 Maurício Chagas da Silva
Maurício Chagas da Silva Egon Campos dos Santos
Egon Campos dos Santos Maicon Pierre Lourenço
Maicon Pierre Lourenço Mateus Pereira Gouvea†
Mateus Pereira Gouvea† Hélio Anderson Duarte*
Hélio Anderson Duarte*- Grupo de Pesquisa em Química Inorgânica Teórica, Departamento de Química, Instituto de Ciências Exatas (ICEx), Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
The electronic, structural, and mechanical properties of the modified imogolites have been investigated using self consistent charge-density functional-tight binding method with “a posteriori” treatment of the dispersion interactions (SCC-DFTB-D). The zigzag (12,0) imogolite has been used as the initial structure for the calculations. The functionalization of the inner surface of (12,0) imogolite nanotubes (NTs) by organosilanes and by heat treatment leading to the dehydroxylation of the silanols were investigated. The reaction of the silanols with the trimethylmethoxysilanes is favored and the arrangement of the different substitutions leads to the most symmetrical structures. The Young moduli and band gaps (BGs) are slightly decreased. However, the dehydroxylation of the silanol groups in the inner surface of the imogolite leads to the increase of the Young moduli and a drastic decrease of the BG to about 4.4 eV. It has been shown that the degree of the dehydroxylation can be controlled by heat treatment and tune the BG, eventually, leading to a semiconductor material with well-defined NT structure.
Introduction
Imogolite, halloysite, and chrysotile are an important class of nanostructured clay mineral. They have been envisaged for several applications such as gas storage (Azzouz, 2012; Assima et al., 2013; Cavallaro et al., 2014), separation (Li et al., 2014; Murali et al., 2014; Zhong et al., 2014), controlled delivery systems (Tu et al., 2013; Wang et al., 2013; Lun et al., 2014; Lvov et al., 2014; Rao et al., 2014), composites (Tham et al., 2014; Peng et al., 2015; Xu et al., 2015), and catalysis (Machado et al., 2013; Gomez et al., 2014). They are natural nanotubes (NTs) and can be easily synthesized and modified (Price et al., 2001; Shchukin et al., 2005, 2006; Shchukin and Mohwald, 2007; Veerabadran et al., 2007; Lvov et al., 2008; Alhuthali and Low, 2013; Wang and Huang, 2013). Halloysite (Bates et al., 1950a) and chrysotile (Bates et al., 1950b; Whittaker, 1956) are polydisperse and multi-walled with diameters in the range of 15–100 nm and length of 500 nm up to mm range. Halloysite, Al2Si2O5(OH)4, is formed by a sheet of gibbsite [Al(OH)3] in the inner part and a layer of silicate outside and chrysotile, Mg3Si2O5(OH)4, is formed by a sheet of tridymite (SiO2) in the inner part and a sheet of brucite [Mg(OH)2] in the outer part of the NT. Imogolite (Cradwick et al., 1972) is very distinct from the other natural NTs. It is monodisperse, single-walled, with a well-defined diameter of 2.3 nm and its length is typically in the range around 100 nm. The strain energy curve defined as the energy to bend the ideal lamellar structure to form the NT has been used to explain the monodispersity of imogolite (Guimaraes et al., 2007). Actually, this curve presents a minimum explaining its well-defined diameter. Its formula Al2Si(OH)4O3 is formed by a gibbsite sheet in the inner part of the NT and a silanol group in the center of the hexagons of the gibbsite. The mismatch of the bond distances explains the bending and NT formation of the imogolite, which results the minimum in the strain energy curve. Carbon NTs and all other inorganic NTs present strain energy curves that decrease asymptotically to 0 with the increase of the diameter (Tenne et al., 1992).
Furthermore, it has been shown that the silanol group can be replaced by Ge(OH)4 to form aluminogermanate imogolite-like (img-Ge) NTs with very similar structure but with larger diameter (Wada and Wada, 1982; Levard et al., 2008, 2009, 2010, 2011). It has been reported that img-Ge is easily synthesized and it is possible to control its diameter and the formation of single-walled or double-walled NTs during the syntheses (Thill et al., 2012a,b; Yucelen et al., 2012). It is expected that the electronic and mechanical properties are also affected by modifying its structure. Actually, it has been shown by SCC-DFTB calculations that different imogolite-like NTs based on phosphate, phosphite, arsenate, and arsenite substituents of the silanol can be synthesized if the pH is adequately controlled (Guimaraes et al., 2013). However, the syntheses of these NTs remain to be achieved.
Actually, single-walled NTs (SWNTs) have been considered important building blocks in the development of materials with enhanced properties. Therefore, imogolite and img-Ge are becoming an important target in the development of nanotechnology, since it has been shown to be easily modified externally and internally. As it has been suggested elsewhere (Qi et al., 2008; Kang et al., 2011, 2014; Ma et al., 2011, 2012), the outer surface modification of the SWNT can improve its compatibility with other materials and liquid phase and the inner surface modification can be used for molecular recognition, nanoreactors, and size selective catalysis. The main strategy is normally covalently immobilizing organic functional groups in the inner surface as it was made by Kang et al. (2011). They have modified the inner surface by the reaction of organosilane with the silanol groups of imogolite. The modified NT was detailed characterized by powder X-ray diffraction (XRD), thermogravimetric analysis (TGA), transmission electron microscopy (TEM), solid state NMR, nitrogen physisorption, and water adsorption. It was estimated that 35% of the silanols were substituted in the inner surface of the imogolite. Another modification is based on the dehydroxylation of the inner surface of imogolite by heat treatment up to 450°C (Kang et al., 2010). The process leads to the modification of the inner surface, which has been characterized in detail by FTIR, TEM and XRD, NMR, and TGA techniques.
In spite of the importance of these achievements, the structural and mechanical properties of modified NTs are very difficult to be evaluated. In the present work, SCC-DFTB-D calculations have been performed for investigating the effect of the inner surface modification of imogolite NTs in their electronic, structural, and mechanical properties.
Materials and Methods
Electronic Structure Calculations
The electronic structure calculations of the modified imogolites were carried out using the self consistent charge-density functional-tight binding (SCC-DFTB) method (Porezag et al., 1995; Seifert et al., 1996; Elstner et al., 1998) as implemented in the DFTB+ software package (Aradi et al., 2007). This is an approximate density functional theory method that uses a minimal, localized, and confined atomic basis set and tight-binding-like approximations to the Hamiltonian (Oliveira et al., 2009). The linear combination of atomic orbitals (LCAO) ansatz is used:
The overlap matrix and the Fock-like matrix are described by the Eqs 2 and 3:
where μ and v refers to the minimal basis sets centered on the A and B nuclei. The effective potential is defined according to Eq. 4.
