 Ilknur Erucar
Ilknur Erucar Seda Keskin
Seda Keskin- 1Department of Natural and Mathematical Sciences, Faculty of Engineering, Ozyegin University, Istanbul, Turkey
- 2Department of Chemical and Biological Engineering, Koc University, Istanbul, Turkey
Metal organic frameworks (MOFs) have emerged as great alternatives to traditional nanoporous materials for CO2 separation applications. MOFs are porous materials that are formed by self-assembly of transition metals and organic ligands. The most important advantage of MOFs over well-known porous materials is the possibility to generate multiple materials with varying structural properties and chemical functionalities by changing the combination of metal centers and organic linkers during the synthesis. This leads to a large diversity of materials with various pore sizes and shapes that can be efficiently used for CO2 separations. Since the number of synthesized MOFs has already reached to several thousand, experimental investigation of each MOF at the lab-scale is not practical. High-throughput computational screening of MOFs is a great opportunity to identify the best materials for CO2 separation and to gain molecular-level insights into the structure–performance relationships. This type of knowledge can be used to design new materials with the desired structural features that can lead to extraordinarily high CO2 selectivities. In this mini-review, we focused on developments in high-throughput molecular simulations of MOFs for CO2 separations. After reviewing the current studies on this topic, we discussed the opportunities and challenges in the field and addressed the potential future developments.
Introduction
We have witnessed the very quick growth of metal organic frameworks (MOFs) in the last two decades. MOFs are crystalline materials composed of metal nodes connected with organic linkers to create highly porous structures. They have exceptional physical properties such as very large surface areas [the highest reported one with 6,411 m2/g (Grunker et al., 2014)], high pore volumes (1–4 cm3/g), a large variety of pore sizes, and good stabilities. MOFs have been used in a wide range of chemical applications (Mueller et al., 2006) including gas storage and gas separation (Sun et al., 2013; Zornoza et al., 2013; Adatoz et al., 2015; Basdogan and Keskin, 2015), catalysis (Gascon et al., 2014), sensing (Zhu et al., 2013; Muller-Buschbaum et al., 2015), drug storage, and delivery (Della Rocca et al., 2011; Keskin and Kizilel, 2011; Bag et al., 2016). Gas separation has been the most widely examined application since MOFs offer a large variety in their pore sizes, shapes and chemistries. The number of synthesized MOFs has been rapidly increasing and already reached to several thousand as shown in Figure 1A. Existence of large numbers of MOFs generates both an opportunity and a challenge: There are thousands of candidate materials to achieve the target gas separations. On the other hand, it is challenging to identify the best performing MOFs because experimental synthesis, characterization, and testing of a new material for a gas separation application generally take several months.
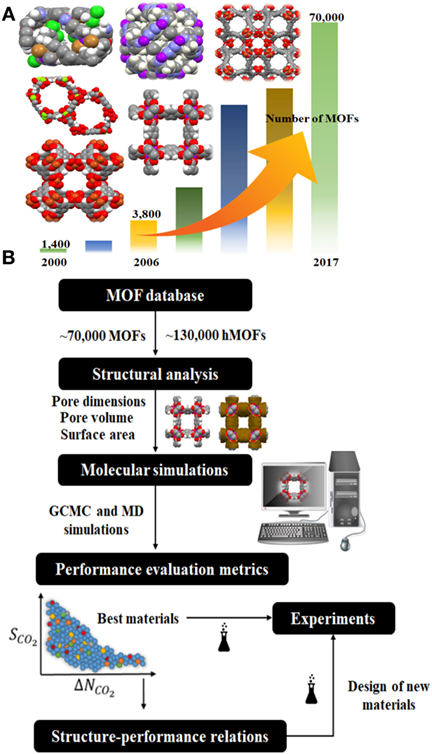
Figure 1. Schematic diagram of (A) the rapid increase in the number of synthesized metal organic frameworks, (B) high-throughput computational screening methodology.
