 Erika Medel
Erika Medel Rubicelia Vargas
Rubicelia Vargas- Departamento de Química, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana-Iztapalapa, México City, Mexico
Non-covalent interactions are fundamental for understanding the chemical behavior of porous materials with guest molecules, which is key for designing new materials. The Quantum Theory of Atoms in Molecules has enabled us to visualize and analyze non-covalent interactions in host-guest systems, particularly with Metal-Organic Frameworks (MOFs) as hosts. Using this tool, we have investigated the adsorption mechanisms of highly polluting gases such as CO and
1 Introduction
Research on Metal-Organic Frameworks (MOFs) has grown significantly since the late 20th century. The exceptional tunability of MOFs makes them unique materials suitable for various applications (Maleki and Taheri-Ledari, 2023), some of the most common being gas adsorption for purposes such as environmental purification, storage, or mixture separation, and drug delivery. Several articles have analyzed these applications and their theoretical studies in detail (Li et al., 2024; Peralta, 2024; Davis et al., 2025).
However, there are still challenges to overcome for the application of MOFs, such as increasing stability in water and designing structures with specific functions and properties (Khafaga et al., 2024). To address these challenges, the main avenue is the modulation of non-covalent interactions. The high efficiency of MOFs is often associated with the combined action of various interactions. It has also been shown that the stability of certain solid materials relies on the effect that hydrogen bonding networks have on the formation and electronic properties of these systems (Rajapaksha et al., 2023).
Physisorption and chemisorption are both well-established mechanisms of molecules sorption in MOFs (Sánchez-Serratos et al., 2016; Petit, 2018). In both, the nature of the metal or ligand plays a crucial role, as these factors significantly influence the intermolecular interactions between guest molecules and the MOF. Non-covalent interactions, such as hydrogen bonding, electrostatic forces, and dispersion forces, (Schneider, 2022), largely determine the interaction energy between gas molecules and the MOF.
The presence of unsaturated metal sites (UMS) (Kökçam-Demir et al., 2020) under dry conditions can promote strong physisorption or even chemisorption. While this can enhance guest uptake, it may also have undesirable effects, such as compromising the structural integrity of the MOF or hindering material recyclability.
Functionalization is an effective strategy for modulating the molecular interactions and consequently the interaction energy in MOFs (Medel et al., 2023b). This can be accomplished by modifying the ligand or altering the pore environment (Mandal et al., 2021; Eddaoudi et al., 2002). In the latter case, polar solvent molecules, such as methanol or water are commonly used. These solvent molecules primarily interact with UMS or with the inorganic metal cluster, to prevent excessive physisorption while maintaining the material’s guest uptake capacity.
Hydrogen bonds, both conventional and unconventional, are widely recognized as the primary intermolecular interactions in various chemical systems (Steiner, 2002). However, there are other types of important intermolecular interactions that require further exploration (Hobza et al., 2010; Schneider, 2022). These include dihydrogen bonds (Grabowski, 2013), H
We have employed theoretical and computational chemistry to investigate the nature of non-covalent interactions involved in gas adsorption within MOFs. The analysis of electron density critical points provides valuable insights into the strength and nature of these interactions (Johnson et al., 2010), framed within the Quantum Theory of Atoms in Molecules (QTAIM) (Bader, 1990). The electron density, being an observable property, can be determined either experimentally or through theoretical methods. In our group, the electron density is calculated using Density Functional Theory (DFT) (Parr, 1983) within the Kohn–Sham (Kohn and Sham, 1965; Hohenberg and Kohn, 1964) framework. The subsequent analysis to identify and classify critical points is conducted using GPUAM (Graphics Processing Units for Atoms and Molecules) (Cruz et al., 2019; Hernández-Esparza et al., 2014; Hernández-Esparza et al., 2019), a specialized software developed in-house. Recently, the use of QTAIM and other electron density-based tools for describing systems dominated by intermolecular interactions has increased. However, molecular finite models predominate, which often limits the scope of the methodology and highlights the importance of using periodic calculations (Santibañez and Mendizabal, 2023). QTAIM analysis in these systems presents specific challenges, particularly because atomic basins can adopt complex geometries in crystals to accommodate ring and cage critical points de-la Roza et al. (2009). Using this methodology, we have conducted several studies on the intermolecular interactions in MOFs designed for gas trapping (Lara-García et al., 2019; Sánchez-Bautista et al., 2019; Garrido-Olvera et al., 2019; Barrios-Vargas et al., 2020; Landeros-Rivera et al., 2020; Rivera-Almazo et al., 2021). These investigations have provided a deeper understanding of the mechanisms underlying gas adsorption and the role of non-covalent interactions in these materials.