The right terms are the external potential, usually due to the A nuclei, the Coulomb contribution and the exchange/correlation potential due to the electron density of the atom A, respectively. The electron density is written as a superposition of atom-like densities centered on the nuclei A, as described by the Eq. 5:
From the LCAO approach, the following secular problem is obtained:
An extension of the DFTB, taking into account the first order density fluctuations in a simple but efficient way has been proposed by Elstner et al. (1998) and allows for the explicit treatment of charge-transfer effects. The charges are transferred between the different atoms according to the chemical hardness, which is related to the Hubbard parameter Uα, in a self-consistent manner. The SCC-DFTB total energy is defined according to the Eq. 7:
The γAB = γAB(UA,UB,|RA − RB|) and , where is the valence number of electrons of the isolated atom A and qA is the Mulliken charge calculated using the KS orbitals.
The three center terms in the Hamiltonian are neglected and the two-center contributions can be considered of short range. The integrals arising in the Eq. 3 are tabulated for each pair of atoms normally using the local density approximation (LDA), which are called Slater–Koster parameters. The repulsion energy does not decay to 0 for long interatomic distances. At the SCC-DFTB method, the Erep is fitted to the difference between the DFT energy and Ebnd as a function of the interatomic distance using a suitable reference structure. The Ebnd is defined by the Eq. 8:
Slater–Koster parameters for describing the imogolite (Guimaraes et al., 2007) were used. It has been shown that reliable structural, energetic, and electronic properties are obtained for nanostructured clay minerals (Guimaraes et al., 2007, 2010; Lourenco et al., 2012, 2014; da Silva et al., 2013). “A posteriori” correction to the van der Waals interactions (SCC-DFTB-D) improves the results for organic molecules or non-bonding interactions of organic residues. We have used the approach proposed by Zhechkov et al. (2005). A model study for the intermolecular interactions of methoxy groups showed in Figure S4 in Supplementary Material indicated that the SCC-DFTB-D method provides interaction energies in good agreement with the PBE/aug-cc-pVTZ, with difference of <1 kJ⋅mol−1 as shown in Table S1 in Supplementary Material.
Initial configurations of the modified NTs were based on the optimized structure of the zigzag (12,0) imogolite NT. As it was shown by Guimaraes et al. (2007), this NT is the most stable structure with 13.2 Å of inner diameter and the b lattice parameter of 8.60 Å, as it is shown in Figure 1. We consider a one-dimensional periodic approximation for all systems, setting the periodicity of the systems along y axis and applying vacuum of 100 Å in the other directions, x and z. A converged Monkhorst–Pack sampling 1 × 4 × 1 k-point grid was used in our calculations for all systems.
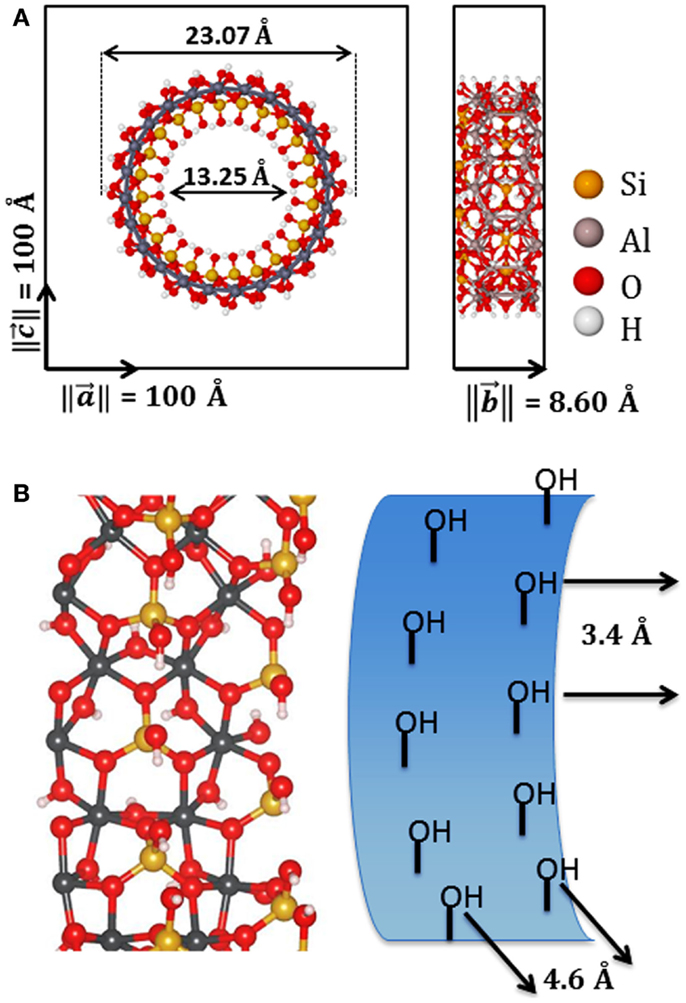
Figure 1. (A) Front and side view of zigzag imogolite (12,0) nanotube unit cell and its lattice parameters; (B) inner surface hydroxyl orientation and main distances between radial and axial inner hydroxyl.
Young Modulus
The elastic mechanical behavior of the NTs was investigated by calculating the Young modulus (Y) in the direction of the periodic axis. The total electronic SCC-DFTB-D energy (E) of a NT system must be related to the strain (ε) applied to this system along the periodic direction, Eqs 9 and 10 and Figure 2. In Eq. 9, lo is the equilibrium unit cell lattice vector size in the direction of the periodic axis (y), l is the new size of the unit cell lattice vector after a small perturbation, and the strain (ε) is a dimensionless unit that reflects how much a system is compressed or stretched in the direction of the periodic axis. In Eq. 10, the total electronic SCC-DFTB-D energy (E) is expanded by Maclaurin series around the equilibrium position of lo or in this case ε = 0. To guarantee the extensivity of the elastic property, the total energy (E) is divided by the equilibrium unit cell volume (Veq), Eq. 10:
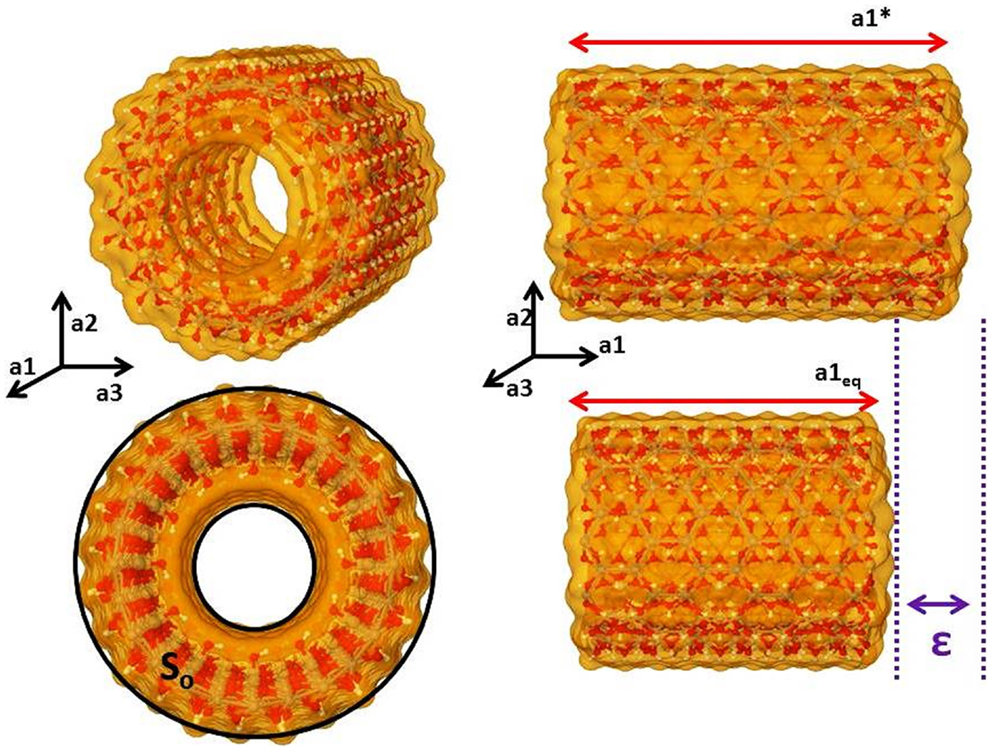
Figure 2. The unit cell of imogolite (12,0) showing the transversal area (So), lattice vector (a1eq), periodic axis (a1, a2, a3), stressed periodic vector (a1*), and the deformation parameter (ε).
