 Boris Agea-Blanco
Boris Agea-Blanco Stefan Reinsch
Stefan Reinsch Ralf Müller
Ralf Müller- 1Division 5.6 “Glass”, Federal Institute for Materials Research and Testing (BAM), Berlin, Germany
- 2IQS School of Engineering, Barcelona, Spain
The manufacture of sintered glasses and glass-ceramics, glass matrix composites, and glass-bounded ceramics or pastes is often affected by gas bubble formation. Against this background, we studied sintering and foaming of barium silicate glass powders used as SOFC sealants using different powder milling procedures. Sintering was measured by means of heating microscopy backed up by XPD, differential thermal analysis, vacuum hot extraction (VHE), and optical and electron microscopy. Foaming increased significantly as milling progressed. For moderately milled glass powders, subsequent storage in air could also promote foaming. Although the powder compacts were uniaxially pressed and sintered in air, the milling atmosphere significantly affected foaming. The strength of this effect increased in the order Ar ≈ N2 < air < CO2. Conformingly, VHE studies revealed that the pores of foamed samples predominantly encapsulated CO2, even for powders milled in Ar and N2. Results of this study thus indicate that foaming is caused by carbonaceous species trapped on the glass powder surface. Foaming could be substantially reduced by milling in water and 10 wt% HCl.
Introduction
Glass powders are widely used in fabricating sintered glass, sintered glass-ceramics, glass matrix composites, and glass-bonded ceramics or pastes when lower fabrication or processing temperatures, gas-tight seals, or complex shapes are required (Rabinovich, 1985; Schiller et al., 2008; Müller and Reinsch, 2012). One problem often addressed within this context is concurrent crystallization and sintering (Müller, 1994; German, 1996; Prado and Zanotto, 2002; Pascual and Duran, 2003; Prado et al., 2003a,b, 2008). This effect is most pronounced in sintered glass-ceramics, where a large crystal fraction is desired and rapid crystallization starts from the powder surface (Müller and Reinsch, 2012).
Many practical applications of sintered glass-ceramics, however, rely on slowly crystallizing glasses. Such glasses are, for instance, used for low temperature co-fired ceramics (Imanaka, 2005), paste glasses (Hwang et al., 2002), or SOFC sealants (Fergus, 2005; Gross et al., 2005). In the case of slow crystallization, however, another problem may arise. Due to the low glass viscosity required for joining and gas-tight sealing or obtaining a desired crystallinity, gas bubble formation, and related sample swelling (“foaming”) often occur, even when organic aids are not used in powder processing.
Thus, Lucchini et al. (1983) observed an increased bubble formation with increasing glass volume fraction for sodium and calcium lead silicate glass-bonded barium hexaferrites and attributed this effect to glass volatilization. Pore formation was also observed in porcelain stoneware tiles (Leonelli et al., 2001) and lead borosilicate glass frits (Hwang et al., 2002). In the latter case, effusing oxygen or water, physically or chemically adsorbed to the glass powder surface during manufacturing and storage, has been supposed to be potential foaming sources. However, neither dry quenching of the molten glass nor using vacuum-dried B2O3 for glass melting could reduce foaming. Lara et al. (2004) observed foaming during sintering and crystallization treatments of Ca, Mg, and Zn alumosilicate glass powders for SOFC sealing to be most pronounced in Ca alumosilicate glass powders (up to 30% silhouette area increase in heating microscopy experiments). The authors discussed the formation of crystals of lower density and/or gas evolution during crystallization for causing the observed foaming. More recently, foaming was mainly attributed to pore coalescence during over-firing (Lim et al., 2006), obviously assuming an encapsulated sintering atmosphere as the major foaming source. Due to the decreased sintering pressure of larger pores, less gas is forced to dissolve into the glass melt, and low viscosity allows easy bubble growth. An analogous explanation was given by Kim et al. (2007) for foaming of lead-free Bi2O3–B2O3–SiO2 solder glass powders and by Müller et al. (2009) for LTCC model glass powders. Undesired porosity was also observed during sintering ashes for porcelain tile production, which was attributed to “some boiling and trapped gas effects” (Fernandes and Ferreira, 2007).
The aim of this paper was to study the effect of glass powder milling on foaming of barium disilicate glass powder compacts. The glass powders under study have been dry-milled for different times in different atmospheres, including argon, nitrogen, air, and carbon dioxide, or wet-milled in water and 10 wt% HCl, uniaxially pressed and sintered in air. Densification and foaming was studied by means of heating microscopy backed up by XRD, Differential thermal analysis (DTA), vacuum hot extraction (VHE), and microscopy.
Experimental
Materials
The present study was undertaken on a commercial barium disilicate glass with minor additions of B2O3, Al2O3, and ZnO used for SOFC sealing (Kerafol, 2010). The maximum particle size of the as-received commercial glass frit was limited to <2 mm by manual crushing in a metal mortar and sieving.
In order to reach an appropriate offset particle size for the milling experiments, pre-milling was performed in air using a planetary ball mill (Fritsch Pulverisette 5, Fritsch, Idar-Oberstein, Germany) loaded with two 500-ml corundum jars (∅in ≈ 102 mm, h = 78 mm). Each jar was filled with six corundum balls (∅ ≈ 20 mm) and about 250 g of the glass frit. Milling was done for 15 min at 320 rpm (maximum speed of supporting disk) (FRITSCH GmbH, 1987). No milling aids were used. Afterwards, a mechanical sieve (Analysette 3 PRO, Fritsch, Idar-Oberstein, Germany) was used to reduce the particle size distribution to 40–250 μm (“p0” in Table 1).
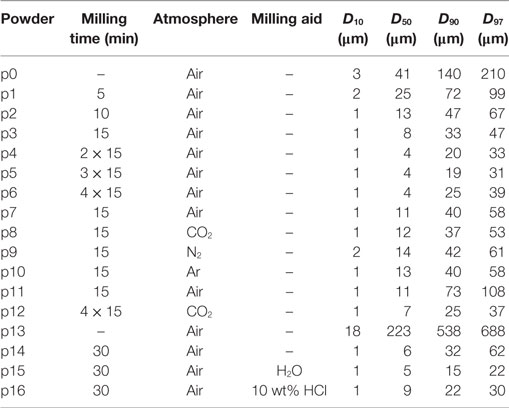
Table 1. Particle size of glass powders after different milling (p0: pre-milled).
Milling was performed in a planetary ball mill equipped with two 25-ml corundum grinding jars (∅in ≈ 33 mm, h = 45 mm; Planetary micro mill Pulverisette 7, Fritsch, Idar-Oberstein, Germany) (FRITSCH GmbH, 1995). The jars were filled with four corundum balls (∅ ≈ 12 mm) and ≈8 g of glass powder. In order to provide a controlled milling atmosphere within these grinding jars, special ring seals were used. These rings were made of steel equipped with top and bottom silicone seals and a gas inlet capillary, which could be closed by a shuttle valve mounted on top of the rotating jar holder.
