 Hesham A. M. Gomaa1*
Hesham A. M. Gomaa1* Asmaa M. Atta2
Asmaa M. Atta2 Mohamed E. Shaker1
Mohamed E. Shaker1 Sami I. Alzarea1
Sami I. Alzarea1 Tariq G. Alsahli1
Tariq G. Alsahli1 Eid Alatwi1
Eid Alatwi1 Arafa Musa3Fatma A. M. Mohamed4Hayat Ali Alzahrani5Aliaa M. Mohassab6
Arafa Musa3Fatma A. M. Mohamed4Hayat Ali Alzahrani5Aliaa M. Mohassab6 Alshaimaa Abdelmoez7
Alshaimaa Abdelmoez7 Stefan Bräse8*
Stefan Bräse8* Bahaa G. M. Youssif7*
Bahaa G. M. Youssif7*- 1Department of Pharmacology, College of Pharmacy, Jouf University, Sakaka, Aljouf, Saudi Arabia
- 2Pharmaceutical Chemistry Department, Faculty of Pharmacy, Badr University in Cairo (BUC), Badr City, Cairo, Egypt
- 3Department of Pharmacognosy, College of Pharmacy, Jouf University, Sakaka, Aljouf, Saudi Arabia
- 4Department of Clinical Laboratory Sciences, College of Applied Medical Sciences at Al-Qurayyat, Jouf University, Al-Qurayyat, Saudi Arabia
- 5Applied Medical Science College, Medical Laboratory Technology Department, Northern Border University, Arar, Saudi Arabia
- 6Department of Medicinal Chemistry, Faculty of Pharmacy, Minia University, Minia, Egypt
- 7Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Assiut University, Assiut, Egypt
- 8Institute of Biological and Chemical Systems, IBCS-FMS, Karlsruhe Institute of Technology, Karlsruhe, Germany
Introduction: A novel series of thiazole-based derivatives 11a-f and 12a-f was developed, synthesized, and tested for antiproliferative activity as dual EGFR/VEGFR-2 inhibitors, antioxidants, and antibacterial agents.
Methods: The structures of the new compounds 11a-f and 12a-f were validated using NMR spectroscopy and elemental microanalysis. The antiproliferative activity of 11a-f and 12a-f was tested against a panel of four cancer cell lines using MTT assay.
Results and Discussion: Compounds 11d and 11f had the highest antiproliferative activity, with GI50 values of 30 and 27 nM, respectively, making them more potent than erlotinib (GI50 = 33 nM). Inhibitory studies for EGFR and VEGFR-2 demonstrated that compounds 11d and 11f were the most potent derivatives with dual inhibitory activity. Furthermore, compounds 11d and 11f exhibited significant antioxidant activity at 10 μM, with radical scavenging activity of 71% and 73%, respectively, compared to the reference Trolox (78%). Moreover, compounds 11a-f and 12a-f exhibit significant inhibitory activity against E. coli DNA gyrase, with compounds 11b, 11e, and 12b displaying the highest inhibitory efficacy, yielding IC50 values of 182, 190, and 197 nM, respectively, in comparison to the reference novobiocin (IC50 = 170 nM). Compounds 11b and 11e have significant antibacterial efficacy against both Gram-positive and Gram-negative bacterial strains, as demonstrated by a twofold serial dilution experiment. They exhibit similar efficacy against S. aureus, E. coli, and P. aeruginosa, demonstrating more potency than ciprofloxacin, however displaying reduced effectiveness against B. subtilis compared to ciprofloxacin.
1 Introduction
Nitrogen-containing heterocyclic molecules are crucial in drug development, with around 75% of FDA-approved small-molecule medicines comprising one or more nitrogen-based heterocycles (Al-Wahaibi et al., 2024a; Mohamed et al., 2024). Thiazole, also known as 1,3-thiazole, is an azole heterocyclic moiety that includes one sulfur and one nitrogen atom at positions one and 3, respectively. Their varied biological activity is evidenced by numerous clinically authorized thiazole-containing drugs exhibiting various pharmacological effects. Most of these compounds are 2,4-disubstituted thiazole derivatives, with just a limited number being 2,5-disubstituted or 2,4,5-trisubstituted thiazoles (Arshad et al., 2022; Alateeq et al., 2024; Abdelazeem et al., 2017; Abdel-Aziz et al., 2021; Abdel-Aziz et al., 2022). Several medicines having antibacterial, antiparkinsonian, antithrombotic, antifungal, antiulcer, anti-inflammatory, anticancer, antiparasitic, and antigout action contain one thiazole moiety in their structure (Figure 1) (Jadhav et al., 2021; Chugh et al., 2022).

Figure 1. Structures of some clinically approved thiazole-based drugs.
Enzyme inhibition has recently been recognized as a vital and important target method for tumor treatment (Liu et al., 2024; Wang et al., 2021). Thiazole-based derivatives in cancer therapy have been shown to efficiently inhibit many enzymes and enzymatic pathways, including tyrosine kinase inhibitors, B-RAF enzyme inhibitors, and microtubule function suppression (Rana et al., 2023; Sharma et al., 2020). Several risk factors contribute to molecular variations or mutations in critical proteins, such as tyrosine kinases, initiating carcinogenesis. These tyrosine kinases play crucial roles in proper cell proliferation, differentiation, metabolism, migration, and cell-cycle regulation by phosphorylating tyrosine residues in proteins (Theivendren et al., 2021; Pang et al., 2022). Conversely, receptor tyrosine kinases (RTKs) are high-affinity cell surface receptors that facilitate transmembrane signalling and play a transformative role in a variety of malignancies (Batool et al., 2023; Qi et al., 2024). The FDA (Food and Drug Administration) has approved numerous medications to treat cancer caused by activated RTKs (Zhong et al., 2021; Kumar et al., 2024).
Epidermal Growth Factor Receptor (EGFR) is a type of membrane RTK that is overexpressed in many cancers. Cancer progression is intimately tied to EGFR TK signal transmission, therefore blocking receptor activation can effectively stop tumor growth (Al-Wahaibi et al., 2023a; Al-Wahaibi et al., 2024b). VEGFR-2 (Vascular Endothelial Growth Factor Receptor) is a RTK that can promote angiogenesis (Marzouk et al., 2020). VEGFR-2, a member of the VEGFR family, regulates the proliferation of blood vessels in tumors and is required to form solid tumors. Blocking VEGFR-2 has been proposed as a potential method to reduce angiogenesis (Mahmoud et al., 2023; Mahmoud et al., 2024).
EGFR and VEGFR-2 have been identified as viable therapeutic targets in cancer treatment. They are essential in signalling networks that regulate tumor cell angiogenesis, motility, differentiation, and proliferation (Al-Wahaibi et al., 2024a; Xie et al., 2018). EGFR and VEGFR-2 often share similar signalling pathways in a complicated network of interconnections. Inhibiting EGFR can diminish VEGF synthesis and obstruct angiogenesis. Simultaneously increasing VEGFR-2 expression may ultimately result in resistance to EGFR treatments. Thus, the concurrent inhibition of both EGFR and VEGFR-2 has arisen as an effective cancer therapeutic strategy, producing a synergistic impact (Liu Z.-L. et al., 2023; Liu X.-J. et al., 2023).
Bacterial infections, caused by Gram-positive or Gram-negative bacteria, are the predominant form of infections contracted in hospitals or by the general populace (Al-Wahaibi et al., 2024c; Al-Wahaibi et al., 2024d). Furthermore, bacteria have acquired resistance to almost all presently utilized antibiotics due to their prolonged, extensive, and improper application, intensifying the issue (Al-Wahaibi et al., 2024e). Each year, drug-resistant infections kill approximately 0.7 million people worldwide, a figure that might rise to 10 million by 2050 if current trends continue (Agrawal and Patel, 2024). Consequently, it is critical to speed up the development of new antibacterials that are highly efficient against both susceptible and resistant pathogens.
DNA gyrase, a topoisomerase II enzyme, modifies the topology of DNA. It has two parts, called GyrA and GyrB, that are important for separating two DNA strands and starting a process called negative supercoiling during DNA replication (Abdel-Aziz et al., 2023; Al-Wahaibi et al., 2021). Antibacterial medications that particularly target DNA gyrase work through two mechanisms: gyrase poisoning, as reported with Ciprofloxacin, and inhibiting the ATP binding site, as demonstrated in Novobiocin (Tiz et al., 2019). Because of its profound relevance, DNA gyrase has emerged as a fascinating target for developing antibacterial medications.
The prevalence of cancer-related mortality as well as new cases associated with treatment or chronic infections highlight the link between infection and cancer. Infectious agents including bacteria and viruses cause approximately 2 million new cancer cases (Seong et al., 2025; Portela et al., 2025). Individuals with persistent infections are more likely to develop cancer because their immune systems are unable to battle both the pathogen and the development of cancer cells (Woo et al., 2025). This weakness may also result from overly aggressive cancer treatments such as chemotherapy, radiotherapy, and surgical resection, which make patients susceptible to pathogenic infections. Furthermore, chronic infection causes inflammation, which contributes to the development of cancer (Zolfi et al., 2025).
Considering these precedents, this report presents an efficient synthesis of a new series of thiazole-based derivatives hybridized with coumarin or benzofuran (11a-f and 12a-f, Figure 2), in line with our research objectives of developing innovative methodologies for synthesizing heterocyclic systems with promising pharmacological properties (Al-Wahaibi et al., 2023b; Al-Wahaibi et al., 2023c; Aly et al., 2024; Aly et al., 2023; Elbastawesy et al., 2015; El-Sheref et al., 2023; Frejat F. O. A. et al., 2022). Furthermore, we investigated the biological activities of the newly synthesized compounds, which have potential anticancer, antibacterial, and antioxidant effects.
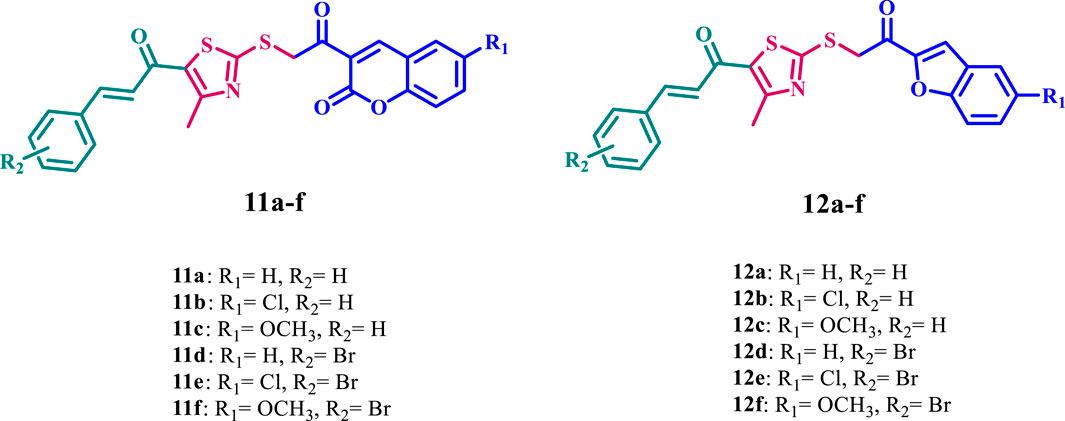
Figure 2. Structures of new compounds 11a-f and 12a-f.