In the equilibrium position, Eq. 10 can be approximated by Eq. 11 if considering E(ε = 0) as the 0 potential reference for the system and taking into account that in the equilibrium position the first derivative of the energy related to the strain must be 0. Hence, the second derivative will be related to the elastic force constant of the NT along the periodic direction (kaxi), Eq. 12. Taking strain transformations that maintain the unit cell volume (Veq) constant, the Young modulus for the NT was defined by the Eqs 12 and 13:
The protocol used in this work was to apply different strain factors from −0.5 to 0.5% in the unit cell periodic vector along the axis, relaxing the atomic position in each strain, and then obtaining a polynomial parabolic energetic curve, Eq. 14 and Figure 3. Comparing Eqs 13 and 14, the Y is related to the third polynomial coefficient (a2) of a polynomial fit curve, Eq. 15. By our approximation, the coefficients a0 and a1 were very close to 0:
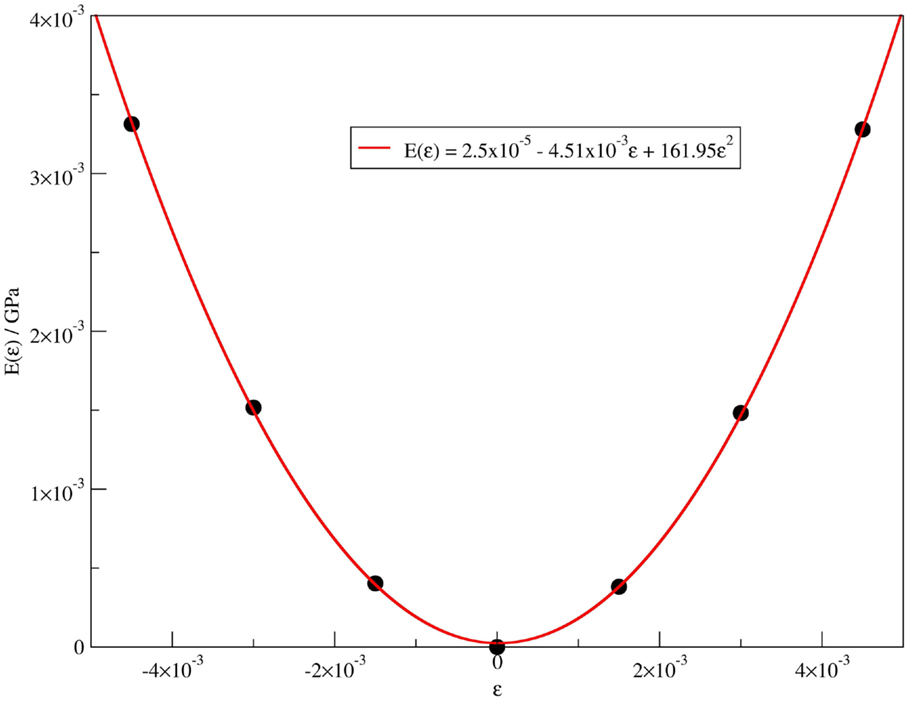
Figure 3. Polynomial quadratic fit to the energy vs. strain data for imogolite (12,0) and fitted equation.
Systems like carbon or imogolite NT is quite simple to define the equilibrium unit cell volume (Veq) using a transversal sector area of the NT (So) and the unit cell periodic vector length, Figure 2. Besides, in a simple NT the volume (Veq) is So times the lattice periodic vector length. This approach has been used to calculate the Young moduli of the clay mineral NTs (Guimaraes et al., 2007, 2010, 2013; Lourenco et al., 2014). However, in the present work, due to the inner modifications, the unit cell volume cannot be approached by this simply assumption. The volume of the unit cell was computed by numerical integration of the molecular region defined inside the unit cell. Monte Carlo integration strategies are the most powerful and less time consuming algorithm used to get molecular volumes compared to discrete numerical volume integrator. The van der Waals radius is used to establish a region of molecular volume. More details about the molecular volume estimates can be found in the Supplementary Material.
Results and Discussion
Imogolite Structures
The (12,0) imogolite NT has been predicted to be the most stable structure at SCC-DFTB-D level of theory with internal diameter about 13.25 Å (Guimaraes et al., 2007; Lourenco et al., 2014). DFT calculations have been performed at the B3LYP level indicating that (10,0) imogolite is the most stable (Demichelis et al., 2010). It is important to highlight that it has been shown recently that the diameter of the imogolite can be controlled in some extent depending of the electrolyte used in the synthesis (Yucelen et al., 2012). The Figure 1B shows the structural parameters of the inner side of the NTs. There are 24 silanol groups in each unit cell of (12,0), arranged in two alternate radial lines. Hydroxyls in the same radial line have distances of 3.4 Å and, in between different radial lines, of 4.6 Å. The silanol groups are easily modified by reaction with organosilanes or dehydroxylation by heat treatment releasing water molecules.
Functionalization with Methyltrimethoxysilane [CH3Si(OCH3)3]
Figure 4 shows the representation of the reaction that was carried out by Kang et al. (2011). The CH3Si(OCH3)3 molecule reacts with the silanol groups leading to methanol formation and the functionalized inner surface. Initially, the unit cell was modified with only one CH3Si(OCH3)3 to form monosubstituted (η1-img), bisubstituted (η2-img), and trisubstituted (η3-img). For the η2-img, silanol groups from the same radial line or from neighboring radial lines (axial) were calculated, respectively. The η3-img was calculated taking into account two hydroxyls in the same radial line and one from the neighbor hydroxyl radial line (see Figure 5). Table 1 shows the reaction energy per unit cell for the condensation of CH3Si(OCH3)3 with the inner surface silanols leading to the four different products. The distance of 4.6 Å between the hydroxyls in different radial lines leads to bonding stress for the axial η2-img and η3-img. The η1-img and radial η2-img are energetically more favored with reaction energy of −118.2 and −127.3 kJ⋅mol−1(unit cell)−1. The difference is about 10 kJ⋅mol−1(unit cell)−1 favoring the radial η2-img. It is important to note that these calculations were performed at gas phase. The solvation energy and the entropy change, due to the formation of methanol, are expected to modify the reaction energy estimates. However, the relative stability is well characterized by the SCC-DFTB-D method.