With the experience gained from studying non-covalent interactions in the adsorption of pollutant gases by MOFs, we have extended our research to another critical application of MOFs: their use as Drug Delivery Systems (DDS) (Medel et al., 2023b; Medel et al., 2023a). Drug delivery systems (DDS) refer to formulations or devices designed to distribute therapeutic substances throughout the body, improving their efficacy and helping to reduce side effects (Jain, 2008). Over the past decade, the number of publications proposing MOFs as DDSs has increased. However, some crucial features in the development of these systems remain a challenge, such as controlled drug release to avoid sudden release peaks and protection of guest molecules.
In this context, we have examined the role of pore functionalization in biocompatible MOFs (BioMOFs) (Tibbetts and Kostakis, 2020), which can be also bioinspired. In the development of these systems, in addition to biocompatibility, key features such as controlled drug release to avoid sudden bursts, and the protection of guest molecules are of utmost importance. In these characteristics, intermolecular interactions play a crucial role, influencing the stability, efficiency, and functionality of the DDS (He et al., 2021; Wang et al., 2020; Kumar et al., 2020).
In this paper, we review our contributions to two key topics: gas adsorption in MOFs and the use of BioMOFs as DDS. This review highlights the importance of characterizing non-covalent interactions in the precise design of these materials. Additionally, we emphasize the potential of theoretical and computational chemistry of periodic systems to effectively contribute to understanding the encapsulation mechanisms in MOFs and the potential it has to study other types of materials such as Hydrogen-Bonded Organic Frameworks (HOF) and Covalent Organic Framework (COFs). Furthermore, we present new theoretical results in which we propose the use of a BioMOF as a DDS for phenylethylamine derivatives.
2 Non-covalent interactions in MOFs
2.1 Gas adsorption
Environmental gases represent a major problem worldwide. Their complex composition hinders their adsorption; however, due to the tunability and adsorptive properties of MOFs, they are favorable candidates for this application (Wang et al., 2025).
In this topic, we elucidated the mechanism behind the enhancement of
Building on the study of pollutant gas adsorption by InOF-1, we investigated CO capture with this MOF and identified two key interactions:
From a different perspective, some practical applications of MOFs require the activation of unsaturated metal sites (UMS), as these sites are often occupied by Lewis-Base (LB) solvent molecules. Common methods for removing LB solvent molecules and activating the UMS typically involve harsh conditions as supercritical
The study focused on the HKUST-1 MOF, which exhibits a specific coordination between Cu–Cu paddlewheel nodes and the oxygen atoms of the ligand. In this structure, all UMS at the Cu centers, where LB solvent molecules can bind, are oriented toward the pore, making them accessible to guest molecules. To better understand the experimental findings of the proposed technique, DFT computations and electron density analysis were conducted. The DFT results showed strong agreement with our experimental observations, further validating the approach (Díaz-Ramírez et al., 2025).
2.2 Drug delivery systems
The use of biomolecules as ligands for metal bonding has given rise to a new class of MOFs, known as BioMOFs, with improved biocompatibility and specific functionality (Nabipour et al., 2020). Although the biomedical use of BioMOFs is still in its early stages, reports indicate superior characteristics of BioMOFs compared to conventional bioorganic or inorganic systems (Cai et al., 2019).