The newly synthesized compounds were examined for their impact on cell viability in a normal cell line to evaluate their safety profile. The antiproliferative efficacy of novel compounds was evaluated against a panel of four different cancer cell lines. The most effective compounds were assessed for their dual inhibition of EGFR and VEGFR-2, in addition to their antioxidant properties. All newly synthesized compounds were evaluated as DNA gyrase inhibitors, and the most effective derivatives were examined for their antibacterial activity against Gram-positive bacteria (S. aureus and B. subtilis) and Gram-negative bacteria (E. coli and P. aeruginosa).
2 Results and discussion
2.1 Chemistry
Scheme 1 highlights the schematic pathway employed for synthesizing the target compounds 11a-f and 12a-f. The synthesis was initiated with the condensation of 2-hydroxy benzaldehyde derivatives 2a-b with ethyl acetoacetate in the presence of piperidine, employing ethanol as the solvent. The reaction was performed at 0°C for 24 h to yield 3-acetyl-2H-chromen-2-one derivatives 3a-b (Sahu et al., 1996). Following this, the bromination of 3a-b was conducted using N-bromosuccinimide (NBS) and p-toluene sulfonic acid (PTSA) in acetonitrile under reflux for 12 h, resulting in the formation of 3-(2-bromoacetyl)-2H-chromen-2-ones 4a-b (Kakkar et al., 2018).
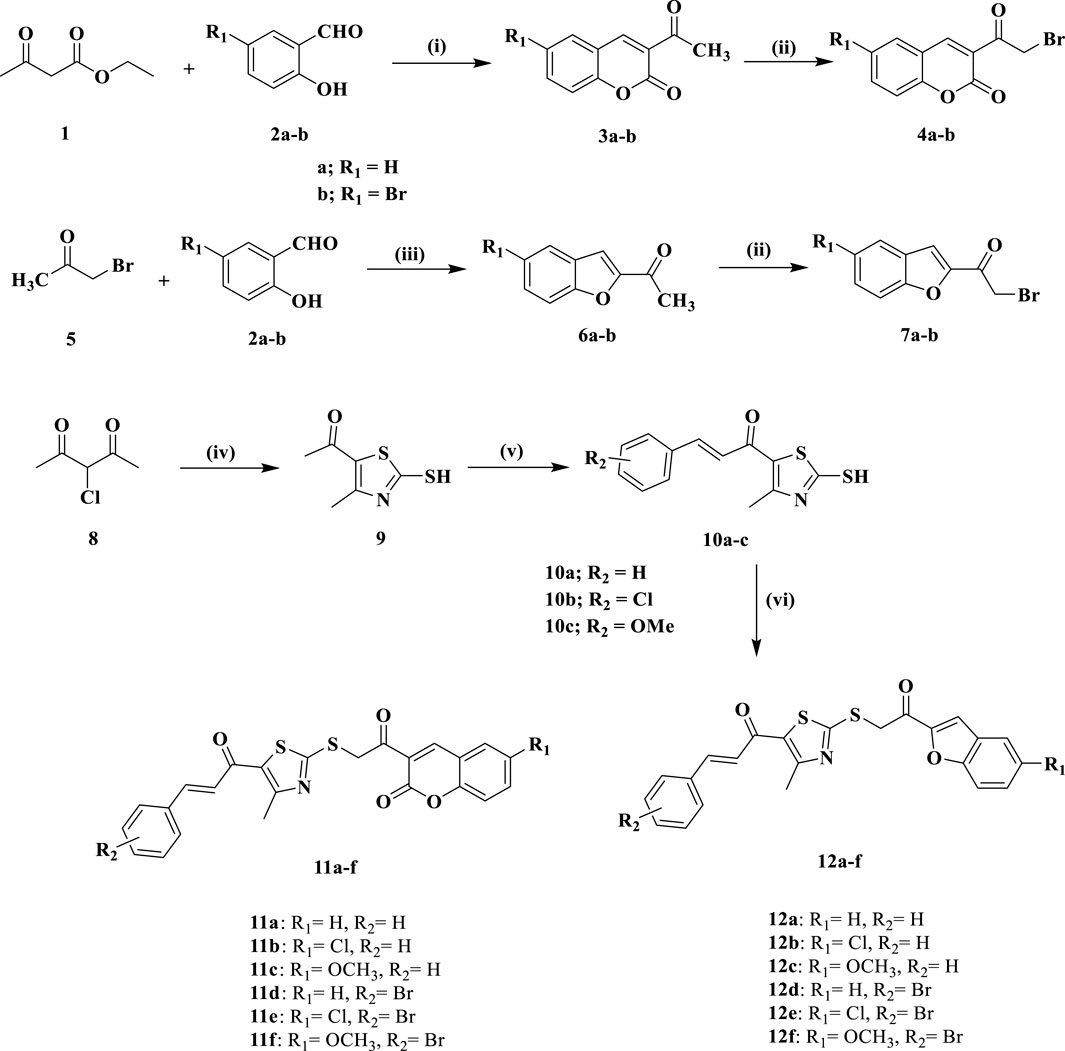
Scheme 1. Synthesis of new compounds 11a-f and 12a-f.
Similarly, compounds 7a-b were synthesized by condensing compounds 2a-b with bromoacetone five in ethanolic KOH at 0°C for 4 h to yield compounds 6a-b (Chidan Kumar et al., 2015), which were then brominated with a mixture of NBS and PTSA to yield the benzofuran derivatives 7a-b (Kakkar et al., 2018). The synthesis of chalcone intermediates 10a-c is depicted in Scheme 1, commencing with the cyclization of 3-chloropentane-2,4-dione (8) into 1-(2-mercapto-4-methylthiazol-5-yl)ethan-1-one (9) via reacting with ammonia and carbon disulfide in absolute ethanol (Hashem et al., 2024). The acetyl group of the thiazole ring in 9 then participates in a Claisen-Schmidt condensation reaction with various substituted benzaldehydes in ethanol under basic conditions, yielding chalcone derivatives 10a-c (Hashem et al., 2024). The thiazole chalcones 10a-c are subjected to S-alkylation with 3-(2-bromoacetyl)-2H-chromen-2-ones 4a-b or benzofuran derivatives 7a-b, utilizing sodium carbonate and sodium iodide in acetone at room temperature for 6 h. This reaction gives the novel compounds 11a-f and 12a-f in substantial yields.
Reagents and conditions: (i) Piperidine, EtOH, 0°C, 24 h; (ii) NBS, PTSA.H2O, acetonitrile, reflux, 12 h; (iii) KOH, EtOH, 0°C, 4 h; (iv) CS2, NH3, ethanol, 20°C, 6 h; (v) appropriate aromatic aldehyde, 60% NaOH, EtOH, 0°C, 18 h; (vi) 4a-b/7a-b, Na2CO3, NaI, acetone, r.t., 5 h.
The structures of the target compounds 11a-f and 12a-f were confirmed by 1H NMR, 13C NMR, and elemental analysis. The 1H NMR spectra exhibited characteristic peaks, including a singlet for the methylene group at δ 4.92–4.98 ppm, a singlet for the methyl protons at δ 2.55–2.61 ppm, the chalcone moiety displays distinct resonances, with the α-proton at δ 7.32–7.63 ppm and the β-proton at δ 7.48–7.77 ppm. In some cases, these signals were well-defined, whereas in others, they overlapped with aromatic protons. The aromatic protons corresponding to the coumarin, benzofuran, and benzene rings appeared in their expected region of δ 6.5–8.5 ppm, while the characteristic singlet for the C5 proton of the coumarin moiety was observed at a higher chemical shift (8.73–8.81 ppm). The 13C NMR spectra further supported the structural elucidation, revealing distinctive signals for the carbonyl moiety of the chalcone moiety at δ 180–183 ppm, the methylene carbon at δ 38–39 ppm, the methyl carbon at δ 16–19 ppm, and the methoxy carbon at approximately δ 55 ppm.
As a representative example, compound 11f exhibited well-defined NMR spectral features. In the 1H NMR spectrum, the C5 proton of the coumarin core resonated as a singlet at δ 8.73 ppm, whereas the α-proton of the chalcone moiety appeared at δ 7.63 ppm and the β-proton at δ 7.72 ppm, both as doublets. The methylene (-CH2-) group was observed as a singlet at δ 4.92 ppm, while the methoxy group exhibited a singlet at δ 3.77 ppm. The methyl group attached to the thiazole ring was also detected as a singlet at δ 2.59 ppm, aligning with its expected chemical shift. The 13C NMR spectrum of 11f further confirmed its structure, displaying a carbonyl signal for the chalcone moiety at δ 193.48 ppm and another carbonyl signal adjacent to the coumarin ring at δ 182.10 ppm. The methoxy carbon was observed at δ 55.94 ppm, the methylene carbon at δ 38.11 ppm, and the methyl carbon at δ 19.03 ppm. These chemical shifts were entirely consistent with the proposed structure, confirming the successful synthesis of 11a-f and 12a-f.
2.2 Biology
2.2.1 Evaluation of cell viability effect
To assess the viability impact of new targets 11a-f and 12a-f, the human mammary gland epithelial (MCF-10A) normal cell line was used. The MTT test was applied to assess the cell viability of 11a-f and 12a-f following 4 days of incubation with MCF-10A cells (El-Sherief et al., 2019; Ramadan et al., 2020). Table 1 indicates that none of the analyzed compounds exhibited cytotoxicity since all compounds maintained over 90% cell viability at a concentration of 50 µM.
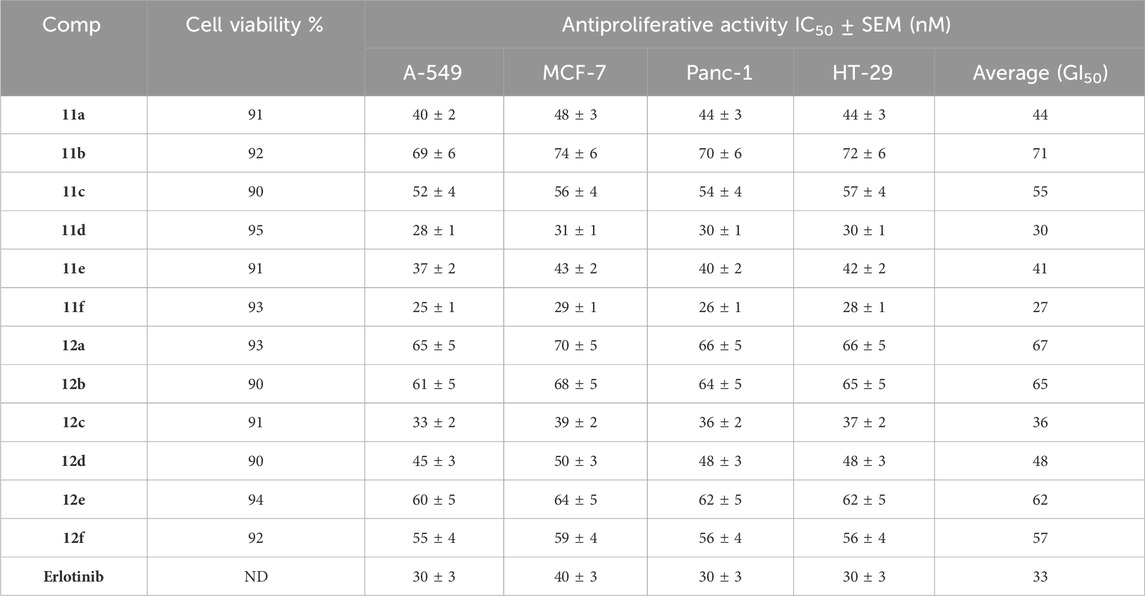
Table 1. Cell viability assay and IC50 values of compounds 11a-f and 12a-f against four cancer cell lines.