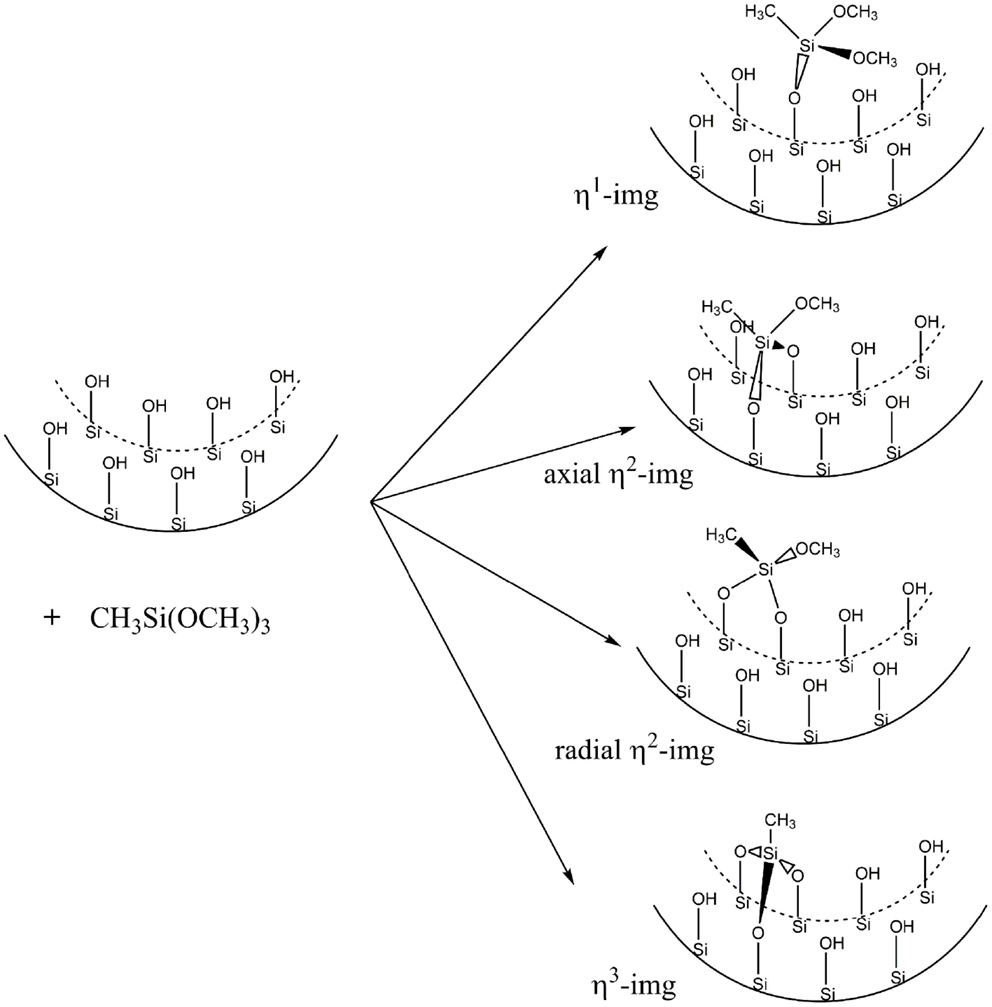
Figure 4. Inner surface representations for the modified zigzag (12,0) imogolite NT.

Figure 5. The optimized structures of the substituted (12,0) imogolite.

Table 1. Reaction energies for the condensation of CH3Si(OCH3)3 with the inner surface silanols leading to different products.
Kang et al. (2011) reported solid 29Si NMR data for the studied reaction and suggested that three products are obtained when the imogolite NTs are treated with the CH3Si(OCH3)3 leading to about 24–38% of the substituted silanols. Our results indicate that radial η2-img is the most stable (hereafter only η2-img) and it will be used in the subsequent calculations.
The condensation reaction was investigated for up to 8 η2-img substitutions, i.e., up to 16 (a fraction of 66%) silanol groups per unit cell was modified. Initially, for two substitutions all possible arrangements were calculated. The orientation of the methyl and methoxy groups in the silane can be arranged in different manner with respect to each methylmethoxysilane (CH3SiOCH3). Table S2 in Supplementary Material shows the relative energies of the different configurations. The difference of the energy is not larger than 3.5 kJ⋅mol−1(unit cell)−1 and the most stable was found for methoxy–methoxy, i.e., the two methoxy oriented toward each other. This result is probably due to the possible hydrogen bond interaction between the two groups.
Concerning the configuration of the silane groups in the unit cell, different possibilities can be envisaged. For the two substitutions, it was calculated all possibilities and the relative energies are very small, not larger than 3 kJ⋅mol−1(unit cell)−1. The most stable configuration is the one that the two groups are the most distant as possible from each other. This is expected since the methyl groups can contribute to the van der Walls repulsion destabilizing the system. We have also calculated all possibilities for the three substitutions and this is also the case, the configuration that keeps the CH3SiOCH3 groups the most distant is the most stable. For the other systems, we have calculated the most stable structures keeping in mind that the methoxy groups must be oriented to each other and the organosilanes are in a configuration that leads to the largest distance from each other.
Table 2 shows the condensation reaction energies for the different number of η2-img substitutions, their band gaps (BGs) and mechanical properties. The subsequent condensation reactions increasing the fraction of silanols substituted increase slightly the reaction energy, in about 8 kJ⋅mol−1(unit cell)−1. The optimized geometries of the most stable substituted imogolite are shown in Figure 5 indicating that the NT is just slightly deformed upon the functionalization with organosilane. These results indicate the presence of the organosilane does not affect the reaction energy of the neighbor silanols. It is well known that SCC-DFTB-D calculations overestimate the BG energies; however, it can offer the general tendency of the system studied. It indicates that the BG is slightly decreased by 1 eV with the presence of the substituents. This is expected since at the Fermi level, the electronic states correspond to the lone electron pairs of the oxygen. The equilibrium volume of the unit cell is increased by about 61 Å3 per substitution. This indicates that the modification of the inner part of the NTs does not change their main structural characteristics. The axial elastic constant and the Young’s modulus are related to the stiffness of the NT with respect to the axial deformations. The estimated values indicate that these two properties are just slightly modified leading to a small decrease of the Young’s modulus.
