 Naresh Thota
Naresh Thota Jianwen Jiang
Jianwen Jiang- Department of Chemical and Biomolecular Engineering, National University of Singapore, Singapore
Amphiphilic materials can assemble into a wide variety of morphologies and have emerged as a novel class of candidates for drug delivery. Along with a large number of experiments reported, computational studies have also been conducted in this field. At an atomistic/molecular level, computations can facilitate quantitative understanding of experimental observations and secure fundamental interpretation of underlying phenomena. This review summarizes the recent computational studies on amphiphilic copolymers and peptides for drug delivery. Atom-resolution and time-resolved insights are provided from bottom-up to microscopically elucidate the mechanisms of drug loading/release, which are indispensable in the rational screening and design of new amphiphiles for high-efficacy drug delivery.
Introduction
Among the conventional methods for cancer treatment, surgery physically removes tumor in human body, chemotherapy uses medicines to destroy cancer cells, and radiation exerts high-energy beam to kill or decline the growth of cancer cells (Kumar and Kumar, 2014). Nevertheless, chemotherapy and radiation also kill healthy cells. To overcome this limitation, targeted and sustained drug delivery has been proposed (Chen and Liu, 2012). Targeted delivery can effectively inhibit the growth of cancer cells and reduce damage to healthy cells; meanwhile, sustained release offers several advantages such as less frequent administration and better compliance (Natarajan et al., 2014). In targeted and sustained delivery, drug carriers play an indispensable role. Over the last decade, there has been considerable interest in the development of new materials as drug carriers (Hubbell and Chikoti, 2012). Particularly, amphiphilic materials have emerged as a novel class of candidates for drug delivery (Shimixu et al., 2005). They have the ability to assemble into a wide variety of morphologies such as micelles, vesicles and fibers. As illustrated in Figure 1, drugs can be loaded in these morphologies, transported into cells, and released upon a stimulus (e.g., by altering temperature, pH, or ionic strength).
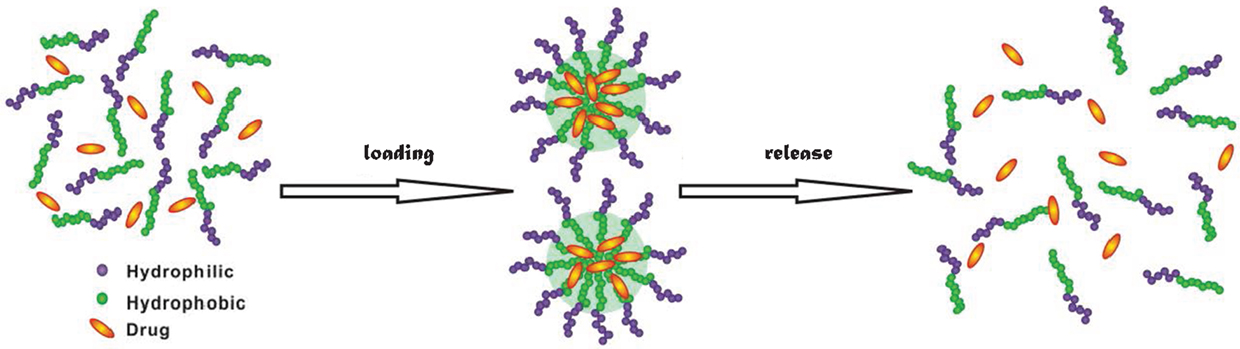
Figure 1. Drug loading and release in amphiphilic materials.
The loading, stability, and release profiles of drugs in amphiphilic materials depend largely on the morphologies assembled, which in turn are governed by material structure and functionality. Low molecular weight surfactants were initially tested for drug delivery; however, they have insufficient micellization capacity and tend to rapidly dissociate in blood (i.e., kinetically unstable) (Lawrence, 1994; Uchegbu and Vyas, 1998; Drummond and Fong, 1999). Alternatively, amphiphilic copolymers (Meng et al., 2009; Tyrrell et al., 2010; Tanner et al., 2011; Li et al., 2012) and peptides (Tian et al., 2012; Hosseinkhani et al., 2013) have been under extensive studies, primarily by experiments. Meanwhile, computational studies have also been reported to secure quantitative interpretation of experimental results and provide microscopic insights from bottom-up (Ahmad et al., 2010; Loverde, 2014). There are enormous synthetic and natural amphiphilic materials, thus it is a formidable task to experimentally test all materials for drug delivery. In this aspect, computations are substantially more economical and feasible to screen and identify ideal candidates. Furthermore, new materials for drug delivery can be rationally designed through computations and subsequently tested by experiments.
This review summarizes the recent representative computational studies for drug delivery in amphiphilic copolymers and peptides. First, the computational methodology commonly used in this field is briefly described. Then, the computational studies of copolymer and peptides are presented and discussed, respectively. Finally, the major concluding remarks are highlighted with a few perspectives for future studies.
Methodology
Most of the computational studies for drug delivery use molecular dynamics (MD) simulations. Following the Newton’s second law, a MD simulation mimics the natural pathway of molecular motion to sample successive configurations. At a given temperature, the initial velocities of molecules are assigned by the Maxwell–Boltzmann distribution. The interactions between molecules at each time are computed, and then the equations of motion are solved numerically with an appropriate time step to update the velocities and positions for the next successive steps (Allen and Tildesley, 1987; Frenkel and Smit, 2002). In classical MD simulations, molecular interactions are described by a force field with certain functional forms and parameters. For polymer and peptides, the widely adopted force fields are AMBER (Cornell et al., 1995), OPLS (Jorgensen et al., 1996), and CHARMM (MacKerell et al., 1998).
Drug loading and release occur in a long-time scale, usually in the order of several microseconds or longer. Currently, fully atomistic (FA) simulations are too expensive to mimic such phenomena for large systems. With this regard, coarse-grained (CG) simulations are more prevailed, in which the number of degrees of freedom is reduced (see Figure 2) and thus the simulations can be accelerated. In the literature, several CG models have been developed on the basis of structure (Go and Taketomi, 1978), statistical potential (Miyazawa and Jernigan, 1985), elastic network (Tama et al., 2002), etc. Particularly, Marrink and coworkers developed the MARTINI model for lipids, surfactants with a few CG bead types (Marrink et al., 2004, 2007), and further extended to biomolecules with more classifications of bead types (Monticelli et al., 2008). The MARTINI model uses four to one mapping, i.e., four heavy atoms/molecules are collectively mimicked as a single bead for linear molecules. Then, four types of interaction sites are considered namely polar (P), non-polar (N), apolar (C), and charged (Q). Within the four types, subtypes are further divided on the basis of hydrogen-bonding capability (d: donor, a: acceptor, da: both and O: none) and degree of polarity (from 1 being lowest polarity to 5 being highest polarity). The interactions between different beads contain non-bonded and bonded terms. The non-bonded includes Lennard–Jones and electrostatic potentials, while the bonded includes stretching, bending, and torsional potentials. The potentials were parameterized in a systematic way to reproduce partitioning free energies between polar and apolar phases for a large number of compounds. The grouping in the MARTINI model reduces the number of atoms in simulations and increases the time scale by three- to fourfold compared with FA simulations. It has been demonstrated that the MARTINI model can well describe peptides, lipids, membranes, etc. Nevertheless, the MARTINI model is associated with several limitations: at a low temperature, it may cause rapid freezing and nucleation of water; the pairwise electrostatic interactions beyond 1.2 nm are not incorporated, which may affect the properties of a system with a large number of charged beads; the current model parameterization prevents examining the transition of secondary structure.
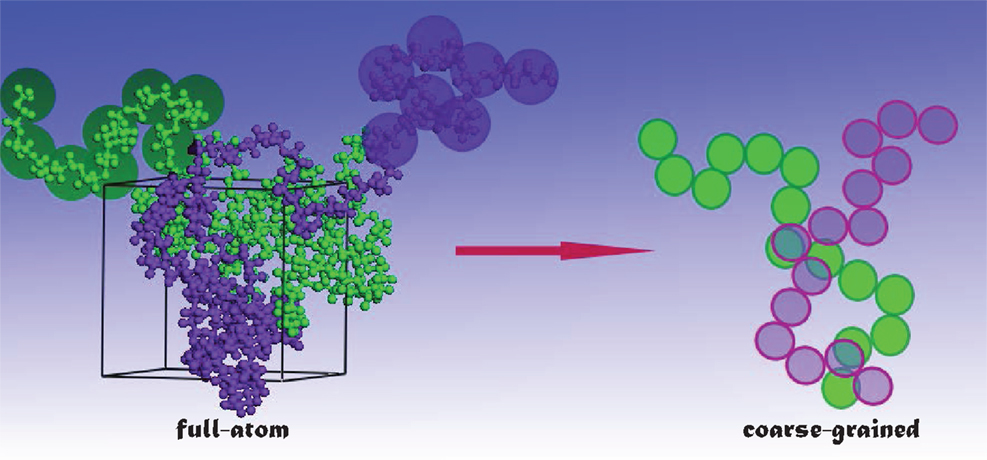
Figure 2. From full-atom to coarse-grained modeling.
Another CG method often used in this filed is dissipative particle dynamics (DPD), which is a higher level CG representation introduced by Hoogerbrugge and Koelman (1992) and Koelman and Hoogerbrugge (1993) and modified by Español and Warren (1995). In a DPD simulation, the system is represented by a set of soft particles (beads). The soft and short-ranged interactions include four terms: bond stretching between connected particles, conservative repulsive interaction for excluded volume effect, dissipative interaction for viscous drag between moving particles, and random interaction for stochastic impulse among particles. The amplitude of these interactions is dictated by the Fluctuation–Dissipation theorem. A distinguishing feature of DPD method is that momentum is conserved between dissipative particles and thus it is able to simulate hydrodynamic time and length scales much larger than those by conventional MD. However, a major limitation of DPD method is the unclear physical time and length scales that are actually simulated, and it is difficult to link DPD to microscopic dynamics. Other limitations include: the size of dissipative particles can be freely tuned and it is to a certain extent ambiguous; no unique way exists to estimate the interaction parameters and the parameters are not transferable. Despite the above limitations, both DPD and CG simulations can fairly well describe the equilibrium thermodynamic and structural properties, and have been widely used to investigate amphiphilic materials (Sevink et al., 2013; Li et al., 2014).