2.2.2 Antiproliferative assay
The MTT assay was employed to assess the antiproliferative effects of targets 11a-f and 12a-f on four human cancer cell lines: colon cancer (HT-29), pancreatic cancer (Panc-1), lung cancer (A-549), and breast cancer (MCF-7), with erlotinib serving as a reference compound (Mahmoud et al., 2023; Alshammari et al., 2022). Table 1 presents each compound’s median inhibitory concentration (IC50) and average IC50 (GI50) for each compound against the four cancer cell lines.
Results from Table 1 revealed that compounds 11a-f and 12a-f exhibited notable antiproliferative efficiency, with GI50 values between 27 nM and 71 nM relative to the reference Erlotinib (GI50 = 33 nM), and all evaluated compounds demonstrated greater sensitivity towards the lung cancer (A-549) cell line compared to the other cell lines assessed. Furthermore, coumarin-based compounds (11a-f) exhibit greater reactivity than benzofuran counterparts (12a-f). Compounds 11a, 11d, 11e, 11f, 12c, and 12d exhibited the most potent antiproliferative activity, with GI50 values of 44, 30, 41, 27, 36, and 48 nM, respectively, rendering compounds 11d and 11f more potent than erlotinib (GI50 = 33 nM).
Compound 11f (R1 = 4-OMe, R2 = Br, coumarin-based) had the highest efficacy among all synthesized derivatives, demonstrating a GI50 value of 27 nM, which is 1.2-fold more active than erlotinib (GI50 = 33 nM) against the four tested cancer cell lines.
Compound 11f exhibited more potency than erlotinib against the four evaluated cancer cell lines. It demonstrated optimal efficacy against lung cancer (A-549) and breast cancer (MCF-7) cell lines. Compound 11f exhibited IC50 values of 25 nM for A-549 and 29 nM for MCF-7, demonstrating 1.2- and 1.4-fold more potency than erlotinib (IC50 values = 30 and 40 nM, respectively).
The substitution pattern at the fourth position of the phenyl group in the chalcone moiety (R1), along with the type of substitution in the phenyl groups of the coumarin or benzofuran moieties (R2), significantly influences the antiproliferative activity of compounds 11a-f and 12a-f. For example, compounds 11d (R1 = H, R2 = Br, coumarin-based) and 11e (R1 = Cl, R2 = Br, coumarin-based), which share identical structural characteristics with compound 11f but possess different R1 substituents, exhibited diminished antiproliferative activity. Compounds 11d and 11e had GI50 values of 30 and 41 nM, respectively, which are 1.1- and 1.5-fold lower than compound 11f. The data reveal that the electron-donating group (OMe) is more tolerant of activity than the electron-withdrawing group, with the order of activity (R1) being OMe > H > Cl.
Compound 11c (R1 = 4-OMe, R2 = H, coumarin-based), which features an unsubstituted coumarin ring, demonstrated a marked decrease in antiproliferative efficacy. Compound 11c showed a GI50 value of 55 nM, demonstrating a twofold inferior potency to that of compound 11f. This result demonstrates the importance of the bromine atom in the coumarin moiety for antiproliferative action. Moreover, compound 12f (R1 = 4-OMe, R2 = Br, benzofuran-based), which shares the same structure as 11f but incorporates a benzofuran moiety in place of coumarin, exhibited a significant reduction in antiproliferative efficacy. Compound 12f exhibited a GI50 value of 57 nM, indicating it is 2.1-fold less effective than compound 11f, illustrating that the coumarin moiety is superior for antiproliferative activity than the benzofuran moiety.
A similar principle can be used to compare compounds 11d (R1 = H, R2 = Br, coumarin-derived) and 11e (R1 = Cl, R2 = Br, coumarin-derived), both of which are coumarin derivatives, to compounds 12d (R1 = H, R2 = Br, benzofuran-derived) and 12e (R1 = Cl, R2 = Br, benzofuran-derived), both of which are benzofuran derivatives. Compound 12d showed a GI50 value of 48 nM, which was 1.6-fold less effective than the coumarin-based derivative 11d (GI50 = 30 nM), while compound 12e demonstrated a GI50 value of 62 nM, which was 1.5-fold less efficient than its coumarin counterpart, derivative 11e (GI50 = 41 nM).
2.2.3 Evaluation of EGFR inhibitory activity
The most effective antiproliferative compounds, 11a, 11d, 11e, 11f, and 12c, were evaluated for their capacity to inhibit EGFR utilizing the EGFR-TK assay (Al-Wahaibi et al., 2022). The findings are presented in Table 2.

Table 2. IC50 values of compounds 11a, 11d, 11e, 11f, and 12c against EGFR and VEGFR-2.
The results of this assay correspond with the antiproliferative assay findings, indicating that compounds 11d (R1 = H, R2 = Br, coumarin-based) and 11f (R1 = OMe, R2 = Br, coumarin-based) are the most effective derivatives of EGFR inhibitors, exhibiting IC50 values of 76 ± 3 and 71 ± 3 nM, respectively, surpassing the potency of the reference drug Erlotinib (IC50 = 80 ± 5 nM). Compounds 11d and 11f were the most effective derivatives exhibiting antiproliferative effects. Compounds 11e (R1 = Cl, R2 = Br, coumarin-based) and 12c (R1 = OMe, R2 = H, benzofuran-based) exhibited significant anti-EGFR activity, with IC50 values of 89 ± 3 nM and 83 ± 3 nM, respectively. In contrast compound 11a showed the lowest potency as an EGFR inhibitor, with an IC50 value of 93 ± 4 nM, compared to Erlotinib (IC50 = 80 nM). The results indicate that compounds 11d and 11f have significant antiproliferative activity and may serve as EGFR inhibitors.
2.2.4 Evaluation of VEGFR-2 inhibitory action
Compounds 11a, 11d, 11e, 11f, and 12c were evaluated for their capacity to inhibit VEGFR-2, with Sorafenib serving as the control agent (Al-Wahaibi et al., 2024a). The results are displayed as IC50 values in Table 2. The findings indicated that the examined compounds had moderate to good VEGFR-2 inhibitory activity, with IC50 values between 2.90 and 5.20 µM, compared to Sorafenib, which had an IC50 value of 0.17 µM. In all cases, the evaluated compounds exhibit a potency 17-fold inferior to the reference Sorafenib. Compound 11f (R1 = OMe, R2 = Br, coumarin-based), the most effective antiproliferative and EGFR inhibitor, also exhibited the highest potency as a VEGFR-2 inhibitor, with an IC50 value of 2.90 ± 0.010 µM. Based on the in vitro experiments, we infer that compound 11f exhibits significant antiproliferative activity and functions as a dual inhibitor of EGFR and VEGFR-2, necessitating structural modifications to enhance its efficacy.
2.3 Evaluation of antioxidant activity
Antioxidant agents have become essential in medicine due to their extensive preventative and therapeutic usage across many diseases. Free radicals are integral to cancer, cardiovascular diseases, autoimmune illnesses, and age-related issues, prompting novel medical strategies (Goyal et al., 2025). The scavenging of stable free radicals by DPPH (2,2-diphenyl-1-picrylhydrazyl) (El-Sheref et al., 2023) was employed to assess the possible antioxidant capabilities of compounds 11d, 11f, and 12c, with Trolox serving as a reference, Table 3. The assay was performed at three distinct concentrations of the examined compounds (100, 50, and 10 µM).
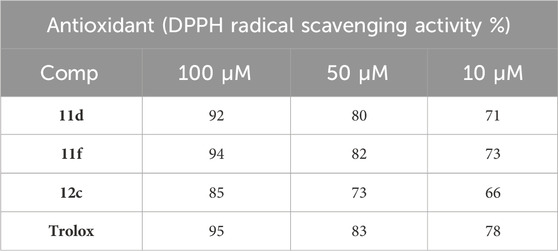
Table 3. Antioxidant activity of compounds 11d, 11f, and 12c.
Compounds 11d and 11f, derivatives of coumarin, had substantial antioxidant activity at 10 μM, scavenging DPPH radicals by 71% and 73%, respectively, compared to Trolox (78%). Moreover, compounds 11d and 11f exhibited comparable radical scavenging activity to Trolox at concentrations of 100 and 50 μM, respectively (Table 3). Conversely, compound 9, a benzofuran derivative, was identified as the least active derivative regarding antioxidant activity, underscoring the significance of the coumarin moiety for such action. The results suggested that compounds 11d and 11f may be considered effective antiproliferative agents with antioxidant properties.
2.4 Evaluation of antimicrobial activity
2.4.1 E. coli DNA gyrase inhibitory action
A supercoiling experiment was performed to assess the inhibitory efficacy of compounds 11a-f and 12a-f against E. coli DNA gyrase (Abdel-Aziz et al., 2023). Results are presented as residual activity (RA) of the enzyme at 1 μM of compounds or IC50 values for compounds with RA < 50% as presented in Table 4.
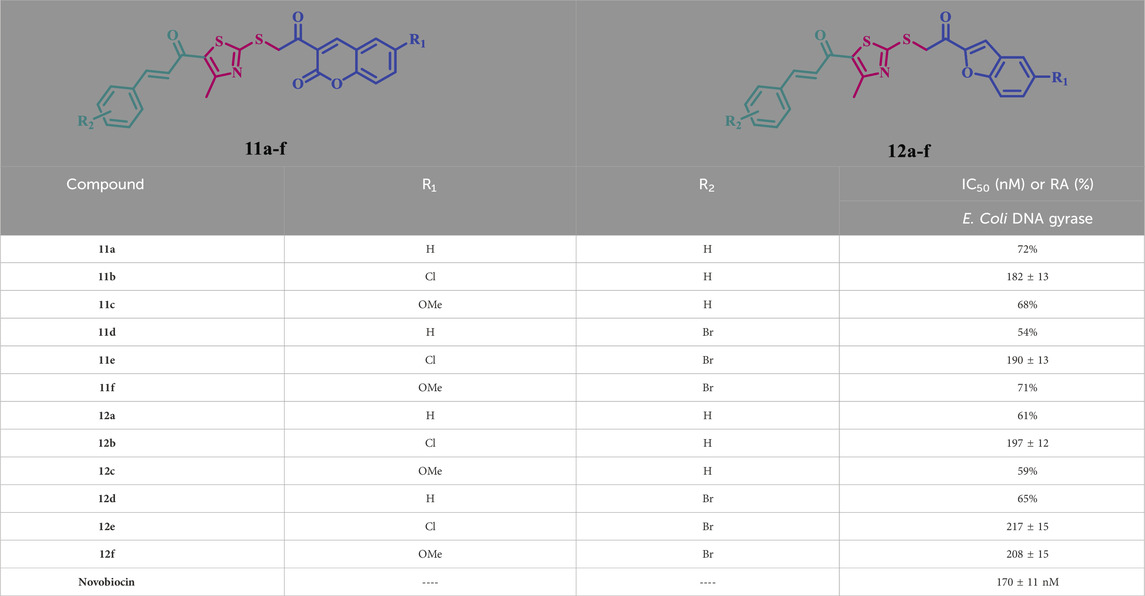
Table 4. IC50 values of compounds 11a-f and 12a-f against E. Coli DNA gyrase.