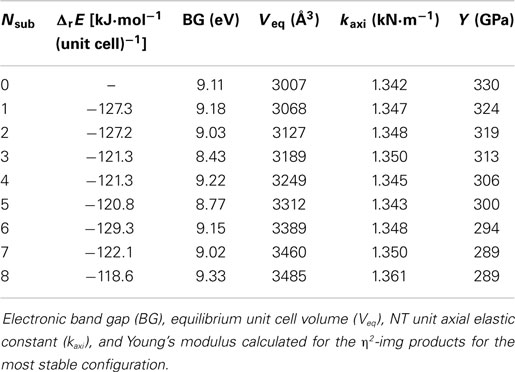
Table 2. Mean values and SD of the reaction energy per number of substitution (Nsub).
Dehydroxylation of Imogolite
Kang et al. (2010) also showed that upon heat treatment above 300°C imogolite undergoes dehydroxylation, which is only partly reversed upon rehydrating conditions. The 29Si and 27Al NMR experiments have been carried out to show that about 73% of the silanol groups were dehydroxylated to form SiO2 as shown in Figure 6. The gibbsite structure in the outer surface is not damaged with partial dehydroxylation. Therefore, the modification of the inner surface of the imogolite in a selective manner was successfully achieved by Kang et al. (2010).
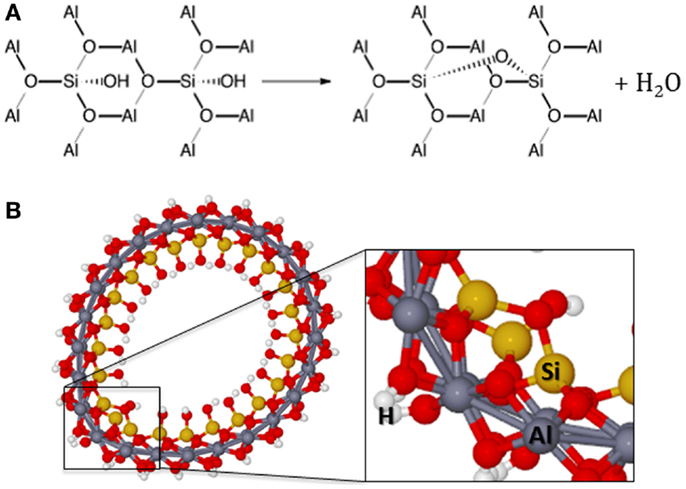
Figure 6. (A) Dehydroxylation reaction scheme in the inner surface. (B) Structure of the first dehydroxylation of the imogolite.
The dehydroxylation will lead to the formation of the Si–O–Si bonds. The Si–Si distance in the inner side of the imogolite is about 4.27 Å between silicon atoms of the same radial silanols and 4.79 Å between silicon atoms of different radial silanols. Therefore, dehydroxylation takes place between the radial silicon atoms. The Si–OH bond distance is about 1.67 Å and the strain in the Si–O–Si bonding will be very large between silicon atoms of different radial silanols. Actually, even for the radial silicon atoms, it is expected that the strain in the Si–O–Si bonding will cause deformation of the NT. It was investigated the dehydroxylation of up to 50% of the silanol groups, which means about six dehydroxylations per unit cell of the (12,0) imogolite. The optimized structure for the first dehydroxylation (n = 1) presents Si–O–Si bond distance of about 1.80 Å, which is about 0.12 Å larger than the Si–O–Al bond distance. Furthermore, the Si–O–Si angle is estimated to be about 118.6°, elongated from the ideal tetrahedral angle of 109.5° but 9° smaller than the mean value for the Si–O–Al angle. Figure 6 shows the structure of the first dehydroxylation of the imogolite.
The second dehydroxylation can occur in different position of the unit cell. An extensive study has been performed to verify, which is the most favored site for the second (n = 2) dehydroxylation. Table S3 in Supplementary Material shows the relative energies of the different sites. The most stable is the one that leads to the most symmetric structure with the second dehydroxylation occurring in the opposite side of the NT but in the same radial silanols line as shown in Figure 7.

Figure 7. Optimized structures of the dehydroxylated (12,0) imogolite.
Table 3 shows the reaction energy for the dehydroxylations in the same radial line and the respective BGs and Young moduli. As it can be seen in Figure 7, the respective hydroxylations lead to the deformation of the NT. The cylindrical structure is recovered with the subsequent dehydroxylations. The reaction energy of the dehydroxylation varies from 484 to 538 kJ⋅mol−1 per hydroxylation. One could argue that the dehydroxylations in the different radial silanols would lead to a more favored structure. Table S3 in Supplementary Material shows a comparison of the two possibilities. The dehydroxylations in the same radial silanols lead to a structure that is more favored. For the structure with six dehydroxylations, the difference is about 40 kJ⋅mol−1 per dehydroxylation favoring the structure shown in Figure 7.
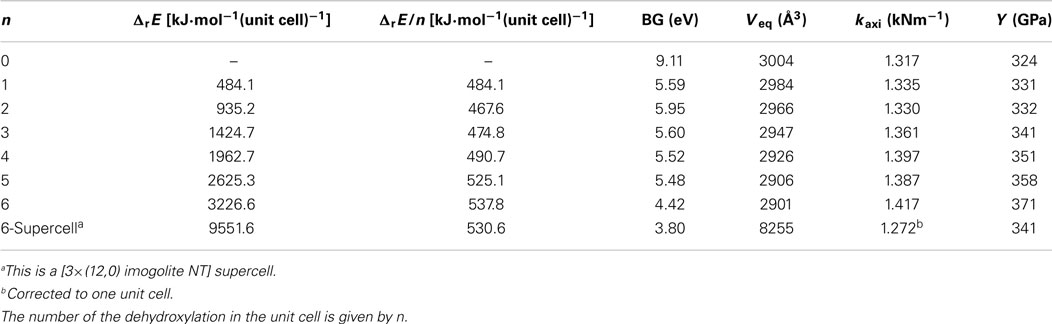
Table 3. Reaction energy of the dehydroxylation of imogolite in the same radial line.
The Young modulus is increased from 324 to 371 GPa, as expected, since the Si–O–Si bound lead to a more rigid structure. However, the BG is drastically decreased from 9.1 to about 4.4 eV. The band structures of the different dehydroxylated structures are shown in Supplementary Material. It is important to highlight that this is an upper bound for the BG. At the B3LYP/86-21G* level of theory using helical symmetry, the BG energy was predicted for the imogolite to be about 7.2 eV (Demichelis et al., 2010). This value must be contrasted with the 9.1 eV of the SCC-DFTB-D calculations. This means that with the heat treatment of the imogolite, one can control the BG energy and, eventually, produce a semiconductor material with well-defined NT structure.
One could argue that the favored dehydroxylations in the same radial silanols of the unit cell are an artifact. It is expected that the dehydroxylations occur in alternate radial silanols in such way that the inner side structure of the NT will be more homogeneous. Therefore, a supercell containing three unit cells was constructed with alternate dehydroxylations as shown in Figure 8. The results are indicated in Table 3. The helical hydrogen bonding inside of the structure contributes to its stabilization. The BG energy is decreased to about 3.80 eV indicating that the material can be actually a semiconductor if one takes into account the SCC-DFTB-D method overestimates the BG of about 2–4 eV with respect to the DFT.