Amphiphilic Copolymers
Consisting of chemically different blocks, amphiphilic copolymers can assemble into a wide variety of morphologies (Mai and Eisenberg, 2012). In an aqueous solution, the hydrophobic blocks form a core or an inner layer, while the hydrophilic blocks constitute a shell or an outer layer. The morphologies may be affected by numerous factors such as polymer concentration, hydrophobic/hydrophilic ratio, end-group, and solvent content (Hayward and Pochan, 2010). Toward the development of stable drug carriers, Rösler et al. reviewed the approaches to modify chemical structures or physically attach auxiliary agents onto copolymers (Rösler et al., 2001). Gaucher et al. discussed the synthesis of copolymers and the issues of micelle formation, characterization, and stability related to drug retention and targeting properties (Gaucher et al., 2005). The effects of polymeric nanostructure shape on drug delivery were comprehensively summarized by Venkataraman et al. (2011). Recent advances in the design and development of copolymers with cleavable linkers for intracellular drug delivery were highlighted by Wei et al. (2013).
While most experimental studies for drug delivery in copolymers are focused on loading efficiency and release profile, computations offer insightful microscopic details such as molecular interactions between drugs and carriers, equilibrium and dynamic properties as listed in Table 1. From FAMD simulations, Patel et al. evaluated the interactions and solubilities of two drugs (fenofibrate and nimodipine) in a series of micelle-forming poly(ethylene oxide)-b-poly(caprolactone) (PEO-b-PCL) with different combinations of blocks. The predicted solubilities were in good agreement with experimental data and better than the predictions by group contribution method (Patel et al., 2008). Two hydrophobic drugs namely Cucurbitacin B (CuB) and Cucurbitacin I (CuI) were further examined by them in PEO-b-PCL at various ratios of PCL/PEO (0.5, 1, and 2). With increasing PCL/PEO ratio, both drugs showed an increase in solubility. This was attributed to the increase in favorable polar interactions and to the formation of additional hydrogen bonds between drugs and PCL rather than to the increase in hydrophobic characteristics (Patel et al., 2009). In their subsequent study, PCL was branched and linked to PEO as PEO-b-3PCL and tested for various drug encapsulations. Containing multiple hydrogen bond acceptors and donors, Cucurbitacin in the branched copolymer was found to be more soluble than in the linear counterpart; but fenofibrate and nimodipine were less soluble in the branched copolymer due to the absence of hydrogen bond donors and the clustering of hydrogen bond acceptors. This study suggested that multi-hydrophobic copolymers could potentially increase drug loading for hydrophobic drugs with evenly distributed multiple hydrogen bond donors and acceptors (Patel et al., 2010).

Table 1. Drug delivery in amphiphilic copolymers.
A combination of MD simulation and docking calculation was employed to predict the compatibility between three hydrophobic model drugs (curcumin, paclitaxel, and vitamin D3) and a triblock copolymer poly(ethylene oxide)-desaminotyrosyl-tyrosine octyl esters-poly(ethylene oxide) (PEG–DTO–PEG). The binding energies of drug-polymer complexes were evaluated by a rapid grid-based method. The grid size varied between 64 × 64 × 64 Å and 126 × 126 × 126 Å using grid spaces between 0.375 and 0.5 Å. Among three algorithms namely genetic algorithm, Lamarkian genetic algorithm, and simulated annealing, the Lamarkian genetic algorithm was found to yield the strongest binding affinity. The energetically favorable conformation was attributed to strong H-bonding and π–π interactions between drug and polymer. However, vitamin D3 exhibited the highest loading even with less strong H-bonding and π–π interactions. Thus, the study revealed the significant role of subtle structural characteristics (e.g., size, flexibility, and rigidity) in drug loading and suggested the computations of drug-polymer pairs would be a powerful prescreening tool in the development and optimization of new drug delivery systems (Costache et al., 2009). Based on MD simulation, Kasimova et al. proposed a method to potentially measure drug loading capability in a polymer micelle. As shown in Figure 3, the method was based on the simulation of a molecular system where polymer, drug, and water were tightly compacted together, thus reproducing the interior of a real micelle. Then the Flory–Huggins interaction parameters were calculated and used to estimate drug solubility. The simulation data were validated on four drugs (cyclosporin A, griseofulvin, ketoconazole, and quercetin dehydrate) with different physical–chemical properties in PEG-hexPLA. A linear relationship was observed between the solubility and mass fraction for the four drugs. This method could be further utilized to estimate drug incorporation efficiency upon structural modification of polymer chains (Kasimova et al., 2012).
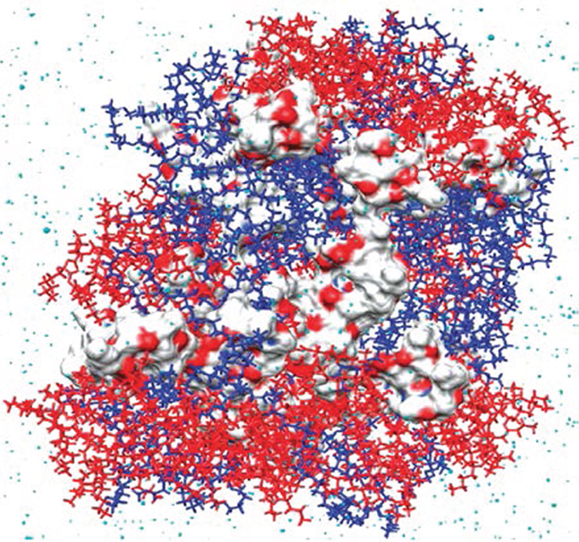
Figure 3. Snapshot of cyclosporin A (in van der Waals surface) and PEG–hexPLA (PEG in blue and hexPLA in red) mixture (Kasimova et al., 2012). Reproduced with permission from American Chemical Society.
The above computational studies reported drug solubility, miscibility, and compatibility in different copolymers. While these equilibrium properties are useful to determine drug loading, dynamic information is also important. Toward this end, Razavilar et al. examined the diffusion of hydrophobic CuB in linear and branched PEO-b-PCL at different amounts of water. They found the diffusivity was double in linear PEO-b-PCL (~2.5 × 10−8 cm2/s) compared with that in branched PEO-b-3PCL (~1.3 × 10−8 cm2/s). The slower dynamics in branched copolymer was attributed to a cumulative effect of stronger hydrogen bonding and lower swelling. Moreover, the diffusivity was found to be insensitive to water amount (Razavilar and Choi, 2014).
DPD method has been commonly applied to simulate drug loading and delivery in amphiphilic copolymers. Guo et al. investigated paclitaxel (PTX) loading in poly(ethylene oxide)-b-poly(l-lactide) (PEO–PLLA) and poly(ethylene oxide)-b-poly(d-lactide) (PEO–PDLA). It was found that the polymers chains assembled around PTX to form core/shell structured fibers, with a PTX/PLA core and a PEO shell. The distribution of PTX could be affected by the chirality and length of PLA. For PEO-b-PDLA and PEO-b-PLLA systems, PLA and PTX were distributed homogeneously in the fiber core; however, PTX was more populated in the inner core for stereocomplex formulation. The simulation results agreed well with measured data (Guo et al., 2009). Subsequently, they simulated drug diffusion into a micelle core. When represented by a short and branched chain, drug exhibited easier diffusion. With increasing hydrophobic block length of polymer, higher loading efficiency was found. While increasing polymer concentration would also lead to higher loading, different morphologies like fiber-like structures might form due to increased polymer–polymer interactions. Drug structure, hydrophobic block length and drug-polymer compatibility were identified to be the key factors governing drug loading efficiency and distribution (Guo et al., 2012b).
Posocco et al. reported a systematic DPD simulation study on structural and physical factors affecting the assembly of (d-l)-PLA/PEO di/triblock copolymers for Nifedipine loading. The morphological phase diagrams were predicted for racemic PLA and PEO copolymers with AB and ABA architectures. Scaling laws for micellar dimensions were used to derive micellar characteristics such as aggregation number and degree of hydration. As shown in Figure 4, the morphology of PLA–PEO was observed to be spherical at a low Nifedipine loading (4%) but switched to columnar shape at a high loading (22%). With increasing Nifedipine loading in PLA–PEO–PLA (3–7%), the morphology rearranged from sphere to bilayer. Such loading-dependent morphology transition suggested the maximum drug loading could be accommodated by the copolymers (Posocco et al., 2010).
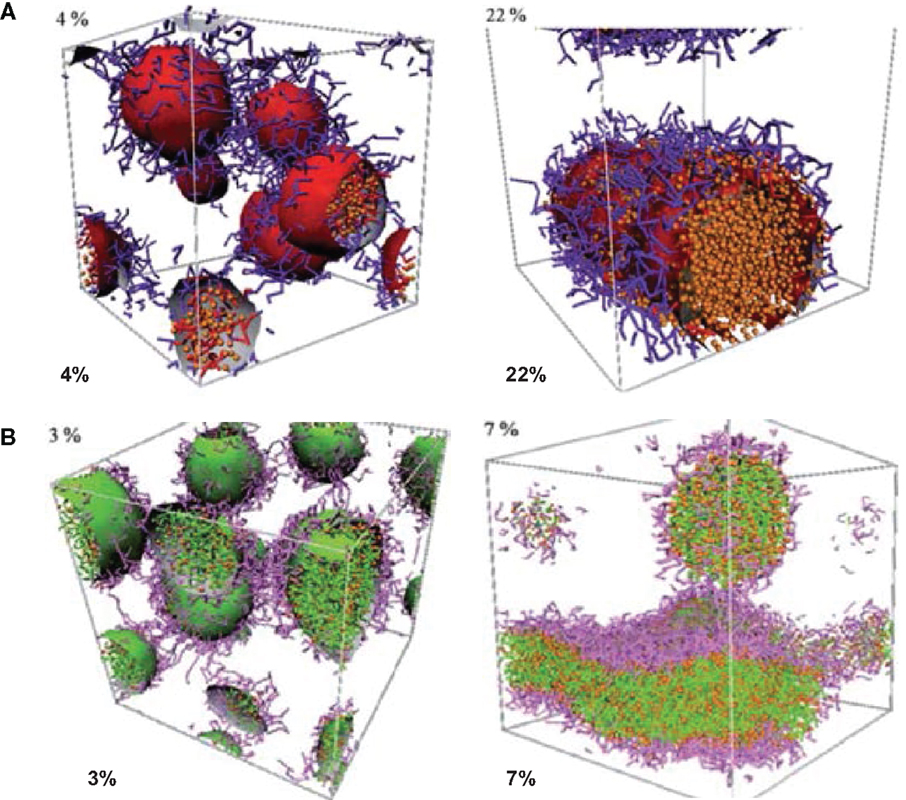
Figure 4. Morphologies of (A) PLA–PEO with 4 and 22% Nifedipine loading (B) PLA–PEO–PLA with 3 and 7% Nifedipine loading. PLA: red, PEO: purple, Nifedipine: orange (Posocco et al., 2010). Reproduced with permission from the Royal Society of Chemistry.