Phenylethylamine (PHEA) derivatives give rise to a wide variety of compounds related to drugs and neuroreceptors (Khan et al., 2012). Therefore, studying the encapsulation of this molecule in BioMOFs is highly relevant to advancing the understanding and design of Drug Delivery Systems (DDS). So, we investigated the encapsulation of PHEA and its derivative (Figure 1), the neuroreceptor dopamine (DA), in SU-101 BioMOFs using computational chemistry methods. Additionally, we explored the functionalization of these systems with
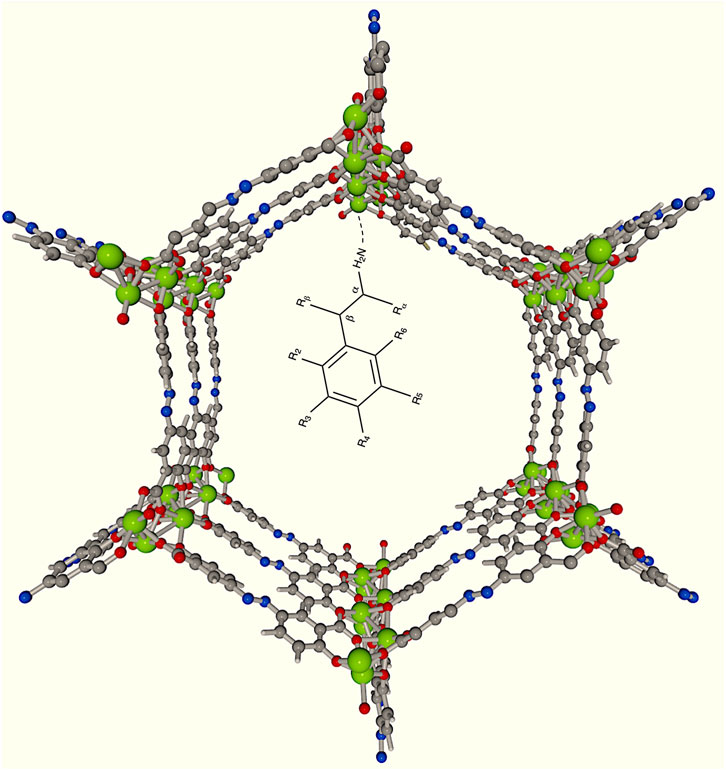
Figure 1. Schematic representation of the encapsulation of PHEA and its possible derivatives in a
SU-101 BioMOFs are bioinspired MOFs that are biocompatible. They are highly stable, functionalizable, and have great potential for drug delivery, as they remain unchanged at wide pH ranges, from 2 to 14. In addition, they have been exposed to simulated physiological conditions with favorable results and have a particle size suitable for biological applications (Grape et al., 2020). BioMOF SU-101 are composed of ellagic acid as ligands, these molecules are antioxidants, and the node is formed by Bi metal, which interacts with different oxygens of the MOF structure and presents an unsaturated metal site where guest molecules may interact. Thus, our results indicate that both PHEA and DA form a Bi
To study the effect of pore functionalization we started by including a water molecule with a theoretical stochastic method (García et al., 2019). This molecule formed a Bi
Finally, functionalization with a MeOH molecule caused DA to shift slightly toward the UMS and realign the
We explored the DDS design with