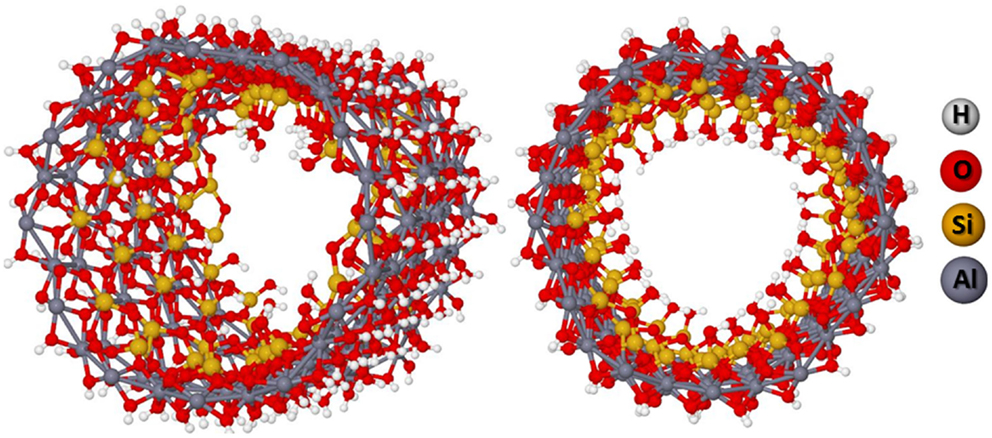
Figure 8. (12,0) NT supercell with 50% of dehydroxylation leading to the helical hydrogen bonding network.
Final Remarks
Imogolite is becoming a target material for developing advanced materials with tuned properties. It has been reported that functionalization of the inner and outer surface have been successfully achieved by modifying its properties toward gas adsorption, reactivity, and as component of composites. However, the electronic, structural, and mechanical properties of such functionalized materials have not been detailed investigated. The present study provides insights about these properties and how they are modified with respect to the functionalization of the inner surface of the imogolite NTs. The methyltrimethoxysilane functionalization of the inner surface has been calculated for different level of substitution. The results indicate that the methyltrimethoxysilane prefers the η2-img sites involving silanols that are in the same radial line in the unit cell. Different substitutions occur to provide the most symmetrical structure. The arrangement of the methoxy groups between silanes in the different radial lines is more favored when they are directed to each other leading to hydrogen bonding. The BGs and the Young moduli are slightly decreased with the functionalization.
The thermal treatment of the imogolite leads to its dehydroxylation releasing water. This is a more severe modification since the oxo groups bridging the silicon atoms of the imogolite structure change the nature of the bonding of the system. The different dehydroxylations are favored leading to the most symmetrical structure as expected. The Young moduli are increased by 50 GPa with the dehydroxylations and the reaction energy increases with the presence of dehydroxylated silanols. The dehydroxylation reaction energy varies from 484 to 538 kJ⋅mol−1 indicating that the degree of the dehydroxylation can, in principle, be controlled by heat treatment. The dehydroxylation leads to a drastic decrease of the BG energy from 9.1 eV (imogolite) to 4.4 eV (with six dehydroxylations per unit cell). It is important to note that the SCC-DFTB-D provides an upper bound of the BG energy since it overestimates it. Indeed, it is expected for this system the BG is at least 3 eV smaller, taking into account the comparison between the SCC-DFTB-D and B3LYP calculations for the ideal imogolite. A supercell containing three unit cells with six dehydroxylations each were calculated leading to a helical hydrogen bonding network inside of the NT and a BG of 3.8 eV. Therefore, it seems the dehydroxylation can, eventually, lead to a semiconductor material. Actually, depending on the level of the dehydroxylation one could tune the BG energy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MS would like to thank Prof. Dr. Dennis R. Salahub and Prof. Dr. Sergei Noskov of the University of Calgary for the facilities during the writing of the present manuscript. We would like to thank the Brazilian funding agencies: Conselho Nacional para o Desenvolvimento Cientifico e Tecnológico (CNPq); Coordenação de Aperfeiçoamento de Pessoal do Ensino Superior (CAPES); and, Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG). This work is also supported by the National Institute of Science and Technology for Mineral Resources, Water and Biodiversity (INCT-ACQUA).
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fmats.2015.00016/abstract
References
Alhuthali, A., and Low, I. M. (2013). Water absorption, mechanical, and thermal properties of halloysite nanotube reinforced vinyl-ester nanocomposites. J. Sci. Mater. 48, 4260–4273. doi: 10.1007/s10853-013-7240-x
Aradi, B., Hourahine, B., and Frauenheim, T. (2007). DFTB+, a sparse matrix-based implementation of the DFTB method. J. Phys. Chem. A 111, 5678–5684. doi:10.1021/jp070186p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Assima, G. P., Larachi, F., Beaudoin, G., and Molson, J. (2013). Dynamics of carbon dioxide uptake in chrysotile mining residues – effect of mineralogy and liquid saturation. Int. J. Greenh. Gas Control 12, 124–135. doi:10.1016/j.ijggc.2012.10.001
Azzouz, A. (2012). Achievement in hydrogen storage on adsorbents with high surface-to-bulk ratio – prospects for Si-containing matrices. Int. J. Hydrogen Energy 37, 5032–5049. doi:10.1016/j.ijhydene.2011.12.024
Bates, T. F., Hildebrand, F. A., and Swineford, A. (1950a). Morphology and structure of endellite and halloysite. Am. Mineral. 35, 463–484.