Computational methods play a critical role in screening large number of MOFs in a time-effective manner to identify the most promising materials (Colon and Snurr, 2014). Predicting gas separation potential of a MOF using computer simulations is significantly faster and cheaper than doing the corresponding experiments. Once the best candidates are identified by simulations, experimental efforts can be directed to these materials. Molecular simulations have been successful in providing information about gas adsorption, diffusion, and separation in MOFs (Jiang et al., 2011). Grand canonical Monte Carlo (GCMC) simulations accurately predict adsorption of various gases in MOFs and molecular dynamics (MD) simulations are used to compute gas diffusion in MOFs (Keskin et al., 2009). Readers are directed to several excellent book chapters and review articles for discussion of computer simulations of MOFs (Jiang et al., 2011; Jiang, 2012a,b, 2014). Once the correct computational models are chosen to represent MOF-gas interactions, molecular-level insights that cannot be obtained via experiments can be gained from simulations. Molecular simulation studies recently focused on CO2 separation given the importance of research for clean energy technologies, and examined several types of MOFs to report their CO2 selectivities (Nalaparaju et al., 2015; Zhang et al., 2015).
Due to the rapid increase in the number of synthesized MOFs, improved molecular simulations techniques and increased computational powers, recent studies have focused on “high-throughput” simulations of MOFs where thousands of different structures are screened to identify the top materials for CO2 separations. In this mini-review, we first summarized the recent literature on high-throughput molecular simulations of MOFs for CO2 separations. We then focused on structure–performance relations that can be produced from computer simulations of MOFs which can lead to design and development of new MOFs having extraordinarily good CO2 separation performances. We finally discussed the opportunities and challenges of using high-throughput molecular simulations for MOFs, addressed the open gaps and suggested possible future directions in this research field.
Large-Scale Screening of MOFs
High-throughput computational screening studies identify the best MOF candidates for a target application in a reasonable time in addition to describing the characteristic features of the best materials using quantitative structure–property relationships (QSPR). The schematic diagram for the large-scale computational screening of MOFs is given in Figure 1B. First a database is constructed to screen MOFs and their structural properties such as pore size, surface area are calculated using computational techniques (Willems et al., 2012). GCMC and MD simulations are performed to obtain adsorption and diffusion of CO2 in MOFs, respectively. Data obtained from molecular simulations are used to calculate several performance evaluation metrics of MOFs. For example, GCMC output is used to compute adsorption selectivity, working capacity, regenerability, which are critical metrics in describing the potential of a MOF adsorbent for CO2 separations (Bae and Snurr, 2011). MOFs are finally ranked based on these metrics and the most promising materials are identified for further experimental testing. The QSPR analysis is performed either for all MOFs or only for the promising ones to get insights into design of new materials with predetermined structural properties that can give better CO2 separation performance.
Table 1 summarizes the large-scale computational screening studies of MOFs, in which at least hundreds of materials are examined, for CO2 separations. Removing CO2 from natural gas (CO2/CH4), from power plant flue gas (CO2/N2), from petroleum refineries (CO2/H2), and from water (CO2/H2O) were investigated in these studies. There are two MOF databases, synthesized MOFs (real) taken from the Cambridge Structural Database (Allen, 2002) and hypothetical MOFs (hMOFs) which are computationally generated from the libraries of metals and organic linkers (Wilmer et al., 2012a,b,c). For example, Watanabe and Sholl (2012) initially screened >30,000 synthesized MOFs from the CSD and finally studied 359 MOFs that have appropriate pore sizes for CO2/N2 separations. Snurr’s group (Chung et al., 2014) started with 20,000 MOFs, excluded highly disordered materials and ended up with having 4,764 MOFs. In this way, they constructed a very useful database CoRE MOF (computation-ready experimental MOFs), to be used in molecular simulations. MOFs with zero accessible surface areas were discarded from the CoRE MOF and remaining 2,054 MOFs were used in simulations for CO2/H2O separation (Li et al., 2016). Most of the studies have focused on adsorption-based CO2 separations using GCMC. In order to understand membrane-based gas separation potential of MOFs, MD simulations should be performed to compute CO2 permeabilities through the MOF membranes. In contrast to GCMC simulations, MD simulations are computationally expensive, therefore they are rare. Jiang’s group (Qiao et al., 2016a) used the CoRE MOF database to study both adsorption-based and membrane-based CO2/N2 and CO2/CH4 separations using molecular simulations. The same group also screened the hMOF database using combination of GCMC and MD simulations for membrane-based CO2 separation (Qiao et al., 2016b).