Milling progress was studied in air starting from glass powder p0 (Table 1). Glass powders were milled for 5–60 min at 766 rpm (maximum speed of supporting disk) (FRITSCH GmbH, 1995). Milling was temporarily stopped for 30 min after 15 min of milling to prevent overheating. Starting with a large decrease of D50 during the first milling stage, the decrease in particle size progressively slowed down to level out at ≈4 μm. At 60-min dry-milling, slightly increased D90 and D97 values indicate progressive particle agglomeration (p1–p6).
Milling in controlled atmospheres (p7–p10) was carried out for 15 min also at 766 rpm (FRITSCH GmbH, 1995). In this case, the sealed milling jars were evacuated to <20 mbar and re-filled with CO2, N2, and Ar (99.99% purity, Air Liquide™, Germany) to 105 Pa (1 bar). Evacuation and re-filling was repeated five times to minimize the amount of residual air. A liquid nitrogen trap was used to increase N2 purity. For studying the effect of powder storage in air after milling, the glass frit was crushed to <1000 μm (Jaw Crusher, Retsch BB51), sieved to 200–1000 μm, and milled in air (p11) and CO2 (p12) as described.
Wet-milling started from a glass frit crushed using the same jaw-crusher: about 400 g of starting glass frit was repeatedly crushed using a gap width decreasing from 1000 to 200 μm in 200-μm steps. The last crushing step (200 μm gap width) was repeated three times (p13). Milling jars were filled with 8 g of this powder (p13) and milled for 30 min as for milling in controlled atmospheres. For powder p14, dry-milling was carried out in ambient air as a reference. Powders p15 and p16 were wet-milled in 8-g pure water and 10 wt% HCl (diluted from fuming HCl 37 wt%, Merck, Germany), respectively. Pure water was supplied by a MilliQ® device (Merck, Germany). Wet-milled powders were dried for 2 days at 120°C in air before sintering.
For the study of sintering and microstructural evolution, cylindrical powder compacts were uniaxially pressed in air at 60 MPa (green dimensions: ∅ ≈ 5 mm, h ≈ 2 mm, and m ≈ 0.1 g) without organic aids. After milling, the powder was stored in a closed HDPE box for between 2 and 4 days before uniaxial pressing. During that time, no significant effect on sintering and foaming could be observed.
For the study of gas release from the powder surface and foamed porosity, powder compacts of cylindrical shape were uniaxially pressed at 105 MPa (∅ ≈ 5 mm, h ≈ 1 mm, and m ≈ 0.050 g) without binders or other aids and then cut down using a scalpel to pieces of ≈9–10 mg. These pieces had been stored in a HDPE container before being studied by VHE.
Methods
Glass viscosity, ηG, was measured by means of rotational concentric cylinder viscometry (VT550, Haake, Erlangen, Germany) for η < 105 Pa s. The glass transition temperature Tg = 649 ± 3°C and the coefficient of thermal expansion CTE25–400°C = 7.6 × 10−6 K−1 were determined with a horizontal dilatometer (402 E, Netzsch, Selb, Germany; bulk glass bars, 25 mm × 5 mm × 5 mm). The viscosity data obtained in pascal second and degree Celsius could be approximated with logη = −2.77 + 2644/(T − 480) within Δ logη ≈ ±0.02 accuracy.
Glass density, ρG = 3.61 g/cm3, was measured from glass bars by means of Archimedes’ principle. A Mastersizer 2000 (Malvern Instruments, Wocestershire, U.K.) was used for particle size measurement. Particle agglomeration was minimized by dispersing ≈10 mg of glass powder in a 0.003M Na4P2O7 solution and subsequent ultra-sonic treatments (1–5 min). The bulk density of green and sintered specimens was obtained from their sample geometry (heating microscope) and weight.
The microstructure of powder compacts heated to selected temperatures and quenched in air was studied from polished cross-sections by means of environmental scanning electron microscopy (ESEM-FEG, Philips-XL 30, Eindhoven, Netherlands) or by an optical microscope (JENAPOL, Carl Zeiss Jena, Jena, Germany). Green powder compacts were embedded in synthetic resin for preparing cross-sections. Porosity was measured by image analysis of optical micrographs using the software Image C (Aquinto AG, Berlin, Germany).
Crystallization was studied by means of X-ray diffraction (Philips PW 1710, Eindhoven, Netherlands) using copper Kα with λ = 1.5418 Å, in Bragg–Brentano symmetry. Data were collected for 2θ = 5–80° in steps of 0.02°/s. Diffraction patterns were analyzed using EVA 15.1 software (Bruker-AXS, Karlsruhe, Germany) and compared to the JCPDS database (JCPDS, 2009, International Center for Diffraction Data). DTA–MS runs were performed in air at 5 K/min (15 K/min below 500°C) using ≈25-mg pieces of powder compacts and Pt crucibles (TAG 24, Setaram, Caluire, France). The DTA device was coupled with a mass spectrometer (Balzers Quadstar 421, Balzers, Liechtenstein) by a heated (180°C) quartz glass capillary. In this way, evolved gases were simultaneously recorded in multiple ion detection (MID) modus.
Gas release from green and sintered powder compacts was studied by means of VHE with mass spectrometer evolved gas detection (QMA4005, Balzers Instruments, Balzers, Liechtenstein). VHE analysis was performed in vacuum (10−4–10−5 mbar) during heating at 20 K/min using the MID mode (Müller et al., 2005).
Shrinkage of cylinder-shaped powder compacts during heating at 5 K/min (<500°C, 15 K/min) was examined using a heating microscope (Leitz, Wetzlar, Germany) with optical data acquisition (Hesse Prüftechnik, Osterode, Germany). Shrinkage and foaming are presented in terms of the silhouette area change, sA = ΔA/A0, where A0 is the initial sample silhouette area of the green compact.
Results
Shrinkage and Foaming
Figure 1 shows the silhouette area change, sA, for powders milled in air for different milling times (p1–p6). Sintering of the pre-milled powder (p0) starts at ≈710°C, and final densification is attained at ≈800°C. With increasing milling time, i.e., with decreasing particle size, the onset and saturation stage of sintering shift to a lower temperature by ≈20 and ≈30 K, respectively, while the attainable maximum densification tends to increase. Beyond maximum densification, progressive foaming is evident, which is strongly promoted by prolonged milling.
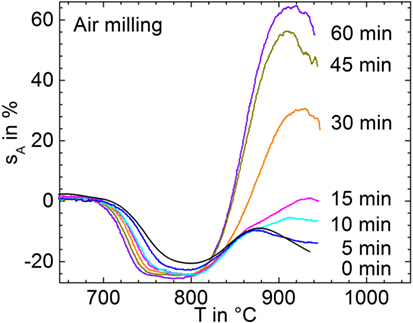
Figure 1. Silhouette area change, sA, versus temperature for glass powders milled in air during heating at 5 K/min (p1–p6). Curve labels: milling time in minutes, 0 min indicates the pre-milled powder (p0). Adapted from Agea Blanco et al. (2015).