Compounds 11a-f and 12a-f show notable inhibitory activity against E. coli DNA gyrase, with IC50 values ranging from 182 to 208 nM, compared to the reference novobiocin, which has an IC50 value of 170 nM. The evaluated compounds exhibited lower potency in all cases than the reference novobiocin. Compounds 11b, 11e, and 12b demonstrated the most significant inhibitory activity against E. coli DNA gyrase, with IC50 values of 182, 190, and 197 nM, respectively. Compound 11b (R1 = Cl, R2 = H, coumarin-based) exhibited the highest efficacy as a DNA gyrase inhibitor, with an IC50 value of 182 nM, which is 1.1-fold less effective than the reference novobiocin, which has an IC50 value of 170 nM. Compound 11e (R1 = Cl, R2 = Br, coumarin-based) exhibited the second highest activity as a DNA gyrase inhibitor, with an IC50 value of 190 nM, demonstrating equipotency with compound 11b, so underscoring the significance of halogen atoms in the inhibitory action against DNA gyrase.
Compound 12b, a benzofuran-based derivative (R1 = Cl, R2 = H), possesses identical structural characteristics to compound 11b, but includes a benzofuran moiety instead of a coumarin moiety. It exhibits an IC50 value of 197 nM, indicating reduced potency compared to compound 11b (IC50 = 182 nM). The data reaffirmed the significance of the coumarin moiety in the efficacy of these compounds as antiproliferative and antibacterial agents. Compounds 12e and 12f, both benzofuran derivatives, exhibited significant inhibitory activity against DNA gyrase, with IC50 values of 217 and 208 nM, respectively, approximately 1.2-fold less effective than compound 11b.
2.4.2 Antibacterial activity
The antibacterial efficacy of compounds 11b, 11e, and 12b was evaluated against Gram-positive bacteria (S. aureus and B. subtilis) and Gram-negative bacteria (E. coli and P. aeruginosa). Table 5 presents the MICs (nM) of these compounds against the evaluated bacteria, utilizing ciprofloxacin as the reference drug, determined through a twofold serial dilution approach on a 96-well microtiter plate (Frejat F. O. et al., 2022).
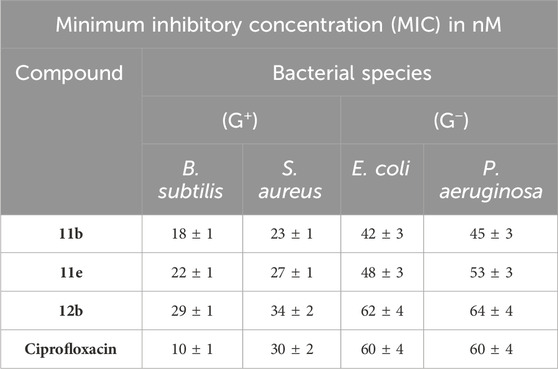
Table 5. MIC values of compounds 11b, 11e, and 12b against four bacterial species.
Compound 11b (R1 = Cl, R2 = H, coumarin-based) had the highest potency among the compounds evaluated, with MIC values of 23, 42, and 45 nM against S. aureus, E. coli, and P. aeruginosa, respectively. It exhibited superior efficacy to ciprofloxacin against the examined species but had a MIC value of 18 nM against B. subtilis, which is 1.8-fold less efficient than ciprofloxacin (MIC = 10 nM). Compound 11e (R1 = Cl, R2 = Br, coumarin-based) demonstrated the second highest efficacy. The MIC values were similar to those of compound 11b against S. aureus and E. coli, as shown in Table 5. Nonetheless, it was 2.2 times less efficacious than ciprofloxacin against B. subtilis. Ultimately, the benzofuran-based derivative, compound 12b (R1 = Cl, R2 = H), exhibited the lowest potency among the derivatives, demonstrating MIC values inferior to those of ciprofloxacin against all tested species. The observations indicate that compounds 11b and 11e are effective antibacterial agents with a broad spectrum of activity against Gram-positive and Gram-negative species, potentially functioning as DNA gyrase inhibitors.
2.5 Molecular docking
The molecular docking studies were conducted to rationalize the in vitro potency of the most active compound (11f) against three distinct biological targets: EGFR, VEGFR-2, and E. coli DNA gyrase. Docking was performed using Auto-Dock Vina (Sharma et al., 2025), and the docking poses were visualized and analyzed with Discovery Studio Visualizer (Rustagi et al., 2025). Initially, docking protocols were validated by redocking the cocrystallized ligands into the corresponding protein active sites to ensure accuracy and reliability. For EGFR, the crystal structure with PDB code 5D41 (Bhanja and Patra, 2025) was utilized. Redocking of the native ligand, EAI001, yielded a binding affinity of −9.3 kcal/mol and an RMSD of 1.1 Å, confirming the validity of the docking conditions. The superimposition between the redocked and cocrystallized ligand is depicted in Figure 3.
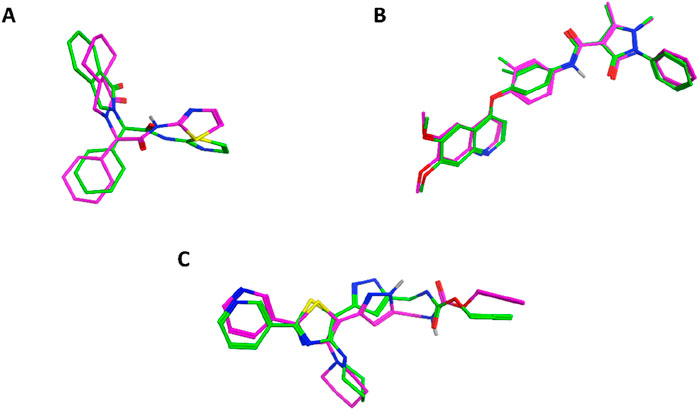
Figure 3. Superimposition of redocked (magenta) and cocrystallized (green) ligands in the active sites of (A) EGFR (5D41), (B) VEGFR-2 (3U6J), and (C) E. coli DNA gyrase (3G7E).
Subsequently, docking of compound 11f into the allosteric site of EGFR, previously identified as the binding pocket for EAI001, demonstrated a binding affinity of −8.5 kcal/mol. Detailed interaction analysis revealed that the coumarin ring of 11f engaged in significant hydrophobic interactions with Leu718, Leu844, and Ala743. Additionally, the thiazole moiety formed hydrophobic contacts with Ala743 and Lys745, alongside a pi-sulfur interaction with Met790 and a sulfur-X interaction with Asp855. The benzene ring of 11f further stabilized the binding through hydrophobic interactions with Met766, Leu858, and Leu788. Notably, the methoxy group appeared to contribute to the compound’s enhanced potency by participating in hydrophobic interactions with Leu747, Ile759, and Leu788, potentially improving binding affinity compared to other derivatives. A summary of these interactions is presented in Figure 4.
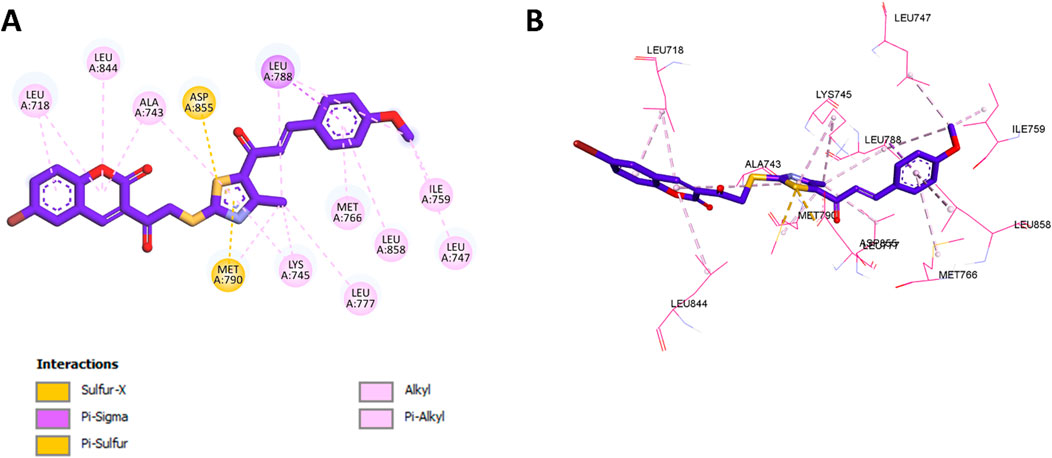
Figure 4. (A) 2D interaction diagram and (B) 3D binding mode of 11f within the allosteric site of EGFR.
For VEGFR-2, docking was conducted using the crystal structure with PDB code 3U6J (Reang et al., 2023), which represents the ATP-binding site of the kinase. Validation through redocking of the cocrystallized ligand yielded a binding affinity of −7.2 kcal/mol and an RMSD of 0.512 Å, confirming the accuracy of the docking protocol. The superimposition of the redocked and cocrystallized ligand is also shown in Figure 3. Docking of compound 11f into VEGFR-2 produced a binding affinity of −8.9 kcal/mol, with several notable interactions observed, as presented in Figure 5. The coumarin moiety of 11f exhibited hydrophobic interactions with Leu889, Val899, and Cys1045 and formed a pi-donor hydrogen bond with Asp1046. Additionally, the bromine substituent on the coumarin ring engaged in hydrophobic interaction with Ile888. The carbonyl groups of 11f formed classical hydrogen bonds with key residues Cys919 and Lys868, further stabilizing the complex. Moreover, the thiazole ring contributed to the binding through hydrophobic interactions with Leu1035, Val848, Ala866, Phe1047, and Leu840.
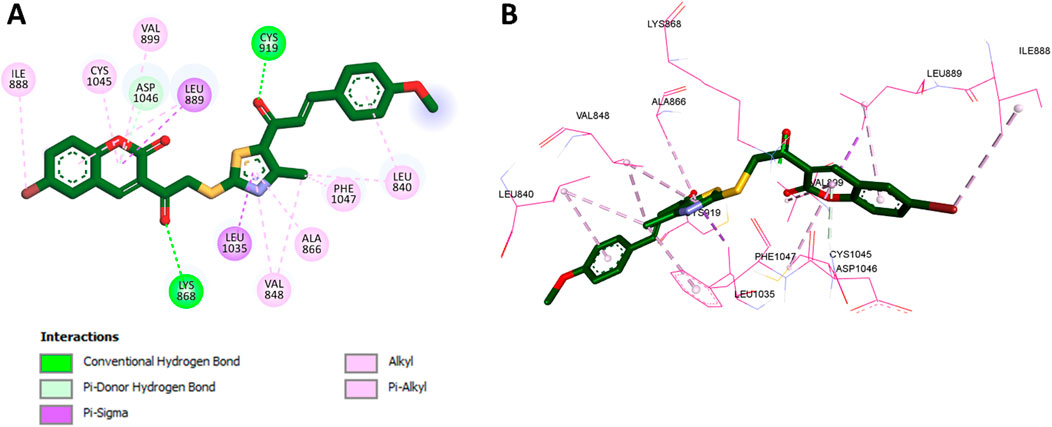
Figure 5. (A) 2D interaction diagram and (B) 3D binding mode of 11f within the VEGFR-2 ATP-binding site.
