Babaoglu, K., Qi, J., White, S. W., et al. (2009). Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model 49 (2), 444–460. doi:10.1021/ci800293n
Hunter, J. C., Gurbani, D., Ficarro, S. B., Carrasco, M. A., Lim, S. M., Choi, H. G., et al. (2014). In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. 111(24), 8895–8900. doi:10.1073/pnas.1404639111
Ishikawa, T., Seto, M., Banno, H., Kawakita, Y., Oorui, M., Taniguchi, T., et al. (2011). Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/Epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold. J. Med. Chem. 54 (23), 8030–8050. doi:10.1021/jm2008634
Jain, A. N. (2008). Bias, reporting, and sharing: computational evaluations of docking methods. J. Comput. Aided Mol. Des. 22 (3-4), 201–212. doi:10.1007/s10822-007-9151-x
Keter, F. K. G., Nell, I. A., Zyl, M., and Darkwa, W. E. (2014). Phosphinogold(I) dithiocarbamate complexes: effect of the nature of phosphine ligand on anticancer properties inorg. Chem 53 (4), 2058–2067. doi:10.1021/ic4025926
Kim, J. H., Ofori, S., Parkin, S., Vekaria, H., Vekaria, H., Sullivan, P. G., and Awuah, S. G. (2021). Anticancer gold(III)-bisphosphine complex alters the mitochondrial electron transport chain to induce in vivo tumor inhibition. Chem. Sci. 12, 7467–7479. doi:10.1039/D1SC01418H
Kim, J. H. R., Parkin, E., and Awuah, S. (2019). Gold(I/III)-Phosphine complexes as potent antiproliferative agents. Sci. Rep. 9 (1), 12335. doi:10.1038/s41598-019-48584-5
Li, J., Abel, R., Zhu, K., Cao, Y., Zhao, S., and Friesner, R. A. (2011). The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins 79 (10), 2794–2812. doi:10.1002/prot.23106
Lin, X., Murray, J. M., Rico, A. C., Wang, M. X., Chu, D. T., Zhou, Y., et al. (2006). Discovery of 2-pyrimidyl-5-amidothiophenes as potent inhibitors for AKT: synthesis and SAR studies. Bioorg. & Med. Chem. Lett., 16(16), 4163–4168. doi:10.1016/j.bmcl.2006.05.092
Liu, J., Li, S., Wang, Q., Feng, Y., Xing, H., Yang, X., et al. (2024). Sonrotoclax overcomes BCL2 G101V mutation–induced venetoclax resistance in preclinical models of hematologic malignancy. Blood 143 (18), 1825–1836. doi:10.1182/blood.2023019706
Löwe, J., Li, H., Downing, K. H., and Nogales, E. (2001). Refined structure of αβ-tubulin at 3.5 Å resolution11Edited by I. A. Wilson. J. Mol. Biol. 313(5), 1045–1057. doi:10.1006/jmbi.2001.5077
Massie, C. E., Mills, I. G., and Lynch, A. G. (2017). The importance of DNA methylation in prostate cancer development. J. Steroid Biochem. Mol. Biol. 166, 1–15. doi:10.1016/j.jsbmb.2016.04.009
Mohd Kamal, K., Ghazali, A. R., Ab Mutalib, N. S., Abu, N., Chua, E. W., and Masre, S. F. (2024). The role of DNA methylation and DNA methyltransferases (DNMTs) as potential biomarker and therapeutic target in non-small cell lung cancer (NSCLC). Heliyon, 10(19), e38663. doi:10.1016/j.heliyon.2024.e38663
Muddagoni, N., Bathula, R., Dasari, M., and Potlapally, S. (2021). Homology modeling, virtual screening, Prime- MMGBSA, AutoDock-identification of inhibitors of FGR protein. Biointerface Res. Appl. Chem. 11, 11088–11103. doi:10.33263/BRIAC114.1108811103
Nag, S., Zhang, X., Srivenugopal, K. S., Wang, M. H., Wang, W., and Zhang, R. (2014). Targeting MDM2-p53 interaction for cancer therapy: are we there yet? Curr. Med. Chem. 21(5), 553–557. doi:10.2174/09298673113206660325