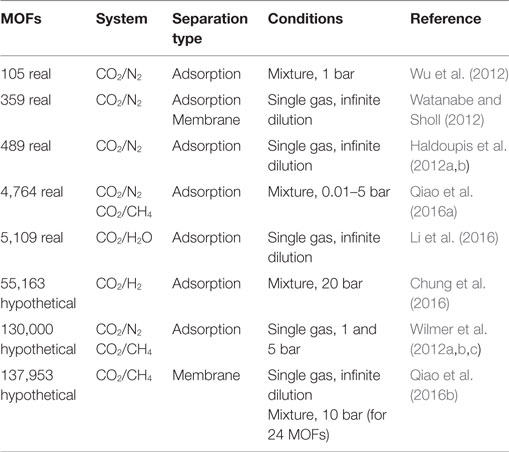
Table 1. Large-scale screening of MOFs for CO2 separations.
Adsorption and diffusion data of gas mixtures are required to assess potential of the MOFs because gases naturally exist as mixtures in separation processes. Molecular simulations of gas mixtures are time-consuming especially if the number of materials to be studied is high. Therefore, several simulations listed in Table 1 used “single gas” adsorption data to evaluate the mixture separation potential of MOFs. However, assessing mixture separation performance of MOFs using pure gas adsorption data can be misleading because materials suggested to be promising based on pure gas data are likely to underperform in real applications under the presence of gas mixtures since interactions between multiple gases strongly change the adsorbent’s behavior (Basdogan et al., 2015). Another assumption of several simulation studies is “infinite dilution” condition in which molecular simulations were performed at very low pressures to significantly reduce the computational time. However, the operating condition pressure of CO2 separation applications in industry are generally well above 1 bar.
The first QSPR model was established for adsorption-based CO2/N2 separation using 105 MOFs and results showed that increasing the difference of isosteric heat of adsorption of gases with decreasing porosity is a useful approach to improve CO2/N2 selectivities of MOFs (Wu et al., 2012). Structure–property relationships which include pore size, surface area, pore volume were also studied for hMOFs considering separation of CO2 from CH4 and N2 (Wilmer et al., 2012a,b,c). It was concluded that although none of the evaluation metrics such as selectivity, working capacity, are perfect predictors of CO2 separation performance, the analysis of relations between these metrics and structural properties provides several hints for future design of porous materials. Molecular simulations were recently performed to screen 55,163 hMOFs for CO2/H2 separation and a genetic algorithm including optimization methods was used to identify the optimum physical properties of hMOFs which showed the highest separation performance (Chung et al., 2016). The limiting pore diameter and pore size distribution were reported as the key factors that affect CO2 permeation of hMOF membranes (Qiao et al., 2016b).
Conclusion and Future Prospective
MOF Databases
The hMOF library has been specifically useful in providing structure–performance relations due the large structural functionality of the materials but practical synthesizability of hMOFs is still not fully confirmed. Using the real MOF and hMOF databases to get a better understanding of synthesis of new MOFs with desired properties is crucial. A pore recognition approach was recently developed to quantify similarity of pore structures and materials were classified using topological data analysis (Lee et al., 2017). With this approach, MOFs with similar pore geometries were identified and materials that are similar to top performing structures were screened for CH4 storage. Future studies focusing on CO2 separation will be useful since the shape of the pores plays an essential role in the CO2 separation. Computational crystal structure prediction is a new research area for MOFs. A highly porous solid was recently identified using energy–structure–function maps that describe the possible structures that are available to a candidate molecule and both CH4 storage capacity and C3H8/CH4 selectivity were predicted using the molecular structure as the only input (Pulido et al., 2017). Similar studies on accurate predictions of crystal structures for efficient CO2 separations will be very interesting. The energy–structure–function maps could be even used to guide the experimental discovery of MOFs with high CO2 selectivity.