Figure 2 illustrates the shrinkage and foaming behavior of glass powders milled for 15 min in controlled atmospheres (p7–p10). Sintering starts at ≈700°C for all powders and undergoes an initial deceleration at ≈760°C. The weak shrinkage between 760 and 800°C, best visible for the N2 milled powder, might be caused by viscous settling of the sample to the substrate under the effect of gravity. Except for milling in CO2, the shrinkage maximum occurs at 800°C beyond which a progressive increase of sA due to foaming occurs. This foaming is most pronounced for milling in CO2, where it already starts at ≈770°C. This finding gives clear evidence that foaming can be affected by the milling atmosphere and that CO2 is most efficient in this context.
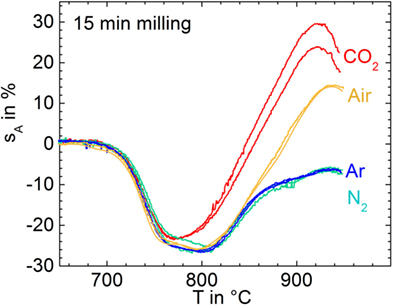
Figure 2. Silhouette area change, sA, versus temperature for glass powders milled for 15 min in different atmospheres (p7–p10) during heating at 5 K/min. Two experiments are shown for each condition in order to illustrate reproducibility. Adapted from Agea Blanco et al. (2015).
Figure 3 shows shrinkage and foaming of glass powders milled for 15 min in air (p11) and stored for different exposure time in air before uniaxial pressing and sintering. It is clearly seen that even short storage (1 day) substantially promoted foaming. This result indicates that gas uptake during storage in the ambient atmosphere can promote foaming.
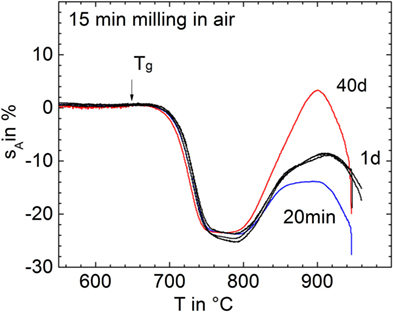
Figure 3. Silhouette area change, sA, versus temperature for glass powders milled for 15 min in air (p11) and subsequently stored in air for different times (curve labels) before sintering at 5 K/min. Arrow: Tg = 649°C. Adapted from Agea Blanco et al. (2015).
Figure 4 illustrates the evolution of porosity during sintering for the powder milled for 15 min in N2. The minimum porosity occurs at 795°C, corresponding to a maximum densification stage in Figure 2. At 875°C, foaming is clearly detectable. The foaming maximum in Figure 2 occurred at ≈940°C. Conformingly, the largest porosity in Figure 4 is seen for the cross-section of the sample heated to 950 and 1000°C. No crystals could be observed even for the sample heated to 1000°C. Conformingly, X-ray diffraction patterns of the powder milled for 15 min in N2 (p9), heated to 940°C, and quenched in air did not reveal the presence of crystalline phases (not shown).
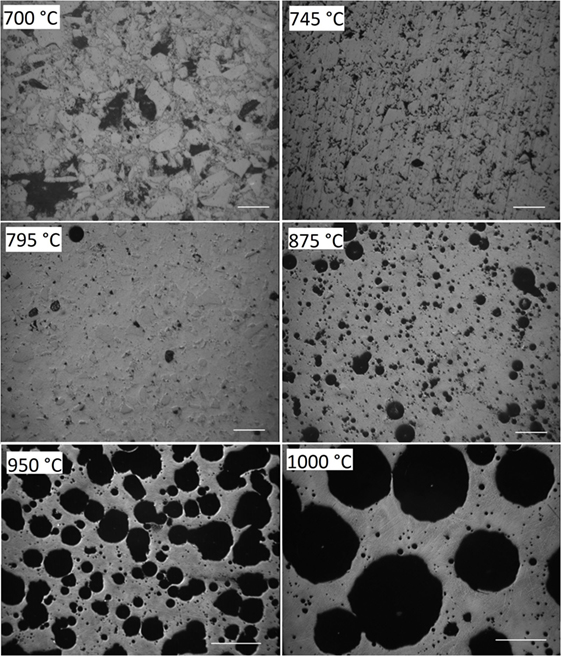
Figure 4. Optical micrographs of powder compacts (15 min milling in N2, p9) heated at 5 K/min to the temperatures indicated and quenched in air. Diamond polished cross-sections. Bars = 100 μm (700–875°C) and 500 μm (950, 1000°C). Porosity: 48% (700°C), 12% (745°C), 4% (795°C), 20% (875°C), 52% (950°C), and 53% (1000°C).
Figure 5 illustrates the VHE–MS degassing behavior of green powder compacts milled for 15 min in different atmospheres. Below the onset of gas bubble bursting (<800°C), water (m/e = 18) is the most prominent degassing species. Degassing of surface-adsorbed water occurs between 50 and 350°C, as previously seen for other silicate glass powders (Müller et al., 2005). This effect causes a broad weakly structured degassing peak, which is similar for all powders. The flat degassing curve between 400 and 600°C mainly reflects the VHE blank value of water. Water degassing is newly accelerated when the temperature approaches Tg (649°C, arrows) and then decreases due to sintering >700°C, indicating that the water degassing mechanism is not exhausted but delayed by sintering. The second most intensive degassing below the onset of bubble bursting is that of CO2 (m/e = 44) followed by CO (m/e = 28). The related mass number is probably not caused by N2 because of the quite different degassing characteristics of N (m/e = 14).