For E. coli DNA gyrase, docking studies were performed using the crystal structure with PDB code 3G7E (Sofi et al., 2025), which represents the ATP-binding site of the GyrB subunit. Redocking of the native ligand resulted in a binding affinity of −6.7 kcal/mol and an RMSD of 1.126 Å, confirming the suitability of the docking settings. The superimposition of the redocked and cocrystallized ligand is included in Figure 3. Docking of compound 11b, which exhibited the highest in vitro potency against DNA gyrase, resulted in a binding affinity of −9.3 kcal/mol. The coumarin moiety of 11b was found to establish extensive hydrophobic interactions with Val120, Val43, Ile78, Val167, Leu132, and Ala47, along with a pi-sulfur interaction with Met95. The thiazole ring of 11b further contributed hydrophobic interactions with Ile78 and Pro79. Notably, the sulfur atom adjacent to the thiazole moiety formed a classical hydrogen bond with Gly77. In addition, both the phenyl ring and the chlorine substituent in 11b engaged in hydrophobic interactions with Ala53. A comprehensive summary of these interactions is depicted in Figure 6.
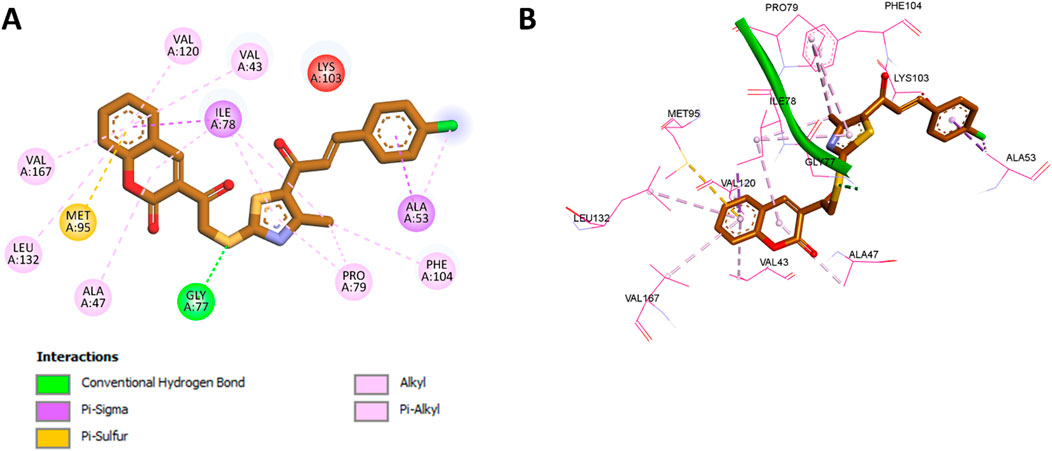
Figure 6. (A) 2D interaction diagram and (B) 3D binding mode of 11b within the ATP-binding site of the GyrB subunit.
3 Conclusion
In this study, we designed, synthesized, and biologically evaluated a novel series of thiazole-based derivatives (11a-f and 12a-f) as multi-targeted inhibitors with potent antiproliferative, antioxidant, and antibacterial activities. Compounds 11d and 11f exhibited the strongest antiproliferative effects (GI50 = 30 and 27 nM, respectively), surpassing Erlotinib, through dual EGFR/VEGFR-2 inhibition. Structure-activity relationship analysis highlighted the critical role of the coumarin moiety and electron-donating substituents in enhancing activity. Additionally, 11d and 11f demonstrated robust antioxidant potential. This dual antiproliferative and antioxidant profile is particularly valuable, as oxidative stress plays a significant role in cancer pathogenesis and progression. Furthermore, 11b, 11e, and 12b exhibited potent antibacterial activity, inhibiting E. coli DNA gyrase and displaying broad-spectrum efficacy. The multi-target profile of these derivatives underscores their potential as versatile therapeutic agents against cancer and bacterial infections. Further studies are required to assess their in vivo efficacy, pharmacokinetics, and toxicity. Future research will focus on optimizing VEGFR-2 inhibition while retaining the compounds’ anticancer, antioxidant, and antibacterial properties.
In summary, these thiazole-based derivatives represent a promising class of multi-targeted inhibitors with significant potential for the development of new therapeutic agents addressing both cancer and bacterial infections, two major global health challenges. The dual EGFR/VEGFR-2 inhibition mechanism, coupled with antioxidant and antibacterial properties, positions these compounds as versatile candidates for further development in medicinal chemistry and drug discovery pipelines.
4 Experimental
4.1 Chemistry
General Details: Refer to Appendix A.
4.1.1 General procedure for the synthesis of target compounds (11a-f) and (12a-f)
A mixture of thiazole chalcones 10a-c (1 mmol) and the corresponding coumarins 4a-b or benzofurans 7a-b (1 mmol) in acetone containing sodium carbonate (1.5 mmol, 0.159 g) and sodium iodide (2 mmol, 0.3 g) was stirred at room temperature for 5 h. After the reaction was complete, the mixture was refrigerated overnight. The precipitate was filtered, washed with distilled water, and recrystallized using ethanol.
4.1.1.1 (E)-3-(2-((5-Cinnamoyl-4-methylthiazol-2-yl)thio)acetyl)-2H-chromen-2-one (11a)
Yellow powder; 0.312 g, 70% yield; m.p. 215°C–217°C; 1H NMR (500 MHz, DMSO-d6) δ 8.81 (s, 1H, coumarin-C4-H), 8.02–7.98 (m, 2H, Ar-H), 7.76 (d, J = 15.3 Hz, 1H, chalcone-β C=H), 7.66–7.58 (m, 5H, Ar-H & chalcone-α C=H), 7.39–7.31 (m, 3H, Ar-H), 4.95 (s, 2H, CH2), 2.60 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 190.33, 181.78, 166.60, 164.28, 159.32, 155.06, 145.08, 138.06, 137.05, 134.06, 132.07, 130.84, 130.09, 126.06, 125.11, 124.38, 122.83, 120.13, 119.32, 116.85, 38.45, 18.81. Anal. Calc. (%) for C24H17NO4S2: C, 64.41; H, 3.83; N, 3.13. Found: C, 64.33; H, 3.87; N, 3.17.
4.1.1.2 (E)-3-(2-((5-(3-(4-Chlorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)acetyl)-2H-chromen-2-one (11b)
Yellow powder; 0.366 g, 76% yield; m.p. 222°C–223°C; 1H NMR (500 MHz, DMSO-d6) δ 8.81 (s, 1H, coumarin-C4-H), 8.02–7.97 (m, 2H, Ar-H), 7.72 (d, J = 15.4 Hz, 1H, chalcone-β C=H), 7.66–7.59 (m, 3H, Ar-H & chalcone-α C=H), 7.54 (d, J = 8.2 Hz, 2H, Ar-H), 7.43 (d, J = 8.4 Hz, 2H, Ar-H), 4.95 (s, 2H, CH2), 2.61 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 191.02, 180.88, 166.56, 163.87, 157.18, 153.19, 147.11, 141.15, 137.16, 133.47, 131.68, 130.30, 129.04, 126.04, 125.33, 124.62, 121.89, 120.85, 119.44, 116.97, 38.68, 19.25. Anal. Calc. (%) for C24H16ClNO4S2: C, 59.81; H, 3.35; N, 2.91. Found: C, 59.74; H, 3.39; N, 2.84.
4.1.1.3 (E)-3-(2-((5-(3-(4-Methoxyphenyl)acryloyl)-4-methylthiazol-2-yl)thio) acetyl)-2H-chromen-2-one (11c)
Yellow powder; 0.329 g, 69% yield; m.p. 229°C–231°C; 1H NMR (500 MHz, DMSO-d6) δ 8.80 (s, 1H, coumarin-C4-H), 8.01–7.98 (m, 2H, Ar-H), 7.72 (d, J = 15.3 Hz, 1H, chalcone-β C=H), 7.66–7.62 (m, 2H, Ar-H & chalcone-α C=H), 7.61–7.59 (m, 2H, Ar-H), 7.58 (s, 1H, Ar-H), 6.92 (d, J = 8.4 Hz, 2H, Ar-H), 4.94 (s, 2H, CH2), 3.79 (s, 3H, OCH3), 2.58 (s, 3H, thiazole-CH3); 13C NMR (100 MHz, DMSO-d6) δ 192.16, 180.16, 166.18, 160.61, 156.76, 153.26, 150.68, 143.56, 138.33, 137.03, 134.17, 131.25, 130.55, 127.31, 125.69, 124.07, 123.11, 122.19, 119.22, 118.58, 55.92, 38.43, 19.02. Anal. Calc. (%) for C25H19NO5S2: C, 62.88; H, 4.01; N, 2.93. Found: C, 62.76; H, 4.13; N, 3.87.
4.1.1.4 (E)-6-Bromo-3-(2-((5-cinnamoyl-4-methylthiazol-2-yl)thio)acetyl)-2H-chromen-2-one (11d)
Yellow powder; 0.405 g, 77% yield; m.p. 255°C–257°C; 1H NMR (500 MHz, DMSO-d6) δ 8.73 (s, 1H, coumarin-C4-H), 8.22 (s, 1H, Ar-H), 7.97 (d, J = 8.9 Hz, 1H, Ar-H), 7.77–7.74 (m, 2H, Ar-H & chalcone-βC = H), 7.63 (d, J = 15.4 Hz, 1H, chalcone-αC = H), 7.59 (dd, J = 6.9, 2.4 Hz, 2H, Ar-H), 7.39–7.31 (m, 3H, Ar-H), 4.93 (s, 2H, CH2), 2.59 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 190.55, 180.84, 169.14, 164.58, 158.37, 151.95, 145.48, 144.16, 140.91, 140.33, 137.05, 134.46, 132.17, 129.87, 129.29, 128.27, 124.76, 120.54, 119.24, 116.94, 37.18, 18.73. Anal. Calc. (%) for C24H16BrNO4S2: C, 54.67; H, 3.06; N, 2.66. Found: C, 54.58; H, 3.15; N, 2.57.
4.1.1.5 (E)-6-Bromo-3-(2-((5-(3-(4-chlorophenyl)acryloyl)-4-methylthiazol-2-yl)thio)acetyl)-2H-chromen-2-one (11e)
Yellow powder; 0.398 g, 71% yield; m.p. 236°C–237°C; 1H NMR (500 MHz, DMSO-d6) δ 8.73 (s, 1H, coumarin-C4-H), 8.22 (d, J = 2.3 Hz, 1H, Ar-H), 7.90 (dd, J = 8.9, 2.4 Hz, 1H, Ar-H), 7.80 (d, J = 8.5 Hz, 2H, Ar-H), 7.62 (d, J = 15.5 Hz, 1H, chalcone-β C=H), 7.47 (dd, J = 8.7, 2.5 Hz, 3H, Ar-H), 7.34 (d, J = 15.5 Hz, 1H, chalcone-α C=H), 4.93 (s, 2H, CH2), 2.59 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 190.57, 181.22, 171.59, 160.41, 155.56, 148.87, 144.02, 138.80, 135.91, 134.31, 132.84, 132.10, 131.13, 130.19, 128.72, 124.24, 121.66, 118.65, 117.19, 113.43, 37.84, 18.14. Anal. Calc. (%) for C24H15BrClNO4S2: C, 51.40; H, 2.70; N, 2.50. Found: C, 51.28; H, 2.76; N, 2.44.