Force Fields
The main and perhaps the most important input of molecular simulations of MOFs is the force field (FF) that describes physical and chemical interactions between gases and MOFs. At the early stages of molecular simulation of MOFs, FFs specific to gas-MOF interactions were developed using quantum-level calculations (Sagara et al., 2004; Bordiga et al., 2005). These computationally demanding calculations are not easily applicable to large number of materials. Therefore, generic FFs, Universal Force Field (Rappe et al., 1992), and Dreiding (Mayo et al., 1990) have been used for simulation of gas adsorption and diffusion in MOFs (Keskin et al., 2009; McDaniel et al., 2015). Molecular simulations employing either UFF or Dreiding showed good agreement with the experimentally measured gas uptake data of MOFs, validating the usage of generic FFs for MOFs (Colon and Snurr, 2014). The CO2 adsorption isotherms of hMOFs were recently computed using both the UFF and an ab initio FF (McDaniel et al., 2015). Significant quantitative differences between the CO2 uptakes predicted by the generic FF and the ab initio FF were reported. Studies examining the impact of using different types of FFs on the predicted CO2 separation performances of MOFs are therefore strongly needed.
Charge Assignment Methods
In order to capture the electrostatic interactions between CO2 and MOFs, partial charges should be assigned to each atom in the framework. There is a variety of methods for extracting atomic charges from the results of quantum mechanical calculations but performing these quantum-level calculations for thousands of MOFs is not feasible. Several approximate methods which have a compromise between time efficiency and the rigor have been developed. The charge equilibration method (Ramachandran et al., 1996) extended charge equilibration method (Wilmer et al., 2012a,b,c) and periodic charge equilibration method (PQeq) (Haldoupis et al., 2012a,b) were used for MOFs. Comparison between PQeq and high quality point charges derived from quantum chemistry for the top performing materials showed a considerable disagreement in the calculated CO2/N2 selectivities although the PQeq charges were shown to give a quick estimate about the potential of the material (Haldoupis et al., 2012a,b). A recent study examined the impact of charge assignment methods on the high-throughput computational screening of MOFs for CO2/H2O separations and found that majority of the top MOFs ranked based on CO2 selectivities are identical regardless of the charge assignment method (Wei et al., 2017). These initial results suggested that studies examining the impact of the charge assignment method on the ranking of MOFs for other CO2 separations are needed.
Flexible MOFs
Molecular simulations should be performed for multiple materials on time scales shorter than the same materials can be assessed experimentally. When thousands of MOFs are screened in high-throughput molecular simulations, rigid framework assumption is used because it saves a significant computational time. Studies showed that including lattice flexibility does not make any important change in the gas adsorption results of MOFs that have pore sizes larger than the gas molecules (Greathouse and Allendorf, 2008; Perez-Pellitero et al., 2010; Haldoupis et al., 2012a,b). On the other hand, MOF flexibility can be important for diffusion of large gas molecules in the MOFs having narrow windows (Chokbunpiam et al., 2013; Verploegh et al., 2015). It was recently shown that flexibility has an important role in MD simulations of MOF membranes (Erucar and Keskin, 2016). Considering flexibility of the framework made a negligible effect on the gas permeability and selectivity of the MOFs having large pores but more pronounced changes were seen in gas permeabilities of the materials having narrow pores. Therefore, once the potential value of a MOF has been demonstrated for CO2 separations using high-throughput molecular simulations, further studies such as flexible simulations should be performed to increase the precision of initial assessment at least for the promising materials and this is currently an open research area.
Membrane Simulations
Several experiments showed that MOFs can be highly CO2 selective membranes (Adatoz et al., 2015). Considering the experimental challenges in fabricating thin-layer membranes from new materials and long time requirements of membrane testing, identification of promising MOF membranes using computer simulations will greatly contribute in directing experimental efforts. Recent molecular simulation studies indicated that several MOFs outperform well-known polymer membranes in terms of selectivity for CH4/H2 separations (Erucar and Keskin, 2016). It is very possible that there are many more MOFs that can outperform current membrane-based CO2 separation technologies. Understanding diffusion mechanisms of gas mixtures in MOFs using the MD simulations will be very useful not only to study MOF membranes but also to provide the knowledge of molecular transport of gases in MOFs which is strongly required for the development of MOF devices in other chemical applications such as catalysis and sensing.