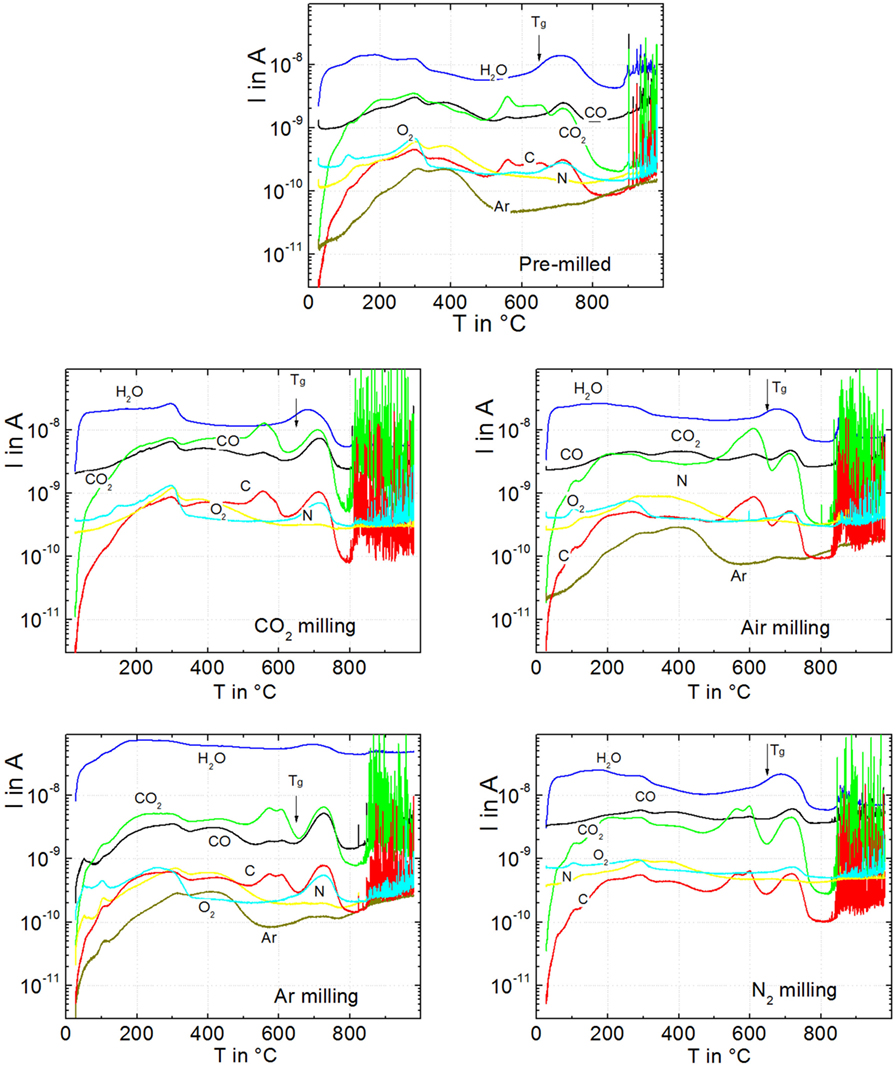
Figure 5. Degassing of green powder compacts during heating at 20 K/min for pre-milling (p0), milling in air (p7), in CO2 (p8), in N2 (p9), and in Ar (p10). Degassing activity is presented in terms of respective ion currents, I, versus temperature, T. Sample mass: ≈9 mg. Arrows: Tg = 649°C. Adapted from Agea Blanco et al. (2015).
During bubble bursting (spikes >800°C), CO2 is the dominant species followed by C (m/e = 12), which is expected to occur as a fragment of CO2. This finding indicates that CO2 degassing is less exhausted below sintering than that of other volatiles. In contrast to CO2, Ar (m/e = 40) and N2 (m/e = 14) did not significantly contribute to bubble bursting even for the powders milled in these atmospheres. The CO2-milled powder shows the most intensive CO2 degassing during bubble bursting confirming the observed ordering of foaming activity in Figure 2. Confirmingly, integrating the CO2 ion currents (m/e = 44) for each of the 9-mg samples over the temperature range of foaming (800–1000°C) yields 6.0, 3.8, 3.1, 2.9, and 0.2 μA min g−1 for milling in CO2, Air, Ar, N2, and the pre-milled powder, respectively. Furthermore, bubble bursting starts at ≈800°C (logη/Pas = 5.4) when milled in CO2, while for milling in Ar, N2, and air, bubble bursting starts at ≈840°C (logη/Pas = 4.6). This effect resembles the similar trend in onset temperatures of foaming in Figure 2. The onset temperature of bubble bursting is controlled by viscosity and bubble pressure. The almost identical sintering observed among all samples in Figure 2 indicates that the milling atmosphere has a negligible effect on glass viscosity. The early onset of foaming for milling in CO2 should, therefore, mainly reflect increased bubble pressure.
Enhanced CO2, CO, and C degassing is evident around 300°C, at ≈500–650°C, and at ≈650–800°C below the onset of foaming. Since it is obviously delayed by sintering, the latter peak at ≈650–800°C is the most likely source of foaming. It is worth noting that the onset of this degassing peak nicely correlates with the glass transition temperature (arrows in Figure 5). Similar degassing patterns were observed for the diffusion-limited release of water dissolved during glass melting at ambient pressure (Müller et al., 2005). However, it is seen from the much less intensive bubble bursting of pre-milled powders that this effect should have minor impact on the extensive bubble bursting of milled powders. Instead, because of the large effect of particle size on foaming (Figure 1), this observation might indicate diffusive degassing from the near-surface region of the powder.
Figure 6 compares CO2 degassing, silhouette area change, and DTA curves of a glass powder milled for 1 h in CO2 (p12). The prolonged milling time was required in order to overcome the lower gas detection limit of the DTA–MS device used here. Besides CO2, no other volatiles were detectable. Figure 6 gives clear evidence that CO2 is the dominant foaming source even during heating in air and ambient pressure. The broad structureless degassing peak (Tmax at ≈300°C) further indicates a pronounced continuous release of CO2 during heating and that sintering only traps a rather small fraction of its initial amount. Nevertheless, this small amount obviously causes substantial foaming until the foamed sample starts to collapse at >880°C via gas bubble bursting (spikes). In contrast to Figure 5, no pronounced increase of degassing activity is seen just above Tg. The DTA curve reveals a weak endothermic shoulder at Tg, a distinct endothermic shoulder within the temperature range of shrinkage and a broad pronounced peak nicely correlating with foaming.
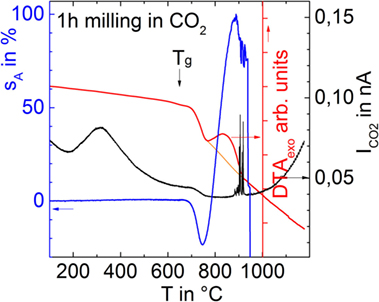
Figure 6. Silhouette area change (sA), MS degassing current for CO2 , and DTA of a glass powder milled for 1 h in CO2 (p12) during heating in ambient air at 5 K/min.
Figure 7 compares sintering and foaming when milled for 30 min in air (p13), water (p14), and 10 wt% HCl (p15). Sintering starts at ≈700°C in all cases. Wet-milled powders exhibit slightly increased area shrinkage, which may at least partially reflect different powder compact green densities due to possibly altered glass surface properties (ρ0 = 62, 55, and 55% for p14, p15, and p16, respectively). Furthermore, shrinkage was slightly decelerated for the powder milled in HCl, although its particle size does not significantly differ from powders p14 and p15 (Table 1). This effect is accompanied by a shift of the foaming onset by 30 K to 820°C. Both observations hint of a possible increase of viscosity. The latter effect could be caused by increased glass viscosity (e.g., related to aqueous Ba dissolution from the glass surface), the presence of rigid inclusions (e.g., BaCl2 precipitates), or by both phenomena.
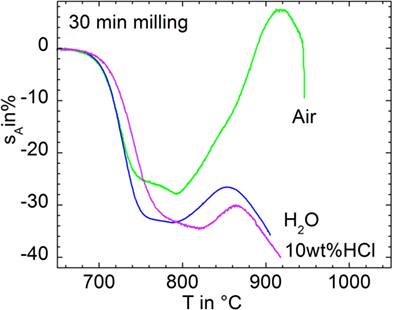
Figure 7. Silhouette area change, sA, versus temperature for 30 min milled in air (p14), water (p15), and 10 wt% HCl (p16) during heating at 5 K/min.