The most stable geometry obtained with the PHEA molecule as a guest exhibits an alkaline earth interaction, Mg
2.3 Systematic study of phenylethylamine family
Building on the previous study, in which various PHEA and DA geometries were analyzed within
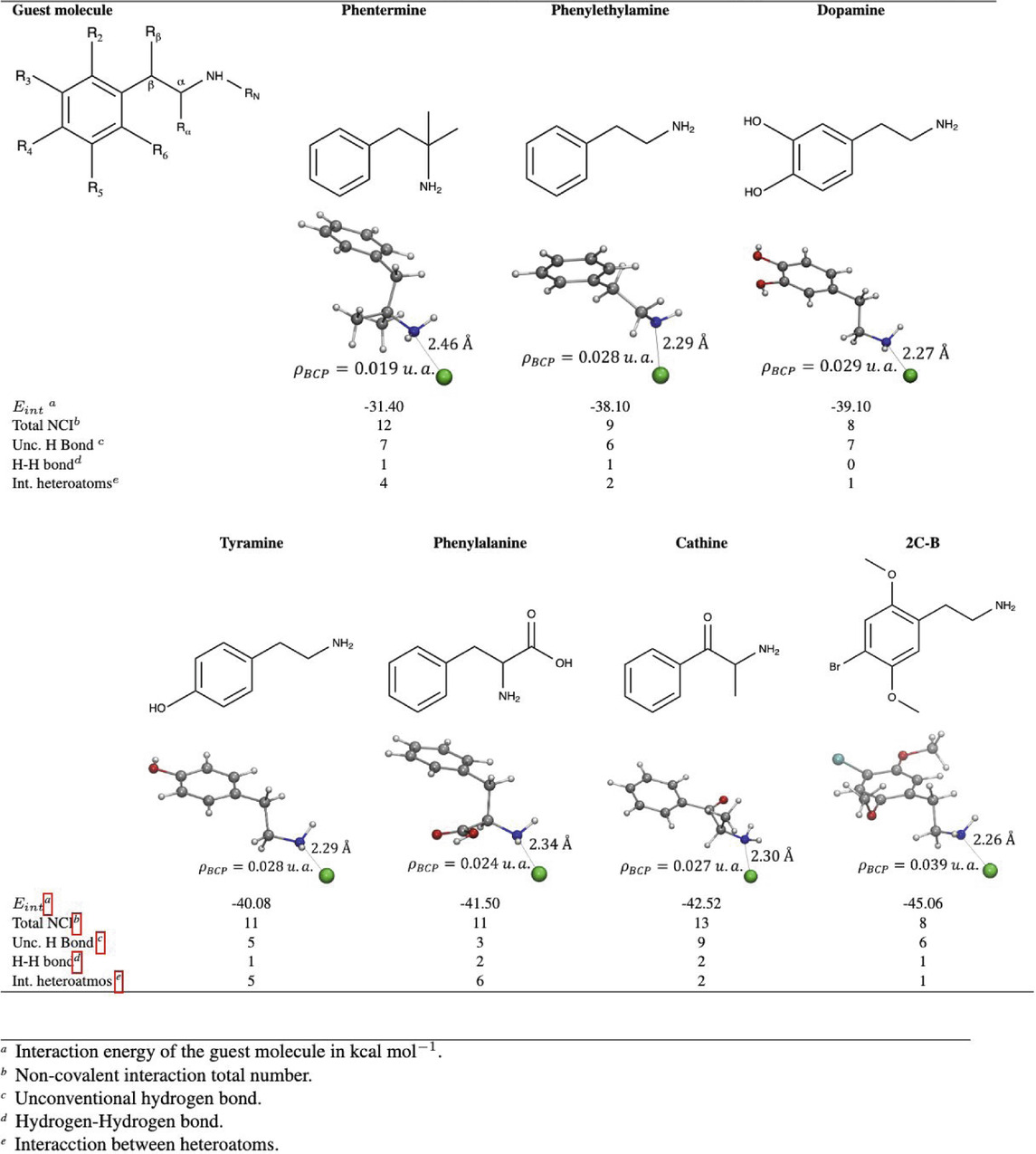
Table 1. Interaction energy and non-covalent interactions determined using QTAIM, Guest@Mg2(olz) systems. The colors of the atoms are as follows: gray C, red O, white H, blue N, cyan Br and green Mg.
The molecules with functional groups most similar to PHEA are phentermine (PHE) and dopamine (DA). They present similar
The
On the other hand, for phenylalanine and cathine the highest percentage contribution to the
Based on our analysis, it is shown that phentermine, dopamine, tyramine, cathine and 2C-B could be encapsulated in
These analysis shows that molecules without substituents near the amine, such as dopamine and tyramine, present the highest contribution to the
3 Discussion
We have shown, through theoretical and computational chemistry calculations, that it is possible to tune the interaction energy by functionalizing the pores of MOFs and BioMOFs.
The methodology we have followed, both in previous studies and in the present one, allows us to analyze the effect of pore functionalization as well as the substituent effect on the guest molecule concerning its interaction energy with the host. This methodology offers several possibilities for further exploring functionalization in the pores or the structure of MOFs. We believe that these theoretical tools and methods can contribute to the rational design of drug carriers, as well as materials for adsorbing pollutant gases.
