 José Jesús Benítez1*
José Jesús Benítez1* José Alejandro Heredia-Guerrero2
José Alejandro Heredia-Guerrero2 Susana Guzmán-Puyol2
Susana Guzmán-Puyol2 Markus J. Barthel3
Markus J. Barthel3 Eva Domínguez4
Eva Domínguez4 Antonio Heredia4
Antonio Heredia4
- 1Instituto de Ciencia de Materiales de Sevilla (ICMS), Spanish Research Council (CSIC) and University of Seville, Seville, Spain
- 2Smart Materials, Nanophysics, Fondazione Istituto Italiano di Tecnologia (IIT), Genova, Italy
- 3Drug Discovery and Development Department, Fondazione Istituto Italiano di Tecnologia (IIT), Genova, Italy
- 4Instituto de Hortofruticultura Subtropical y Mediterránea (IHSM), Spanish Research Council (CSIC) and University of Malaga, Malaga, Spain
Free-standing polyesters films from mono and polyhydroxylated fatty acids (C16 and C18) have been obtained by non-catalyzed melt-condensation polymerization in air at 150°C. Chemical characterization by Fourier transform infrared spectroscopy and 13C Magic Angle Spinning Nuclear Magnetic Resonance (13C MAS–NMR) has confirmed the formation of the corresponding esters and the occurrence of hydroxyl partial oxidation, which extent depends on the type of hydroxylation of the monomer (primary or secondary). Generally, polyester films obtained are hydrophobic, insoluble in common solvents, amorphous and infusible as revealed by X-ray diffraction and differential scanning calorimetry. In ω-polyhydroxy acids, esterification reaction with primary hydroxyls is preferential and, therefore, the structure can be defined as linear with variable branching depending on the amount of esterified secondary hydroxyls. The occurrence side oxidative reactions like the diol cleavage are responsible for chain cross-linking. Films are thermally stable up to 200–250°C though this limit can be extended up to 300°C in the absence of ester bonds involving secondary hydroxyls. By analogy with natural occurring fatty polyesters (i.e., cutin in higher plants), these polymers are proposed as biodegradable and non-toxic barrier films or coatings to be used, for instance, in food packing.
Introduction
The use of plastics in the packaging industry is massive and solidly implanted and, despite the increasing tendency to recycle, their disposal is causing a serious and global environmental problem. This is originated by the fact that, in contact with the soil, most of hydrocarbon-based materials have degradation periods of decades and beyond. Furthermore, in the case of food containers, the toxicity associated to the release of some additives like bisphenol A from the plastic matrix or the protective internal coating is becoming a notorious health concern (NPT-CERHR Monograph on the Potential Human Reproductive and Developmental Effects of Bisphenol A, 2008).
In Nature, higher plants have solved the “packing problem” by wrapping their fruits, leaves, stems, and seeds with a cuticle, a continuous extracellular protective membrane essentially composed of a polymeric polyester skeleton supporting other components like external and embedded waxes, phenolic compounds, and polysaccharides (Pollard et al., 2008). In aerial parts, such polymeric framework is the cutin, and it constitutes an abundant renewable stock representing up to 1200 kg/crop hectare (Heredia, 2003). The inherent characteristics of biopolymer cutin (Domínguez et al., 2011), i.e., non-toxicity, biodegradability, and availability, are very attractive features to overcome the aforementioned drawbacks of current packaging materials and, consequently, the design and obtaining of cutin inspired polymers is proposed as an alternative. The insolubility and infusibility of natural cutin impedes its direct processing and, therefore, the strategy comprises the chemical degradation of the biopolymer, the isolation of the monomers, and the reconstruction of the polyester under synthetic conditions. Chemically, cutin can be described as an amorphous polymeric network of ester bonded C16 and C18 polyhydroxylated fatty acids (Baker and Holloway, 1970; Kolattukudy, 1981; Walton, 1990). Thus, the reaction involved is an esterification between the carboxylic acid and the alcohol groups that is typically performed by polycondensation in an organic media using an acidic surfactant as a promoter or a metallic catalyst (Bawn and Huglin, 1962; Saam, 1998; Heredia-Guerrero et al., 2009; Liu et al., 2011; Zhang et al., 2011). However, to avoid the use of organic solvents as well as aromatic and heavy metal catalysts jeopardizing the non-toxicity of the final product, we have used the direct, non-catalyzed synthesis from molten precursors in air. Conceptually, this is a quite simple method for preparing thin films or coatings that could be easily scaled up to large production processes at a very low cost. Particularly, and, if conceived as internal food container coatings, the preservation of the insolubility and infusibility of natural cutin is desirable to resist the sterilization protocols and the chemical attack of edible fluids.
Thus, the aim of this article is the characterization of the materials resulting from the non-catalyzed melt-polycondensation of cutin representative C16 monomers like 16-hydroxy, 10,16-dihydroxy and 9,10,16-trihydroxypalmitic acids, as well as other reference C18 polyhydroxyacids to state the effect of the hydroxylation degree in the physical and chemical properties of the polyhydroxyesters obtained. Results will constitute a reference for designing a synthesis of non-toxic and biodegradable protective films or coatings based on the valorization of residues like peels of large scale production fruits.
Materials and Methods
Products
16-Hydroxypalmitic acid (Aldrich, 98%) (HPA), 12-hydroxystearic acid (Aldrich, 99%) (HSA), 9,10-dihydroxystearic (Sigma-Aldrich, 99%) (diHSA), and aleuritic (DL-threo-9,10,16-trihydroxypalmitc, Fluka, 93.8%) (triHPA) were commercially available and used as received. The product named as 10,16-dihydroxyhexadecanoic acid (diHPA) actually refers to an extract obtained from tomato mature green cuticles (Luque et al., 1995). Cuticles were isolated by incubation for 5 days in a 2% pectinase/0.2% cellulose solution at pH = 3.6 and treated with chloroform:methanol (3/1, v/v) for 3 h to remove waxes. Dewaxed cuticles were depolymerized by saponification in 1% KOH/methanol for 6 h under refluxing conditions. The filtrate was neutralized with HCl 1N and extracted with diethyl ether. The solid contains about 82% (w/w) of 10(9),16-dihydroxyhexadecanoic acids and minor amounts of other diacids and hydroxyacids (Baker and Holloway, 1970; del Río and Hatcher, 1998; Kosma et al., 2010). The chemical structure of monomers and polyester nomenclature used along this article are summarized in (Figure 1).
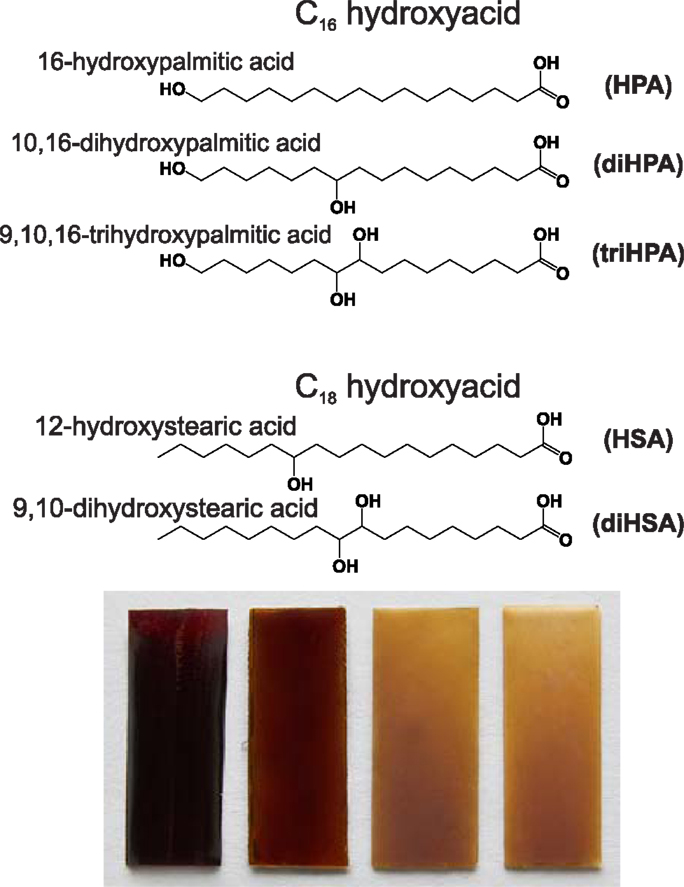
Figure 1. Chemical structure of monomers, nomenclature used and polyester appearance.