Simulation Conditions
As discussed in Table 1, most of the high-throughput simulations were performed at dilute conditions considering single gases. Future studies should focus on molecular simulations of CO2 mixtures under practical operating conditions although this requires significant computation power and time. Presence of water vapor in the flue gas stream must be considered in studies focusing of CO2/N2 separation because water can adversely affect the adsorption of CO2 and N2 by competing for adsorption sites and even by affecting the MOF’s stability. Considering the role of impurities such as water vapor and contaminants such as SOx in CO2 separations is a relatively new research area for MOFs (Li et al., 2015) and more studies will be very useful. Finally, it is important to mention that MOFs are generally simulated as perfect, defect-free structures. However, point defects in MOF structures play an important role in the gas storage and separation performances of MOFs. Sholl and Lively (2015) recently discussed the challenges and opportunities of defects present in MOFs. It was mentioned that increased CO2 uptakes and mesopore formation have been noted from linker vacancies. These may have implications for adsorption and membrane-based gas separation applications since the mesopores can provide low-resistance diffusion pathways through MOF crystals (Choi et al., 2011). Addressing the challenges and opportunities in the defect engineering of MOF using simulation is a challenging task and more studies are required in this area.
Guiding Experiments
Current molecular simulations generally focused on the relations between CO2 selectivity and structural properties. Although this information is useful, it is not straightforward for experimentalists to design new materials with predescribed pore sizes or surface areas. It is much easier to design MOFs based on predefined building blocks, metals, and organic linkers. A recent work showed that MOFs containing lanthanides provide the best performance for CO2/CH4 separation whereas MOFs with alkali metals have the worst separation performance (Qiao et al., 2016a,b). Therefore, studies focusing on the relation between CO2 selectivity and the type of metal and/or organic linker of the MOF will be very useful for guiding the experiments. Another recent work (Zhang et al., 2017) suggested that molecular design of new MOFs with better CO2 capture properties by synergizing multiscale modeling from molecular simulation to breakthrough prediction is possible. This initial study will motivate the future research on multiscale modeling of existing MOFs to accelerate the development of more useful materials for CO2 separation applications.
We aimed to discuss the open gaps in molecular simulations of MOFs for CO2 separation. The key contribution of high-throughput molecular simulations is to accelerate the innovation in materials research and development of new MOFs that can lead to efficient CO2 separation technologies. We believe that new computational algorithms and strong collaborations between chemists, materials scientists, computer scientists, and engineers will fasten this research area.
Author Contributions
SK and IE wrote the submitted mini-review. They both searched the current literature, addressed the research gaps, and discussed the potential future developments in the field to provide a brief and comprehensive review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, QY, and handling editor declared their shared affiliation.
Funding
SK acknowledges ERC-2017-Starting Grant. This study has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (ERC-2017-Starting Grant, grant agreement No 756489-COSMOS).
References
Adatoz, E., Avci, A. K., and Keskin, S. (2015). Opportunities and challenges of MOF-based membranes in gas separations. Sep. Purif. Technol. 152, 207–237. doi: 10.1016/j.seppur.2015.08.020