Most notably, however, Figure 7 gives clear evidence that foaming has been substantially reduced by means of wet-milling. Thus, foaming caused a silhouette area increase of ΔsA = 35% for the 30 min dry-milled powder (p14), whereas ΔsA = 7% and even 4% were evident for the powders milled in water (p15) and HCl (p16), respectively. Table 1 indicates that this effect is not just feigned by a deviating particle size distribution as D10 and D50 values are quite similar. Decreased D90 and D97 data for water milling may reflect a reduced agglomeration tendency.
Water- versus dry-milling experiments, similar to that presented in Figure 7, have also been conducted repeatedly using various milling materials including Cr–Ni steel, Si3N4, and WC. In all cases, a substantial reduction of foaming was observed. The use of steel milling jars in wet-milling could reduce foaming from ΔsA = 48% (dry-milling) to ΔsA = 9%. Similar ratios (ΔsA = 52–34% versus ΔsA = 5–8%) occurred for Al2O3, Si3N4, and WC. Furthermore, it was found that for milling in corundum jars and 10 wt% HCl, prolonged storage up to 12 days did not significantly influence ΔsA, which scattered between 5 and 6% for all applied storage times.
Discussion
As discussed in previous literature, different sources may contribute to foaming: gases encapsulated within the closed pore volume of the powder compact (Lim et al., 2006; Kim et al., 2007; Müller et al., 2009), gases adsorbed onto the glass powder surface (Hwang et al., 2002), glass volatilization (Lucchini et al., 1983), or effusing oxygen from the glass bulk (Hwang et al., 2002). Due to the strong effect of particle size on foaming activity (Figures 1 and 5), the latter two mechanisms should not dominate foaming in the present study.
Gas Trapped in the Foaming Sample
In order to reveal to what extent encapsulated and adsorbed gases may contribute to foaming in the present study, the amount of gas trapped in the maximal foamed sample, nMax, was estimated first. The 15-min N2-milled powder was used for this estimation as an optimum between detectable foaming and minor bubble bursting activity below the foaming maximum. Nevertheless, nMax can only provide a lower limit of this amount.
(i) As an initial approach for estimating nMax, the porosity, P, of foamed samples was measured from cross-section micrographs of powder compacts heated to different temperatures and quenched in air, by means of image analysis. Respective values are shown in Figure 8 (right ordinate, gray circles). Maximal porosity scatters around 52% (0.52). nMax can be estimated from this value with Eq. 1, where P is the porosity, V the sample volume, P⋅V the pore volume, R the ideal gas constant, T the temperature, and p the internal pressure:
p was assumed to be ≈105 Pa (1 bar) due to the low viscosity expected at the foaming maximum and because the majority of pore radii, r, were much greater than 50 μm. For such condition and a surface energy of γ ≈ 0.3 J m−2 (estimated with SciGlass 6.5 software), the Laplace pressure PL ≈ 2γ/r (German, 1996) should be small (<0.12 × 105 Pa) and was therefore neglected. Based on P ≈ 0.52, nMax ≈ 16 × 10−8 mol was found.
(ii) A more average value of P can be calculated from the linear isotropic shrinkage, si = (ΔV/V0)−3, where V0 is the initial sample volume. si is related to P according to Eq. 2 (Winkel et al., 2012)
where ρ and ρ0 denote the relative density and the relative green density, respectively. In order to calculate si from the measured silhouette area change data, sA, the current sample volume, V, has to be inferred from the sample silhouette area, A. In that case, a regular sample shape must be adopted. Up to 800°C, the sample shape could be reasonably approximated as a cylinder. Its volume is given by Eq. 3 as follows (Sieber, 1980):
where d, h, and A (where A = d⋅h) represents the diameter and the height of the cylinder and its silhouette area, respectively. Above 850°C, heating microscopy data showed that samples underwent substantial rounding resembling more a hemisphere (not shown). The volume of a hemisphere can be related to its sample silhouette according to Eq. 4 (Sieber, 1980)
where d refers to the diameter of the hemisphere and A = (1/8)πd2 to its silhouette. Above 920°C, the sample shape rather resembles a spherical cap. Its volume is given by Eq. 5 as follows (Sieber, 1980):
In this case, d denotes the base radius of the cap.
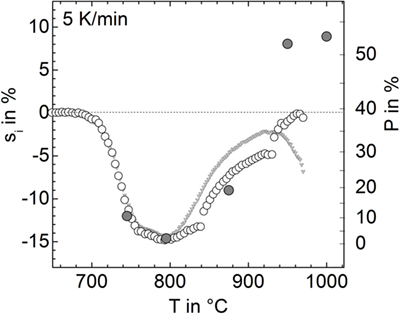
Figure 8. Linear isotropic shrinkage, si (open circles, left ordinate) and respective porosity, P (gray circles, right ordinate) versus temperature for the powder milled for 15 min in nitrogen (p9). For calculating si from sA, a different sample shape has been adopted: cylinder (T < 840°C), hemisphere (840–930°C), and spherical cap (930–980°C). Gray triangles: silhouette area shrinkage (see Figure 1), sA, rescaled by 1/2. Gray circles: porosity measured from cross-sectional micrographs.
The linear isotropic shrinkage calculated in this way is shown in Figure 8 (open circles) in terms of si (left ordinate) and P (right ordinate). Below 840°C, si clearly resembles the measured silhouette area shrinkage rescaled by 1/2. Above 800°C, silhouette area shrinkage significantly deviates from si. The discontinuities in si at 840 and 930°C represent a rough measure of the calculation errors caused by the incorrect, sample-shape assumptions. Among the large variety of error sources, the loss of rotational symmetry of the sample during foaming seems to be most important. Nevertheless, estimating nMax from shrinkage data indicates similar volumes of the maximal foamed sample and the green powder compact, i.e., similarity of the respective porosity P ≈ 0.39. Thus, as a rough measure, nMax ≈ 12 × 10−8 mol could be estimated from Eq. 1.
Encapsulated Gas
The amount of gas encapsulated within the closing porosity during sintering, nEnc, was roughly estimated assuming that porosity suddenly closes at ρ = 0.8 (German, 1996). The number of gas moles encapsulated therein was approximated from Eq. 1, estimating V from the measured sample weight (94 mg), the relative compact density (ρ = 0.8) and the glass density (ρG = 3.61 g/cm3). This estimation results in nEnc ≈ 8 × 10−8 mol.
Comparing nEnc ≈ 8 × 10−8 mol with nMax ≈ 12–16 × 10−8 mol suggests that gases physically encapsulated within the porosity of sintered compacts may actually contribute to foaming. However, since mainly CO2 is released during bubble bursting and only traces of N2 and Ar were detected even for the powders milled in these atmospheres (Figure 5), the powder atmosphere must have changed after milling.
Moreover, this estimation was done for the sample milled for 15 min in N2. As seen from Figure 1, prolonged milling significantly increases foaming activity. Since the amount of encapsulated gas, i.e., the volume of 20% porosity, should not be affected by particle size and keeping in mind that nMax provides only a lower limit, this observation gives clear evidence that the encapsulated sintering atmosphere does not significantly contribute to foaming in this case. Instead, foaming seems to be clearly caused by species located at the glass powder surface.