Polyester Synthesis by Non-Catalyzed Melt-Condensation Polymerization
For this procedure, about 150 mg of solid monomer were placed on open carbon doped Teflon molds (30 mm ×10 mm and 1 mm deep) and heated in air inside a natural air convention oven at 150°C for 16–24 h. After cooling for several hours at room conditions, pale yellow/brown rubbery free-standing films with a thickness of about 300–350 μm can be easily separated from the mold. In the case of poly HSA, a very viscous product is obtained. To prepare samples under reduced pressure, the oven was pumped for 12 h at 25 mbar.
Polymer Synthesis by DBSA Assisted Polycondensation
As a reference for the samples obtained by the previous method, polyesters of HPA, diHPA, and triHPA have been prepared by low-temperature polycondensation in similar conditions as those reported (Saam, 1998). Typically, about 200–250 mg of monomer, 80 mg of dodecylbenzenesulfonic acid (DBSA), and 5 mL of toluene were introduced in a 50-mL flask. The mixture was briefly heated to about 100°C under continuous stirring until solids were dissolved, and then cooled to 80°C. Minor losses of organic solvent by evaporation were corrected and, in these conditions, the product began to precipitate in about 20 min forming a solid phase at the bottom of the flask. After 3 h, the reaction was stopped and the solid was filtered, washed sequentially three times with toluene, chloroform, and methanol and dried at room temperature for 24 h. A continuous layer adapting to the bottom of the flask was obtained only when triHPA was used.
Independently on the preparation method, solids obtained are quite insoluble in common solvents such as water, chloroform, toluene, dimethyl sulfoxide (DMSO), and light alcohols. Only poly HPA and poly HSA were found to be soluble in some of these solvents.
Characterization
Transmission infrared spectra of samples were collected in a Fourier transform infrared spectroscopy (FTIR) spectrometer (FT/IR-6200, JASCO) equipped with a DTGS detector and using KBr pellets containing 1.5% (w/w) of sample. Spectra were recorded in the 4000–600 cm−1 range at 4 cm−1 resolution and accumulating 50 scans.
Solid state 13C MAS–NMR proton decoupling single-pulse spectra of polymers were obtained with a Bruker Avance DRX-400 spectrometer using a magnetic field of 9.36 T and equipped with a multinuclear probe. Minced samples were packed in 4 mm Ø zirconia rotors and spun at 10 KHz. The spectra were acquired at a frequency of 100.61 MHz, using a π/6 pulse width of 2.5 μs and a pulse space of 10 s to ensure full relaxation and to allow quantitative analysis from peak areas. The chemical shifts are reported in ppm referenced to tetramethylsilane.
Differential scanning calorimetry thermograms were acquired with a DSC Q20 (TA Instruments) from −60 to 150°C under dry nitrogen flow (50 mL/min) at 10°C/min. Accurately weighed small pieces (about 3.5–4 mg) were cut from the films and stabilized at 55% RH in a glove box for 5 days. After stabilization, samples were packed in hermetic aluminum pans inside the glove box and a pin hole was made immediately before running the DSC experiment. Samples were first cooled to −60°C and then a heating–cooling–heating cycle was performed. The glass transition temperature (Tg) is obtained from the second heating using the inflection method.
Water uptake was calculated from the desorption peak in the first heating. With values well below 1% (w/w), this method was found to be more accurate than weighing.
X-ray diffraction (XRD) patterns were obtained at the CITIUS X-ray laboratory (University of Seville, Spain) using a Bruker D8 Advance instrument equipped with a Cu Kα radiation source operating at 40 kV and 30 mA. XRD patterns were obtained in the 3–70° 2θ-range with a step size of 0.015° and a time step of 0.1° 2θ/min.
Thermal stability of polyesters was monitored with a SDT Q600 TGA/DSC analyzer (TA Instruments). Samples (about 6 mg) were heated from RT to 500°C at 5°C/min under N2 flow (100 mL/min).
Tensile measurements were done with a MTS Criterion 42 machine equipped with a 10-N load cell. Rectangular uniform pieces (7 mm × 20 mm) and typically 300–350 μm thick were brought to rupture at a constant deformation rate of 0.2 mm/min at room environmental conditions. Stress values were calculated using the specimen cross-section under no applied load and the Young’s modulus from the initial slope of the stress–strain curves. Experiments were repeated at least five times and values averaged.
Insoluble fraction was calculated considering the weight loss of minced samples (about 200 mg) placed inside a glass microfiber extraction thimbles and immersed in dimethyl sulfoxide (DMSO) at 90°C for 16 h under continuous magnetic stirring.
Gel-permeation chromatography (GPC) measurements were carried out on an Agilent 1260 Infinity quaternary LC system using two PLGel 5-μm MIXED-C columns at 25°C and a refractive index detector. Tetrahydrofuran (THF) was used as eluent at 1 mL/min flow rate. The system was calibrated with Agilent EasyVial PS standards. Due to the very low solubility of most of these polyhydroxyesters in THF, only consistent results can be obtained for poly HPA and poly HSA.
Results
Chemical Analysis of Polyhydroxyesters Obtained by Non-Catalyzed Melt Polycondensation in Air
In every case, ester formation is clearly stated from the carbonyl 13C MAS–NMR peak around 174 ppm (Garbow and Stark, 1990; Zlotnik-Mazori and Stark, 1998; Ahmed et al., 2003; Pandey et al., 2010; Arrieta-Baez et al., 2011; Liu et al., 2011) (Figure 2) and the characteristic FTIR ν(C=O) around 1730 cm−1 and ν(OC–O–C) bands at 1240 and 1170 cm−1 (Bellamy, 1975) (Figure 3).
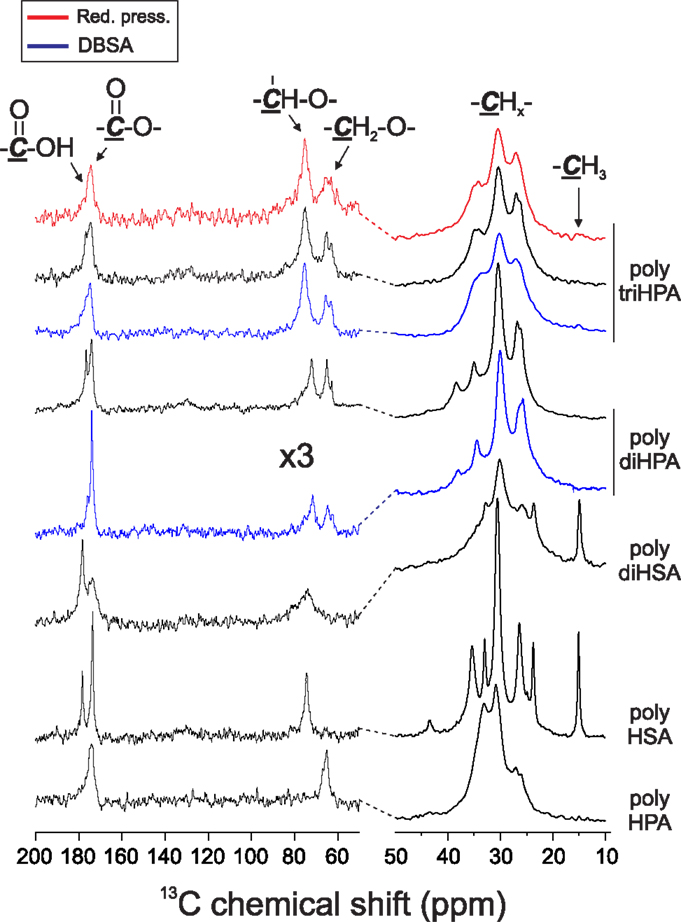
Figure 2. Solid state 13C MAS-NMR spectra of polyesters. For clarity, the intensity of the 50–200 ppm region is multiplied by a factor of 3.