Allen, F. H. (2002). The cambridge structural database: a quarter of a million crystal structures and rising. Acta Crystallogr. B 58, 380–388. doi:10.1107/S0108768102003890
Bae, Y. S., and Snurr, R. Q. (2011). Development and evaluation of porous materials for carbon dioxide separation and capture. Angew. Chem. Int. Edit. 50, 11586–11596. doi:10.1002/anie.201101891
Bag, P. P., Wang, D., Chen, Z., and Cao, R. (2016). Outstanding drug loading capacity by water stable microporous MOF: a potential drug carrier. Chem. Commun. 52, 3669–3672. doi:10.1039/c5cc09925k
Basdogan, Y., and Keskin, S. (2015). Simulation and modelling of MOFs for hydrogen storage. Crystengcomm 17, 261–275. doi:10.1039/C4CE01711K
Basdogan, Y., Sezginel, K. B., and Keskin, S. (2015). Identifying highly selective metal organic frameworks for CH4/H2 separations using computational tools. Ind. Eng. Chem. Res. 54, 8479–8491. doi:10.1021/acs.iecr.5b01901
Bordiga, S., Vitillo, J. G., Ricchiardi, G., Regli, L., Cocina, D., Zecchina, A., et al. (2005). Interaction of hydrogen with MOF-5. J. Phys. Chem. B 109, 18237–18242. doi:10.1021/jp052611p
Choi, K. M., Jeon, H. J., Kang, J. K., and Yaghi, O. M. (2011). Heterogeneity within order in crystals of a porous metal–organic framework. J. Am. Chem. Soc. 2011, 11920–11923. doi:10.1021/ja204818q
Chokbunpiam, T., Chanajaree, R., Saengsawang, O., Reimann, S., Chmelik, C., Fritzsche, S., et al. (2013). The importance of lattice flexibility for the migration of ethane in ZIF-8: molecular dynamics simulations. Microporous Mesoporous Mater. 174, 126–134. doi:10.1016/j.micromeso.2012.12.047
Chung, Y. G., Camp, J., Haranczyk, M., Sikora, B. J., Bury, W., Krungleviciute, V., et al. (2014). Computation-ready, experimental metal-organic frameworks: a tool to enable high-throughput screening of nanoporous crystals. Chem. Mater. 26, 6185–6192. doi:10.1021/cm502594j
Chung, Y. G., Gomez-Gualdron, D. A., Li, P., Leperi, K. T., Deria, P., Zhang, H. D., et al. (2016). In silico discovery of metal-organic frameworks for precombustion co2 capture using a genetic algorithm. Sci. Adv. 2, 1–9. doi:10.1126/sciadv.1600909
Colon, Y. J., and Snurr, R. Q. (2014). High-throughput computational screening of metal-organic frameworks. Chem. Soc. Rev. 43, 5735–5749. doi:10.1039/c4cs00070f
Della Rocca, J., Liu, D. M., and Lin, W. B. (2011). Nanoscale metal-organic frameworks for biomedical imaging and drug delivery. Acc. Chem. Res. 44, 957–968. doi:10.1021/ar200028a
Erucar, I., and Keskin, S. (2016). Computational assessment of MOF membranes For CH4/H2 separations. J. Membr. Sci. 514, 313–321. doi:10.1016/j.memsci.2016.04.070
Gascon, J., Corma, A., Kapteijn, F., and Xamena, F. X. L. I. (2014). Metal organic framework catalysis: Quo vadis? ACS Catal. 4, 361–378. doi:10.1021/cs400959k
Greathouse, J. A., and Allendorf, M. D. (2008). Force field validation for molecular dynamics simulations of IRMOF-1 and other isoreticular zinc carboxylate coordination polymers. J. Phys. Chem. C 112, 5795–5802. doi:10.1021/jp076853w
Grunker, R., Bon, V., Muller, P., Stoeck, U., Krause, S., Mueller, U., et al. (2014). A new metal-organic framework with ultra-high surface area. Chem. Commun. 50, 3450–3452. doi:10.1039/c4cc00113c
Haldoupis, E., Nair, S., and Sholl, D. S. (2012a). Finding MOFs for highly selective CO2/N2 adsorption using materials screening based on efficient assignment of atomic point charges. J. Am. Chem. Soc. 134, 4313–4323. doi:10.1021/ja2108239
Haldoupis, E., Watanabe, T., Nair, S., and Sholl, D. S. (2012b). Quantifying large effects of framework flexibility on diffusion in MOFs: CH4 and CO2 in ZIF-8. Chemphyschem 13, 1–4. doi:10.1002/cphc.201200529
Jiang, J. W. (2012a). “Metal-organic frameworks for CO2 capture: what are learned from molecular simulations,” in Coordination Polymers and Metal Organic Frameworks, eds O. Ortiz, and L. Ramirez (Nova Science Publishers), 225–247.