Bates, T. F., Sand, L. B., and Mink, J. F. (1950b). Tubular crystals of chrysotile asbestos. Science 111, 512–513. doi:10.1126/science.111.2889.512
Cavallaro, G., Lazzara, G., Milioto, S., Palmisano, G., and Parisi, F. (2014). Halloysite nanotube with fluorinated lumen: non-foaming nanocontainer for storage and controlled release of oxygen in aqueous media. J. Colloid Interface Sci. 417, 66–71. doi:10.1016/j.jcis.2013.11.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cradwick, P. D., Wada, K., Russell, J. D., Yoshinag, N., Masson, C. R., and Farmer, V. C. (1972). Imogolite, a hydrated aluminum silicate of tubular structure. Nat. Phys. Sci. 240, 187. doi:10.1038/physci240187a0
da Silva, M. C., dos Santos, E. C., Lourenco, M. P., and Duarte, H. A. (2013). Structural, mechanical and electronic properties of nano-fibriform silica and its organic functionalization by dimethyl silane: a SCC-DFTB approach. J. Mol. Model. 19, 1995–2005. doi:10.1007/s00894-012-1583-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demichelis, R., Noel, Y., D’Arco, P., Maschio, L., Orlando, R., and Dovesi, R. (2010). Structure and energetics of imogolite: a quantum mechanical ab initio study with B3LYP hybrid functional. J. Mater. Chem. 20, 10417–10425. doi:10.1039/c0jm00771d
Elstner, M., Porezag, D., Jungnickel, G., Elsner, J., Haugk, M., Frauenheim, T., et al. (1998). Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 58, 7260–7268. doi:10.1103/PhysRevB.58.7260
Gomez, L., Hueso, J. L., Ortega-Liebana, M. C., Santamaria, J., and Cronin, S. B. (2014). Evaluation of gold-decorated halloysite nanotubes as plasmonic photocatalysts. Catal. Commun. 56, 115–118. doi:10.1016/j.catcom.2014.07.017
Guimaraes, L., Enyashin, A. N., Frenzel, J., Heine, T., Duarte, H. A., and Seifert, G. (2007). Imogolite nanotubes: stability, electronic, and mechanical properties. ACS Nano 1, 362–368. doi:10.1021/nn700184k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guimaraes, L., Enyashin, A. N., Seifert, G., and Duarte, H. A. (2010). Structural, electronic, and mechanical properties of single-walled halloysite nanotube models. J. Phys. Chem. C 114, 11358–11363. doi:10.1021/jp100902e
Guimaraes, L., Pinto, Y. N., Lourenco, M. P., and Duarte, H. A. (2013). Imogolite-like nanotubes: structure, stability, electronic and mechanical properties of the phosphorous and arsenic derivatives. Phys. Chem. Chem. Phys. 15, 4303–4309. doi:10.1039/c3cp44250k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, D.-Y., Brunelli, N. A., Yucelen, G. I., Venkatasubramanian, A., Zang, J., Leisen, J., et al. (2014). Direct synthesis of single-walled aminoaluminosilicate nanotubes with enhanced molecular adsorption selectivity. Nat. Commun. 5, 3342. doi:10.1038/ncomms4342
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kang, D. Y., Zang, J., Jones, C. W., and Nair, S. (2011). Single-walled aluminosilicate nanotubes with organic-modified interiors. J. Phys. Chem. C 115, 7676–7685. doi:10.1021/jp2010919
Kang, D. Y., Zang, J., Wright, E. R., McCanna, A. L., Jones, C. W., and Nair, S. (2010). Dehydration, dehydroxylation, and rehydroxylation of single-walled aluminosilicate nanotubes. ACS Nano 4, 4897–4907. doi:10.1021/nn101211y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levard, C., Masion, A., Rose, J., Doelsch, E., Borschneck, D., Dominici, C., et al. (2009). Synthesis of imogolite fibers from decimolar concentration at low temperature and ambient pressure: a promising route for inexpensive nanotubes. J. Am. Chem. Soc. 131, 17080–17081. doi:10.1021/ja9076952
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levard, C., Masion, A., Rose, J., Doelsch, E., Borschneck, D., Olivi, L., et al. (2011). Synthesis of Ge-imogolite: influence of the hydrolysis ratio on the structure of the nanotubes. Phys. Chem. Chem. Phys. 13, 14516–14522. doi:10.1039/c1cp20346k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levard, C., Rose, J., Masion, A., Doelsch, E., Borschneck, D., Olivi, L., et al. (2008). Synthesis of large quantities of single-walled aluminogermanate nanotube. J. Am. Chem. Soc. 130, 5862. doi:10.1021/ja801045a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Levard, C., Rose, J., Thill, A., Masion, A., Doelsch, E., Maillet, P., et al. (2010). Formation and growth mechanisms of imogolite-like aluminogermanate nanotubes. Chem. Mater. 22, 2466–2473. doi:10.1021/cm902883p
Li, C. P., Wang, J. Q., Luo, X., and Ding, S. J. (2014). Large scale synthesis of Janus nanotubes and derivative nanosheets by selective etching. J. Colloid Interface Sci. 420, 1–8. doi:10.1016/j.jcis.2013.12.062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lourenco, M. P., de Oliveira, C., Oliveira, A. F., Guimaraes, L., and Duarte, H. A. (2012). Structural, electronic, and mechanical properties of single-walled chrysotile nanotube models. J. Phys. Chem. C 116, 9405–9411. doi:10.1021/jp301048p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lourenco, M. P., Guimaraes, L., da Silva, M. C., de Oliveira, C., Heine, T., and Duarte, H. A. (2014). Nanotubes with well-defined structure: single- and double-walled imogolites. J. Phys. Chem. C 118, 5945–5953. doi:10.1021/jp411086f
Lun, H. L., Ouyang, J., and Yang, H. M. (2014). Natural halloysite nanotubes modified as an aspirin carrier. RSC Adv. 4, 44197–44202. doi:10.1039/c4ra09006c
Lvov, Y., Aerov, A., and Fakhrullin, R. (2014). Clay nanotube encapsulation for functional biocomposites. Adv. Colloid Interface Sci. 207, 189–198. doi:10.1016/j.cis.2013.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lvov, Y. M., Shchukin, D. G., Mohwald, H., and Price, R. R. (2008). Halloysite clay nanotubes for controlled release of protective agents. ACS Nano 2, 814–820. doi:10.1021/nn800259q
Ma, W., Kim, J., Otsuka, H., and Takahara, A. (2011). Surface modification of individual imogolite nanotubes with alkyl phosphate from an aqueous solution. Chem. Lett. 40, 159–161. doi:10.1246/cl.2011.159
Ma, W., Yah, W. O., Otsuka, H., and Takahara, A. (2012). Surface functionalization of aluminosilicate nanotubes with organic molecules. Beilstein J. Nanotechnol. 3, 82–100. doi:10.3762/bjnano.3.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Machado, G. S., Ucoski, G. M., de Lima, O. J., Ciuffi, K. J., Wypych, F., and Nakagaki, S. (2013). Cationic and anionic metalloporphyrins simultaneously immobilized onto raw halloysite nanoscrolls catalyze oxidation reactions. Appl. Catal. A Gen. 460, 124–131. doi:10.1016/j.apcata.2013.04.014
Murali, R. S., Padaki, M., Matsuura, T., Abdullah, M. S., and Ismail, A. F. (2014). Polyaniline in situ modified halloysite nanotubes incorporated asymmetric mixed matrix membrane for gas separation. Sep. Purif. Technol. 132, 187–194. doi:10.1016/j.seppur.2014.05.020