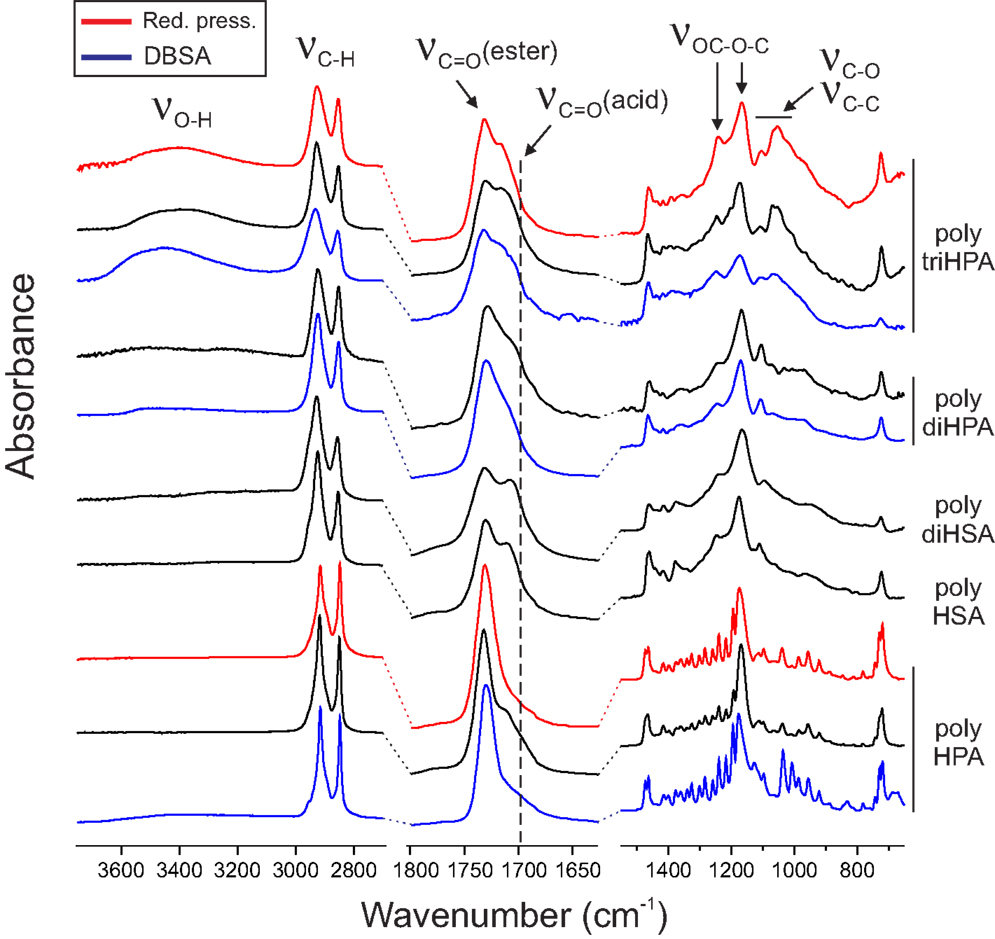
Figure 3. FTIR spectra of polyhydroxyesters.
A detailed analysis of obtained polyesters is given below:
Poly HPA
In poly HPA (Figure 2), single 13C MAS–NMR peaks at 174.0 ppm (–COO–CH2–) and 65.3 ppm (–COO–CH2–) reveal a very high esterification degree. This is confirmed by a very weak contribution of free acid at 9 ppm in the corresponding 1H MAS–NMR spectrum (not shown). A residual non-reacted hydroxyl population can also be detected from the weak signal at 61.8 ppm (HO–CH2–). On the other side, the quantitative analysis of the 13C MAS–NMR data shows an excess of carbonyls and a deficiency of singly bonded (C–O) when compared to the nominal composition of the polyester (Table 1). This is the result of the partial oxidation of hydroxyl groups to ketones and –COOH species as presumed from a weak MAS–NMR signal at 43.4 ppm (–CH2–COR) and confirmed from 13C NMR in solution (Figure S1 in Supplementary Material). Such oxidation by-products are not observed in samples prepared under reduced pressure. 13C NMR data also detect the absence of etherification and dehydration reactions in both preparation conditions.
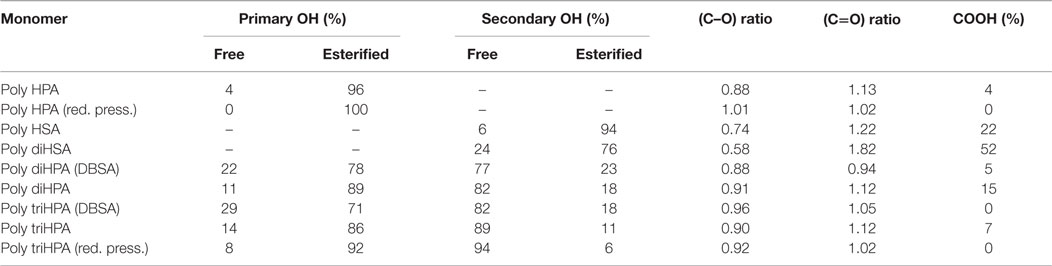
Table 1. Chemical composition from 13C MAS–NMR.
FTIR data, (Figure 3), confirm the formation of the polyester by the characteristic peaks at 1732 and 1169 cm−1 (Liu et al., 2011) and the low intensity of hydroxyls (3600–3200 cm−1 band). The broad shoulder around 1715 cm−1 is assigned to carbonyls in a loosely packed polyester phase as well as to ester groups interacting with residual hydroxyls and to loosely associated carboxyls (Heredia-Guerrero et al., 2014). Additionally, a weak absorption at 1775 cm−1 is assigned to peroxyester (R–CO–O–O–R′) species (Davison, 1951) as an oxidation by-product.
When prepared under reduced pressure (red trace in Figure 3), the FTIR spectrum reveals a reduction of peroxy species due to the weaker exposure to oxygen.
Poly HSA
The esterification of the secondary hydroxyls is characterized by 13C MAS–NMR peaks at 173.5 (–COO–CH<) and 74.4 ppm (–COO–CH<) (Figure 2). The presence of free acid (13C 178.3 ppm and 1H ~9 ppm) is notorious, and there is little indication of free hydroxyl groups (no appreciable contribution on the higher chemical shift side of the 74.4 ppm peak (–CH2–CHOH–CH2–) and no aliphatic signal at 38 ppm (–CH2–CHOH–CH2–). Based on the quantification of 13C MAS–NMR spectra (Table 1), there is a clear deficiency of (C–O) and an excess of (C=O) species. These data reveal that secondary hydroxyl oxidation is noticeable. Indeed, the intermediate ketone is clearly detected by the 43.5 ppm 13C MAS–NMR peak (–CH2–CO–CH2–) in the solid and by the corresponding 211.9 ppm signal (–CH2–CO-CH2–) after dissolving in CDCl3 (not shown).