Jiang, J. W. (2012b). Recent development of in silico molecular modeling for gas and liquid separations in metal-organic frameworks. Curr. Opin. Chem. Eng. 1, 138–144. doi:10.1016/j.coche.2011.11.002
Jiang, J. W. (2014). Molecular simulations in metal-organic frameworks for diverse potential applications. Mol. Simulat. 40, 516–536. doi:10.1080/08927022.2013.832247
Jiang, J. W., Babarao, R., and Hu, Z. Q. (2011). Molecular simulations for energy, environmental and pharmaceutical applications of nanoporous materials: from zeolites, metal-organic frameworks to protein crystals. Chem. Soc. Rev. 40, 3599–3612. doi:10.1039/c0cs00128g
Keskin, S., and Kizilel, S. (2011). Biomedical applications of metal organic frameworks. Ind. Eng. Chem. Res 50, 1799–1812. doi:10.1021/ie101312k
Keskin, S., Liu, J., Rankin, R. B., Johnson, J. K., and Sholl, D. S. (2009). Progress, opportunities, and challenges for applying atomically detailed modeling to molecular adsorption and transport in metal-organic framework materials. Ind. Eng. Chem. Res. 48, 2355–2371. doi:10.1021/ie800666s
Lee, Y., Barthel, S. D., Dlotko, P., Moosavi, S. M., Hess, K., and Smit, B. (2017). Quantifying similarity of pore-geometry in nanoporous materials. Nat. Commun. 8, 1–8. doi:10.1038/ncomms15396
Li, P. W., Chen, J. H., Zhang, J. Y., and Wang, X. L. (2015). Water stability and competition effects toward CO2 adsorption on metal organic frameworks. Sep. Purif. Rev. 44, 19–27. doi:10.1080/15422119.2014.884507
Li, S., Chung, Y. G., and Snurr, R. Q. (2016). High-throughput screening of metal-organic frameworks for CO2 capture in the presence of water. Langmuir 32, 10368–10376. doi:10.1021/acs.langmuir.6b02803
Mayo, S. L., Olafson, B. D., and Goddard, W. A. (1990). DREIDING: a generic force field for molecular simulations. J. Phys. Chem. 94, 8897–8909. doi:10.1021/j100389a010
McDaniel, J. G., Li, S., Tylianakis, E., Snurr, R. Q., and Schmidt, J. R. (2015). Evaluation of force field performance for high-throughput screening of gas uptake in metal-organic frameworks. J. Phys. Chem. C 119, 3143–3152. doi:10.1021/jp511674w
Mueller, U., Schubert, M., Teich, F., Puetter, H., Schierle-Arndt, K., and Pastré, J. (2006). Metal organic frameworks-prospective industrial applications. J. Mater. Chem. 16, 626–636. doi:10.1039/B511962F
Muller-Buschbaum, K., Beuerle, F., and Feldmann, C. (2015). MOF based luminescence tuning and chemical/physical sensing. Microporous Mesoporous Mater. 216, 171–199. doi:10.1016/j.micromeso.2015.03.036
Nalaparaju, A., Khurana, M., Farooq, S., Karimi, I.A., and Jiang, J.W. (2015). CO2 capture in cation-exchanged metal-organic frameworks: holistic modeling from molecular simulation to process optimization. Chem. Eng. Sci. 124, 70–78. doi:10.1016/j.ces.2014.09.054
Perez-Pellitero, J., Amrouche, H., Siperstein, F. R., Pirngruber, G., Nieto-Draghi, C., Chaplais, G., et al. (2010). Adsorption of CO2, CH4, and N2 on zeolitic imidazolate frameworks. Chem. Eur. J. 16, 1560–1571. doi:10.1002/chem.200902144
Pulido, A., Chen, L. J., Kaczorowski, T., Holden, D., Little, M. A., Chong, S. Y., et al. (2017). Functional materials discovery using energy-structure-function maps. Nature 543, 657–664. doi:10.1038/nature21419
Qiao, Z., Zhang, K., and Jiang, J. (2016a). In silico screening of 4764 computation-ready, experimental metal-organic frameworks for CO2 separation. J. Mater. Chem. A 4, 2105–2114. doi:10.1039/C5TA08984K
Qiao, Z. W., Peng, C. W., Zhou, J., and Jiang, J. W. (2016b). High-throughput computational screening of 137953 metal-organic frameworks for membrane separation of A CO2/N2/CH4 Mixture. J. Mater. Chem. A 4, 15904–15912. doi:10.1039/C6TA06262H
Ramachandran, S., Lenz, T. G., Skiff, W. M., and Rapper, A. K. (1996). Toward an understanding of Zeolite Y as a cracking catalyst with the use of periodic charge equilibration. J. Phys.Chem. 100, 5898–5907. doi:10.1021/jp952864q