Adsorbed Gases
In order to check the potential effect of adsorbed gases on foaming, the respective amount of adsorbed gas, nA, was roughly estimated assuming a mono-atomic layer of nitrogen molecules remaining stable until sintering. The specific powder surface area, as = 0.42 m2/g, was taken from PSD analysis as the average from all 15 min milling experiments. The diameter of the nitrogen molecule, , was set at 0.34 nm (Doremus, 1973). The total powder surface area, S = 3.7 × 10−2 m2 was then obtained from the measured sample mass, m ≈ 9.4 × 10−2 g. Assuming that one nitrogen molecule occupies an area of , nA ≈ 70 × 10−8 mol of nitrogen molecules can be adsorbed onto the total sample surface area. It is interesting to note that nA is about 10 times larger than nMax emphasizing the potentially strong effect of surface desorption on foaming. Taking into account that multiple physisorbed layers or multilayers grown from chemisorption and reorganization or chemical reaction processes can occur (Bhushan, 2013), this conclusion seems even more reasonable.
For physically adsorbed gases, as is expected for N2 and Ar, this conclusion however, is questionable because of low thermal stability (Bhushan, 2013). Thus, Figure 5 shows that N2 and Ar do not significantly contribute to foaming even for milling in these atmospheres and that the respective broad degassing peak is exhausted at around 500°C. Low desorption temperatures are also known for CO2. Thus, complete degassing of physically adsorbed CO2 was found <250°C for silica surfaces (Antonini and Hochstra, 1972) and <120°C for η-Al2O3 (Morterra et al., 1977).
Much higher desorption temperatures can be expected for chemisorbed gases. Water is known to strongly interact with silicate glass surfaces. Thus, a significant concentration of 1.2 OH groups per square nanometer was found even after high vacuum annealing at 700°C (5.2 OH/nm2 at 23°C) (Dunken, 1981). Conformingly, Figure 5 shows that water is the dominant degassing species below the onset temperature of foaming at ≈800°C.
Nevertheless, bubble bursting, i.e., foaming, is dominated by CO2. This gives clear evidence that CO2 adsorption results in carbonaceous species being thermally stable at least up to the temperature at which the open porosity of sintering powder compacts is closed (≈750°C). It is reasonable to assume that the carbonaceous species, most likely carbonates, may provide sufficient thermal stability. Thus, the decomposition temperature of BaCO3 in air is found at ≈900°C (Liptay, 1976), BaCO3 species at Pt/BaO/γ-Al2O3 surfaces remain stable up to 500°C in vacuum (Epling et al., 2008), and pronounced CO2 degassing from silica glass-fiber surfaces between 500 and 700°C has been attributed to ≡Si–O–C(O)–O–Si≡ (Eremenko et al., 1991). Further, Cerruti and Morterra (2004) reported carbonate species stable up to 800°C at the surface of bioactive glass powders. It is also known that intensive milling of alkaline earth metasilicates can yield remarkable CO2 uptake (Kalinkin and Kalinkina, 2010) and that even grinding of Na2Si2O5 glass in air can cause detectable amounts of NaCO3 at the surface (Baker et al., 1995). Conclusions on the nature of the dominant carbonaceous species, however, cannot be drawn from the present study. Due to the expected small concentration (≈60 ppm CO2 would result from nMax = 12 × 10−8 mol, mSample ≈ 92 mg, and ρGlass ≈ 3.61 g/cm3 as discussed above for Figure 8), XRD, DTA, and ATR-FTIR studies conducted so far have not yet revealed any reliable hint of the nature of the carbonaceous species responsible for foaming found in the present study.
Dissolved Gases
The detection of these carbonaceous species might be even more complicated as adsorbed carbonaceous species could partially diffuse into the glass forming a layer of near-surface dissolved carbonates during heating. Such an effect seems reasonable as Figure 6 indicates a large amount of carbonaceous surface species even during progressive heating. In this way, desorption of these species can be accompanied by simultaneous inward diffusion. Once desorption is complete, the dissolved carbonaceous species may provide a further source of CO2 degassing. Such a possibility might be indicated by Figure 5 in as much as the onset of the degassing activity at ≈650–800°C nicely correlates with glass transition temperature (Tg). This latter mechanism is most likely responsible for foaming as it is obviously retarded by sintering. Similar VHE degassing patterns could be attributed to the diffusive release of bulk water (Müller et al., 2005). CO2 dissolution in oxide glasses was comprehensively studied by Brooker et al. (2001) who detected like species in different local environments, albeit at higher pressure and temperature (0.2–2.7 GPa, 1175–1700°C) and not for boron-containing glasses. CO2 solubility of 0.11 and 0.28 wt% was reported for natural rhyolytic melts at 1100°C for 200 and 500 MPa, respectively (Tamic et al., 2001). Although much lower solubility should be expected for 1 bar, increased local pressure can be expected during sintering and healing of the mechanically damaged near-surface region of milled glass powders.
Foaming Mitigation Strategies
As shown above, encapsulated gas can contribute to the foaming of coarse glass powders. In this case, vacuum sintering seems to be the only foaming mitigation strategy. On the other hand, gases with better mobility could help to enhance diffusive degassing from closed porosity below the onset of foaming.
As clearly shown in the present study and known from common experience, gases trapped on the powder surface can essentially contribute to foaming. Hence, the use of coarser glass particles is the most obvious and probably the most frequently applied strategy in reducing sinter foaming.
Beyond this, however, the present study (sintering at ≈700–800°C) indicates that foaming is dominated by carbon gases, even when organic aids have not been used in powder processing, and that foaming can be affected by the milling atmosphere, milling aids, and powder storage. These observations can be reasonably explained assuming that gas uptake occurs at the freshly fractured and highly damaged glass powder surface as a relaxation mechanism and that carbonaceous species are preferentially adsorbed as a result of adsorption–desorption phenomena during milling or later storage.
In this sense, further foaming mitigation strategies could be based on minimizing the possible uptake of ambient CO2 during powder processing. Among these strategies, powder processing in a controlled atmosphere or vacuum seems to be the most obvious but probably most expensive method. Alternatively, less CO2 uptake can be achieved by means of blocking active (i.e., the most basic) glass surface sites, which are most likely related to BaO.
The significant decrease of foaming activity attained by water milling presented in Figure 7 indicates that this blocking can be made with water. Figure 5 shows that water does not contribute much to foaming, although it is the most prominent degassing species below the onset of sintering. This advantage probably reflects its better diffusivity. It should be noted, however, that this strategy relies on sufficiently high sintering temperatures. For lower sintering temperatures, water and even less stable species could also contribute to foaming. Additional BaCO3 formation during water milling as a result of aqueous dissolution of Ba from the glass surface and CO2 from the ambient atmosphere does not seem very likely since a pH value >12 is required for BaCO3 formation at 1 bar (Brookins, 1988) and pH <11 was measured for milling in water in the present study.