In the FTIR spectrum, both the ester bands (1731, 1246 and 1175 cm-1) and the low concentration of free hydroxyls are confirmed (Figure 3). The high concentration of free acid is characterized by the broadening on the lower wavenumber side (~1700 cm−1) of the carbonyl stretching and by the definition of the peak at 1712 cm−1 (ester carbonyl perturbed by –COOH). As in poly HPA, a small amount of per-oxidation is evidenced by the weak band around 1775 cm–1.
Gel-permeation chromatography results are consistent with the low polymerization degree of poly HSA and reveal the presence of oligomers containing few monomeric units (n = 1–5) (Table 2). However, esterification degree of hydroxyls is as high as in poly HPA (Table 1). The combination of both results, i.e., low Mw and high percentage of esterified hydroxyls suggest intra-esterification and the formation of cyclic oligomeric structures in poly HSA.
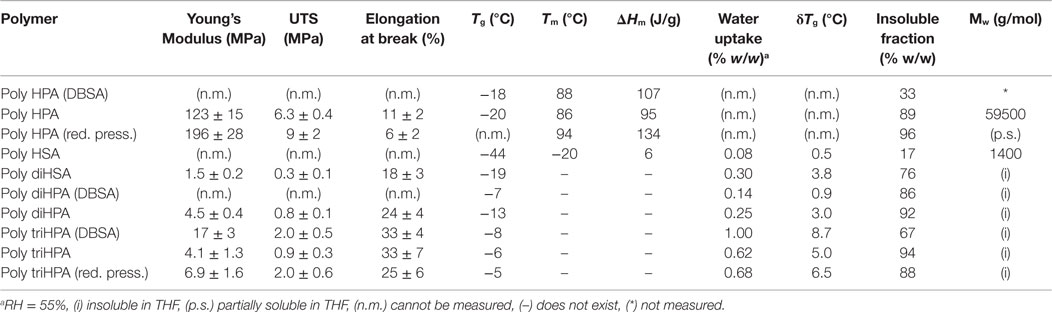
Table 2. Physical parameters of polyhydroxyesters obtained.
Poly diHSA
13C MAS–NMR spectrum of poly diHSA (Figure 2) displays broad ester (173.6 ppm) and (C–O) peaks, which is interpreted as a variety of chemical configurations. Thus, vicinal –OH groups may remain free (~76 ppm) or esterify (one, doublet at 74 and 81 ppm; both, single peak at 76 ppm) and consequently no reliable quantitative chemical information can be directly extracted. However, the most relevant findings are the strong presence of free acid (sharp peak at 178.3 ppm, about 52% of carbonyls) and the intense reduction of (C–O) bonds (Table 1). As in the previous case, this is the consequence of hydroxyl oxidation to carboxylic acid.
FTIR data (Figure 3) support NMR information and detect the ester formation (bands at 1731, 1236 and 1166 cm-1) and the reduced population of free hydroxyl groups (weak broad bands around 3500 and 3250 cm−1). The broadening of the carbonyl stretching and the development of 1708 and 1690 cm−1 components are the consequence of the more heterogeneous chemical environment of carbonyl groups and the perturbation exerted by the considerable population of free acid.
Poly diHPA
Poly diHPA is the only polyester of the series that has been prepared from a hydroxyacid extract mostly composed of 10,16 dihydroxypalmitic acid but containing other acids and hydroxyacids from the C18 and C16 families. For that reason, data in Table 1 are referenced to the monomeric mixture composition. Poly diHPA is slightly deficient in (C–O) and enriched in carbonyl groups, which suggests that hydroxyls are partially oxidized to acid.
The 13C MAS–NMR spectrum (Figure 2) evidences the ester formation (174.1 ppm) and the presence (~15%) of free acid (13C 176 ppm and 1H 9.6 ppm). The (C–O) signal reveals that most of primary hydroxyls are esterified (Saam, 1998; Deshmukh et al., 2003, 2005) (65.1 ppm,–COO–CH2–) and only about 11% of them remain as non-reacted (62.9 ppm, –CH2OH). On the other side, the 72.2 ppm peak (–CH2–CHOH–CH2–) indicates that secondary hydroxyls are mostly intact. Indeed, by comparing the total amount of ester carbonyls and the esterified primary hydroxyls and by quantifying the aliphatic peak at 38.4 ppm (–CH2–CHOH–CH2–), we have calculated that only about 18% of secondary hydroxyls have reacted.
As in previous cases, FTIR confirms the formation of the ester by the characteristic (C=O) and (C–O–C) stretchings (Figure 3). It also detects the presence of non-reacted hydroxyl groups (broad bands around 3500 and 3250 cm−1). The carbonyl peak (1729 cm−1) is accompanied by a shoulder at 1712 cm−1 that gathers the contribution of both –COOH and –OH groups hydrogen bonding ester carbonyls.
Poly triHPA
Poly triHPA is obtained from the most hydroxylated acid (aleuritic acid) of the series and the 13C MAS–NMR is displayed in Figure 2. The dominant ester peak (174.6 ppm) is accompanied by a free acid fraction (–CH2–COOH, 176.7 ppm and –CH2–COOH, 34.9 ppm) that it is estimated in about 7%. Esterification is mainly with primary hydroxyls (65.3 ppm) leaving a non-reacted (63.1 ppm) population of about 14%. Most of secondary hydroxyls (75.2 ppm) are preserved though a small peak ~84 ppm can be considered as the result of mono esterification of the vicinal diol moiety. From the area of this peak, and the difference between the ester carbonyls and esterified primary hydroxyls, the fraction of esterified secondary –OH groups is calculated to be ~11%. As the others polyesters of the series, poly triHPA is enriched in (C = O) and deficient in (C–O) suggesting a small hydroxyl oxidation.
Fourier transform infrared spectroscopy (Figure 3) reveals the excess of non-reacted hydrogen bonded hydroxyls with broad ν(O–H) bands at 3465 and 3375 cm−1. Such hydroxyl surplus is also perturbing the carbonyl vibration causing a broad peak at 1717 cm−1 accompanying the ester vibration at 1731 cm−1. In addition to the (C–O–C) stretching in ester group at 1173 cm−1, there is a doublet (1069 and 1053 cm−1) that we assign to the (C–O) stretching of vicinal secondary hydroxyls.
As a reference for the non-catalyzed polycondensation in air, the reaction has been performed under reduced pressure (25 mbar). The polyester obtained after 12 h (Figures 2 and 3, red traces) is chemically quite similar to the one prepared in air. 13C MAS–NMR displays an ester peak at 174.4 ppm with some broadening at higher chemical shift but no evidence of free acid is detected. The lack of free acid is also confirmed from the absence of the aliphatic signal ~35 ppm (–CH2–COOH) and the narrowing on the low wavenumber side of the carbonyl stretching. Most of primary (92%) and about 6% of secondary hydroxyls are esterified and the (C–O)/(C=O) signal ratio suggests no hydroxyl oxidation. The (C–O) ratio <1 is likely due to a very mild dehydration favored by pumping. The higher esterification degree is supported by both the weakening of both the hydroxyl band around 3400 cm−1 and the shoulder at 1717 cm−1.
Chemical Analysis of Polyhydroxyesters Obtained by DBSA Promoted Polycondensation
As reference for the non-catalyzed process, the polycondensation reaction has also been performed in the presence of a hydrophilic acidic agent such as DBSA (dodecylbenzenesulfonic acid). The analysis is basically restricted to diHPA and triHPA because their potential technical interest considering the abundance of their natural sources.