By integrating FAMD and DPD simulations, Luo and Jiang examined the loading and release of camptothecin (CPT) in poly(β-amino ester)–methyl ether capped poly (ethylene glycol) (PAE–PEG). FAMD was utilized to estimate the Flory–Huggins interaction parameters and miscibility of binary components. On this basis, DPD was applied to examine the micellization of PAE–PEG, CPT loading and release. As illustrated in Figure 5, CPT loading in PAE-PEG was governed by adsorption-growth-micellization mechanism. CPT was initially adsorbed on the small clusters assembled by polymer chains, then the clusters grew in size, and finally CPT was loaded into large-size micelles. The loading efficiency was predicted as 0.84, close to experimental measured 0.82. Upon protonation, CPT was released from micelles/vesicles by swelling-demicellization-releasing mechanism. Initially, the micelles were swollen rapidly and PAEH chains moved out from the hydrophobic core, then the swollen micelles demicellized and CPT moved out, and finally the entire micelle was dissociated into free polymer chains and CPT (Luo and Jiang, 2012).
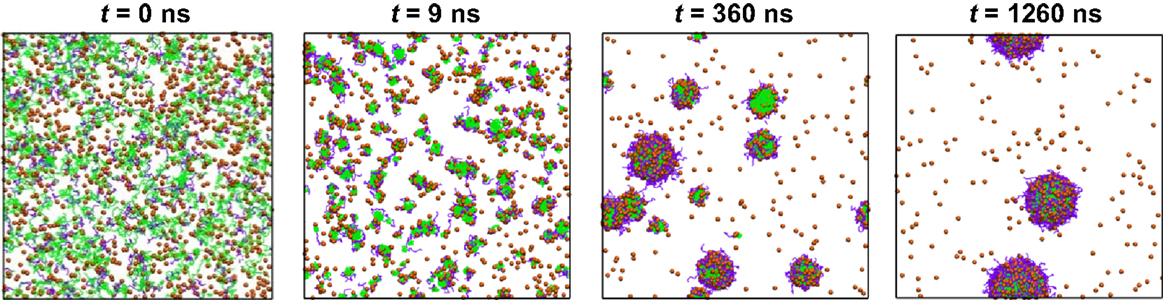
Figure 5. Dynamics process of CPT loading. CPT is shown as orange sphere, PAE and PEG are in green and purple (Luo and Jiang, 2012). Reproduced with permission from Elsevier.
Other copolymers were also explored for drug delivery. Nie et al. applied DPD simulation to examine doxorubicin (DOX) in a four-armed star triblock poly(ϵ-caprolactone-b-poly (2-(diethylamino) ethyl methacrylate)-b-poly(poly-(ethylene glycol) methyl ether methacrylate) [4AS-PCL-b-PDEAEMA-b-PPEGMA). The assembled core-mesosphere-shell micelles with three layers could load DOX at the core and the core/mesosphere interface. Upon increasing DOX content from 1 to 7%, the micelle shape changed and its shell was partially open for water to access. Due to the stretching of pH-sensitive PDEAEMA, cracks were observed in the blank micelles at pH = 5, which might facilitate drug release. However, the drug-loaded micelles did not exhibit any drug release at different pH values. This was attributed to the fact that stirring is usually applied in experiment but not incorporated in the simulation (Nie et al., 2013). They also investigated the effectiveness of drug distribution in pH-sensitive AlBmCn copolymers with different lengths of hydrophilic block, pH-sensitive block and hydrophobic block. From DPD simulation, it was demonstrated that the formation of firework-like three layered structure (spherical core, pH-sensitive layer, and shell) was crucial to drug release. As illustrated in Figure 6, drug loaded in the pH-sensitive layer was easier to release with a much faster rate than in the core; drug release in the pH-sensitive layer exhibited a remarkable rise upon increasing the length of B blocks. The mechanically propelled effect due to the extension of pH-sensitive blocks had a significant contribution to drug release. Moreover, it was revealed that tuning hydrophilic chain length would promote micelle stability and increasing hydrophobic chain length resulted in different mechanisms for drug release (Nie et al., 2014). The same group further combined DPD simulation and experiment to examine DOX loading and release in 4-miktoarm star polymers namely poly(ε-caprolactone)2-[poly(2-(diethylamino)ethylmethacrylate-b-poly(poly(ethylene glycol) methyl ether methacrylate)]2 [(PCL)2(PDEA-b-PPEGMA)2] with different block ratios. The polymer formed spherical micelles with PCL core, pH-sensitive PDEA mesosphere and PPEGMA shell. The increase in hydrophobic block length showed higher DOX loading from 9 to 19%. Upon increasing concentration, DOX was initially loaded in the mesosphere then diffused into the core. A swelling-demicellization-releasing mechanism was suggested for the release process. As also observed by experiment, the amount of DOX released at pH = 5.0 was larger than at pH = 7.4 and 6.0. The study demonstrated that the miktoarm star copolymers could be promising for the controlled and sustained release of DOX (Lin et al., 2014).
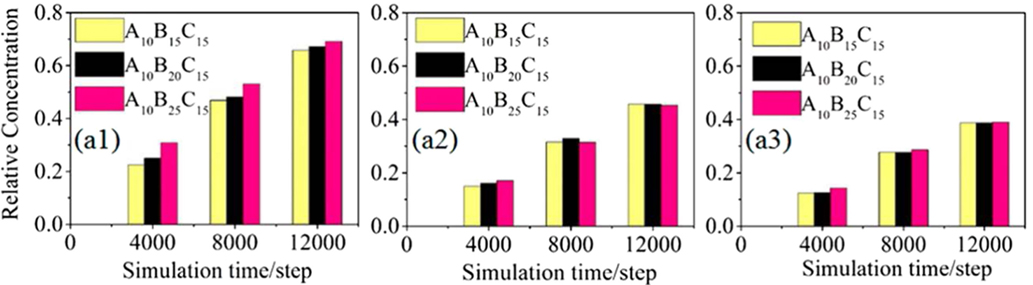
Figure 6. Relative concentrations of drugs in (a1) the pH-sensitive layer, (a2) both of the pH-sensitive layer and the core, and (a3) the core. The drugs had different compatibilities with the micelles composing different lengths of pH-sensitive B blocks (Nie et al., 2014). Reproduced with permission from American Chemical Society.
A multiscale simulation in conjunction with experiment was adopted by Jiang et al. to investigate the loading and structure detail of PTX in poly(ethylene glycol)-b-dendritic oligo(cholic acid) (PEGxCAy, x = 2K, 3K, 5K and y = 4, 6, 8). The systems were mimicked by a CG model described by the MARTINI force field. The hydrophobic-hydrophilic balance was demonstrated to be crucial for forming stable carrier and effective drug loading, and losing balance would lead to less stable micelles with limited drug loading capacity or precipitation. Figure 7A shows the structure of PTX–PEG5KCA8 loaded with 17% (w/w%) PTX at the end of 1.5 μs simulation. PTX was encapsulated within the amphiphilic envelope of CA groups. The size distribution of PTX-loaded micelles shown in Figure 7B was not uniform, as also observed in dynamic light scanning measurement. The CG systems were then reversely mapped to FA representations, and hydrogen-bonds were found to form between PTX and micelles (Jiang et al., 2015).
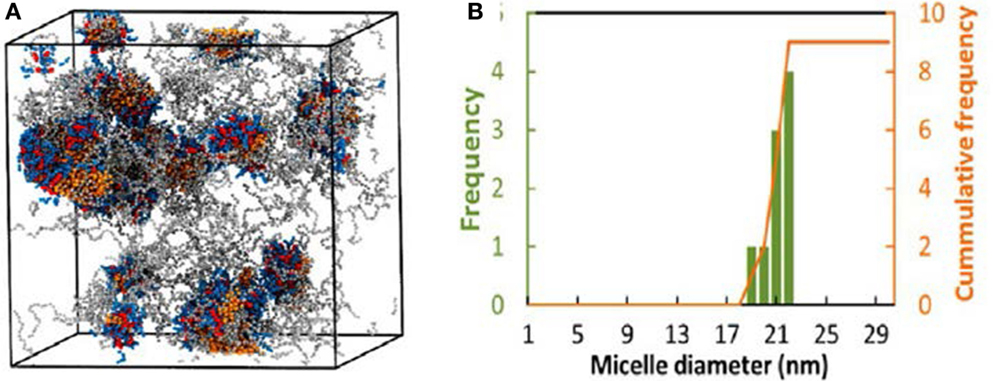
Figure 7. (A) PTX–PEG5KCA8 with 17% PTX loading at 1.5 μs. Colors: PTX (orange), PEG (gray), Cholic acid (blue) and lysine (red). (B) Size distribution of PTX–PEG5KCA8 (Jiang et al., 2015). Reproduced with permission from American Chemical Society.
A handful of simulation studies were targeted to elucidate the delivery of drug-loaded carriers into lipid membranes. Srinivas et al. examined the delivery of hydrophilic model drugs assisted by a patchy polymer across a lipid vesicle. The drugs were mimicked as unconnected beads and loaded in patch-forming polymer micelles composed of two types of blocks (AB and CB). In the first stage, micelle–vesicle interactions were evaluated and the full absorption of micelles was observed without the shape change of vesicle. In the second stage, the interaction between drug-loaded micelle and vesicle was evaluated, and the vesicle reorganized in accommodating the micelles. As shown in Figure 8, the drugs were successfully transported across the vesicle, released, and distributed into the vesicle. The simulation revealed that hydrophilic drugs can be delivered by polymer assisted pathway, which is insightful for experimentalists to develop better carriers for hydrophilic drugs (Srinivas et al., 2013). Alternatively, Nicola et al. performed simulation for the delivery of Pluronic micelles across DPPC lipid bilayer. In accord with experimental observation, Pluronic chains were found to release from the micelles to the bilayer and thus the micelle size was changed. In the presence of drug inside the micelle core, the release process was significantly affected due to the interplay of drug-micelle and micelle-bilayer interactions, and the micelle size increased sharply by slightly altering drug hydrophobicity. This study provides detailed understanding of fundamental mechanism underlying drug delivery (Nicola et al., 2014).