Alternatively, foaming was substantially reduced by milling in 10 wt% HCl (Figure 7). The decreased shrinkage rate and the simultaneously increased foaming onset temperature indicate increased glass viscosity, at least near to the powder surface. Such phenomenon could be caused by Ba dissolution from the glass surface. It is reasonable to assume that this effect would allow the formation of BaCl2, which is thermally stable up to 963°C (melting point) (Merck, 2015). In this sense, Ba dissolution from the glass surface and the formation of thermally stable Ba salts may provide another strategy for blocking active CO2 adsorption sites. In acidic aqueous solutions, CO2 solubility is also very limited (Acker et al., 1999).
Another foaming mitigation strategy could be utilizing increased sintering temperatures. This is due to the fact that foaming intensity is controlled by the temperature relationships between densification (TS), decomposition of foaming species (TD), and crystallization (TC). Most pronounced foaming is expected for TS < TD < TC. For TD ≪ TS surface-adsorbed foaming agents largely decompose and escape via open pores. For TD ≈ TS, this degassing process is retarded by closing porosity, which later yields gas bubble formation and foaming. Due to this general relationship, foaming probability seems to increase with decreasing sintering temperature as even less stable surface-adsorbed species (e.g., water or less stable carbonaceous species as indicated by Figure 5) may contribute to foaming. On the other hand, a higher sintering temperature can, for example, affect redox equilibria of the glass melt, increase oxygen mobility, and could cause oxygen degassing as a new foaming source. For most practical cases, however, a substantial change of glass transition temperature does not seem to be an applicable foaming mitigation strategy.
For a given glass, however, foaming of coarse powders is expected to be less intense not only due to the fact that a less specific surface will adsorb less foaming species as discussed above but also due to the more complete degassing, which can occur until porosity is closed. On the other hand, sintering is controlled by the effective viscosity of powder compacts. Hence, dispersed rigid particles can also decrease the shrinkage rate (Müller et al., 2007; Müller and Reinsch, 2012) and therefore provide more complete surface desorption. In a similar sense, glasses with a less temperature-dependent viscosity, causing a broader temperature range of sintering, should be less prone to foaming.
For TD > TC, foaming can be retarded by the presence of crystals, which hinder gas bubble coalescence and growth due to the increased effective viscosity of the crystal bearing melt. If full densification is not reached (TC ≈ TS), the remaining open porosity will allow full degassing without foaming even when TS < TD.
However, our final conclusions on how to mitigate sinter foaming, beyond the rather speculative discussion presented here, require much deeper understanding of the potential foaming mechanism and may largely depend on given experimental or processing conditions.
Conclusion
Sintering and foaming of barium silicate glass powder compacts were studied for different milling times and atmospheres. Foaming was strongly promoted by progressive milling and was affected by the milling atmosphere where foaming activity increased in the order Ar ≈ N2 < air < CO2. For moderately milled glass powders, subsequent storage in air could also promote foaming. Degassing studies revealed that foaming is mainly driven by CO2 even for powders milled in Ar and N2. Only traces of these species were detected during bubble bursting of compacts for which glass powder milling was performed within these atmospheres. This observation, together with respective estimation of the possible amount of gas encapsulated within the closing porosity during densification, suggests that an encapsulated milling atmosphere does not dominate foaming. Instead, a potentially large effect on foaming of stable surface-adsorbed carbonaceous species was indicated by enhanced CO2 degassing during foaming. In this sense, milling in water and weak HCl acid could substantially reduce foaming.
Author Contributions
RM: acquisition and design of the work, data interpretation, literature review, partial manuscript draft and revising, and final approval. SR: conception of experimental work, thermo-analytical measurements and data interpretation, literature review, partial manuscript draft, and final approval. BA-B: conception of experimental work, other experimental work, data analysis, literature review, partial manuscript draft, and final approval. All authors agree to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge experimental support by our colleagues M. Gaber (vacuum hot extraction), R. Sojref (glass powder preparation and characterization), I. Feldmann (SEM), and D. Nicolaides (XRD). We finally acknowledge financial support by Erasmus Lifelong Learning Programs for BA-B (MINISTERIO DE EDUCACIÓN, CULTURA Y DEPORTE # 11015347).
References
Acker, K., Barnes, I., Becker, K.-H., Crutzen, P. J., Hüttl, R., and Kalaß, D. (1999). Atmospheric Environmental Research: Critical Decisions between Technological Progress and Preservation of Nature, 1st Edn. Heidelberg, Germany: Springer, 186.
Agea Blanco, B., Blaess, C., Reinsch, S., Brauer, D., and Müller, R. (2015). Sintering and foaming of barium and calcium silicate glass powders. Addition. Conf. 2015, 000031–000037. doi: 10.4071/CICMT-TA21
Antonini, J. F., and Hochstra, G. (1972). Surface states of pristine silica surfaces: II. UHV studies of the CO2 adsorption-desorption phenomena. Surf. Sci. 32, 665–686. doi:10.1016/0039-6028(72)90193-8
Baker, G. J., Greaves, G. N., Surman, M., and Oversluizen, M. (1995). An oxygen XAFS study of sodium disilicate glass surfaces. Nucl. Instrum. Methods Phys. Res. Sect. B-Beam Interact. Mat. Atoms 97, 375–382. doi:10.1016/0168-583X(95)00332-0
Bhushan, B. (2013). Solid Surface Characterization. Introduction to Tribology, 2nd Edn. West Sussex: John Wiley & Sons.
Brooker, R. A., Kohn, S. C., Holloway, J. R., and McMillan, P. F. (2001). Structural controls on the solubility of CO2 in silicate melts: part II: IR characteristics of carbonate groups in silicate glasses. Chem. Geol. 174, 241–254. doi:10.1016/S0009-2541(00)00318-1
Brookins, G. D. (1988). Eh-pH Diagrams for Geochemistry, 1st Edn. Berlin, Heidelberg: Springer-Verlag.
Cerruti, M., and Morterra, C. (2004). Carbonate formation on bioactive glasses. Langmuir 20, 6382–6388. doi:10.1021/la049723c
Dunken, H. H. (1981). “Chemische Reaktionen an Glasoberflächen,” in Physikalische Chemie der Glasoberfläche (Leipzig: VEB Deutscher Verlag für Grundstoffindustrie), 262–338.
Epling, W. S., Peden, C. H. F., and Szanyi, J. (2008). Carbonate formation and stability on a Pt/BaO/γ-Al2O3 NOx storage/reduction catalyst. J. Phys. Chem. C 112, 10952–10959. doi:10.1021/jp712180q
Eremenko, G. O., Larichev, M. N., and Leypunsky, I. O. (1991). “Thermal desorption mass-spectrometric (TDMS) analysis of the carbon dioxide desorption from the glass fiber surface,” in MICC 90, ed. I.N.e.a. Fridlyander (Moscow: Elsevier Science Publishers Ltd.).
Fergus, J. W. (2005). Sealants for solid oxide fuel cells. J. Power Sources 147, 46–57. doi:10.1016/j.jpowsour.2005.05.002
Fernandes, H. R., and Ferreira, J. M. F. (2007). Recycling of chromium-rich leather ashes in porcelain tiles production. J. Eur. Ceram. Soc. 27, 4657–4663. doi:10.1016/j.jeurceramsoc.2007.03.037
German, R. M. (1996). Sintering Theory and Practice. New York, Chichester, Brisbane, Toronto, Singapore: John Wiley & Sons, Inc.