4.1.1.6 (E)-6-Bromo-3-(2-((5-(3-(4-methoxyphenyl)acryloyl)-4-methylthiazol-2-yl)thio)acetyl)-2H-chromen-2-one (11f)
Yellow powder; 0.456 g, 82% yield; m.p. 232°C–235°C; 1H NMR (500 MHz, DMSO-d6) δ 8.73 (s, 1H, coumarin-C4-H), 8.10 (d, J = 2.4 Hz, 1H, Ar-H), 7.97 (d, J = 8.9 Hz, 1H, Ar-H), 7.75 (dd, J = 8.9, 2.5 Hz, 1H, Ar-H), 7.72 (d, J = 15.3 Hz, 1H, chalcone-β C=H), 7.63 (d, J = 15.4 Hz, 1H, chalcone-α C=H), 7.58 (d, J = 8.0 Hz, 2H, Ar-H), 6.93 (d, J = 8.7 Hz, 2H, Ar-H), 4.92 (s, 2H, CH2), 3.77 (s, 3H, OCH3), 2.59 (s, 3H, thiazole-CH3); 13C NMR (100 MHz, DMSO-d6) δ 193.48, 182.10, 166.58, 159.74, 152.98, 144.81, 143.56, 139.65, 139.26, 136.44, 135.40, 132.16, 130.56, 129.58, 127.68, 125.70, 124.10, 122.17, 119.90, 114.66, 55.94, 38.11, 19.03. Anal. Calc. (%) for C25H18BrNO4S2: C, 53.96; H, 3.26; N, 2.52. Found: C, 53.84; H, 3.37; N, 2.57.
4.1.1.7 (E)-1-(2-((2-(Benzofuran-2-yl)-2-oxoethyl)thio)-4-methylthiazol-5-yl)-3-phenylprop-2-en-1-one (12a)
Yellow powder; 0.281 g, 67% yield; m.p. 219°C–221°C; 1H NMR (500 MHz, DMSO-d6) δ 8.12 (s, 1H, Ar-H), 7.73 (d, J = 15.3 Hz, 1H, chalcone-β C=H), 7.68 (d, J = 8.0 Hz, 1H, Ar-H), 7.62 (d, J = 8.4 Hz, 2H, Ar-H), 7.53 (d, J = 8.0 Hz, 1H, Ar-H), 7.48 (d, J = 15.3 Hz, 1H, chalcone-α C=H), 7.48–7.43 (m, 5H, Ar-H), 4.98 (s, 2H, CH2), 2.55 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 183.56, 179.86, 167.21, 162.02, 151.27, 144.53, 142.87, 140.32, 138.59, 136.45, 135.09, 133.22, 130.88, 129.63, 127.97, 126.08, 124.40, 119.92, 118.90, 38.49, 16.74. Anal. Calc. (%) for C23H17NO3S2: C, 65.58; H, 4.08; N, 3.34. Found: C, 65.66; H, 4.02; N, 3.29.
4.1.1.8 (E)-1-(2-((2-(Benzofuran-2-yl)-2-oxoethyl)thio)-4-methylthiazol-5-yl)-3-(4-chlorophenyl)prop-2-en-1-one (12b)
Yellow powder; 0.349 g, 77% yield; m.p. 215°C–216°C; 1H NMR (500 MHz, DMSO-d6) δ 8.11 (s, 1H, Ar-H), 7.85 (d, J = 7.8 Hz, 1H, Ar-H), 7.78 (d, J = 8.6 Hz, 2H, Ar-H), 7.72 (d, J = 8.4 Hz, 1H, Ar-H), 7.61 (d, J = 15.5 Hz, 1H, chalcone-β C=H), 7.54 (t, J = 7.2 Hz, 1H, Ar-H), 7.46 (d, J = 8.5 Hz, 2H, Ar-H), 7.36 (t, J = 7.2 Hz, 1H, Ar-H), 7.32 (d, J = 15.5 Hz, 1H, chalcone-α C=H), 4.97 (s, 2H, CH2), 2.54 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 183.41, 181.80, 168.48, 158.39, 156.21, 150.98, 142.51, 136.05, 133.48, 132.49, 131.18, 129.55, 127.29, 125.37, 124.75, 123.78, 117.27, 115.62, 112.76, 37.84, 18.73. Anal. Calc. (%) for C23H16ClNO3S2: C, 60.85; H, 3.55; N, 3.09. Found: C, 60.94; H, 3.67; N, 3.13.
4.1.1.9 (E)-1-(2-((2-(Benzofuran-2-yl)-2-oxoethyl)thio)-4-methylthiazol-5-yl)-3-(4-methoxyphenyl)prop-2-en-1-one (12c)
Yellow powder; 0.386 g, 86% yield; m.p. 227°C–229°C; 1H NMR (500 MHz, DMSO-d6) δ 8.11 (s, 1H, Ar-H), 7.71 (d, J = 9.7 Hz, 1H, Ar-H), 7.59 (d, J = 8.6 Hz, 2H, Ar-H), 7.52 (d, J = 7.8 Hz, 1H, Ar-H), 7.48 (d, J = 15.9 Hz, 1H, chalcone-β C=H), 7.44–7.38 (m, 2H, Ar-H & chalcone-α C=H), 7.30–7.25 (m, 1H, Ar-H), 6.93 (d, J = 8.7 Hz, 2H, Ar-H), 4.96 (s, 2H, CH2), 3.76 (s, 3H, OCH3), 2.54 (s, 3H, thiazole-CH3); 13C NMR (100 MHz, DMSO-d6) δ 184.09, 181.77, 169.79, 165.26, 158.74, 154.58, 152.29, 151.26, 142.90, 142.21, 137.02, 130.53, 129.51, 127.97, 124.08, 123.41, 119.47, 114.68, 112.75, 55.92, 37.55, 16.45. Anal. Calc. (%) for C24H19NO4S2: C, 64.12; H, 4.26; N, 3.12. Found: C, 64.19; H, 4.33; N, 3.04.
4.1.1.10 (E)-1-(2-((2-(5-Bromobenzofuran-2-yl)-2-oxoethyl)thio)-4-methylthiazol-5-yl)-3-phenylprop-2-en-1-one (12d)
Yellow powder; 0.438 g, 88% yield; m.p. 245°C–246°C; 1H NMR (500 MHz, DMSO-d6) δ 7.94 (t, J = 2.0 Hz, 1H, Ar-H), 7.84 (d, J = 2.1 Hz, 1H, Ar-H), 7.72 (d, J = 15.9 Hz, 1H, chalcone-β C=H), 7.60–7.54 (m, 4H, Ar-H), 7.47 (d, J = 15.9 Hz, 1H, chalcone-α C=H), 7.39–7.31 (m, 3H, Ar-H), 4.97 (s, 2H, CH2), 2.54 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 183.71, 181.49, 164.94, 163.59, 159.07, 155.83, 154.22, 150.02, 141.96, 138.34, 131.86, 130.26, 128.93, 127.03, 126.06, 119.21, 117.88, 116.64, 112.34, 37.16, 16.72. Anal. Calc. (%) for C23H16BrNO3S2: C, 55.43; H, 3.24; N, 2.81. Found: C, 55.39; H, 3.27; N, 2.79.
4.1.1.11 (E)-1-(2-((2-(5-bromobenzofuran-2-yl)-2-oxoethyl)thio)-4-methylthiazol-5-yl)-3-(4-chlorophenyl)prop-2-en-1-one (12e)
Yellow powder; 0.362 g, 68% yield; m.p. 251°C–252°C; 1H NMR (500 MHz, DMSO-d6) δ 8.10 (d, J = 1.9 Hz, 1H, Ar-H), 8.05 (s, 1H, Ar-H), 7.79 (d, J = 8.5 Hz, 2H, Ar-H), 7.74 (d, J = 8.9 Hz, 1H, Ar-H), 7.69 (dd, J = 8.9, 2.0 Hz, 1H, Ar-H), 7.62 (d, J = 15.5 Hz, 1H, chalcone-β C=H), 7.47 (d, J = 8.5 Hz, 2H, Ar-H), 7.33 (d, J = 15.5 Hz, 1H, chalcone-α C=H), 4.97 (s, 2H, CH2), 2.54 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 183.09, 179.21, 164.94, 157.79, 155.17, 153.90, 148.03, 143.85, 140.30, 139.32, 138.33, 133.47, 131.85, 127.02, 126.13, 124.75, 123.76, 121.50, 116.94, 37.57, 16.44. Anal. Calc. (%) for C23H15BrClNO3S2: C, 51.84; H, 2.84; N, 2.63. Found: C, 51.91; H, 2.89; N, 2.59.
4.1.1.12 (E)-1-(2-((2-(5-Bromobenzofuran-2-yl)-2-oxoethyl)thio)-4-methylthiazol-5-yl)-3-(4-methoxyphenyl)prop-2-en-1-one (12f)
Yellow powder; 0.417 g, 79% yield; m.p. 238°C–239°C; 1H NMR (500 MHz, DMSO-d6) δ 7.94 (t, J = 2.4 Hz, 1H, Ar-H), 7.84 (d, J = 2.1 Hz, 1H, Ar-H), 7.58 (d, J = 8.6 Hz, 3H, Ar-H), 7.55 (dd, J = 8.5, 2.1 Hz, 1H, Ar-H), 7.48 (d, J = 15.9 Hz, 1H, chalcone-β C=H), 7.41 (d, J = 15.9 Hz, 1H, chalcone-α C=H), 6.93 (d, J = 8.7 Hz, 2H, Ar-H), 4.96 (s, 2H, CH2), 3.76 (s, 3H, OCH3), 2.58 (s, 3H, thiazole-CH3); 13C NMR (100 MHz, DMSO-d6) δ 183.71, 181.49, 165.27, 157.82, 156.21, 146.82, 138.04, 136.73, 134.13, 133.17, 130.25, 129.60, 128.26, 127.31, 123.42, 122.18, 119.89, 119.22, 117.25, 55.99, 37.84, 16.14. Anal. Calc. (%) for C24H18BrNO4S2: C, 54.55; H, 3.43; N, 2.65. Found: C, 54.64; H, 3.40; N, 2.67.
4.2 Biology
4.2.1 Assay for cell viability
The viability effects of 11a-f and 12a-f on the human mammary gland epithelial (MCF-10A) normal cell line were evaluated using the MTT test (El-Sherief et al., 2019; Ramadan et al., 2020). Refer to Appendix A for additional information.
4.2.2 Antiproliferative assay
The MTT assay was employed to assess the antiproliferative efficacy of 11a-f and 12a-f against four human cancer cell lines, utilizing Erlotinib as a reference control (Mahmoud et al., 2023; Alshammari et al., 2022). Dose-response assays determined the IC50 values for the novel compounds. We derived the reported data from a minimum of two separate experiments, each consisting of three repetitions per concentration. Appendix A provides experimental details.
4.2.3 Assay for EGFR inhibitory action
The most effective antiproliferative compounds, 11a, 11d, 11e, 11f, and 12c, were evaluated for their capacity to inhibit EGFR utilizing the EGFR-TK assay (Al-Wahaibi et al., 2022). Refer to Appendix A for more details.
4.2.4 Assay for VEGFR-2 inhibitory action
Compounds 11a, 11d, 11e, 11f, and 12c were assessed for their ability to inhibit VEGFR-2, using sorafenib as the control agent (Al-Wahaibi et al., 2024a). The outcomes are presented as IC50 values. Appendix A outlines more experimental details.
4.2.5 Antioxidant assay
The scavenging of stable free radicals by DPPH (El-Sheref et al., 2023) was employed to assess the antioxidant activities of compounds 11d, 11f, and 12c, with Trolox serving as a reference. The assay was performed at three different concentrations of the examined compounds (100, 50, and 10 µM). Appendix A contains more details.