An important pending task is including solvent effects, specially in DDS, where explicit interactions strongly influence drug release. Although challenging, finite models based on the geometries with the methodology presented here could be used with QM/MM methods or surface periodic models combined with molecular dynamics may prove useful.
From our perspective, it is possible to test the potential of a MOF or a BioMOF as a host, depending on the application, before conducting experiments. A new perspective is to test Hydrogen-bonded and Covalent Organic Framework (HOFs and COFs), such as DDS and for water treatment, respectively. In this way, the type of theoretical calculations performed in our studies can guide the design of specific materials for specific applications. HOFs are promising materials for biomedical applications due to their excellent biocompatibility, low toxicity, and high flexibility. Highly stable COFs are excelling in contaminant adsorption and heterogeneous catalysis. In HOFs, the ligands are assembled by hydrogen bonds, making the analysis of non-covalent interactions crucial to understanding these materials. While, in addition to stability, COFs present highly functionalizable ligands, which motivates us to test new horizons of functionalization.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
EM: Conceptualization, Formal Analysis, Investigation, Methodology, Writing – review and editing. RV: Conceptualization, Formal Analysis, Investigation, Methodology, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The discounted publication fee was funded by Universidad Autónoma Metropolitana.
Acknowledgments
We thank to the Laboratorio de Supercómputo y Visualización en Paralelo at the Universidad Autónoma Metropolitana-Iztapalapa for access to their computer facilities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1579977/full#supplementary-material
References
Alkorta, I., Elguero, J., and Frontera, A. (2020). Not only hydrogen bonds: other noncovalent interactions. Crystals 10, 180. doi:10.3390/cryst10030180
Barrios-Vargas, L. J., Ruiz-Montoya, J. G., Landeros-Rivera, B., Álvarez, J. R., Alvarado-Alvarado, D., Vargas, R., et al. (2020). Confined benzene within InOF-1: contrasting CO2 and SO2 capture behaviours. Dalton Trans. 49, 2786–2793. doi:10.1039/c9dt04667d
Becke, A. D. (1993). A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 98, 1372–1377. doi:10.1063/1.464304
Cai, H., Huang, Y. L., and Li, D. (2019). Biological metal–organic frameworks: structures, host–guest chemistry and bio-applications. Coord. Chem. Rev. 378, 207–221. doi:10.1016/j.ccr.2017.12.003
Civalleri, B., Zicovich-Wilson, C. M., Valenzano, L., and Ugliengo, P. (2008). B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 10, 405–410. doi:10.1039/b715018k
Cruz, J. C., Hernández-Esparza, R., Álvaro, V.-M., Vargas, R., and Garza, J. (2019). Implementation of the molecular electrostatic potential over graphics processing units. J. Chem. Inf. Model. 59, 3120–3127. doi:10.1021/acs.jcim.8b00951
Davis, S., Athira, E., and Rajan, V. K. (2025). Density functional theory to decrypt metal-organic framework-a review. Comput. Mater. Sci. 247, 113537. doi:10.1016/j.commatsci.2024.113537
de-la Roza, A. O., Blanco, M. A., Pendás, A. M., and Luaña, V. (2009). Critic: a new program for the topological analysis of solid-state electron densities. Comput. Phys. Commun. 180, 157–166. doi:10.1016/j.cpc.2008.07.018
Díaz-Ramírez, M. L., Park, S. H., Rivera-Almazo, M., Medel, E., Peralta, R. A., Ibarra, I. A., et al. (2025). Gas-flow activation of MOFs: unlocking efficient catalysis through dynamic bonding. Chem. Sci. 5, 2581–2588. doi:10.1039/d4sc07011a
