 Carlo Massobrio
Carlo Massobrio Evelyne Martin2
Evelyne Martin2 Mauro Boero
Mauro Boero Assil Bouzid
Assil Bouzid Guido Ori
Guido Ori- 1Institut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), University of Strasbourg, CNRS UMR 7504, Strasbourg, France
- 2Université de Lille, CNRS, Centrale Lille, ISEN, Université Valenciennes, UMR 8520-IEMN, Lille, France
- 3Chaire de Simulation à l'Échelle Atomique (CSEA), École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
The inclusion of dispersion (van der Waals, vdW) forces in first-principles modeling of disordered chalcogenides is analyzed and critically discussed in view of their impact on the atomic structure. To this purpose we considered the case of glassy GeTe4. We selected a vdW correction (termed vdWG hereafter) introduced by Grimme (2006) and, as an alternative, the approach (termed vdWW hereafter) based on the Wannier functions formalism (Silvestrelli, 2008). It appears that a strategy based on the update of the vdW interactions due to changes in the electronic structure during the dynamical evolution (i.e., the vdWW one) provides results clearly different from those obtained in the vdWG framework. By keeping in mind that the nature of the present results is preliminary and reflect a trend to be confirmed, we draw attention to the different levels of agreement with experimental data obtained with the two vdW schemes employed.
1. Introduction
Chalcogenide glasses have a tremendous range of potential applications, as they can be found in sensitive media for optical recording, re-writable discs, nonvolatile memory devices, and optical materials (lasers, fiber optics, and optical lenses for infrared transmission) (Boolchand and Bresser, 2001; Huang et al., 2004; Kato et al., 2006; Le Roux et al., 2012). Their non-trivial atomic structure has long been a challenge for measurements, stimulating the application of realistic theoretical approaches at the density functional theory (DFT) level. Quite recently, several contributions in this area have been devoted to the impact of dispersion forces on first-principles molecular dynamics (FPMD) descriptions of disordered chalcogenide materials (Micoulaut, 2013; Micoulaut et al., 2014, 2017; Bouzid et al., 2015b; Lampin et al., 2017). We recall that dispersion forces are not included in the original Kohn-Sham DFT formalism, calling for the addition of appropriate corrections bound not to alter the first-principle character of the model description. In the case of disordered network-forming chalcogenides, dispersion forces are more likely to play a role in GexTe1−x than in GexSe1−x systems. This is exemplified, for instance, by the behavior of glassy GeTe4 (g-GeTe4), proved to be much more sensitive to dispersion forces (Micoulaut, 2013; Bouzid et al., 2015b) than g-GeSe4 or g-GeS4 (Chaker et al., 2018b). Compelling evidence on the impact of vdW forces for Te-rich ternary glasses has been also collected for Ga10Ge15Te75 (Chaker et al., 2018a). The above results have been obtained by exploiting the vdW scheme due to Grimme (vdWG), in particular in the version named D2 (Grimme, 2006). This approach has an extended record of reliability, resulting from its capability to account properly for the action of dispersion forces in a large variety of systems. As such, at a first sight, one might tend to conclude that unambiguous information exists on how the atomic structure of disordered chalcogenides is affected by the inclusion of dispersion forces.
However, a word of caution is in order to put these findings in the right perspective. Indeed, the Grimme formalism is not built on any update of the vdW interactions due to changes in the electronic structure occurring during the dynamical evolution. This is somewhat awkward in light of the strong interplay between electronic structure and ionic positions inherent in FPMD. A revealing example was provided by the case of liquid GeSe2, for which an alternative description was employed for the dispersion forces, the vdWW one (Lampin et al., 2017). In this case the electronic structure, obtained within the selected DFT-GGA (generalized gradient approximation) framework, is used to compute the maximally localized Wannier functions (MLWF) on which the van der Waals scheme is based.1 Considering liquid GeSe2, we found that the MLWF approach reproduces existing first-principles atomic structures while the vdWG one is less performing in the case of the Ge-Ge pair correlation function (Lampin et al., 2017). Turning to glassy GeSe4 and glassy GeS4, the two vdW approaches were found to agree to a large extent, exceptions again occurring for some moderate differences in the intensity of the peaks in the Ge-Ge pair correlation function (Chaker et al., 2018b).