4.2.6 DNA gyrase inhibitory assay
A supercoiling experiment was performed to assess the inhibitory efficacy of compounds 11a-f and 12a-f against E. coli DNA gyrase (Abdel-Aziz et al., 2023). Results are presented as RA of the enzyme at 1 μM of compounds or IC50 values for compounds with RA < 50%. Refer to Appendix A for more information.
4.2.7 Antibacterial assay and MIC calculations
The antibacterial efficacy of compounds 11b, 11e, and 12b was evaluated against Gram-positive bacteria (S. aureus and B. subtilis) and Gram-negative bacteria (E. coli and P. aeruginosa). The MICs (nM) of the tested compounds against the evaluated bacteria were determined through a twofold serial dilution approach on a 96-well microtiter plate (Frejat F. O. et al., 2022). See Appendix A for more details.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
HG: Software, Funding acquisition, Writing – original draft, Data curation. AA: Methodology, Software, Writing – original draft. MS: Investigation, Writing – original draft, Resources. SA: Investigation, Writing – original draft, Formal Analysis, Visualization. TA: Validation, Software, Writing – original draft, Data curation. EA: Software, Data curation, Resources, Writing – review and editing. AM: Conceptualization, Writing – review and editing, Formal Analysis. FM: Formal Analysis, Visualization, Conceptualization, Methodology, Writing – review and editing. HA: Formal Analysis, Methodology, Writing – review and editing, Validation, Visualization. AM: Methodology, Validation, Investigation, Software, Writing – original draft, Formal Analysis, Writing – review and editing, Visualization. AA: Investigation, Data curation, Writing – review and editing, Validation, Visualization, Writing – original draft, Formal Analysis, Methodology. SB: Writing – review and editing, Supervision. BY: Resources, Conceptualization, Investigation, Writing – review and editing, Data curation, Visualization, Validation, Writing – original draft, Formal Analysis, Software, Supervision, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Deanship of Graduate Studies and Scientific Research at Jouf University under grant No. (DGSSR-2024-01-01014). The authors also acknowledge support from the KIT-Publication Fund of the Karlsruhe Institute of Technology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1595997/full#supplementary-material
References
Abdelazeem, A. H., El-Saadi, M. T., Said, E. G., Youssif, B. G., Omar, H. A., and El-Moghazy, S. M. (2017). Novel diphenylthiazole derivatives with multi-target mechanism: synthesis, docking study, anticancer and anti-inflammatory activities. Bioorg. Chem. 75, 127–138. doi:10.1016/j.bioorg.2017.09.009
Abdel-Aziz, S. A., Cirnski, K., Herrmann, J., Abdel-Aal, M. A., Youssif, B. G., and Salem, O. I. (2023). Novel fluoroquinolone hybrids as dual DNA gyrase and urease inhibitors with potential antibacterial activity: design, synthesis, and biological evaluation. J. Mol. Struct. 1271, 134049. doi:10.1016/j.molstruc.2022.134049
Abdel-Aziz, S. A., Taher, E. S., Lan, P., Asaad, G. F., Gomaa, H. A., El-Koussi, N. A., et al. (2021). Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives bearing 1, 3-thiazole moiety as novel anti-inflammatory EGFR inhibitors with cardiac safety profile. Bioorg. Chem. 111, 104890. doi:10.1016/j.bioorg.2021.104890
Abdel-Aziz, S. A., Taher, E. S., Lan, P., El-Koussi, N. A., Salem, O. I., Gomaa, H. A., et al. (2022). New pyrimidine/thiazole hybrids endowed with analgesic, anti-inflammatory, and lower cardiotoxic activities: design, synthesis, and COX-2/sEH dual inhibition. Arch. Pharm. 355 (7), 2200024. doi:10.1002/ardp.202200024
Agrawal, A., and Patel, A. K. (2024). Antibiotic resistance profile and detection in ESKAPE pathogens, ESKAPE pathogens: detection, mechanisms and treatment strategies. Springer, 33–77.
Alateeq, A. M., Al-Faiyz, Y. S., McNair, O. D., Wiggins, J. S., and Sayed, A. R. (2024). Synthesis, reaction, and biological activity of thiazoles. Curr. Org. Synth. 22, 481–515. doi:10.2174/0115701794322192240905093650
Alshammari, M. B., Aly, A. A., Youssif, B. G., Bräse, S., Ahmad, A., Brown, A. B., et al. (2022). Design and synthesis of new thiazolidinone/uracil derivatives as antiproliferative agents targeting EGFR and/or BRAFV600E. Front. Chem. 10, 1076383. doi:10.3389/fchem.2022.1076383
Al-Wahaibi, L. H., Amer, A. A., Marzouk, A. A., Gomaa, H. A., Youssif, B. G., and Abdelhamid, A. A. (2021). Design, synthesis, and antibacterial screening of some novel heteroaryl-based ciprofloxacin derivatives as DNA gyrase and topoisomerase IV inhibitors. Pharmaceuticals 14 (5), 399. doi:10.3390/ph14050399
Al-Wahaibi, L. H., Elshamsy, A. M., Ali, T. F., Youssif, B. G., Brase, S., Abdel-Aziz, M., et al. (2024a). Design and synthesis of new dihydropyrimidine derivatives with a cytotoxic effect as dual EGFR/VEGFR-2 inhibitors. ACS omega 9 (32), 34358–34369. doi:10.1021/acsomega.4c01361
Al-Wahaibi, L. H., El-Sheref, E. M., Hammouda, M. M., and Youssif, B. G. (2023a). One-pot synthesis of 1-Thia-4-azaspiro [4.4/5] alkan-3-ones via schiff base: design, synthesis, and apoptotic antiproliferative properties of dual EGFR/BRAFV600E inhibitors. Pharmaceuticals 16 (3), 467. doi:10.3390/ph16030467
Al-Wahaibi, L. H., El-Sheref, E. M., Tawfeek, H. N., Abou-Zied, H. A., Rabea, S. M., Bräse, S., et al. (2024b). Design, synthesis, and biological evaluation of novel quinoline-based EGFR/HER-2 dual-target inhibitors as potential anti-tumor agents. RSC Adv. 14 (45), 32978–32991. doi:10.1039/d4ra06394e
Al-Wahaibi, L. H., Mahmoud, M. A., Alzahrani, H. A., Abou-Zied, H. A., Abdelmoez, A., Youssif, B. G., et al. (2024c). Synthesis, enzyme inhibition, and docking studies of new schiff bases of disalicylic acid methylene-based derivatives as dual-target antibacterial agents. Front. Chem. 12, 1493906. doi:10.3389/fchem.2024.1493906
Al-Wahaibi, L. H., Mahmoud, M. A., Alzahrani, H. A., Abou-Zied, H. A., Gomaa, H. A., Youssif, B. G., et al. (2024d). Discovery of new Schiff bases of the disalicylic acid scaffold as DNA gyrase and topoisomerase IV inhibitors endowed with antibacterial properties. Front. Chem. 12, 1419242. doi:10.3389/fchem.2024.1419242
Al-Wahaibi, L. H., Mahmoud, M. A., Mostafa, Y. A., Raslan, A. E., and Youssif, B. G. (2023b). Novel piperine-carboximidamide hybrids: design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzyme Inhibition Med. Chem. 38 (1), 376–386. doi:10.1080/14756366.2022.2151593
Al-Wahaibi, L. H., Mohammed, A. F., Abdelrahman, M. H., Trembleau, L., and Youssif, B. G. (2023c). Design, synthesis, and biological evaluation of indole-2-carboxamides as potential multi-target antiproliferative agents. Pharmaceuticals 16 (7), 1039. doi:10.3390/ph16071039
Al-Wahaibi, L. H., Mostafa, Y. A., Abdelrahman, M. H., El-Bahrawy, A. H., Trembleau, L., and Youssif, B. G. (2022). Synthesis and biological evaluation of indole-2-carboxamides with potent apoptotic antiproliferative activity as egfr/cdk2 dual inhibitors. Pharmaceuticals 15 (8), 1006. doi:10.3390/ph15081006
Al-Wahaibi, L. H., Rabea, S. M., Mahmoud, M. A., Youssif, B. G., Brase, S., and Abdel-Aziz, S. A. (2024e). Synthesis and antimicrobial evaluation of new 1, 2, 4-triazolo [1, 5-a] pyrimidine-based derivatives as dual inhibitors of bacterial DNA gyrase and DHFR. ACS omega 9 (47), 47261–47273. doi:10.1021/acsomega.4c08365
Aly, A. A., Abdallah, E. M., Ahmed, S. A., Rabee, M. M., Fuhr, O., Ibrahim, M. A., et al. (2024). Synthesis and characterization of new palladium (II) and silver (I) thiosemicarbazone derived by acenaphthenequinone complexes and their antimicrobial activity. Polyhedron 251, 116851. doi:10.1016/j.poly.2024.116851
Aly, A. A., Alshammari, M. B., Ahmad, A., Gomaa, H. A., Youssif, B. G., Braese, S., et al. (2023). Design, synthesis, docking and mechanistic studies of new thiazolyl/thiazolidinylpyrimidine-2, 4-dione antiproliferative agents. Arabian J. Chem. 16 (4), 104612. doi:10.1016/j.arabjc.2023.104612
Arshad, M. F., Alam, A., Alshammari, A. A., Alhazza, M. B., Alzimam, I. M., Alam, M. A., et al. (2022). Thiazole: a versatile standalone moiety contributing to the development of various drugs and biologically active agents. Molecules 27 (13), 3994. doi:10.3390/molecules27133994
Batool, Z., Azfal, A., Liaquat, L., Sadir, S., Nisar, R., Inamullah, A., et al. (2023). Receptor tyrosine kinases (RTKs): from biology to pathophysiology, receptor tyrosine kinases in neurodegenerative and psychiatric disorders. Elsevier, 117–185.
Bhanja, K. K., and Patra, N. (2025). Identification of novel fourth-generation allosteric inhibitors targeting inactive state of EGFR T790M/L858R/C797S and T790M/L858R mutations: a combined machine learning and molecular dynamics approach. J. Phys. Chem. B 129, 3610–3629. doi:10.1021/acs.jpcb.4c07651
Chidan Kumar, C., Then, L. Y., Chia, T. S., Chandraju, S., Win, Y.-F., Sulaiman, S. F., et al. (2015). Benzofuranyl esters: synthesis, crystal structure determination, antimicrobial and antioxidant activities. Molecules 20 (9), 16566–16581. doi:10.3390/molecules200916566
Chugh, V., Pandey, G., Rautela, R., and Mohan, C. (2022). Heterocyclic compounds containing thiazole ring as important material in medicinal chemistry. Mater. Today Proc. 69, 478–481. doi:10.1016/j.matpr.2022.09.150
Elbastawesy, M., Youssif, B., Abdelrahman, M. H., and Hayallah, A. M. (2015). Synthesis and biological evaluation of some new coumarin derivatives as potential antimicrobial, analgesic and anti-inflammatory agents. Der. Pharma. Chem. 7, 337–349.