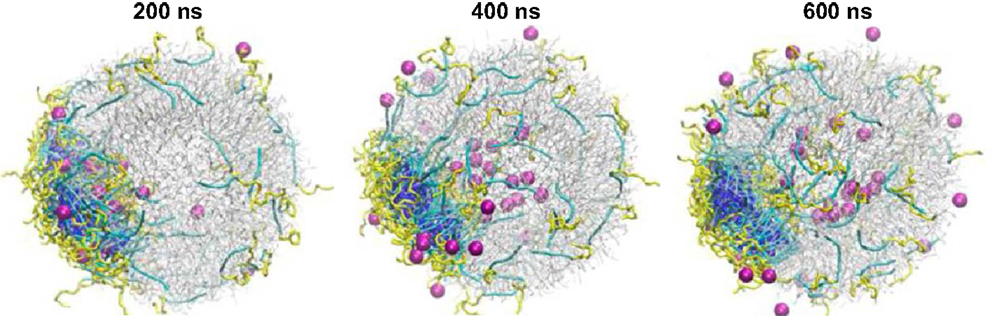
Figure 8. Hydrophilic drug molecules (magenta) transport across the lipid vesicle (gray) assisted by polymer micelles (yellow and blue) and release inside the inner core of the vesicle (Srinivas et al., 2013). Reprinted with permission from American Chemical Society.
Peptides
Some synthetic polymers are cytotoxic to cells and cannot be clinically used. Ideal carriers for drug delivery should possess the characteristics of non-toxic, non-immunogenic, biocompatible, biodegradable, and kinetically stable (Liu et al., 2011). In this context, amphiphilic peptides have emerged as “smart” materials for drug delivery. Composed of hydrophilic and hydrophobic residues, amphiphilic peptides can assemble into various morphologies such as micelles, vesicles, fibers, and hydrogels (Hamley, 2011; Trent et al., 2011). The morphologies are dependent on the ratio of hydrophobic to hydrophilic residues, peptide sequence, concentration, and other factors. Their sizes are usually <100 nm, which is an advantage to prevent from reticuloendothelial system of human body (Cui et al., 2010). More intriguingly, the size, stability, permeability, and elasticity can be fine-tuned by tailoring peptide sequence, length, solution conditions, etc. Amphiphilic peptides have been tested for delivering drug or gene or both, and better therapeutic effects were found on cancer cells or genetic disorders (He et al., 2012; Sundar et al., 2014). Every year, about 17 new peptides enter into clinical tests and about 140 peptides are currently under development (Zhang et al., 2012). With 20 gene-encoded amino acids, it can be envisioned that tremendously large number of peptides would be explored (Ulijn and Smith, 2008; Zhao et al., 2010).
Most computational efforts for amphiphilic peptides have been focused on self-assembly, rather than drug loading/release. Therefore, the studies of self-assembly are also summarized here as listed in Table 2. Jeon et al. simulated the early stages of assembly for diphenylalanine (FF) in both capped and zwitterionic forms, and compared with X-ray crystal structures. It was revealed that electrostatic interactions steered zwitterionic FF into more ordered dimers and trimers, while hydrophobic side-chain interactions played a strong role in determining the structures of large oligomers (Jeon et al., 2013). Simulation as well as experiment was performed by Rissanou et al. to study FF and AA in two solvents (water and methanol). The assembly propensity of FF in water was found to be stronger than in methanol. However, the difference between the two solvents for AA was substantially smaller. The simulation results showed good qualitative agreement with experimental observations (Rissanou et al., 2013). Alternatively, Guo et al. implemented extensive CGMD simulations to probe the assembly of FF. Consistent with previous experiments, nanovesicles and nanotubes were predicted (Figures 9A,B); in addition, new ordered planar bilayers and a rich variety of other vesicle-like structures including toroid, ellipsoid, discoid, and pot-shaped vesicles were also observed. The assembly was revealed to involve the fusion of small vesicles and bilayers at low concentrations but the formation of a bilayer at high concentrations (Guo et al., 2012a). They further predicted that FFF spontaneously assembled into highly compact nanospheres and nanorods (Figures 9C,D). The peptides in nanostructures were predominantly anti-parallel-aligned with the capability to form β-sheets, and no intermediate bilayers existed. From free energy analysis, the assembly of FF and FFF was attributed to the aromatic stacking interactions of side chains; however, the fine morphologies were governed by the interactions of main chains (Guo et al., 2014).
Table 2. Self-assembly and drug delivery in amphiphilic peptides.
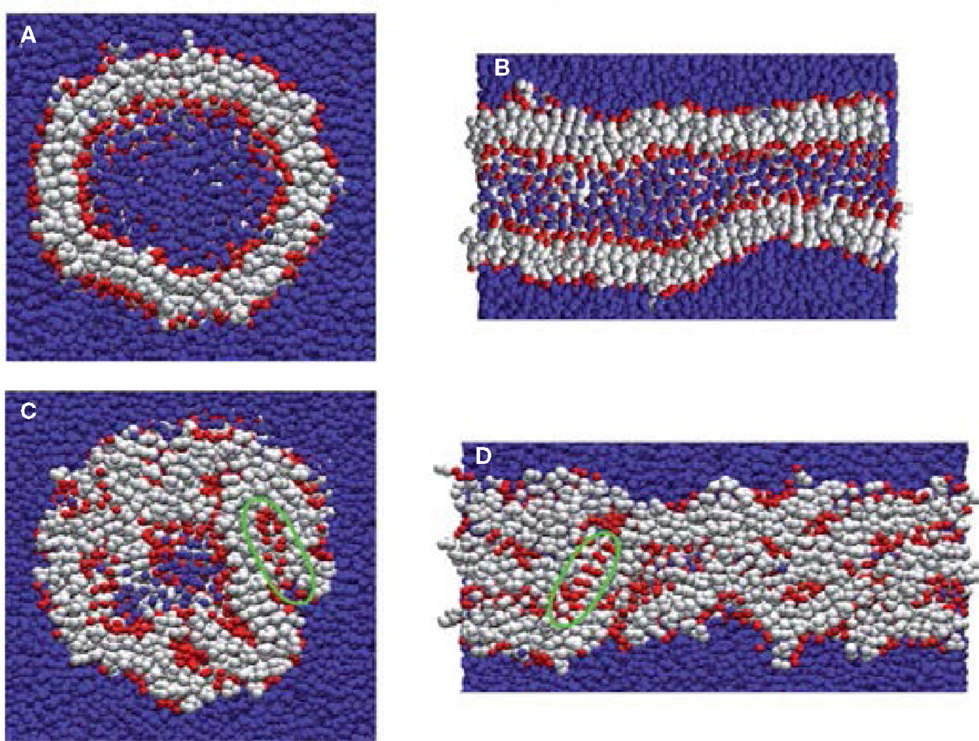
Figure 9. Nanostructures formed by FF and FFF peptides. (A) FF nanovesicle, (B) FF nanotube, (C) FFF nanosphere, and (D) FFF nanorod. Peptide main chain and side chain beads are shown in red and white, respectively. Water beads are shown in blue (Guo et al., 2012a, 2014). Reproduced with permission from American Chemical Society and the Royal Society of Chemistry.
Fluorenylmethoxycarbonyl conjugated peptides were under investigation by a few groups. Mu et al. combined experiment and simulation to examine the assembly of Fmoc-AA. The formation of fibrils with Fmoc groups at the center was observed; nevertheless, the partial exposure of Fmoc groups to water caused the fibrils to aggregate into nanoscale fibers. The radial distribution calculations agreed well with the d-spacings measured by wide angle X-ray scattering for the fibril diameter and π-stacking interactions (Mu et al., 2012). Similarly, Lopez-Perez et al. found Fmoc conjugated tetrapeptides (Fmoc-RGDS and Fmoc-GRDS) retained pre-assembled β-sheet parallel conformations under low concentrations, which were dominated by the interactions among Fmoc units. The study provided a complete microscopic insight into the interactions between Fmoc-peptides comprised within the same sheet or among different sheets (Lopez-Perez et al., 2013).
Using CGMD simulations, Frederix et al. rapidly screened 400 dipeptide combinations of 20 amino acids as a potential precursor for assembly. The candidates showing strong aggregation tendencies were chosen for longer simulations, and good agreement was found by comparing predicted aggregation propensity (AP) scores and supramolecular structures with experimental results known in the literature. Extended simulations on FF showed the formation of a nanotube. This screening protocol allows rapid determination on the capability of peptide assembly (Frederix et al., 2011). Subsequently, they predicted the AP scores of 8000 tripeptides as a function of hydrophobicity. As shown in Figure 10, a weak correlation between AP and hydrophilicity was found. The peptides containing charged K, R, D, and E and hydrogen-bonding T and S were identified with high AP scores. Among these, the top 400 combinations were subject to extended simulations for the dynamics and nanostructure of assembly. Then, a set of design rules were proposed with aromatic residues as the most favorable at positions 2 and 3 in a tripeptide, positive and hydrogen-bonding residues in position 1 and negative residues at position 3. Validated by experiments, the simulation results led to the discovery of tripeptides with capability to form hydrogels. The tools developed would promote the search of peptide sequences for supramolecular properties (Frederix et al., 2015).
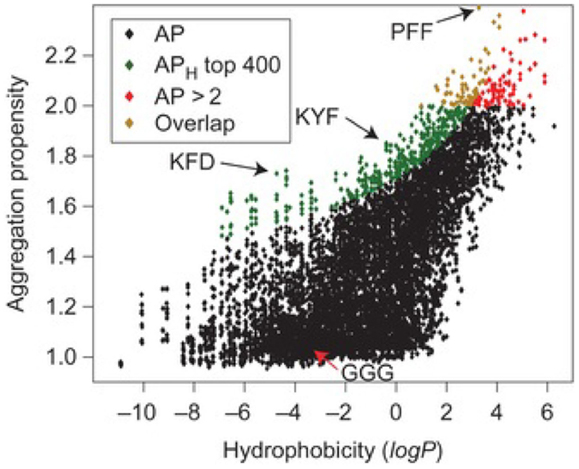
Figure 10. Aggregation propensity (AP) as a function of hydrophobicity for 8,000 tripeptides. Red diamonds represent all tripeptides with AP > 2. Green diamonds represent the top 400 tripeptides from the APH score with the overlapping candidates shown in orange (Frederix et al., 2015). Reproduced with permission from Macmillan Publishers Ltd.
Thota et al. simulated the assembly of a series of short peptides FmDn and FmKn. The peptides consisted of hydrophobic phenylalanine, hydrophilic aspartic acid, and lysine. Within μs-scale duration, FD and FK formed loose polymeric clusters. With increasing the length of Phe residues in FmD and FmK (m = 2–4), larger and more stable micelles were observed to form. FmK and FmD appeared to assemble into quasi-spherical and sheet-like micelles, respectively. In contrast, smaller and less stable micelles were formed with increasing the length of hydrophilic Lys residues in both F3Kn and F6Kn. Interestingly, F3K4 and F6K8 were found to form quasi-spherical micelles with distinct core/shell structure. This suggested the optimal ratio of hydrophobic/hydrophilic residues to be 3/4. Upon increasing this ratio, quasi-spherical micelles tended to become elliptical aggregates; while decreasing this ratio resulted in smaller and less stable micelles. This ratio found is significant in the rational selection of appropriate FmKn peptides to assemble into spherical micelles (Thota et al., 2014).