DBSA Poly diHPA
13C MAS–NMR spectrum of DBSA poly diHPA (Figure 2, blue trace) shows a very sharp peak of ester carbonyl at 174.0 ppm with little contribution (~5%) of free acid (175.8 ppm). As in the non-catalyzed bulk polycondensation homologous, esterification is mostly with primary hydroxyls (64.7 ppm) leaving about 22% of non-reacted groups (62.6 ppm). Esterification with secondary hydroxyls is slightly more extended (23%) using DBSA, as deduced from the broadening of the 75.5 ppm peak at higher chemical shift. It is also visible when comparing the intensities of the 34.5 and 38.1 ppm corresponding to the aliphatic carbon in –CH2–CHO(R)–CH2– and –CH2–CHOH–CH2–, respectively (Figure 2). Though there is a small loss of (C–O) bonds upon formation of the polyester, this is not accompanied by an increment of (C=O). Consequently, the elimination of hydroxyls is not considered to be caused by oxidation but to the migration of the hydroxyl rich species of the monomeric mixture toward the solvent.
When compared to poly diHPA, FTIR of DBSA poly diHPA (Figure 3) shows a sharper carbonyl stretching region with an ester peak at 1733 cm−1 and a weaker shoulder at 1713 cm−1. This is caused by a more reduced population of free acid groups perturbing the carbonyl vibration.
DBSA Poly triHPA
DBSA poly triHPA is characterized as an ester by the 13C MAS–NMR signal at 174.7 ppm (Figure 2, blue trace). Compared to non-catalyzed poly triHPA, the carbonyl signal is broader on the higher chemical shift side but displays no distinctive peak of free acid. Such broadening is due to hydrogen bonding with hydroxyls, which are characterized by the IR band at 3450 cm−1 (Figure 3). Another indication of such hydrogen bonded phase is the shoulder around 1715 cm−1. As in poly triHPA, esterification of primary hydroxyls is preferential (71%) while only 18% of secondary have reacted.
The Structure of Polyhydroxyesters
IR data, i.e., the low frequency and width of the (–CH2–) stretching peaks (2917 and 2850 cm−1), the splitting of the scissoring (1469 and 1465 cm−1) and rocking (729 and 721 cm−1) modes and the presence of progression bands in the 1400–1200 cm−1 (Stein and Sutherland, 1954; Casal et al., 1982) reveal that only poly HPA is crystalline. This is confirmed by XRD (Figure 4) and DSC (Figure 5). Melting temperature (Tm) and enthalpy (ΔHm) values (Table 2) are in good agreement with results reported for poly ω-hydroxyltetradecanoic acid (Liu et al., 2011).
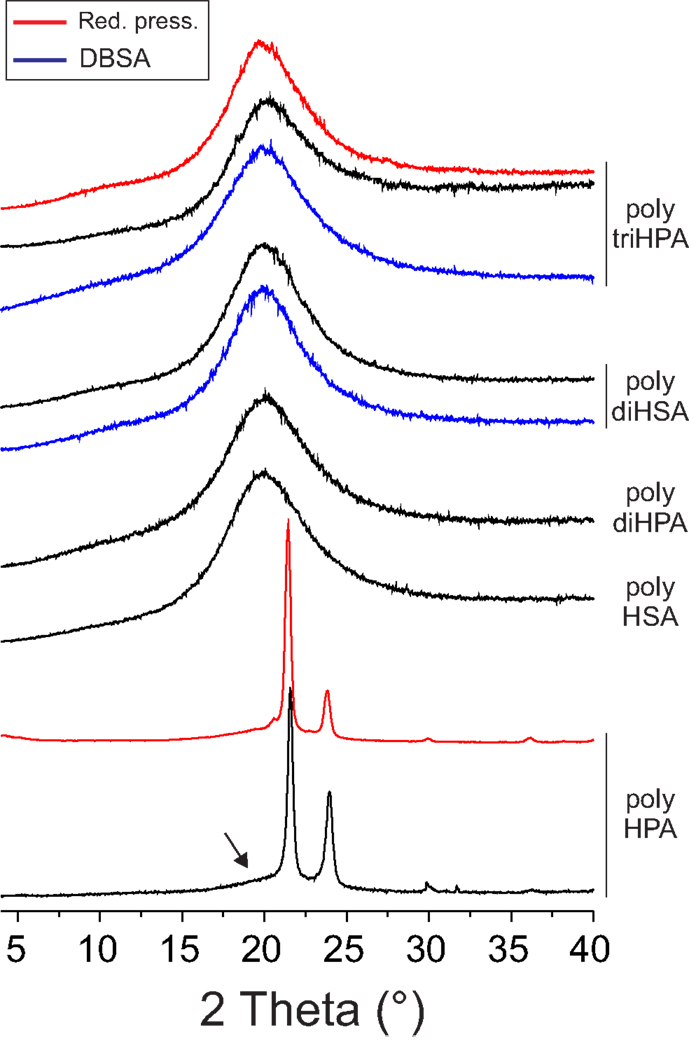
Figure 4. XRD diagrams of polyesters. The broad peak around 20° is typical for amorphous solids. The arrow highlights the formation of an amorphous phase in poly-HPA prepared in air.
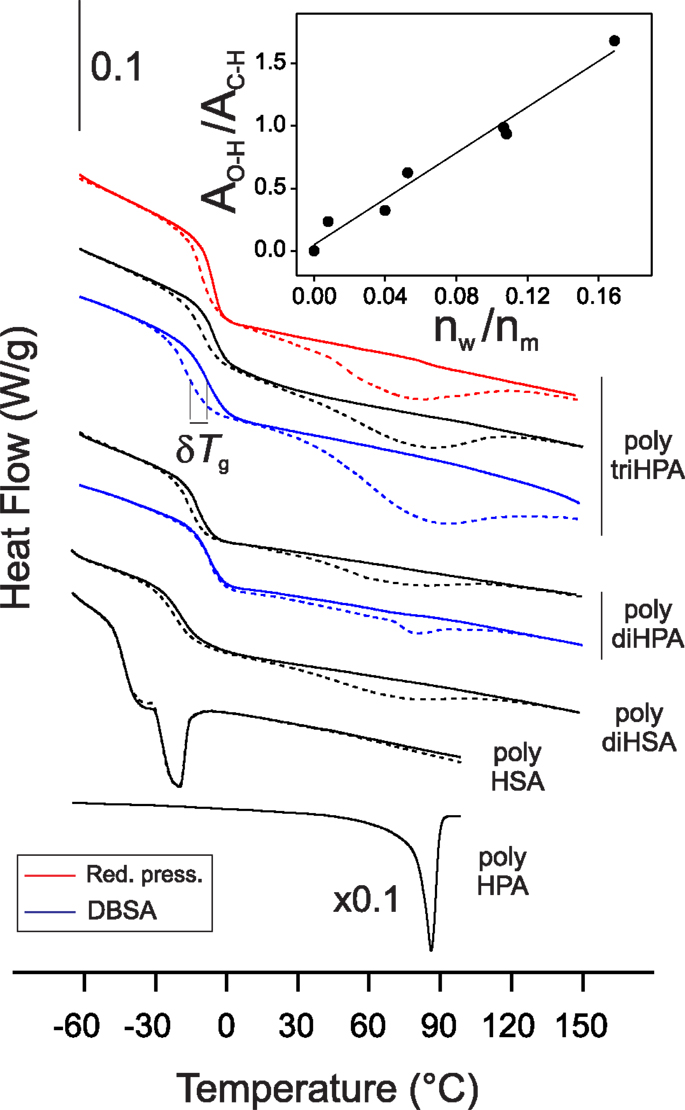
Figure 5. DSC thermograms of polyesters under N2 flow. Dashed lines correspond to samples stabilized at 55% RH and display the desorption of water around 80°C. Solid lines correspond to in situ dried samples. The inset in the upper right corner shows the correlation between the FTIR ν(O–H)/ν(C–H) band area ratio and the amount of adsorbed water (nw) calculated from DSC and referred to the monomer content (nm) (mol/mol).