Nath, P., Datta, A., and Adhikari, S. (2023). Recent advances of metal-based anticancer agents and their in vivo potential against various types of malignancies. In Handbook of Animal Models and its Uses in Cancer Research. Editors: S. Pathak, A. Banerjee, and A. Bisgin (Springer Nature Singapore), 917–943. doi:10.1007/978-981-19-3824-5_47
Ndagi, U. M., Soliman, N., and M, E. (2017). Metal complexes in cancer therapy – an update from drug design perspective. Drug Des. Devel Ther. 11, 599–616. doi:10.2147/DDDT.S119488
Page, N., Wappett, M., O’Dowd, C. R., O’Rourke, M., Gavory, G., Zhang, L., et al. (2022). Identification and development of a subtype-selective allosteric AKT inhibitor suitable for clinical development. Sci. Rep. 12 (1), 15715. doi:10.1038/s41598-022-20208-5
Penning, T. D., Zhu, G.-D., Gong, J., Thomas, S., Gandhi, V. B., Liu, X., et al. (2010). Optimization of phenyl-substituted benzimidazole carboxamide poly(ADP-ribose) polymerase inhibitors: identification of (S)-2-(2-Fluoro-4-(pyrrolidin-2-yl)phenyl)-1H-benzimidazole-4-carboxamide (A-966492), a highly potent and efficacious inhibitor. J. Med. Chem. 53 (8), 3142–3153. doi:10.1021/jm901775y
Puentes-Pardo, J. D., Moreno-SanJuan, S., Casado, J., Escudero-Feliu, J., López-Pérez, D., Sánchez-Uceta, P., et al. (2023). PARP-1 expression influences cancer stem cell phenotype in colorectal cancer depending on p53. Int. J. Mol. Sci 24 (5). doi:10.3390/ijms24054787
Riggio, M., Perrone, M. C., Polo, M. L., Rodriguez, M. J., May, M., Abba, M., et al. (2017). AKT1 and AKT2 isoforms play distinct roles during breast cancer progression through the regulation of specific downstream proteins. Sci. Rep. 7, 44244. doi:10.1038/srep44244
Rubin, E., Shan, K. S., Dalal, S., Vu, D. U. D., Milillo-Naraine, A. M., Guaqueta, D., et al. (2024). Molecular targeting of the human epidermal growth factor receptor-2 (HER2) genes across various cancers. Int. J. Mol. Sci. 25 (2), 1064. doi:10.3390/ijms25021064
Rychahou, P. G., Kang, J., Gulhati, P., Doan, H. Q., Chen, L. A., Xiao, S.-Y., et al. (2008). Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc. Natl. Acad. Sci. 105(51), 20315–20320. doi:10.1073/pnas.0810715105
Sankarganesh, M. R., Revathi, J. D., Solomon, N., and Kumar, R. V. (2019). Gold(III) complex from pyrimidine and morpholine analogue Schiff base ligand: synthesis, characterization, DFT, TDDFT, catalytic, anticancer, molecular modeling with DNA and BSA and DNA binding studies J. Mol. Liq. 294, 111655. doi:10.1016/j.molliq.2019.111655
Schmidtova, S., Kalavska, K., Liskova, V., Plava, J., Miklikova, S., Kucerova, L., et al. (2021). Targeting of deregulated wnt/β-catenin signaling by PRI-724 and LGK974 inhibitors in germ cell tumor cell lines. Int. J. Mol. Sci. 22 (8), 4263. doi:10.3390/ijms22084263
Schulze-Gahmen, U., De Bondt, H. L., and Kim, S.-H. (1996). High-Resolution crystal structures of human cyclin-dependent kinase 2 with and without ATP: bound waters and natural ligand as guides for inhibitor design. J. Med. Chem. 39 (23), 4540–4546. doi:10.1021/jm960402a
Selivanov, V. A. V., Pivtoraiko, T. V., Zeak, V. N., Sukhomlin, J., Trucco, T. & M., Trucco, M., et al. (2011). Reactive oxygen species production by forward and reverse electron fluxes in the mitochondrial respiratory chain. PLoS Comput. Biol. 7 (3), 1001115. doi:10.1371/journal.pcbi.1001115
Sgobba, M., Caporuscio, F., Anighoro, A., Portioli, C., and Rastelli, G. (2012). Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 58, 431–440. doi:10.1016/j.ejmech.2012.10.024