Apart from the short peptides mentioned above, relatively longer peptides were also simulated at various conditions. Emamyari et al. examined the assembly of three ionic peptides (AEAK)4, ((AE)2(AK)2)2 and (AE)4(AK)4 in the presence of a hydrophobic surface. The first two peptides assembled into ribbons regardless of pH value. However, third peptide assembled into ribbons at low and high pH but discs at neutral pH, due to strong intrachain electrostatic interactions among similar charged blocks. These observations were consistent with experimental and theoretical investigations (Emamyari and Fazli, 2014). Thota et al. simulated amphiphilic peptide (AF)6H5K15 (FA32), which was observed to form micelles with Ala and Phe in hydrophobic core, Lys in hydrophilic shell and amphiphilic His at the interface. The assembly process and microscopic structures were analyzed in terms of the number of clusters, the radii of micelle, core and shell, and density profiles. A three-step process was proposed for the assembly: aggregation of small clusters, merging into large clusters, and formation of micelles. With increasing peptide concentration, quasi-spherical micelles changed to elongated shape and micelle size generally increased. The micelle size was predicted to be around 2~4 nm, much smaller than experimentally measured, as attributed to two factors: the simulation was not sufficiently long thus prohibiting the formation of larger micelles; the CG model adopted might not be accurate to describe FA32 (Thota et al., 2013). The derivatives of FA32 were further studied to examine the roles of hydrophobic and hydrophilic residues in assembly. Upon increasing the length of Lys from (AF)6H5K10, (AF)6H5K15 and (AF)6H5K20 to (AF)6H5K25, assembly capability was reduced with the formation of smaller micelles or the presence of individual peptide chains. When Ala was replaced by more hydrophobic Phe in (AF)6H5K15, (AF3)3H5K15, and F12H5K15, larger micelles were formed. With increasing the length of Phe in F4H5K15, F8H5K15, F12H5K15, and F16H5K15, the micelle size increased and the micelles shifted from spherical to fiber-like as shown in Figure 11. This study reveals that assembly capability is reduced by increasing hydrophilicity, whereas increasing hydrophobicity leads to morphology transition (Thota and Jiang, 2014).
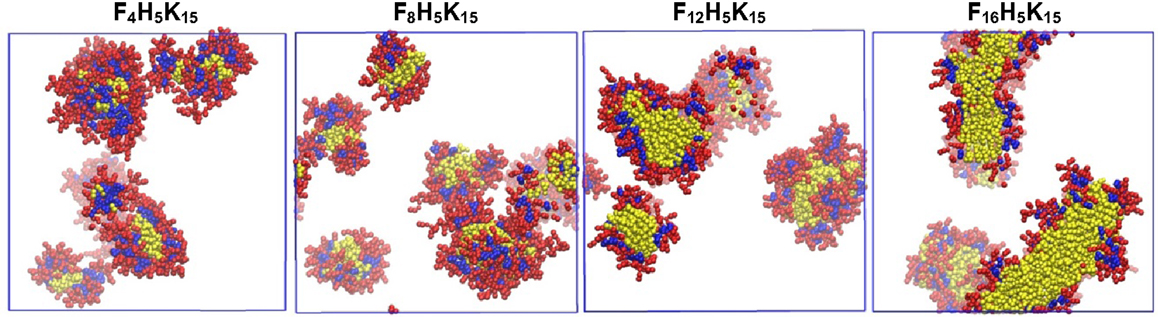
Figure 11. Equilibrium snapshots of F4H5K15, F8H5K15, F12H5K15, and F16H5K15 (Thota and Jiang, 2014). Reprinted with permission from American Chemical Society.
Simulations were also carried out for peptide amphiphiles (i.e., lipid-like peptides). By FAMD simulation starting from a predefined configuration, Schatz and coworkers observed the assembly of C16-SLSLAAAEIKVAV into a cylindrical nanofiber. With a radius of 4.4 nm, the nanofiber was found to contain a broad distribution of secondary structure. The β-sheet population of the SLSL and IKV segments was approximately 25%, as experimentally observed. The stability of nanofiber was attributed to the electrostatic interactions between peptides and sodium ions, as well as the van der Waals interactions between peptides (Lee et al., 2011). The same peptide was simulated by CGMD method from a random configuration. As shown in Figure 12, spherical micelles were initially formed, then aggregated into a three-dimensional network, and finally merged into a nanofiber. Consistent with FAMD simulation, water molecules were excluded from the hydrophobic core (Lee et al., 2012a). Two peptide amphiphiles with variation in the content of valine and alanine ratios (PA1: V2A4E3 and PA2: V4A2E3) were also investigated by them. Though both formed fibers with a size of 8 nm, PA2 showed higher β-sheet content and more hydrogen bonds than PA1 because of higher valine content. The hydrogen bonds might be a cause for the difference in mechanical properties of fibers as observed experimentally (Lee et al., 2012b). Based on a collective assembly coordinate, they further analyzed the free energy profile and mechanism for the assembly of C16-SLSLAAAEIKVAV. The free energy, enthalpy, and entropy differences between the free state and cylindrical nanofiber were found to be −126 kcal/mol, −185 kcal/mol, and −0.19 kcal/(mol⋅K), respectively. It was revealed that the enthalpic driving force for assembly originated from the conformation energy, electrostatic and van der Waals energy between solvent molecules, as well as between solvent and peptide. This study demonstrates the substantially important roles of peptide structural change, solvation, and solvent redistribution in hierarchical assembly (Yu and Schatz, 2013a,b).
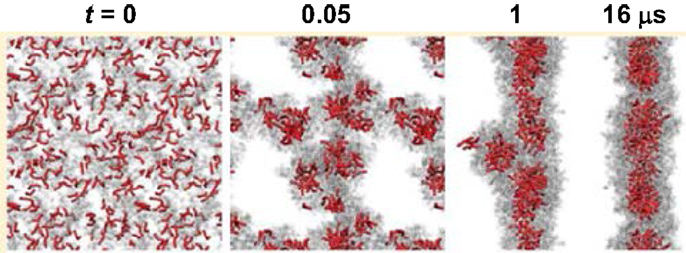
Figure 12. Assembly process of a nanofiber from a random configuration (Lee et al., 2012a). Reprinted with permission from American Chemical Society.
In addition, Nguyen and coworkers developed an extension of the protein intermediate resolution model (ePRIME) in large-scale simulations to examine the effects of electrostatics and temperature in the assembly of 800 peptide amphiphiles (pamitoyl-V3A3E3). Starting from random configurations, they found nanostructures of various sizes and shapes. At optimal conditions, the assembly mechanism for the formation of cylindrical nanofibers was disclosed. A phase diagram between electrostatics and temperature was constructed shown in Figure 13 for the delineating morphological regions in response to external stimuli (Fu et al., 2013). In terms of hydrophobicity, they observed the progressive transition from open network to closed cylindrical structure with increasing hydrophobicity (Fu et al., 2014). Furthermore, they examined solvent effects on peptide assembly. The assembly was found to occur via a multistep process with transient intermediates ultimately leading to stable nanostructures. Different kinetic mechanisms were compared on the basis of solvent-accessible surface area, radius of gyration, relative shape anisotropy, intra/intermolecular interaction, and aggregate size (Fu et al., 2015).
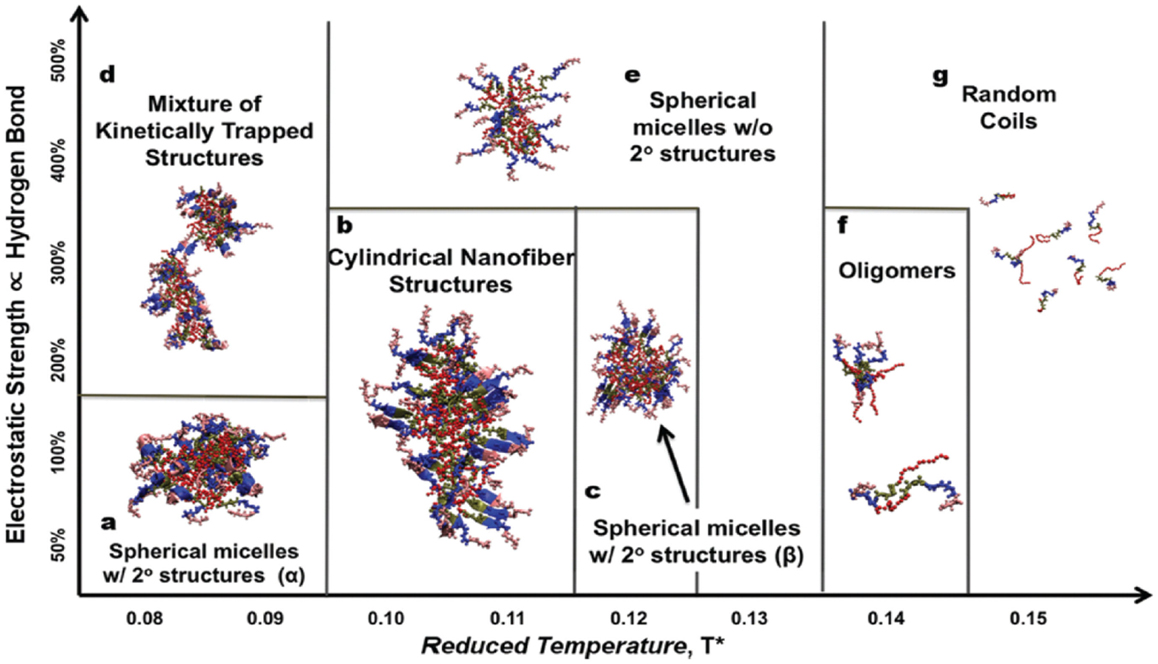
Figure 13. Phase diagram between electrostatics and temperature (Fu et al., 2013). Reprinted with permission from John Wiley and Sons.