The rest of the members of the series are amorphous at room temperature (Davison and Corish, 1955; Snyder et al., 1996) and DSC thermograms display clear vitreous transitions with Tg values generally increasing with the hydroxylation degree of the monomer (Table 2).
Mechanical Properties
Uniaxial tensile parameters of free-standing films obtained are compiled in Table 2. Within the series, crystalline poly HPA is the stiffest and displays a brittle fracture at quite low strain. If compared to values reported for the analogous poly ω-hydroxyl tetradecanoic acid (Liu et al., 2011), it can be deduced that this behavior is likely caused by a low molecular weight polymerization and by partial amorphization, as indicated by the broad peak around 20° in Figure 4. In this sense, self-polycondensation of HPA under reduced pressure is more efficient and yields a higher molecular weight polyester.
The rest of polyesters prepared are amorphous and have quite low Young’s modulus and rupture stress values, as well as moderate (20–30%) elongation at break. They can be defined as viscoelastic materials showing no strain softening or plastic flow regions. Among these polyhydroxyesters, poly triHPA (DBSA) displays the highest tensile parameters, particularly the Young’s modulus. The self-polycondensation of aleuritic acid has been studied in detail in a previous work (Benítez et al., 2014) and it was found that tensile parameters experienced a sudden modification when achieving full esterification. Curiously, poly triHPA (DBSA) values do fit within this gap and we conclude that polycondensation in the organic medium was not completed in the experimental conditions used.
Water Uptake
Hydrophobicity is a desirable feature in barrier polymers to prevent, among others, mechanical modifications by swelling, chemical degradation, and material lixiviation. In this series of polyhydroxyacids, the nominal hydroxyl to acid ratio is equal or above 1 and, therefore, the excess of hydroxyl groups is expected to act as water absorbent sites. To comparatively estimate hydrophobicity, water uptake of films has been evaluated by DSC. In situ sample drying shows two events on the thermogram: the desorption peak around 70–80°C and the Tg shift (δTg) (Figure 5). Both parameters increase with the monomer hydroxylation degree (Table 2). Their good correlation with the normalized area of the FTIR hydrogen bonded hydroxyls (inset in Figure 5, for instance) clearly states that surplus hydroxyls are the origin of polyhydroxyesters surface affinity for water.
Thermal Stability
The thermal stability of a polymer is a very important feature conditioning its potential applications. Moreover, TGA experiments of these polyesters under inert atmosphere were found to be structure sensitive (Figure 6). Thus, TGA patterns of reference monomers (primary HPA and secondary HSA) show two decomposition stages (dashed lines). The low temperature peak (240–250°C) is common and it is mostly associated to the dehydration and decarboxylation processes of the hydroxyacids. Its absence in the polyester series is consistent with spectroscopic data indicating high esterification degrees. On the other side, the second decomposition stage is specific for each type of monohydroxyacid: ~335°C for secondary and ~410°C for primary.
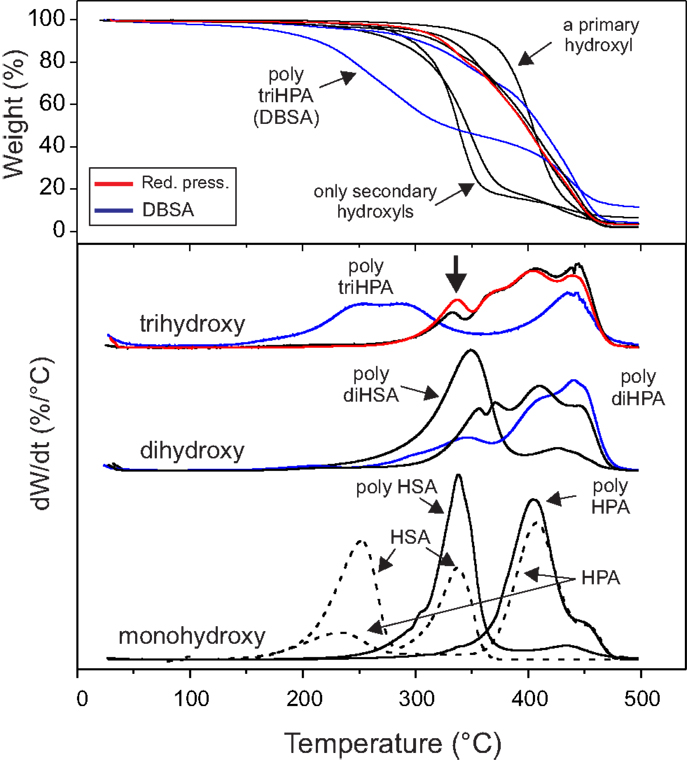
Figure 6. (up) TGA and (bottom) derivative curves of polyesters under N2 flow.
In poly HPA, poly HSA, and poly diHSA it is observed that the thermogravimetric patterns mostly depend on the type of monomer hydroxylation (primary or secondary). Thus, polyesters from exclusively secondary functionalized monomers (poly HSA and poly diHSA) are thermally decomposed at lower temperature (335–350°C) than the one obtained from an exclusively primary hydroxylated precursor (poly HPA) (410°C). The fact that the TGA profile of the polyhydroxyesters overlaps with those of the corresponding monomers, indicates that the primary thermal degradation mechanism in the polymers is the scission of the ester bond into an olefin and a carboxylic acid, followed by their subsequent decomposition (Goldfarb and McGuchan, 1968; Sutton and Tighe, 1973).
In polyhydroxyesters containing both primary and secondary hydroxyls (poly diHPA and poly triHPA), the decomposition peak is mostly displaced toward the high temperature side indicating that esterification with primary hydroxyls is preferential, as already pointed by 13C MAS–NMR.
Poly triHPA (DBSA) is a particular case within the series and shows considerable decomposition starting at 200°C. Since there are no spectroscopic evidences of a significant acid phase responsible for such thermal lability, we consider the possibility of an impurity arising from the DBSA method catalyzing the scission reaction.
Discussion
Experimental results have shown that free-standing polyesters films of C16 and C18 polyhydroxyacids can be prepared by non-catalyzed melt-polycondensation in air. In general, esterification degrees were quite high, particularly when the monomer contains a primary hydroxyl. However, the formation of minor amounts oxidation and dehydration by-products is associated to this preparation procedure. On the other side, the resolution of the 13C MAS–NMR spectra, as well as the overlapping with the (C–O) region, prevents a reliable detection of ether formation. The absence of ether species was only confirmed in poly HPA prepared in air and under reduced pressure (Figure S1 in Supplementary Material).
Consistency of films obtained by non-catalyzed melt-polycondensation in air is found to depend on the type of hydroxylation of the monomer. Thus, products from those containing only secondary hydroxyls are very soft (poly diHSA) or even resinous (poly HSA). On the contrary, films obtained from the ω-hydroxyacid (poly HPA) are crystalline and, mechanically, the most robust within the series. Such a difference is likely due to the balance between inter and intra-estererifiction reactions, leading to the formation of low Mw cyclic structures from exclusively secondary hydroxylated monomers. Thus, free-standing polyesters are better prepared from long-chain hydroxyacids containing primary hydroxyls.
When primary and secondary hydroxyls coexist, experimental data have shown that the formation of the primary ester is preferential. Consequently, their structure can be described as mostly linear with some degree of branching depending on the extent of the esterification with secondary –OH (Figure 7). However, and despite no cross-linking between chains is possible because of the AB (–COOH/–OH) stoichiometry, they are found to be quite insoluble thermosets.