Rappe, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A., and Skiff, W. M. (1992). UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035. doi:10.1021/ja00051a040
Sagara, T., Klassen, J., and Ganz, E. (2004). Computational study of hydrogen binding by metal-organic framework-5. J. Chem. Phys. 121, 12543–12547. doi:10.1063/1.1809608
Sholl, D. S., and Lively, R. P. (2015). Defects in metal-organic frameworks: challenge or opportunity? J. Phys. Chem. Lett. 6, 3437–3444. doi:10.1021/acs.jpclett.5b01135
Sun, Y. B., Wang, L., Amer, W. A., Yu, H. J., Ji, J., Huang, L., et al. (2013). Hydrogen storage in metal-organic frameworks. J. Inorg. Organomet. P 23, 270–285. doi:10.1007/s10904-012-9779-4
Verploegh, R. J., Nair, S., and Sholl, D. S. (2015). Temperature and loading-dependent diffusion of light hydrocarbons in ZIF-8 as predicted through fully flexible molecular simulations. J. Am. Chem. Soc. 137, 15760–15771. doi:10.1021/jacs.5b08746
Watanabe, T., and Sholl, D. S. (2012). Accelerating applications of metal-organic frameworks for gas adsorption and separation by computational screening of materials. Langmuir 28, 14114–14128. doi:10.1021/la301915s
Wei, L., Zizhen, R., Chung, Y., and Li, S. (2017). The role of partial atomic charge assignment methods on the computational screening of metal-organic frameworks for CO2 capture under humid conditions. ChemistrySelect 2, 9458–9465. doi:10.1002/slct.201701934
Willems, T. F., Rycroft, C., Kazi, M., Meza, J. C., and Haranczyk, M. (2012). Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials. Microporous Mesoporous Mater. 149, 134–141. doi:10.1016/j.micromeso.2011.08.020
Wilmer, C. E., Farha, O. K., Bae, Y. S., Hupp, J. T., and Snurr, R. Q. (2012a). Structure-property relationships of porous materials for carbon dioxide separation and capture. Energy Environ. Sci. 5, 9849–9856. doi:10.1039/c2ee23201d
Wilmer, C. E., Kim, K. C., and Snurr, R. Q. (2012b). An extended charge equilibration method. J. Phys. Chem. Lett. 3, 2506–2511. doi:10.1021/jz3008485
Wilmer, C. E., Leaf, M., Lee, C. Y., Farha, O. K., Hauser, B. G., Hupp, J. T., et al. (2012c). Large-scale screening of hypothetical metal-organic frameworks. Nat. Chem. 4, 83–89. doi:10.1038/nchem.1192
Wu, D., Yang, Q. Y., Zhong, C. L., Liu, D. H., Huang, H. L., Zhang, W. J., et al. (2012). Revealing the structure-property relationships of metal-organic frameworks for CO2 Capture from flue gas. Langmuir 28, 12094–12099. doi:10.1021/la302223m
Zhang, K., Nalaparaju, A., and Jiang, J. (2015). CO2 capture in rht metal-organic frameworks: multiscale modeling from molecular simulation to breakthrough prediction. J. Mater. Chem. A. 3, 16327–16336. doi:10.1039/C5TA01866H
Zhang, K., Qiao, Z., and Jiang, J. (2017). Molecular design of zirconium tetrazolate metal–organic frameworks for CO2 capture. Cryst. Growth Des. 17, 543–549. doi:10.1021/acs.cgd.6b01405
Zhu, X., Zheng, H. Y., Wei, X. F., Lin, Z. Y., Guo, L. H., Qiu, B., et al. (2013). Metal-organic framework (MOF): a novel sensing platform for biomolecules. Chem. Commun. 49, 1276–1278. doi:10.1039/c2cc36661d
Keywords: metal organic framework, molecular simulation, CO2 separation, selectivity, adsorption
Citation: Erucar I and Keskin S (2018) High-Throughput Molecular Simulations of Metal Organic Frameworks for CO2 Separation: Opportunities and Challenges. Front. Mater. 5:4. doi: 10.3389/fmats.2018.00004
Received: 01 December 2017; Accepted: 17 January 2018;
Published: 02 February 2018
Edited by:
Dapeng Cao, Beijing University of Chemical Technology, ChinaReviewed by:
Qingyuan Yang, Beijing University of Chemical Technology, ChinaRavichandar Babarao, RMIT University, Australia
Copyright: © 2018 Erucar and Keskin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seda Keskin, c2tlc2tpbkBrdS5lZHUudHI=