Eddaoudi, M., Kim, J., Rosi, N., Vodak, D., Wachter, J., O’Keeffe, M., et al. (2002). Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. SCIENCE 295, 469–472. doi:10.1126/science.1067208
Farha, O. K., Eryazici, I., Jeong, N. C., Hauser, B. G., Wilmer, C. E., Sarjeant, A. A., et al. (2012a). Metal-organic framework materials with ultrahigh surface areas: is the sky the limit? J. Am. Chem. Soc. 134, 15016–15021. doi:10.1021/ja3055639
Farha, O. K., Wilmer, C. E., Eryazici, I., Hauser, B. G., Parilla, P. A., Oneill, K., et al. (2012b). Designing higher surface area metal-organic frameworks: are triple bonds better than phenyls? J. Am. Chem. Soc. 134, 9860–9863. doi:10.1021/ja302623w
García, J. J., Hernández-Esparza, R., Vargas, R., Tiznado, W., and Garza, J. (2019). Formation of small clusters of NaCl dihydrate in the gas phase. New J. Chem. 43, 4342–4348. doi:10.1039/c8nj06315j
Garrido-Olvera, L. P., Sanchez-Bautista, J. E., Alvarado-Alvarado, D., Landeros-Rivera, B., Álvarez, J. R., Vargas, R., et al. (2019). Confined toluene within InOF-1: CO2 capture enhancement. RSC Adv. 9, 32864–32872. doi:10.1039/c9ra05991a
Grabowski, S. J. (2013). Dihydrogen bond and x-h⋯σ interaction as sub-classes of hydrogen bond. J. Phys. Org. Chem. 26, 452–459. doi:10.1002/poc.3109
Grape, E. S., Flores, J. G., Hidalgo, T., Martínez-Ahumada, E., Gutiérrez-Alejandre, A., Hautier, A., et al. (2020). A robust and biocompatible bismuth ellagate MOF synthesized under green ambient conditions. J. Am. Chem. Soc. 142, 16795–16804. doi:10.1021/jacs.0c07525
He, S., Wu, L., Li, X., Sun, H., Xiong, T., Liu, J., et al. (2021). Metal-organic frameworks for advanced drug delivery. Acta Pharm. Sin. B 11, 2362–2395. doi:10.1016/j.apsb.2021.03.019
Hernández-Esparza, R., Álvaro, V.-M., Soriano-Agueda, L. A., Vargas, R., and Garza, J. (2019). Gpus as boosters to analyze scalar and vector fields in quantum chemistry. Int. J. Quantum Chem. 119, 1–13. doi:10.1002/qua.25671
Hernández-Esparza, R., Mejía-Chica, S. M., Zapata-Escobar, A. D., Guevara-García, A., Martínez-Melchor, A., Hernández-Pérez, J. M., et al. (2014). Grid-based algorithm to search critical points, in the electron density, accelerated by graphics processing units. J. Comput. Chem. 35, 2272–2278. doi:10.1002/jcc.23752
Hobza, P., Müller-Dethlefs, K., Chemistry, of, and Great Britain), R. S. (2010). Non-covalent interactions: theory and experiment
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous electron gas. Phys. Rev. 136, B864–B871. doi:10.1103/PhysRev.136.B864
Johnson, E. R., Keinan, S., Mori-Sánchez, P., Contreras-García, J., Cohen, A. J., and Yang, W. (2010). Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506. doi:10.1021/ja100936w
Khafaga, D. S., El-Morsy, M. T., Faried, H., Diab, A. H., Shehab, S., Saleh, A. M., et al. (2024). Metal-organic frameworks in drug delivery: engineering versatile platforms for therapeutic applications. RSC Adv. 14, 30201–30229. doi:10.1039/d4ra04441j
Khan, J. I., Kennedy, T. J., and Christian, J. D. R. (2012). Basic principles of forensic chemistry. Humana Press. doi:10.1007/978-1-59745-437-7
Kohn, W., and Sham, L. J. (1965). Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138. doi:10.1103/physrev.140.a1133
Kökçam-Demir, Ü., Goldman, A., Esrafili, L., Gharib, M., Morsali, A., Weingart, O., et al. (2020). Coordinatively unsaturated metal sites (open metal sites) in metal-organic frameworks: design and applications. Chem. Soc. Rev. 49, 2751–2798. doi:10.1039/c9cs00609e