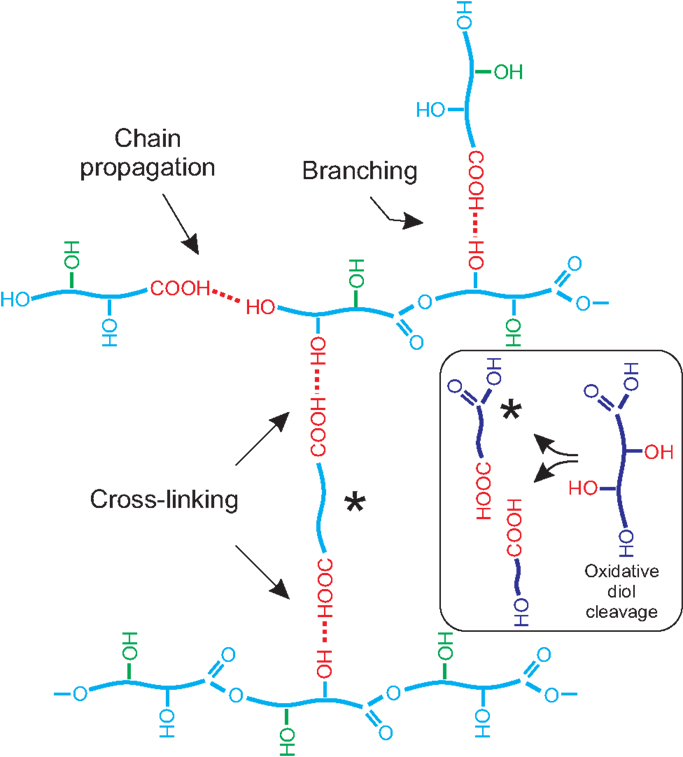
Figure 7. Scheme representing chain propagation, branching, and cross-linking in the structure of a polyhydroxyester resulting from monomers bearing a primary and a secondary hydroxyl (poly diHPA, blue) and one primary and two vicinal secondary hydroxyls (poly triHPA, blue plus green). In this later case, the oxidative vicinal diol cleavage reaction is shown as an inset.
Chemically, polyhydroxyesters obtained by non-catalyzed melt-polycondensation in air are generally deficient in (C–O) bonds and enriched in (C = O) linkages in the form of acid and/or ester. This is the result of hydroxyl oxidation to carbonyl species, which is more intense for secondary hydroxyls, particularly in vicinal configuration, where an oxidative diol cleavage reaction is feasible:
Newly formed carboxylic groups may undergo further esterification with available hydroxyls creating two ester bonds per broken C–C. One of the fragments is a dicarboxylic acid and it is susceptible of generating cross-linking between polymeric chains (Figure 7). This mechanism would explain, for instance, the better consistency of poly diHSA vs. poly HSA as well as its higher insoluble fraction (Table 2).
The same oxidative diol cleavage reaction can also be proposed for poly triHPA. However, the experimental (C=O/C–O) ratio indicates that the oxidation is not as intense as in poly diHSA. The main difference between both monomers is the presence of a primary hydroxyl in the 9,10,16-trihydroxypalmitic acid and we have already confirmed the higher reactivity of primary hydroxyls. Consequently, we propose that the preferential esterification of primary hydroxyls progressively gives rise to a viscous polyester phase that impedes oxygen diffusion to the bulk and reduces the extent of the oxidative diol cleavage in poly triHPA when compared to poly diHSA. The association of cross-linking with the extent of the oxidative diol cleavage and further esterificacion in poly triHPA would explain the increment of the insoluble fraction values in the sense air > reduced pressure > DBSA assisted, as contact with oxygen is reduced.
Poly diHPA is by far the most interesting polyhydroxyester among the series. It has been obtained from a monomeric mixture extracted from tomato cuticles and may represent a stock of about 35,000 t considering only the dry wastes of tomato processing in Europe. The monomeric mixture is mostly composed of positional isomers of the ω-dihydroxyhexadecanoic acid, being the 10,16 the most abundant (Holloway and Deas, 1971). In this molecule, the oxidative cleavage is not possible because of the lack of vicinal diols. However, as biopolyester cutin, synthetic poly diHPA is infusible and quite insoluble, pointing to a cross-linked structure. Our hypothesis is that other minor components are responsible for such properties. The esterification of secondary hydroxyls of different polymeric chains with dioic acids is considered here as the source of cross-linking. Such dioic acid have been detected in the monomeric mixture (~2.5%) (del Río and Hatcher, 1998) or can also be in situ generated by the oxidative cleavage of the small amounts of 9,10,16-trihydroxyhexadecanoic and 9,10,18-trihydroxyoctadecanoic acids (~1.4%). Another option is a partial oxidation of the significant amount of the ω-hydroxyacid (~9.3%) to the dioic acid as observed for poly HPA.
Conclusion
Polycondensation of molten fatty polyhydroxyacids (C16–C18) in the absence of a catalyst is proven to yield generally amorphous, infusible and insoluble free-standing polyester films. The process is relatively slow, as conditioned by the effective elimination of generated water molecules. Esterification is observed to be preferential with primary hydroxyls and, therefore, their structure can be described as linear with variable branching, depending on the extent of the secondary hydroxyl esterification. When conducted in air, partial oxidation of hydroxyl groups takes place, but, despite the harsh preparation conditions used, the oxidation extent is found to be limited by the diminishment of the oxygen diffusion within the viscous bulk as the polyhydroxyester phase is being formed. Hydroxyl oxidation and acid formation is observed to be more intense for secondary groups, particularly, in vicinal diol configuration. Esterification of newly formed dicarboxylic acids with surplus secondary hydroxyls leads to chain cross-linking, which in turn is positive to obtain insoluble, infusible and thermally stable polyhydroxyester films or coatings.