El-Sheref, E. M., Bräse, S., Tawfeek, H. N., Alasmary, F. A., and Youssif, B. G. (2023). Synthesis, antioxidant and antiproliferative actions of 4-(1, 2, 3-Triazol-1-yl) quinolin-2 (1 H)-ones as multi-target inhibitors. Int. J. Mol. Sci. 24 (17), 13300. doi:10.3390/ijms241713300
El-Sherief, H. A., Youssif, B. G., Abdelazeem, A. H., Abdel-Aziz, M., and Abdel-Rahman, H. M. (2019). Design, synthesis and antiproliferative evaluation of novel 1, 2, 4-triazole/schiff base hybrids with EGFR and B-RAF inhibitory activities. Anti-Cancer Agents Med. Chem. 19 (5), 697–706. doi:10.2174/1871520619666181224115346
Frejat, F. O., Cao, Y., Wang, L., Zhai, H., Abdelazeem, A. H., Gomaa, H. A., et al. (2022b). New 1, 2, 4-oxadiazole/pyrrolidine hybrids as topoisomerase IV and DNA gyrase inhibitors with promising antibacterial activity. Arch. Pharm. 355 (7), 2100516. doi:10.1002/ardp.202100516
Frejat, F. O. A., Cao, Y., Zhai, H., Abdel-Aziz, S. A., Gomaa, H. A., Youssif, B. G., et al. (2022a). Novel 1, 2, 4-oxadiazole/pyrrolidine hybrids as DNA gyrase and topoisomerase IV inhibitors with potential antibacterial activity. Arabian J. Chem. 15 (1), 103538. doi:10.1016/j.arabjc.2021.103538
Goyal, S., Thirumal, D., Singh, S., Kumar, D., Singh, I., Kumar, G., et al. (2025). Basics of antioxidants and their importance, antioxidants. Nat’s Def. Against Dis., 1–20. doi:10.1002/9781394270576.ch1
Hashem, H., Hassan, A., Abdelmagid, W. M., Habib, A. G., Abdel-Aal, M. A., Elshamsy, A. M., et al. (2024). Synthesis of new thiazole-privileged chalcones as tubulin polymerization inhibitors with potential anticancer activities. Pharmaceuticals 17 (9), 1154. doi:10.3390/ph17091154
Jadhav, P. M., Kantevari, S., Tekale, A. B., Bhosale, S. V., Pawar, R. P., and Tekale, S. U. (2021). A review on biological and medicinal significance of thiazoles. Phosphorus, Sulfur, Silicon Relat. Elem. 196 (10), 879–895. doi:10.1080/10426507.2021.1945601
Kakkar, S., Kumar, S., Lim, S. M., Ramasamy, K., Mani, V., Shah, S. A. A., et al. (2018). Design, synthesis and biological evaluation of 3-(2-aminooxazol-5-yl)-2 H-chromen-2-one derivatives. Chem. Central J. 12, 130–213. doi:10.1186/s13065-018-0499-x
Kumar, R., Goel, H., Solanki, R., Rawat, L., Tabasum, S., Tanwar, P., et al. (2024). Recent developments in receptor tyrosine kinase inhibitors: a promising mainstay in targeted cancer therapy. Med. drug Discov. 23, 100195. doi:10.1016/j.medidd.2024.100195
Liu, B., Zhou, H., Tan, L., Siu, K. T. H., and Guan, X.-Y. (2024). Exploring treatment options in cancer: tumor treatment strategies. Signal Transduct. Target. Ther. 9 (1), 175. doi:10.1038/s41392-024-01856-7
Liu, X.-J., Zhao, H.-C., Hou, S.-J., Zhang, H.-J., Cheng, L., Yuan, S., et al. (2023b). Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy. Bioorg. Chem. 133, 106425. doi:10.1016/j.bioorg.2023.106425
Liu, Z.-L., Chen, H.-H., Zheng, L.-L., Sun, L.-P., and Shi, L. (2023a). Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 8 (1), 198. doi:10.1038/s41392-023-01460-1
Mahmoud, M. A., Mohammed, A. F., Salem, O. I., Almutairi, T. M., Bräse, S., and Youssif, B. G. (2024). Design, synthesis, and apoptotic antiproliferative action of new 1, 2, 3-triazole/1, 2, 4-oxadiazole hybrids as dual EGFR/VEGFR-2 inhibitors. J. Enzyme Inhibition Med. Chem. 39 (1), 2305856. doi:10.1080/14756366.2024.2305856
Mahmoud, M. A., Mohammed, A. F., Salem, O. I., Rabea, S. M., and Youssif, B. G. (2023). Design, synthesis, and antiproliferative properties of new 1, 2, 3-triazole-carboximidamide derivatives as dual EGFR/VEGFR-2 inhibitors. J. Mol. Struct. 1282, 135165. doi:10.1016/j.molstruc.2023.135165
Marzouk, A. A., Abdel-Aziz, S. A., Abdelrahman, K. S., Wanas, A. S., Gouda, A. M., Youssif, B. G., et al. (2020). Design and synthesis of new 1, 6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: molecular docking and antiproliferative evaluation. Bioorg. Chem. 102, 104090. doi:10.1016/j.bioorg.2020.104090
Mohamed, A. M., Abou-Ghadir, O. M., Mostafa, Y. A., Dahlous, K. A., Bräse, S., and Youssif, B. G. (2024). Design and synthesis of new 1, 2, 4-oxadiazole/quinazoline-4-one hybrids with antiproliferative activity as multitargeted inhibitors. Front. Chem. 12, 1447618. doi:10.3389/fchem.2024.1447618
Pang, K., Wang, W., Qin, J. X., Shi, Z. D., Hao, L., Ma, Y. Y., et al. (2022). Role of protein phosphorylation in cell signaling, disease, and the intervention therapy. MedComm 3 (4), e175. doi:10.1002/mco2.175
Portela, R., Borovnik, K., Costa, C. M., Grilo, I. R., and Pereira, P. M. (2025). High-priority pathogens: where do we stand? in Nanotherapeutics for Infectious Diseases. Jenny Stanford Publishing New York, 93–140.
Qi, Y., Deng, S.-M., and Wang, K.-S. (2024). Receptor tyrosine kinases in breast cancer treatment: unraveling the potential. Am. J. Cancer Res. 14 (9), 4172–4196. doi:10.62347/kivs3169
Ramadan, M., Abd El-Aziz, M., Elshaier, Y. A., Youssif, B. G., Brown, A. B., Fathy, H. M., et al. (2020). Design and synthesis of new pyranoquinolinone heteroannulated to triazolopyrimidine of potential apoptotic antiproliferative activity. Bioorg. Chem. 105, 104392. doi:10.1016/j.bioorg.2020.104392
Rana, R., Kumar, N., Gulati, H. K., Sharma, A., Khanna, A., Badhwar, R., et al. (2023). A comprehensive review on thiazole based conjugates as anti-cancer agents. J. Mol. Struct. 1292, 136194. doi:10.1016/j.molstruc.2023.136194
Reang, J., Sharma, K., Sharma, P. C., Yadav, V., Sharma, V., and Majeed, J. (2023). Discovery of VEGFR inhibitors through virtual screening and energy assessment. J. Biochem. Mol. Toxicol. 37 (5), e23321. doi:10.1002/jbt.23321
Rustagi, V., Gupta, S. R., Singh, A., and Singh, I. K. (2025). Beyond trial and error: leveraging advanced software for Therapeutic discovery. Chem. Biol. Lett. 12 (1), 1251. doi:10.62110/sciencein.cbl.2025.v12.1251
Sahu, S. K., Mishra, A., and Behera, R. K. (1996). Synthesis of thiazole, benzothiazole, oxadiazole, thiadiazole, triazole and thiazolidinone incorporated coumarins. Indian J. Heterocycl. Chem., 91–94.
Seong, H., Izutsu, R., Osaki, M., and Okada, F. (2025). Cancer prevention: past challenges and future directions. Genes Environ. 47 (1), 4. doi:10.1186/s41021-025-00326-y
Sharma, P. C., Bansal, K. K., Sharma, A., Sharma, D., and Deep, A. (2020). Thiazole-containing compounds as therapeutic targets for cancer therapy. Eur. J. Med. Chem. 188, 112016. doi:10.1016/j.ejmech.2019.112016
Sharma, V., Gupta, A., Singh, A., Tyagi, S., Panday, H., Srivastava, S., et al. (2025). Virtual perspectives of sanguinarine on cancer prevention and treatment through molecular dynamic study. Silico Pharmacol. 13 (1), 33. doi:10.1007/s40203-025-00315-7
Sofi, F. A., Masoodi, M. H., and Tabassum, N. (2025). Recent advancements in the development of next-generation dual-targeting antibacterial agents. RSC Med. Chem. doi:10.1039/D4MD00934G
Theivendren, P., Kunjiappan, S., Hegde, Y. M., Vellaichamy, S., Gopal, M., Dhramalingam, S. R., et al. (2021). Importance of protein kinase and its inhibitor: a review, protein kinases: promising targets for anticancer drug research. IntechOpen, 75–100.
Tiz, D. B., Skok, Ž., Durcik, M., Tomašič, T., Mašič, L. P., Ilaš, J., et al. (2019). An optimised series of substituted N-phenylpyrrolamides as DNA gyrase B inhibitors. Eur. J. Med. Chem. 167, 269–290. doi:10.1016/j.ejmech.2019.02.004
Wang, C., Zhang, J., Yin, J., Gan, Y., Xu, S., Gu, Y., et al. (2021). Alternative approaches to target Myc for cancer treatment. Signal Transduct. Target. Ther. 6 (1), 117. doi:10.1038/s41392-021-00500-y
Woo, S., Park, P.-G., An, T., Fatima, M., Moon, Y.-E., Lee, S.-Y., et al. (2025). Mini-review on the therapeutic vaccines targeting chronic infectious diseases: evaluation system of therapeutic vaccines targeting HPV and EBV-related cancers. Hum. Vaccines Immunother. 21 (1), 2457187. doi:10.1080/21645515.2025.2457187
Xie, C., Wan, X., Quan, H., Zheng, M., Fu, L., Li, Y., et al. (2018). Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor-2 inhibitor. Cancer Sci. 109 (4), 1207–1219. doi:10.1111/cas.13536
Zhong, L., Li, Y., Xiong, L., Wang, W., Wu, M., Yuan, T., et al. (2021). Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 6 (1), 201–248. doi:10.1038/s41392-021-00572-w
Keywords: anticancer, antioxidant, antibacterial, EGFR, VEGFR-2
Citation: Gomaa HAM, Atta AM, Shaker ME, Alzarea SI, Alsahli TG, Alatwi E, Musa A, Mohamed FAM, Alzahrani HA, Mohassab AM, Abdelmoez A, Bräse S and Youssif BGM (2025) Design, synthesis, and biological investigation of new thiazole-based derivatives as multi-targeted inhibitors endowed with antiproliferative, antioxidant, and antibacterial properties. Front. Chem. 13:1595997. doi: 10.3389/fchem.2025.1595997
Received: 19 March 2025; Accepted: 18 April 2025;
Published: 06 May 2025.
Edited by:
Marko Jukic, University of Maribor, SloveniaReviewed by:
Mesut Işık, Bilecik Şeyh Edebali University, TürkiyeMeryem Topal, Gümüşhane University, Türkiye
Copyright © 2025 Gomaa, Atta, Shaker, Alzarea, Alsahli, Alatwi, Musa, Mohamed, Alzahrani, Mohassab, Abdelmoez, Bräse and Youssif. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hesham A. M. Gomaa, aGFzb2xpbWFuQGp1LmVkdS5zYQ==; Bahaa G. M. Youssif, Ymd5b3Vzc2lmMkBnbWFpbC5jb20=; Stefan Bräse, YnJhZXNlQGtpdC5lZHU=