Kotzabasaki, M., and Froudakis, G. E. (2018). Review of computer simulations on anti-cancer drug delivery in MOFs. Inorg. Chem. Front. 5, 1255–1272. doi:10.1039/c7qi00645d
Kumar, S., Jain, S., Nehra, M., Dilbaghi, N., Marrazza, G., and Kim, K. H. (2020). Green synthesis of metal–organic frameworks: a state-of-the-art review of potential environmental and medical applications. Coord. Chem. Rev. 420, 213407. doi:10.1016/j.ccr.2020.213407
Landeros-Rivera, B., Ibarra, I. A., Díaz-Ramírez, M. L., Vargas, R., Lara-García, H. A., Garza, J., et al. (2020). A detailed description of the CO molecule adsorbed in InOF-1. Phys. Chem. Chem. Phys. 22, 7969–7974. doi:10.1039/d0cp00579g
Lara-García, H. A., Landeros-Rivera, B., González-Zamora, E., Aguilar-Pliego, J., Gómez-Cortés, A., Martínez, A., et al. (2019). Relevance of hydrogen bonding in CO2 capture enhancement within InOF-1: an energy and vibrational analysis. Dalton Trans. 48, 8611–8616. doi:10.1039/c9dt01266d
Levine, D. J., Runčevski, T., Kapelewski, M. T., Keitz, B. K., Oktawiec, J., Reed, D. A., et al. (2016). Olsalazine-based metal-organic frameworks as biocompatible platforms for H2 adsorption and drug delivery. J. Am. Chem. Soc. 138, 10143–10150. doi:10.1021/jacs.6b03523
Li, D., Yadav, A., Zhou, H., Roy, K., Thanasekaran, P., and Lee, C. (2024). Advances and applications of metal-organic frameworks (MOFs) in emerging technologies: a comprehensive review. Glob. Challenges 8, 2300244. doi:10.1002/gch2.202300244202300244
Ma, L., Jin, A., Xie, Z., and Lin, W. (2009). Freeze drying significantly increases permanent porosity and hydrogen uptake in 4,4-connected metal-organic frameworks. Angew. Chem. - Int. Ed. 48, 9905–9908. doi:10.1002/anie.200904983200904983
Maleki, A., and Taheri-Ledari, R. (2023). Physicochemical aspects of metal-organic Frameworks: a new class of coordinative materials. Springer Nature. doi:10.1007/978-3-031-18675-2
Mandal, S., Natarajan, S., Mani, P., and Pankajakshan, A. (2021). Post-synthetic modification of metal–organic frameworks toward applications. Adv. Funct. Mater. 31, 2006291. doi:10.1002/adfm.202006291
Medel, E., Garza, J., Ibarra, I. A., Martínez, A., and Vargas, R. (2023a). Non-covalent interactions in biocompatible platforms for drug delivery: Mg2(olsalazine) metal-organic framework with phenylethylamine, dopamine and sertraline. Comput. Theor. Chem. 1228, 114265. doi:10.1016/j.comptc.2023.114265
Medel, E., Obeso, J. L., Serrano-Fuentes, C., Garza, J., Ibarra, I. A., Leyva, C., et al. (2023b). Encapsulation of dopamine within su-101: insights by computational chemistry. Chem. Commun. 59, 8684–8687. doi:10.1039/d3cc02304d
Nabipour, H., Mozafari, M., and Hu, Y. (2020). BioMOFs. Elsevier Inc., 321–345. doi:10.1016/b978-0-12-816984-1.00017-2
Oliveira, D. V., Laun, J., Peintinger, M. F., and Bredow, T. (2019). Bsse-correction scheme for consistent Gaussian basis sets of double- and triple-zeta valence with polarization quality for solid-state calculations. J. Comput. Chem. 40, 2364–2376. doi:10.1002/jcc.26013
Osorio-Toribio, G., de, J., Velásquez-Hernández, M., Mileo, P. G., Zárate, J. A., Aguila-Rosas, J., et al. (2020). Controlled transdermal release of antioxidant ferulate by a porous Sc(III) MOF. iScience 23, 101156. doi:10.1016/j.isci.2020.1011562020.101156
Parr, R. G. (1983). Density functional theory. Annu. Rev. Phys. Chem. 34, 631–656. doi:10.1146/annurev.pc.34.100183.003215
Peralta, R. A. (2024). La química detrás de los MOFs: sus grandes aplicaciones. Contactos, Rev. Educ. Ciencias Ing., 69–76.