Oliveira, A. F., Seifert, G., Heine, T., and Duarte, H. A. (2009). Density-functional based tight-binding: an approximate DFT method. J. Braz. Chem. Soc. 20, 1193–1205. doi:10.1590/S0103-50532009000700002
Peng, Q., Liu, M. X., Zheng, J. W., and Zhou, C. R. (2015). Adsorption of dyes in aqueous solutions by chitosan-halloysite nanotubes composite hydrogel beads. Microporous Mesoporous Mater. 201, 190–201. doi:10.1016/j.micromeso.2014.09.003
Porezag, D., Frauenheim, T., Kohler, T., Seifert, G., and Kaschner, R. (1995). Construction of tight-binding-like potentials on the basis of density-functional theory: application to carbon. Phys. Rev. B Condens. Matter 51, 12947–12957. doi:10.1103/PhysRevB.51.12947
Price, R. R., Gaber, B. P., and Lvov, Y. (2001). In-vitro release characteristics of tetracycline HCl, khellin and nicotinamide adenine dineculeotide from halloysite; a cylindrical mineral. J. Microencapsul. 18, 713–722. doi:10.1080/02652040010019532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Qi, X., Yoon, H., Lee, S.-H., Yoon, J., and Kim, S.-J. (2008). Surface-modified imogolite by 3-APS-OsO4 complex: synthesis, characterization and its application in the dihydroxylation of olefins. J. Ind. Eng. Chem. 14, 136–141. doi:10.1016/j.jiec.2007.08.010
Rao, K. M., Nagappan, S., Seo, D. J., and Ha, C. S. (2014). pH sensitive halloysite-sodium hyaluronate/poly(hydroxyethyl methacrylate) nanocomposites for colon cancer drug delivery. Appl. Clay Sci. 9, 33–42. doi:10.1016/j.clay.2014.06.002
Seifert, G., Porezag, D., and Frauenheim, T. (1996). Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA scheme. Int. J. Quantum Chem. 58, 185–192. doi:10.1002/(SICI)1097-461X(1996)58:2<185::AID-QUA7>3.3.CO;2-B
Shchukin, D. G., and Mohwald, H. (2007). Surface-engineered nanocontainers for entrapment of corrosion inhibitors. Adv. Funct. Mater. 17, 1451–1458. doi:10.1002/adfm.200601226
Shchukin, D. G., Sukhorukov, G. B., Price, R. R., and Lvov, Y. M. (2005). Halloysite nanotubes as biomimetic nanoreactors. Small 1, 510–513. doi:10.1002/smll.200400120
Shchukin, D. G., Zheludkevich, M., Yasakau, K., Lamaka, S., Ferreira, M. G. S., and Mohwald, H. (2006). Layer-by-layer assembled nanocontainers for self-healing corrosion protection. Adv. Mater. Weinheim 18, 1672. doi:10.1002/adma.200502053
Tenne, R., Margulis, L., Genut, M., and Hodes, G. (1992). Polyhedral and cylindrical structures of tungsten disulfide. Nature 360, 444–446. doi:10.1038/360444a0
Tham, W. L., Poh, B. T., Ishak, Z. A. M., and Chow, W. S. (2014). Thermal behaviors and mechanical properties of halloysite nanotube-reinforced poly(lactic acid) nanocomposites. J. Therm. Anal. Calorim. 118, 1639–1647. doi:10.1007/s10973-014-4062-2
Thill, A., Guiose, B., Bacia-Verloop, M., Geertsen, V., and Belloni, L. (2012a). How the diameter and structure of (OH)(3)Al2O3SixGe1-xOH imogolite nanotubes Are controlled by an adhesion versus curvature cornpetition. J. Phys. Chem. C 116, 26841–26849. doi:10.1021/jp310547k
Thill, A., Maillet, P., Guiose, B., Spalla, O., Belloni, L., Chaurand, P., et al. (2012b). Physico-chemical control over the single- or double-wall structure of aluminogermanate imogolite-like nanotubes. J. Am. Chem. Soc. 134, 3780–3786. doi:10.1021/ja209756j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tu, J. X., Cao, Z., Jing, Y. H., Fan, C. J., Zhang, C., Liao, L. Q., et al. (2013). Halloysite nanotube nanocomposite hydrogels with tunable mechanical properties and drug release behavior. Compos. Sci. Technol. 85, 126–130. doi:10.1016/j.compscitech.2013.06.011
Veerabadran, N. G., Price, R. R., and Lvov, Y. M. (2007). Clay nanotubes for encapsulation and sustained release of drugs. Nano 2, 115–120. doi:10.1142/S1793292007000441
Wada, S., and Wada, K. (1982). Effects of substitution of germanium for silicon in imogolite. Clays Clay Miner. 30, 123–128. doi:10.1346/ccmn.1982.0300206
Wang, B., and Huang, H.-X. (2013). Effects of halloysite nanotube orientation on crystallization and thermal stability of polypropylene nanocomposites. Polym. Degrad. Stab. 98, 1601–1608. doi:10.1016/j.polymdegradstab.2013.06.022
Wang, Q., Zhang, J. P., and Wang, A. Q. (2013). Alkali activation of halloysite for adsorption and release of ofloxacin. Appl. Surf. Sci. 287, 54–61. doi:10.1016/j.apsusc.2013.09.057
Whittaker, E. J. W. (1956). The structure of chrysotile.2. Clino-chrysotile. Acta Crystallogr. 9, 855–862. doi:10.1107/S0365110X56002473
Xu, W., Luo, B. H., Wen, W., Xie, W. J., Wang, X. Y., and Liu, M. X. (2015). Surface modification of halloysite nanotubes with L-lactic acid: an effective route to high-performance poly(L-lactide) composites. J. Appl. Polym. Sci. 132:41451. doi:10.1002/app.41451
Yucelen, G. I., Kang, D. Y., Guerrero-Ferreira, R. C., Wright, E. R., Beckham, H. W., and Nair, S. (2012). Shaping single-walled metal oxide nanotubes from precursors of controlled curvature. Nano Lett. 12, 827–832. doi:10.1021/nl203880z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhechkov, L., Heine, T., Patchkovskii, S., Seifert, G., and Duarte, H. A. (2005). An efficient a posteriori treatment for dispersion interaction in density-functional-based tight binding. J. Chem. Theory Comput. 1, 841–847. doi:10.1021/ct050065y
Zhong, S., Zhou, C. Y., Zhang, X. N., Zhou, H., Li, H., Zhu, X. H., et al. (2014). A novel molecularly imprinted material based on magnetic halloysite nanotubes for rapid enrichment of 2,4-dichlorophenoxyacetic acid in water. J. Hazard. Mater. 276, 58–65. doi:10.1016/j.jhazmat.2014.05.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: SCC-DFTB, imogolite, functionalization, dehydroxylation, band gap, Young modulus
Citation: da Silva MC, dos Santos EC, Lourenço MP, Gouvea MP and Duarte HA (2015) Structural, electronic, and mechanical properties of inner surface modified imogolite nanotubes. Front. Mater. 2:16. doi: 10.3389/fmats.2015.00016
Received: 23 December 2014; Accepted: 10 February 2015;
Published online: 02 March 2015.
Edited by:
Demircan Canadinc, Koç University, TurkeyReviewed by:
Seda Keskin, Koç University, TurkeyGotthard Seifert, Dresden University of Technology, Germany
Copyright: © 2015 da Silva, dos Santos, Lourenço, Gouvea and Duarte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hélio Anderson Duarte, Grupo de Pesquisa em Química Inorgânica Teórica, Departamento de Química, Instituto de Ciências Exatas (ICEx), Universidade Federal de Minas Gerais, Av. Antonio Carlos, 6627, Bairro Pampulha, Belo Horizonte, Minas Gerais 31.270-901, Brazil e-mail:ZHVhcnRlaEB1Zm1nLmJy
†In memorian.