Comparatively, only few simulation studies have been reported on the loading/release of drugs in amphiphilic peptides. Guo et al. conducted FAMD simulations to calculate solubility parameters and molar volumes, then used DPD method to examine the microstructures of blank/DOX-loaded micelles of cholesterol conjugated oligopeptides His10Arg10 (HR20-Chol). DOX was efficiently loaded in the core of core/shell micelles at pH >6. As illustrated in Figure 14, all the DOX could be loaded in the micelle with most DOX distributed in the cholesterol core and some also inside the histidine middle layer. Upon decreasing pH, the micelle appeared to be swelling from dense conformation and some DOX molecules were not inside the micelle leading to lower loading. The structural transformation at low pH was attributed the protonation of imidazole group in histidine side chains. The results were found to be qualitatively consistent with experiment (Guo et al., 2010). They further applied multiscale simulations to develop structure–performance relationships for DOX loading in HR20-Chol and HR15-Chol. The simulation showed qualitative consistency with in vitro experiment (Guo et al., 2012c). Chen et al. combined experiment and DPD simulation to design polypeptide-based micelles for dual-drug delivery. The micelles were formed by poly(propylene oxide)-b-poly(γ-benzyl-l-glutamate)-b-poly(ethylene glycol) (PPO-b-PBLG-b-PEG) and two drugs, doxorubicin (DOX) and naproxen (Nap). A core-shell-corona structure was observed for the micelles with PPO/Nap, PBLG/DOX and PEG in the core, shell, and corona, respectively. Moreover, the dual-drug-loaded micelles were demonstrated to be biocompatible at normal physiological conditions and retained anti-cancer efficiency (Chen et al., 2014).
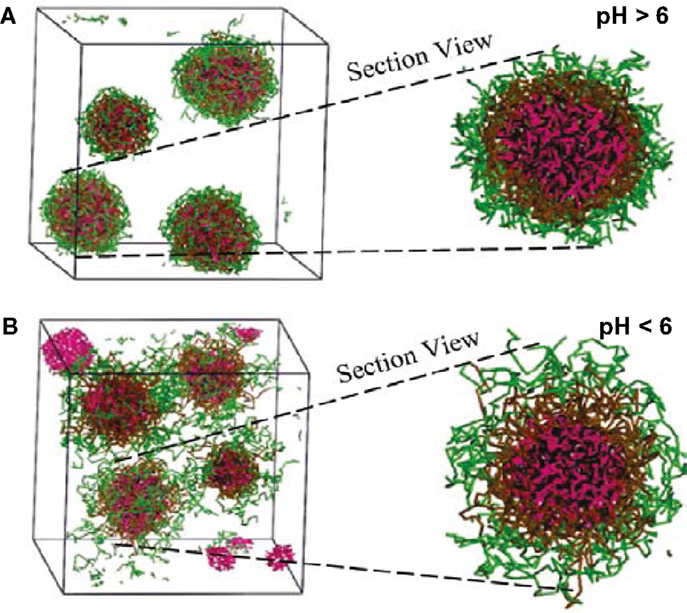
Figure 14. Simulation snapshots of DOX-loaded micelles at (A) pH > 6 and (B) pH < 6 (Guo et al., 2010). Reprinted with permission from American Chemical Society.
Thota et al. conducted CGMD simulation to investigate ibuprofen (IBU) loading/release in FA32 and its derivatives F12H5K15 and F16H5K15. Upon IBU loading in FA32, quasi-spherical core/shell structured micelles were formed. IBU was predominantly located at the core along with Phe and Ala, whereas Lys and His were in the shell and core/shell interface, respectively. Compared with blank FA32, the size of IBU-loaded micelles was larger. With increasing ratio of drug/peptide, the micelles became larger as attributed to increased hydrophobic interactions. In FA32 derivatives, however, IBU loading led to different morphologies; particularly, a well-structured nanofiber was formed in F16H5K15. As shown in Figure 15, IBU release from FA32 micelles upon pH change was found to be slower than from F16H5K15 nanofiber, suggesting the former might be better in controlled release. The simulation study reveals that IBU-loaded morphology can be altered by changing the type of peptide and has a significant effect on IBU release profile (Thota et al., 2015).
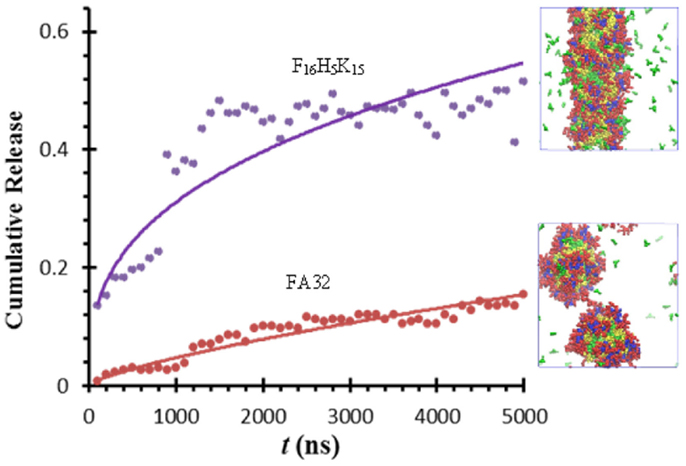
Figure 15. Ibuprofen release from FA32 micelles and F16H5K15 nanofiber (Thota et al., 2015).
Summary and Outlook
In this review, the recent computational studies for drug delivery in amphiphilic copolymers and peptides are summarized. Most studies have used CG models for a wide variety of copolymers (e.g., PEO/PEG, PCL, and PLA based) and peptides (e.g., di-, tri-, short, and long peptides). The effects of chemical structure, hydrophobicity, and hydrophilicity of building blocks, and drug-carrier compatibility are examined. The key factors governing drug loading efficiency and release profile are identified. These microscopic insights are useful to quantitatively understand experimental observations and deeply unravel fundamental issues in drug delivery.
To overcome the long simulation duration involved in drug loading and release, advanced techniques such as metadynamics and targeted MD may be applied in future computational endeavors. By choosing a set of collective variables and reconstructing free energy landscape, metadynamcis method allows for accelerating simulation. However, it is far from trivial to choose appropriate collective variables; additionally, the simulation system tends to reside in high-energy region due to the fluctuation around free energy landscape. By applying a time-dependent geometrical constraint, targeted MD gradually steers a simulation system toward a specified target structure. Nevertheless, equilibrium properties cannot be calculated, the final structure has to be known in prior and the order of events may depend on simulation direction. While simulations with CG models are widely adopted in this field and provide mesoscale insights, the atomistic information of drugs and carriers is not captured. An alternative approach is to mimic the solutes (drugs and carriers) and solvent (water) by atomistic and CG models, respectively. Such a hybrid approach can improve simulation efficiency, meanwhile, provide atom-resolution details. Currently, almost all the simulation studies in this field are focused on a single drug. Experiments have demonstrated that drugs and genes could be simultaneous delivered into cell with better therapeutic effects. Thus, it is intriguing to simulate the loading and delivery of both drugs and genes, and unravel their complex interplay from bottom-up. Furthermore, the transport of drug-loaded carriers through biomembranes into cells is scarcely investigated; however, this transport is of central importance to drug delivery. It is foreseeable that increasingly more computational studies will be carried out to clearly reveal transport mechanisms; thereafter, the entire process of drug delivery including loading, transport, and release can be comprehensively elucidated to facilitate the development of new amphiphilic materials for drug delivery.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to the National University of Singapore and the Ministry of Education of Singapore (R-279-000-353-112) for financial support.
References
Ahmad, S., Johnston, B. F., Mackay, S. P., Schatzlein, A. G., Gellert, P., Sengupta, D., et al. (2010). In silico modelling of drug-polymer interactions for pharmaceutical formulations. J. R. Soc. Interface 7, S423–S433. doi:10.1098/rsif.2010.0190.focus
Allen, M. P., and Tildesley, D. J. (1987). Computer Simulation of Liquids. Oxford: Oxford University Press.
Chen, L., Jiang, T., Cai, C., Wang, L., Lin, J., and Cao, X. (2014). Polypeptide-based smart micelles for dual-drug delivery: a combination study of exepriments and simulations. Adv. Healthc. Mater. 3, 1508–1517. doi:10.1002/adhm.201300638
Chen, Y., and Liu, L. H. (2012). Modern methods for delivery of drugs across the blood–brain barrier. Adv. Drug Deliv. Rev. 64, 640–665. doi:10.1016/j.addr.2011.11.010
Cormier, A. R., Pang, X., Zimmerman, M. I., Zhou, H. X., and Paravastu, A. K. (2013). Molecular structure of RADA16-I designer self-assembling peptide nanofibers. ACS Nano 7, 7562–7572. doi:10.1021/nn401562f
Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M., et al. (1995). A second generation force-field for the simulation of proteins, nucleic-acids, and organic-molecules. J. Am. Chem. Soc. 117, 5179–5197. doi:10.1021/ja00124a002
Costache, A. D., Sheihet, L., Zaveri, K., Knight, D. D., and Kohn, J. (2009). Polymer–drug interactions in tyrosine-derived triblock copolymer nanospheres: a computational modeling approach. Mol. Pharm. 6, 1620–1627. doi:10.1021/mp900114w
Cui, H. G., Webber, M. J., and Stupp, S. I. (2010). Self-assembl of peptide amphiphiles: from molecules to nanostructures to biomaterials. Biopolymers 94, 1–18. doi:10.1002/bip.21328
Drummond, C. J., and Fong, C. (1999). Surfactant self-assembly objects as novel drug delivery vehicles. Curr. Opin. Colloid Interface Sci. 4, 449–456. doi:10.1016/S1359-0294(00)00020-0
Emamyari, S., and Fazli, H. (2014). Ph-dependent self-assembly of EAK16 peptides in the presence of a hydrophobic surface: coarse-grained molecular dynamics simulation. Soft Matter 10, 4248–4257. doi:10.1039/c4sm00307a
Español, P., and Warren, P. (1995). Statistical mechanics of dissipative particle dynamics. Europhys. Lett. 30, 191. doi:10.1209/0295-5075/30/4/001
Frederix, P. W. J. M., Scott, G. G., Abul-Haija, Y. M., Kalafatovic, D., Pappas, C. G., Javid, N., et al. (2015). Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels. Nat. Chem. 7, 30–37. doi:10.1038/nchem.2122
Frederix, P. W. J. M., Ulijn, R. V., Hunt, N. T., and Tuttle, T. (2011). Virtual screening for dipeptide aggregation: toward predictive tools for peptide self-assembly. J. Phys. Chem. Lett. 2, 2380–2384. doi:10.1021/jz2010573
Frenkel, D., and Smit, B. (2002). Understanding Molecular Simulation: From Algorithms to Applications. San Diego, CA: Academic Press.