Stamos, J., Sliwkowski, M. X., and Eigenbrot, C. (2002). Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 277 (48), 46265–46272. doi:10.1074/jbc.M207135200
Steven (2003). Cell signaling and cancer. Cancer Cell 4 (3), 167–174. doi:10.1016/s1535-6108(03)00216-2
Su, B., Zhang, L., Zhuang, W., Zhang, W., and Chen, X. (2021). Knockout of Akt1/2 suppresses the metastasis of human prostate cancer cells CWR22rv1 in vitro and in vivo. J. Cell Mol. Med. 25 (3), 1546–1553. doi:10.1111/jcmm.16246
Tzelepi, V., Logotheti, S., Efstathiou, E., Troncoso, P., Aparicio, A., Sakellakis, M., et al. (2020). Epigenetics and prostate cancer: defining the timing of DNA methyltransferase deregulation during prostate cancer progression. Pathology 52 (2), 218–227. doi:10.1016/j.pathol.2019.10.006
Wang, H., Guo, M., Wei, H., and Chen, Y. (2023). Targeting p53 pathways: mechanisms, structures and advances in therapy. Signal Transduct. Target. Ther. 8 (1), 92. doi:10.1038/s41392-023-01347-1
Yang, H., Rudge, D. G., Koos, J. D., Vaidialingam, B., Yang, H. J., and Pavletich, N. P. (2013). mTOR kinase structure, mechanism and regulation. Nature 497 (7448), 217–223. doi:10.1038/nature12122
Yang, J., Cron, P., Good, V. M., Thompson, V., Hemmings, B. A., and Barford, D. (2002). Crystal structure of an activated Akt/Protein Kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat. Struct. Biol. 9 (12), 940–944. doi:10.1038/nsb870
Yip, H. Y. K., and Papa, A. (2021). Signaling pathways in cancer: therapeutic targets, combinatorial treatments, and new developments. Cells 10 (3), 659. doi:10.3390/cells10030659
Yu, M. Z., Fang, Y., Jehan, M., Zhou, S., and Zhou, W. (2022). Current advances of Nano medicines delivering arsenic trioxide for enhanced tumor therapy. Pharmaceutics 14, 743. doi:10.3390/pharmaceutics14040743
Zarewa, S. A., Binobaid, L., Sulaiman, A. A. A., Sobeai, H. M. A., Alotaibi, M., Alhoshani, A., et al. (2023). Synthesis, Characterization, and anticancer activity of phosphanegold(i) complexes of 3-thiosemicarbano-butan-2-one oxime. Biomedicines 11 (9). doi:10.3390/biomedicines11092512
Zhang, C., Spevak, W., Zhang, Y., Burton, E. A., Ma, Y., Habets, G., et al. (2015). RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526 (7574), 583–586. doi:10.1038/nature14982
Zhang, X., Perez-Sanchez, H., and Lightstone, F. C. (2017). A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Curr. Top. Med. Chem. 17 (14), 1631–1639. doi:10.2174/1568026616666161117112604
Zhang, Z.-M., Liu, S., Lin, K., Luo, Y., Perry, J. J., Wang, Y., et al. (2015). Crystal structure of human DNA methyltransferase 1. J. Mol. Biol. 427 (15), 2520–2531. doi:10.1016/j.jmb.2015.06.001
Keywords: anticancer activity, cytotoxicity, MM-GBSA, molecular docking, structure activity relationship, thiocarbohydrate Phosphinogold(I) complexes
Citation: Mohamed AAK, Asiamah I, Elamin G, Darkwa J and Adokoh CK (2025) In silico molecular studies of Phosphinogold(I) thiocarbohydrate complexes: insights into multi-target anticancer mechanisms. Front. Chem. 13:1533026. doi: 10.3389/fchem.2025.1533026
Received: 22 November 2024; Accepted: 08 May 2025;
Published: 12 June 2025.
Edited by:
Sam P. De Visser, The University of Manchester, United KingdomReviewed by:
Nurul Huda Abd Karim, National University of Malaysia, MalaysiaMing Yueh Tan, Tunku Abdul Rahman University College, Malaysia
Copyright © 2025 Mohamed, Asiamah, Elamin, Darkwa and Adokoh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian K. Adokoh, Y2Fkb2tvaEB1Y2MuZWR1Lmdo