These results drive two considerations. First, the impact of dispersion forces on disordered chalcogenides might differ when adopting different theoretical schemes. This is more unexpected than any dependence on dispersion forces resulting from a mere change in the chemical composition within a given family of compounds. Second, more importantly, the inclusion of such an additional ad hoc part in the Kohn-Sham Hamiltonian can lead to quite different effects when the scheme (the vdWW one) evolves in time with the electronic structure. In view of their higher sensitivity to dispersion forces, it is desirable to consider the case of Te-rich disordered chalcogenides. Glassy GeTe4 is well suited to this purpose due to the availability of results obtained within the vdWG approach. We fulfill this goal with the present study, in which we produce new vdWW data on the structural properties of glassy GeTe4 and we compare them with experiments and the vdWG counterpart available in Bouzid et al. (2015b).
This study shows that the impact of dispersion forces on glassy GeTe4 turns out to be dependent on the recipe employed to model the vdW correction (vdWG vs vdWW schemes). When compared to the case of GexSe1−x chalcogenides, this is indicative of an effect that is stronger when the sensitivity to the inclusion of dispersion forces is higher. The notion of structural properties dependent (even moderately) on the methodology employed to model dispersion forces has to be handled with great care, since it can deeply affect the precise assessment of the atomic structures and bonding properties of glasses.
This paper is organized as follows. In section 2, we provide the methods and computational details of the calculations performed in this work with a special emphasis on the scheme employed to account for the vdW interactions in glassy GeTe4. In section 3, we focus on the total and partial pair correlations functions to show the impact of the different dispersion schemes on the structural properties. In section 4, we underline the novelty, importance and consequences of our findings in the framework of first-principle calculations of disordered systems accounting for vdW corrections.
2. Computational Methods
Our simulations of glassy GeTe4 were performed within the Car-Parrinello (Car and Parrinello, 1985) molecular dynamics (CPMD) scheme as implemented in the CPMD code (Hutter et al., 1995). We employ the exchange functional proposed by Becke (1988) and the correlation functional due to Lee et al. (1988) (BLYP). For the treatment of the core-valence interactions, we adopted a norm-conserving pseudo-potential according to the description of Troullier and Martins (1991). A plane wave basis set is chosen for the representation of the valence electrons with a corresponding energy cutoff of 40 Ry (Bouzid et al., 2015a,b; Lampin et al., 2017). The Brillouin zone integration is restricted to the Γ point.
Keeping in mind that the DFT-D2 formula proposed by Grimme (2006) was employed in a recent investigation (Bouzid et al., 2015b), we turn to an alternative recipe (vdWW) for comparative purposes. It is appropriate in this context to recall the main foundations of this scheme, by referring the reader for details to the original papers thoroughly illustrating this formalism and the connections with the concept of maximally localized Wannier functions (MLWFs) (Wannier, 1937; Silvestrelli et al., 1998; Ikeda and Boero, 2015). Accordingly, one writes the vdW interaction in the standard way (Andersson et al., 1996), namely:
where the subscripts n and l run over all atoms or fragments into which the system is partitioned and f(rnl) is a damping function avoiding double counting of correlations at short range. The C6, nl coefficients can be expressed in terms of the local electronic density ρn(r) as
Within DFT, the applicability of this relationship is not straightforward since the total electronic density ρ(r) cannot be rigorously defined locally. To overcome this difficulty, we resort to the MLWFs wn(r) by rewriting the former definition as
In this case, rnl is the distance between two MLWF centers on pairs of fragments into which the system is partitioned. MLWFs can be calculated on the fly during the CPMD run. In the x-direction (with L the supercell size) the MLWF centers are defined as
with analogous expressions holding for the y and z directions.