Fu, I. W., Markegard, C. B., Chu, B. K., and Nguyen, H. D. (2013). The role of electrostatics and temperature on morphological transitions of hydrogel nanostructures self-assembled by peptide amphiphiles via molecular dynamics simulations. Adv. Healthc. Mater. 2, 1388–1400. doi:10.1002/adhm.201200400
Fu, I. W., Markegard, C. B., Chu, B. K., and Nguyen, H. D. (2014). Role of hydrophobicity on self-assembly by peptide amphiphiles via molecular dynamics simulations. Langmuir 30, 7745–7754. doi:10.1021/la5012988
Fu, I. W., Markegard, C. B., and Nguyen, H. D. (2015). Solvent effects on kinetic mechanisms of self-assembly by peptide amphiphiles via molecular dynamics simulations. Langmuir 31, 315–324. doi:10.1021/la503399x
Gaucher, G., Dufresne, M.-H., Sant, V. P., Kang, N., Maysinger, D., and Leroux, J.-C. (2005). Block copolymer micelles: preparation, characterization and application in drug delivery. J. Control. Release 109, 169–188. doi:10.1016/j.jconrel.2005.09.034
Go, N., and Taketomi, H. (1978). Respective roles of short-range and longrange interactions in protein folding. Proc. Natl. Acad. Sci. U.S.A 75, 559–563. doi:10.1073/pnas.75.2.559
Gudlur, S., Sukthankar, P., Gao, J., Avila, L. A., Hiromasa, Y., Chen, J., et al. (2012). Peptide nanovesicles formed by the self-assembly of branched amphiphilic peptides. PLoS ONE 7:e45374. doi:10.1371/journal.pone.0045374
Guo, C., Luo, Y., Zhou, R., and Wei, G. (2012a). Probing the self-assembly mechanism of diphenylalanine-based peptide nanovesicles and nanotubes. ACS Nano 6, 3907–3918. doi:10.1021/nn300015g
Guo, X. D., Qian, Y., Zhang, C. Y., Nie, S. Y., and Zhang, L. J. (2012b). Can drug molecules diffuse into the core of micelles? Soft Matter 8, 9989–9995. doi:10.1039/c2sm26200b
Guo, X. D., Zhang, L. J., and Qian, Y. (2012c). Systematic multiscale method for studying the structure–performance relationship of drug-delivery systems. Ind. Eng. Chem. Res. 51, 4719–4730. doi:10.1021/ie2014668
Guo, C., Luo, Y., Zhou, R., and Wei, G. (2014). Triphenylalanine peptides self-assemble into nanospheres and nanorods that are different from the nanovesicles and nanotubes formed by diphenylalanine peptides. Nanoscale 6, 2800–2811. doi:10.1039/c3nr02505e
Guo, X. D., Tan, J. P. K., Kim, S. H., Zhang, L. J., Zhang, Y., Hedrick, J. L., et al. (2009). Computational studies on self-assembled paclitaxel structures: templates for hierarchical block copolymer assemblies and sustained drug release. Biomaterials 30, 6556–6563. doi:10.1016/j.biomaterials.2009.08.022
Guo, X. D., Zhang, L. J., Wu, Z. M., and Qian, Y. (2010). Dissipative particle dynamics studies on microstructure of pH-sensitive micelles for sustained drug delivery. Macromolecules 43, 7839–7844. doi:10.1021/ma101132n
Gupta, J., Nunes, C., Vyas, S., and Jonnalagadda, S. (2011). Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations. J. Phys. Chem. B 115, 2014–2023. doi:10.1021/jp108540n
Hamley, I. W. (2011). Self-assembly of amphiphilic peptides. Soft Matter 7, 4122–4138. doi:10.1039/c0sm01218a
Hauser, C. A., Deng, R., Mishra, A., Loo, Y., Khoe, U., Zhuang, F., et al. (2011). Natural tri- to hexapeptides self-assemble in water to amyloid β-type fiber aggregates by unexpected α-helical intermediate structures. Proc. Natl. Acad. Sci.U.S.A. 108, 1361–1366. doi:10.1073/pnas.1014796108
Hayward, R. C., and Pochan, D. J. (2010). Tailored assemblies of block copolymers in solution: it is all about the process. Macromolecules 43, 3577–3584. doi:10.1021/ma9026806
He, C. L., Zhuang, X. L., Tang, Z. H., Tian, H. Y., and Chen, X. S. (2012). Stimuli-sensitive synthetic polypeptide-based materials for drug and gene delivery. Adv. Healthc. Mater. 1, 48–78. doi:10.1002/adhm.201100008
Hoogerbrugge, P. J., and Koelman, J. M. V. A. (1992). Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. Europhys. Lett. 19, 155. doi:10.1209/0295-5075/19/3/001
Hosseinkhani, H., Hong, P. D., and Yu, D. S. (2013). Self-assembled proteins and peptides for regenerative medicine. Chem. Rev. 113, 4837–4861. doi:10.1021/cr300131h
Hubbell, J. A., and Chikoti, A. (2012). Nanomaterials for drug delivery. Science 337, 303–305. doi:10.1126/science.1219657
Jeon, J., Mills, C. E., and Shell, M. S. (2013). Molecular insights into diphenylalanine nanotube assembly: all-atom simulations of oligomerization. J. Phys. Chem. B 117, 3935–3943. doi:10.1021/jp308280d
Jiang, W., Luo, J., and Nangia, S. (2015). Multiscale approach to investigate self-assembly of telodendrimer based nanocarriers for anticancer drug delivery. Langmuir 31, 4270–4280. doi:10.1021/la503949b
Jorgensen, W. L., Maxwell, D. S., and Tiradorives, J. (1996). Development and testing of the opls all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236. doi:10.1021/ja9621760
Kasimova, A. O., Pavan, G. M., Danani, A., Mondon, K., Cristiani, A., Scapozza, L., et al. (2012). Validation of a novel molecular dynamics simulation approach for lipophilic drug incorporation into polymer micelles. J. Phys. Chem. B 116, 4338–4345. doi:10.1021/jp2104819
Koelman, J. M. V. A., and Hoogerbrugge, P. J. (1993). Dynamic simulations of hard-sphere suspensions under steady shear. Europhys. Lett. 21, 363. doi:10.1209/0295-5075/21/3/018
Kumar, N., and Kumar, R. (2014). Nanotechnology and Nanomaterials in the Treatment of Life-Threatening Diseases. Oxford: William Andrew Publishing, 177–246.
Lawrence, M. J. (1994). Surfactant systems: their use in drug delivery. Chem. Soc. Rev. 23, 417–424. doi:10.1039/cs9942300417
Lee, O.-S., Cho, V., and Schatz, G. C. (2012a). Modeling the self-assembly of peptide amphiphiles into fibers using coarse-grained molecular dynamics. Nano Lett. 12, 4907–4913. doi:10.1021/nl302487m
Lee, O.-S., Liu, Y., and Schatz, G. (2012b). Molecular dynamics simulation of β-sheet formation in self-assembled peptide amphiphile fibers. J. Nanopart. Res. 14, 1–7. doi:10.1007/s11051-012-0936-z
Lee, O.-S., Stupp, S. I., and Schatz, G. C. (2011). Atomistic molecular dynamics simulations of peptide amphiphile self-assembly into cylindrical nanofibers. J. Am. Chem. Soc. 133, 3677–3683. doi:10.1021/ja110966y
Li, W., Feng, S. S., and Guo, Y. J. (2012). Tailoring polymeric micelles to optimize delivery to solid tumors. Nanomedicine 7, 1235–1252. doi:10.2217/nnm.12.88
Li, X., Tang, Y. H., Liang, H., and Karniadakis, G. E. (2014). Large-scale dissipative particle dynamics simulations of self-assembled amphiphilic systems. Chem. Commun. 50, 8306–8308. doi:10.1039/c4cc03096f
Lin, W. J., Nie, S. Y., Chen, Q., Qian, Y., Wen, X. F., and Zhang, L. J. (2014). Structure–property relationship of ph-sensitive (PCL)2(PDEA-b-PPEGMA)2 micelles: experiment and DPD simulation. AIChE J. 60, 3634–3646. doi:10.1002/aic.14562
Liu, G. Y., Lv, P. L., Chen, C. J., Liu, X. S., Hu, X. F., and Ji, J. (2011). Biocompatible and biodegradable polymersomes for pH-triggered drug release. Soft Matter 7, 6629–6636. doi:10.1039/c1sm05308f
Lopez-Perez, D. E., Revilla-Lopez, G., Hamley, I. W., and Aleman, C. (2013). Molecular insights into aggregates made of amphiphilic Fmoc-tetrapeptides. Soft Matter 9, 11021–11032. doi:10.1039/c3sm51826d
Loverde, S. M. (2014). Computer simulation of polymer and biopolymer self-assembly for drug delivery. Mol. Simul. 40, 794–801. doi:10.1080/08927022.2014.898118
Loverde, S. M., Klein, M. L., and Discher, D. E. (2011). Nanoparticle shape improves delivery: rational coarse grained molecular dynamics of taxol in worm-like PEG-PCL micelles. Adv. Mater. 24, 3823–3830. doi:10.1002/adma.201103192
Luo, Z. L., and Jiang, J. W. (2012). pH-sensitive drug loading/releasing in amphiphilic copolymer PAE-PEG: integrating molecular dynamics and dissipative particle dynamics simulations. J. Control. Release 162, 185–193. doi:10.1016/j.jconrel.2012.06.027
Mackenzie, R., Booth, J., Alexander, C., Garnett, M. C., and Laughton, C. A. (2015). Multiscale modeling of drug-polymer nanoparticle assembly identifiers parameters influcencing drug encapsulation efficiency. J. Chem. Theory Comput. 11, 2705–2713. doi:10.1021/ct501152a
MacKerell, A. D., Bashford, D., Bellott, M., Dunbrack, R. L., Evanseck, J. D., Field, M. J., et al. (1998). All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616. doi:10.1021/jp973084f
Mai, Y., and Eisenberg, A. (2012). Self-assembly of block copolymers. Chem. Soc. Rev. 41, 5969–5985. doi:10.1039/c2cs35115c
Marrink, S. J., De Vries, A. H., and Mark, A. E. (2004). Coarse grained model for semiquantitative lipid simulations. J. Phys. Chem. B 108, 750–760. doi:10.1021/jp036508g
Marrink, S. J., Risselada, H. J., Yefimov, S., Tieleman, D. P., and De Vries, A. H. (2007). The martini force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824. doi:10.1021/jp071097f
Meng, F. H., Zhong, Z. Y., and Feijen, J. (2009). Stimuli-responsive polymersomes for programmed drug delivery. Biomacromolecules 2, 197–209. doi:10.1021/bm801127d