Gross, S. M., Koppitz, T., Remmel, J., and Reisgen, U. (2005). “Glass-ceramic materials of the system BaO-CaO-SiO2 as sealants for SOFC applications,” in International Conference on Advanced Ceramics and Composites Ceramic Engineering and Science Proceedings, eds N. Bansal, D. Zhu, and W. M. Kriven (Cocoa Beach, FL), 239–245.
Hwang, G. H., Jeon, H. J., and Kim, Y. S. (2002). Physical properties of barrier ribs of plasma display panels: I, formation of pores during sintering of lead borosilicate glass frits. J. Am. Ceram. Soc. 85, 2956–2960. doi:10.1111/j.1151-2916.2002.tb00562.x
Imanaka, Y. (2005). Multilayered Low Temperature Cofired Ceramics (LTCC) Technology. New York: Springer.
Kalinkin, A. M., and Kalinkina, E. V. (2010). The mechanosorption of carbon dioxide by magnesium, calcium, and strontium metasilicates. Russ. J. Phys. Chem. A 84, 1585–1591. doi:10.1134/S0036024410090256
Kerafol. (2010). Glass solder powder type K01 (Product-ID 6.FC0700.DL.00000) Supplied by Kerafol Keramische Folien GmbH Eschenbach Germany.
Kim, B.-S., Lim, E.-S., Lee, J.-H., and Kim, J.-J. (2007). Effect of Bi2O3 content on sintering and crystallization behavior of low-temperature firing Bi2O3-B2O3-SiO2 glasses. J. Eur. Ceram. Soc. 27, 819–824. doi:10.1016/j.jeurceramsoc.2006.04.013
Lara, C., Pascual, M. J., Prado, M. O., and Durán, A. (2004). Sintering of glasses in the system RO-Al2O3-BaO-SiO2 (R=Ca, Mg, Zn) studied by hot-stage microscopy. Solid State Ionics 170, 201–208. doi:10.1016/j.ssi.2004.03.009
Leonelli, C., Bondioli, F., Veronesi, P., Romagnoli, M., Manfredini, T., Pellacani, G. C., et al. (2001). Enhancing the mechanical properties of porcelain stoneware tiles: a microstructural approach. J. Eur. Ceram. Soc. 21, 785–793. doi:10.1016/S0955-2219(00)00266-1
Lim, E. S., Kim, B.-S., Lee, J.-H., and Kim, J.-J. (2006). Effect of BaO content on the sintering and physical properties of BaO-B2O3-SiO2 glasses. J. Non-Cryst. Solids 352, 821–826. doi:10.1016/j.jnoncrysol.2006.01.021
Lucchini, E., Meriani, S., and Slokar, G. (1983). Sintering of glass bonded ceramic barium hexaferrite magnetic powders. J. Sci. Mater. 18, 1331–1334. doi:10.1007/BF01111950
Merck. (2015). Sicherheitsdatenblatt Version 10.13 für Bariumchlorid 99.995 Suprapur®, CAS-Nr. 10361-37-2. Available at: www.merckgroup.com
Morterra, C., Zecchina, A., Coluccia, S., and Chiorino, A. (1977). I.R. spectroscopic study of CO2 adsorption onto η-Al2O3. J. Chem. Soc. Faraday Trans. I 73, 1544–1560. doi:10.1039/f19777301544
Müller, R. (1994). On the kinetics of sintering and crystallization of glass powders. Glastech. Ber-Glass Sci. Technol. 67C, 93–98.
Müller, R., Eberstein, M., Reinsch, S., Schiller, W. A., Deubener, J., and Thiel, A. (2007). Effect of rigid inclusions on sintering of low temperature co-fired ceramics. Phys. Chem. Glass. Eur. J. Glass Sci. Technol. B 48, 259–266.
Müller, R., Meszaros, R., Peplinski, B., Reinsch, S., Eberstein, M., Schiller, W. A., et al. (2009). Dissolution of alumina, sintering, and crystallization in glass ceramic composites for LTCC. J. Am. Ceram. Soc. 92, 1703–1708. doi:10.1111/j.1551-2916.2009.03089.x
Müller, R., Gottschling, P., and Gaber, M. (2005). Water concentration and diffusivity in silicates obtained by vacuum extraction. Glass Sci. Technol. 78, 76–89.
Müller, R., and Reinsch, S. (2012). “Viscous phase silicate processing,” in Processing Approaches for Ceramics and Composites, eds N. Bansal and A. R. Boccaccini (Hoboken, New Jersey, USA: John Wiley & Sons), 75–144.
Pascual, M. J., and Duran, A. (2003). Sintering with concurrent crystallisation of a borosilicate glass. Phys. Chem. Glasses 44, 409–415.
Prado, M. O., Fredericci, C., and Zanotto, E. D. (2003a). Non-isothermal sintering with concurrent crystallization of polydispersed soda-lime-silica glass beads. J. Non-Cryst. Solids 331, 157–167. doi:10.1016/j.jnoncrysol.2003.08.076
Prado, M. O., Zanotto, E. D., and Fredericci, C. (2003b). Sintering polydispersed spherical glass particles. J. Mater. Res. 18, 1347–1354. doi:10.1557/JMR.2003.0185
Prado, M. O., Nascimento, M. L. F., and Zanotto, E. D. (2008). On the sinterability of crystallizing glass powders. J. Non-Cryst. Solids 354, 4589–4597. doi:10.1016/j.jnoncrysol.2008.06.006
Prado, M. O., and Zanotto, E. D. (2002). Glass sintering with concurrent crystallization. Comptes Rendus Chimie 5, 773–786. doi:10.1016/S1631-0748(02)01447-9
Rabinovich, E. M. (1985). Preparation of glass by sintering. J. Sci. Mater. 20, 4259–4297. doi:10.1007/BF00559317
Schiller, W. A., Müller, R., Eberstein, M., Reinsch, S., and Rabe, T. (2008). Sintering of LTCC. cfi/Berichte der DKG 85, 12–17.
Tamic, N., Behrens, H., and Holtz, F. (2001). The solubility of H2O and CO2 in rhyolitic melts in equilibrium with a mixed CO2-H2O fluid phase. Chem. Geol. 174, 333–347. doi:10.1016/S0009-2541(00)00324-7
Keywords: glass powder sintering, milling, foaming, degassing, SOFC
Citation: Agea-Blanco B, Reinsch S and Müller R (2016) Sintering and Foaming of Barium Silicate Glass Powder Compacts. Front. Mater. 3:45. doi: 10.3389/fmats.2016.00045
Received: 15 June 2016; Accepted: 22 September 2016;
Published: 07 October 2016
Edited by:
Wolfram Höland, Ivoclar Vivadent, LiechtensteinReviewed by:
Sylwester Janusz Rzoska, Institute of High Pressure Physics (PAN), PolandJohn Ballato, Clemson University, USA
Copyright: © 2016 Agea-Blanco, Reinsch and Müller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ralf Müller, cmFsZi5tdWVsbGVyQGJhbS5kZQ==