The synthesis of a cutin inspired polyhydroxyester (poly diHPA) is of particular interest since it represents the valorization of the abundant waste coming from the processing of large scale production fruits like tomato.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding is provided by the Spanish Ministerio de Economía y Competitividad under project CTQ2011-24299 and by the Consejería de Economía, Innovación, Ciencia y Empleo of the Andalussian Government (Junta de Andalucía-FEDER) grant TEP-7418. JH-G acknowledges the support of the Marie Curie Intra-European Fellowship (BIOPROTO project).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fmats.2015.00059
References
Ahmed, A., Crawford, T., Gould, S., Ha, Y. S., Hollrah, M., Noor-E-Ain, F., et al. (2003). Synthesis of R- and (S)-10,16-dihydroxyhexadecanoic acid: cutin stereochemistry and fungal activation. Phytochemistry 63, 47–52. doi: 10.1016/S0031-9422(03)00003-7
Arrieta-Baez, D., Cruz-Carrillo, M., Gómez-Patiño, M. B., and Zepeda-Vallejo, L. G. (2011). Derivatives of 10,16-dihydroxyhexadecanoic acid isolated from tomato (solanum lycopersicum) as potential material for aliphatic polyesters. Molecules 16, 4923–4936. doi:10.3390/molecules16064923
Baker, E. A., and Holloway, P. J. (1970). Constituent acids of angiosperm cutins. Phytochemistry 9, 1557–1562. doi:10.1016/S0031-9422(00)85275-9
Bawn, C. E. H., and Huglin, M. B. (1962). The kinetics of the polycondensation of 12-hydroxystearic acid. Polymer 3, 257–262. doi:10.1016/0032-3861(62)90086-1
Benítez, J. J., Heredia-Guerrero, J. A., Guzmán-Puyol, S., Domínguez, E., and Heredia, A. (2014). Polyester films obtained by noncatalyzed melt-condensation polymerization of aleuritic (9,10,16 trihydroxyhexadecanoic) acid in air. J. Appl. Polym. Sci. 132, 1060–1066. doi:10.1002/APP.41328
Casal, H. L., Mantsch, H. H., Cameron, D. G., and Snyder, R. G. (1982). Interchain vibrational coupling in phase II (hexagonal) n-alkanes. J. Chem. Phys. 77, 2825–2830. doi:10.1063/1.444173
Davison, W. H. T. (1951). Infra-red absorption of the carbonyl group. Part I. Diacyl peroxides, per-esters and per-acids. J. Chem. Soc. 2456–2461. doi:10.1039/jr9510002456
Davison, W. H. T., and Corish, P. J. (1955). Infrared spectra and crystallinity. 1. Polyesters. J. Chem. Soc. 2428–2431. doi:10.1039/jr9550002428
del Río, J. C., and Hatcher, P. G. (1998). Analysis of aliphatic biopolymers using thermochemolysis with tetramethylammonium hydroxide (TMAH) and gas chromatography-mass spectrometry. Org. Geochem. 29, 1441–1451. doi:10.1016/S0146-6380(98)00070-9
Deshmukh, A. P., Simpson, A. J., Hadad, C. M., and Hatcher, P. G. (2005). Insights into the structure of cutin and cutan from Agave americana leaf cuticle using HRMAS NMR spectroscopy. Org. Geochem. 36, 1072–1085. doi:10.1016/j.orggeochem.2005.02.005
Deshmukh, A. P., Simpson, A. J., and Hatcher, P. G. (2003). Evidence for cross-linking in tomato cutin using HR-MAS NMR spectroscopy. Phytochemistry 64, 1163–1170. doi:10.1016/S0031-9422(03)00505-3
Domínguez, E., Heredia-Guerrero, J. A., and Heredia, A. (2011). The biophysical design of plant cuticles: an overview. New. Phytol. 189, 938–949. doi:10.1111/j.1469-8137.2010.03553.x
Garbow, J. R., and Stark, R. E. (1990). Nuclear magnetic resonance relaxation studies of plant polyester dynamics. 1. Cutin from limes. Macromolecules 23, 2814–2819. doi:10.1021/ma00212a037
Goldfarb, I. J., and McGuchan, R. (1968). Thermal Degradation of Polyesters. I. Aliphatic Polymers. Technical Report AFML-TR-68-182, Part I. Ohio: Air Force Materials Laboratory, Wright-Patterson Air Force Base.
Heredia, A. (2003). Biophysical and biochemical characteristics of cutin, a plant barrier biopolymer. Biochim. Biophys. Acta. 1620, 1–7. doi:10.1016/S0304-4165(02)00510-X
Heredia-Guerrero, J. A., Benítez, J. J., Domínguez, E., Bayer, I. S., Cingolani, R., Athanassiou, A., et al. (2014). Infrared and Raman spectroscopic features of plant cuticles: a review. Front Plant Sci. 5:1–13. doi:10.3389/fpls.2014.00305
Heredia-Guerrero, J. A., Heredia, A., García-Segura, R., and Benítez, J. J. (2009). Synthesis and characterization of plant cutin mimetic polymer. Polymer 50, 5633–5637. doi:10.1016/j.polymer.2009.10.018
Holloway, P. J., and Deas, A. H. B. (1971). Occurrence of positional isomers of dihydroxyhexadecanoic acid in plant cutins and suberins. Phytochemistry 10, 2781–2785. doi:10.1016/S0031-9422(00)97279-0
Kolattukudy, P. E. (1981). Structure, biosynthesis and biodegradation of cutin and suberin. Ann. Rev. Plant Physiol. 32, 539–567. doi:10.1146/annurev.pp.32.060181.002543
Kosma, D. K., Parsons, E. P., Isaacson, T., Lü, S., Rose, J. K. C., and Jenks, M. A. (2010). Fruit cuticle lipid composition during development in tomato ripening mutants. Physiol. Plant. 139, 107–117. doi:10.1111/j.1399-3054.2009.01342.x
Liu, C., Liu, F., Cai, J., Xie, W., Long, T. E., Turner, S. R., et al. (2011). Polymers from fatty acids: poly(ω-hydroxyl tetradecanoic acid) synthesis and physico-mechanical studies. Biomacromolecules 12, 3291–3298. doi:10.1021/bm2007554
Luque, P., Bruque, S., and Heredia, A. (1995). Water permeability of isolated cuticular membranes: a structural analysis. Arch. Biochem. Biophys. 317, 417–422. doi:10.1006/abbi.1995.1183
NPT-CERHR Monograph on the Potential Human Reproductive and Developmental Effects of Bisphenol A. (2008). Center for the Evaluation of Risks to Human Reproduction, US Department of Health and Human Services, Pub. No. 08-5994.
Pandey, A. K., Nande, S. S., Selukar, B. S., and Garnaik, B. (2010). Synthesis and characterization of novel value added biodegradable poly(aleuritic acid) from renewable resources (shellac) and invertible amphyphilic behaviors in various solvents. e-Polymers 131, 1–12. doi:10.1515/epoly.2010.10.1.1476
Pollard, M., Beisson, F., Li, Y. H., and Ohlrogge, J. B. (2008). Building lipid barriers: biosynthesis of cutin and suberin. Trends Plant Sci. 13, 236–246. doi:10.1016/j.tplants.2008.03.003
Saam, J. C. (1998). Low-temperature polycondensation of carboxylic acids and carbinols in heterogeneous media. J. Polym. Sci. A Polym. Chem. 36, 341–356. doi:10.1002/(SICI)1099-0518(19980130)36:2<341::AID-POLA17>3.3.CO;2-0
Snyder, R. G., Liang, G. L., Strauss, H. J., and Mendelsohn, R. (1996). IR spectroscopy study of the structure and phase behavior of long-chain diacylphosphatidylcholines in the gel state. Biophys. J. 71, 3186–3198. doi:10.1016/S0006-3495(96)79512-7
Stein, R. S., and Sutherland, G. B. B. M. (1954). Effect of intermolecular interactions between CH frequencies on the infrared spectra of n-paraffins and polythene. J. Chem. Phys. 22, 1993–1999. doi:10.1063/1.1739980
Sutton, G. J., and Tighe, B. J. (1973). Poly-α-ester degradation studies. I. Introduction: design and construction of equipment. J. Polym. Sci. A Polym. Chem. 11, 1069–1077. doi:10.1002/pol.1973.170110513
Zhang, S., Lefebvre, H., Tessier, M., and Fradet, A. (2011). Influence of Brønsted acid ionic liquid structure on hydroxyacid polyesterification. Green Chem. 13, 2786–2793. doi:10.1039/c1gc15241f
Keywords: biomimicry, polyhydroxyesters, non-toxic, coatings, waste, valorization
Citation: Benítez JJ, Heredia-Guerrero JA, Guzmán-Puyol S, Barthel MJ, Domínguez E and Heredia A (2015) Polyhydroxyester films obtained by non-catalyzed melt-polycondensation of natural occurring fatty polyhydroxyacids. Front. Mater. 2:59. doi: 10.3389/fmats.2015.00059
Received: 28 May 2015; Accepted: 03 August 2015;
Published: 24 August 2015
Edited by:
Clemens Kilian Weiss, University of Applied Science Bingen, GermanyReviewed by:
Alberto Mariani, University of Sassari, ItalyNágila Maria Pontes Silva Ricardo, Federal University of Ceará, Brazil
Cesar Liberato Petzhold, Federal University of Rio Grande do Sul, Brazil
Copyright: © 2015 Benítez, Heredia-Guerrero, Guzmán-Puyol, Barthel, Domínguez and Heredia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José Jesús Benítez, Instituto de Ciencia de Materiales de Sevilla (ICMS), Americo Vespuccio 49, Isla de la Cartuja, Seville ES-41092, Spain,YmVuaXRlekBpY21zZS5jc2ljLmVz