Miyazawa, S., and Jernigan, R. L. (1985). Estimation of effective interresidue contact energies from protein crystal-structures – quasichemical approximation. Macromolecules 18, 534–552. doi:10.1021/ma00145a039
Mondai, J., and Yethiraj, A. (2012). Effect of secondary structure on the self-assembly of amphiphilic molecules: a multiscale simulation study. J. Chem. Phys. 136, 084902. doi:10.1063/1.3689298
Monticelli, L., Kandasamy, S. K., Periole, X., Larson, R. G., Tieleman, D. P., and Marrink, S. J. (2008). The martini coarse-grained force field: extension to proteins. J. Chem. Theory Comput. 4, 819–834. doi:10.1007/s00894-013-1991-9
Mu, X., Eckes, K. M., Nguyen, M. M., Suggs, L. J., and Ren, P. (2012). Experimental and computational studies reveal an alternative supramolecular structure for fmoc-dipeptide self-assembly. Biomacromolecules 13, 3562–3571. doi:10.1021/bm301007r
Natarajan, J. V., Nugraha, C., Ng, X. W., and Venkataraman, S. (2014). Sustained-release from nanocarriers: a review. J. Control. Release 193, 122–138. doi:10.1016/j.jconrel.2014.05.029
Nicola, A. D., Hezaveh, S., Zhao, Y., Kawakatsu, T., Roccatano, D., and Milano, G. (2014). Micellar drug nanocarriers and biomembranes: how to they interact? Phys. Chem. Chem. Phys. 16, 5093–5105. doi:10.1039/c3cp54242d
Nie, S. Y., Lin, W. J., Yao, N., Guo, X. D., and Zhang, L. J. (2014). Drug release from ph-sensitive polymeric micelles with different drug distributions: insight from coarse-grained simulations. ACS Appl Mater. Interf. 6, 17668–17678. doi:10.1021/am503920m
Nie, S. Y., Sun, Y., Lin, W. J., Wu, W. S., Guo, X. D., Qian, Y., et al. (2013). Dissipative particle dynamics studies of doxorubicin-loaded micelles assembled from four-arm star triblock polymers 4AS-PCL-b-PDEAEMA-b-PPEGMA and their pH-release mechanism. J. Phys. Chem. B 117, 13688–13697. doi:10.1021/jp407529u
Patel, S., Lavasanifar, A., and Choi, P. (2008). Application of molecular dynamics simulation to predict the compatability between water-insoluble drugs and self-associating poly(ethylene oxide)-b-poly(ε-caprolactone) block copolymers. Biomacromolecules 9, 3014–3023. doi:10.1021/bm800320z
Patel, S. K., Lavasanifar, A., and Choi, P. (2009). Roles of nonpolar and polar intermolecular interactions in the improvement of the drug loading capacity of PEO-b-PCL with increasing pcl content for two hydrophobic cucurbitacin drugs. Biomacromolecules 10, 2584–2591. doi:10.1021/bm900512h
Patel, S. K., Lavasanifar, A., and Choi, P. (2010). Molecular dynamics study of the encapsulation capability of a PCL-PEO based block copolymer for hydrophobic drugs with different spatial distributions of hydrogen bond donors and acceptors. Biomaterials 31, 1780–1786. doi:10.1016/j.biomaterials.2009.11.060
Posocco, P., Fermeglia, M., and Pricl, S. (2010). Morphology prediction of block copolymers for drug delivery by mesoscale simulations. J. Mater. Chem. 20, 7742–7753. doi:10.1039/c0jm01301c
Rad-Malekshahi, M., Visscher, K. M., Rodrigues, J. P., De Vries, R., Hennink, W. E., Baldus, M., et al. (2015). The supramolecular organization of a peptide-based nanocarrier at high molecular detail. J. Am. Chem. Soc. 137, 7775–7784. doi:10.1021/jacs.5b02919
Razavilar, N., and Choi, P. (2014). Molecular dynamics study of the diffusivity of a hydrophobic drug cucurbitacin B in pseudo-poly(ethylene oxide-b-caprolactone) micelle environments. Langmuir 30, 7798–7803. doi:10.1021/la500572p
Rissanou, A. N., Georgilis, E., Kasotakis, E., Mitraki, A., and Harmandaris, V. (2013). Effect of solvent on the self-assembly of dialanine and diphenylalanine peptides. J. Phys. Chem. B 117, 3962–3975. doi:10.1021/jp311795b
Rodriguez-Hidalgo, M.-D.-R., Soto-Figueroa, C., and Vicente, L. (2011). Mesoscopic simulation of the drug release mechanism on the polymeric vehicle P(ST-DVB) in an acid environment. Soft Matter 7, 8224–8230. doi:10.1039/c1sm05667k
Rösler, A., Vandermeulen, G. W. M., and Klok, H.-A. (2001). Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev 53, 95–108. doi:10.1016/S0169-409X(01)00222-8
Sevink, G. J. A., Charlaganov, M., and Fraaije, J. G. E. M. (2013). Coarse-grained hybrid simulation of liposomes. Soft Matter 9, 2816–2831. doi:10.1039/c2sm27492b
Shimixu, T., Masuda, M., and Minamikawa, H. (2005). Supramolecular nanotube architectures based on amphiphilic molecules. Chem. Rev. 105, 1401–1443. doi:10.1021/cr030072j
Srinivas, G., Mohan, R. V., and Kelkar, A. D. (2013). Polymer micelle assisted transport and delivery of model hydrophilic components inside a biological lipid vesicle: a coarse-grain simulation study. J. Phys. Chem. B 117, 12095–12104. doi:10.1021/jp405381k
Sundar, S., Chen, Y., and Tong, Y. W. (2014). Delivery of therapeutics and molecules using self-assembled peptides. Curr. Med. Chem 21, 2469–2479. doi:10.2174/0929867321666131212152637
Tama, F., Wriggers, W., and Brooks, C. L. (2002). Explorng global distortions of biological macromolecules and assemblies from low-resolution structural information and elastic network theory. J. Mol. Biol. 321, 297–305. doi:10.1016/S0022-2836(02)00627-7
Tanner, P., Baumann, P., Enea, R., Onaca, O., Palivan, C., and Meier, W. (2011). Polymeric vesicles: from drug carriers to nanoreactors and artificial organelles. Acc. Chem. Res. 44, 1039–1049. doi:10.1021/ar200036k
Thota, N., Hu, Z. Q., and Jiang, J. W. (2015). Ibuprofen loading and release in amphiphilic peptide (AF)6H5K15 and its derivatives: a coarse-grained molecular dynamics simulation study. Mol. Simul.
Thota, N., and Jiang, J. W. (2014). Self-assembly of amphiphilic peptide (AF)6H5K15 derivatives: roles of hydrophilic and hydrophobic residues. J. Phys. Chem. B 118, 2683–2692. doi:10.1021/jp500406p
Thota, N., Luo, Z. L., Hu, Z. Q., and Jiang, J. (2013). Self-assembly of amphiphilic peptide (AF)6H5K15: coarse-grained molecular dynamics simulation. J. Phys. Chem. B 117, 9690–9698. doi:10.1021/jp4059752
Thota, N., Ma, Y. J., and Jiang, J. W. (2014). Molecular insights into the self-assembly of short amphiphilic peptides fmdn and fmkn. RSC Adv. 4, 60741–60748. doi:10.1063/1.3314309
Tian, B., Tao, X., Ren, T., Weng, Y., Lin, X., Zhang, Y., et al. (2012). Polypeptide-based vesicles: formation, properties and application for drug delivery. J. Mater. Chem. 22, 17404–17414. doi:10.1039/c2jm31806g
Trent, A., Marullo, R., Lin, B., Black, M., and Tirrell, M. (2011). Structural properties of soluble peptide amphiphile micelles. Soft Matter 7, 9572–9582. doi:10.1016/j.sbi.2009.07.008
Tyrrell, Z. L., Shen, Y., and Radosz, M. (2010). Fabrication of micellar nanoparticles for drug delivery through the self-assembly of block copolymers. Prog. Polym. Sci. 35, 1128–1143. doi:10.1016/j.progpolymsci.2010.06.003
Uchegbu, I. F., and Vyas, S. P. (1998). Non-ionic surfactant based vesicles (niosomes) in drug delivery. Int. J. Pharm. 172, 33–70. doi:10.1016/S0378-5173(98)00169-0
Ulijn, R. V., and Smith, A. M. (2008). Designing peptide based nanomaterials. Chem. Soc. Rev. 37, 664–675. doi:10.1039/b609047h
Velichko, Y. S., Stupp, S. I., and de La Cruz, M. O. (2008). Molecular simulation study of peptide amphiphile self-assembly. J. Phys. Chem. B 112, 2326–2334. doi:10.1021/jp074420n
Venkataraman, S., Hedrick, J. L., Ong, Z. Y., Yang, C., Ee, P. L. R., Hammond, P. T., et al. (2011). The effects of polymeric nanostructure shape on drug delivery. Adv. Drug Deliv. Rev. 63, 1228–1246. doi:10.1016/j.addr.2011.06.016
Wei, H., Zhuo, R.-X., and Zhang, X. Z. (2013). Design and development of polymeric micelles with cleavable linkers for intracellular drug delivery. Prog. Polym. Sci. 38, 503–535. doi:10.1016/j.progpolymsci.2012.07.002
Yu, T., and Schatz, G. C. (2013a). Free energy landscape for peptide amphiphile self-assembly: stepwise versus continuous assembly mechanisms. J. Phys. Chem. B 117, 14059–14064. doi:10.1021/jp409305e
Yu, T., and Schatz, G. C. (2013b). Free energy profile and mechanism of self-assembly of peptide amphiphiles based on a collective assembly coordinate. J. Phys. Chem. B 117, 9004–9013. doi:10.1021/jp404835q
Zhang, X. X., Eden, H. S., and Chen, X. (2012). Peptides in cancer nanomedicine: drug carriers, targeting ligands and protease substrates. J. Control. Release 159, 2–13. doi:10.1016/j.jconrel.2011.10.023
Zhao, X., Pan, F., Xu, H., Yaseen, M., Shan, H., Hauser, C. A., et al. (2010). Molecular self-assembly and applications of designer peptide amphiphiles. Chem. Soc. Rev. 39, 3480–3498. doi:10.1039/b915923c
Keywords: copolymers, peptides, morphology, loading, release, computation
Citation: Thota N and Jiang J (2015) Computational amphiphilic materials for drug delivery. Front. Mater. 2:64. doi: 10.3389/fmats.2015.00064
Received: 15 July 2015; Accepted: 28 September 2015;
Published: 14 October 2015
Edited by:
Ulrich Kleinekathöfer, Jacobs University Bremen, GermanyReviewed by:
Sergei Noskov, University of Calgary, CanadaPaolo Ruggerone, University of Cagliari, Italy
Copyright: © 2015 Thota and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianwen Jiang, Y2hlampAbnVzLmVkdS5zZw==