Petit, C. (2018). Present and future of MOF research in the field of adsorption and molecular separation. Curr. Opin. Chem. Eng. 20, 132–142. doi:10.1016/j.coche.2018.04.004
Rajapaksha, H., Augustine, L. J., Mason, S. E., and Forbes, T. Z. (2023). Guiding principles for the rational design of hybrid materials: use of DFT methodology for evaluating non-covalent interactions in a uranyl tetrahalide model system. Angew. Chem. - Int. Ed. 135, e202305073. doi:10.1002/anie.202305073
Rivera-Almazo, M., Díaz-Ramírez, M. L., Hernández-Esparza, R., Vargas, R., Martínez, A., Martis, V., et al. (2021). Identification of the preferential CO and SO2 adsorption sites within NOTT-401. Phys. Chem. Chem. Phys. 23, 1454–1463. doi:10.1039/d0cp04668j
Sánchez-Bautista, J. E., Landeros-Rivera, B., Jurado-Vázquez, T., Martínez, A., González-Zamora, E., Balmaseda, J., et al. (2019). CO2 capture enhancement for InOF-1: confinement of 2-propanol. Dalton Trans. 48, 5176–5182. doi:10.1039/c9dt00384c
Sánchez-Serratos, M., Álvarez, J. R., González-Zamora, E., and Ibarra, I. A. (2016). Porous coordination polymers (pcps): new platforms for gas storage. J. Mexican Chem. Soc. 60, 43–57. doi:10.29356/jmcs.v60i2.72
Santibañez, D., and Mendizabal, F. (2023). Understanding lead and mercury adsorption by post-synthetically modified linkers in UiO-66 MOF. a computational theoretical study. Mol. Simul. 49, 481–488. doi:10.1080/08927022.2023.2171073
Schneider, H. J. (2022). Noncovalent interactions: a brief account of a long history. J. Phys. Org. Chem. 35, e4340. doi:10.1002/poc.4340
Steiner, T. (2002). The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 41, 48–76. doi:10.1002/1521-3773(20020104)41:1<48::aid-anie48>3.0.co;2-uCDC.2015.7402694
Tibbetts, I., and Kostakis, G. E. (2020). Recent bio-advances in metal-organic frameworks. Molecules 25, 1291. doi:10.3390/molecules25061291molecules25061291
Tosso, R. D., Parravicini, O., Zarycz, M. N. C., Angelina, E., Vettorazzi, M., Peruchena, N., et al. (2020). Conformational and electronic study of dopamine interacting with the 2 dopamine receptor. J. Comput. Chem. 41, 1898–1911. doi:10.1002/jcc.26361
Wang, H., Jiang, Y., Han, R., Liu, Q., Liu, C., and Yan, Z. (2025). Metal-organic frameworks for low-concentration gases adsorption under ambient conditions: characterization, modification, processing, shaping and applications. Coord. Chem. Rev. 531, 216464. doi:10.1016/j.ccr.2025.216464
Keywords: MOFs, non-covalent interactions, gas adsorption, BioMOF, DDS, phenylethylamine derivatives, QTAIM, DFT
Citation: Medel E and Vargas R (2025) Non-covalent interactions in MOFs: a quantum approach to gas adsorption and molecular encapsulation. Front. Chem. 13:1579977. doi: 10.3389/fchem.2025.1579977
Received: 19 February 2025; Accepted: 19 May 2025;
Published: 06 June 2025.
Edited by:
Elisa Michelini, University of Bologna, ItalyReviewed by:
Mary Cano-Sarabia, Catalan Institute of Nanoscience and Nanotechnology (CIN2), SpainHéctor Martínez-Pérez-Cejuela, University of Valencia, Spain
Copyright © 2025 Medel and Vargas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rubicelia Vargas, cnZhcmdhc0BpenQudWFtLm14