During our first-principles molecular dynamics runs of glassy GeTe4 in the vdWW framework, the temperatures of the ions and of the fictitious electronic degrees of freedom were controlled via Nosé-Hoover thermostats (Nosé, 1984; Hoover, 1985; Blöchl and Parrinello, 1992). An integration step of 5 au (0.168 fs) ensured an optimal control of the conserved quantities all along the molecular dynamics trajectories.
As in Bouzid et al. (2015b) our system is made of 215 atoms (43 Ge, 172 Te) in a cubic supercell of edge equal to 19.24 Å. By taking as initial set of positions one configuration extracted from the trajectory produced in Bouzid et al. (2015b), a temporal cycle was implemented to lose memory of this starting condition. The initial system was brought at an ionic temperature of T = 900 K in 88 ps and equilibrated at that temperature during about 50 ps. Then, the temperature was lowered to T = 300 K for 35 ps to conclude with an additional run of 30 ps exploited for the structural analysis.
In what follows, we concentrate on the total pair correlation function gT(r) obtained as a weighted linear combination of the three partial pair correlation functions gαβ(r), where α and β denote the chemical species considered (Ge, Te). Without resorting to an exhaustive report of structural properties, our purpose is to highlight a few, striking observations of the behaviors recorded in the absence (NovdW data hereafter) and in the presence of dispersion forces, these latter expressed in two different forms (vdWW, present results, vdWG Bouzid et al., 2015b).
3. Results: Total and Partial Pair Correlation Function
The results presented in Bouzid et al. (2015b) showed that the best comparison with the available experimental data for g-GeTe4 is the one obtained through the BLYP exchange-correlation functional when used in conjunction with the vdW forces. In particular, the total pair correlation function was closer to experiments than when using the scheme due to Perdew, Burke and Ernzerhof (PBE) (Perdew et al., 1996, 1997). The resulting network is made by a dominant tetrahedral arrangement with a small number of coordination defects. In that paper, two kinds of conclusions were drawn. First, the results confirmed the importance of accounting for vdW dispersion forces when modeling Ge-Te chemical bonding in disordered binary chalcogenides. Second (more important since not dependent on the specific dispersion correction employed) we noticed that the account of vdW forces is less crucial that a change in the exchange-correlation functional (from PBE to BLYP in this specific case). The results contained in Figure 1 challenge the first set of conclusions referred to above. Four sets of data are reported, namely experiments (Kaban et al., 2003) and simulations without (NovdW) and with [vdWG (Bouzid et al., 2015b), vdWW (present work)] dispersion forces. It appears that the vdWW approach lies very close to the NovdW data, thereby underlying an artificial effect inherent in the use of the vdWG recipe.
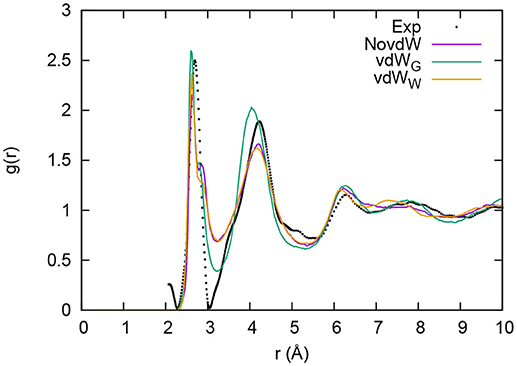
Figure 1. Total correlation function of g-GeTe4. Results with no inclusion of vdW interactions (purple line) and with inclusion of vdW interactions following the Grimme scheme (vdWG, green line) (as reported by Bouzid et al., 2015b) are compared to those including van der Waals interaction in the vdWW formalism (orange line). Experiments are from Kaban et al. (2003).
To corroborate the above findings in terms of atomic arrangements between the different pairs of atoms, we resort to the partial pair correlation functions. These results are reported in Figure 2.
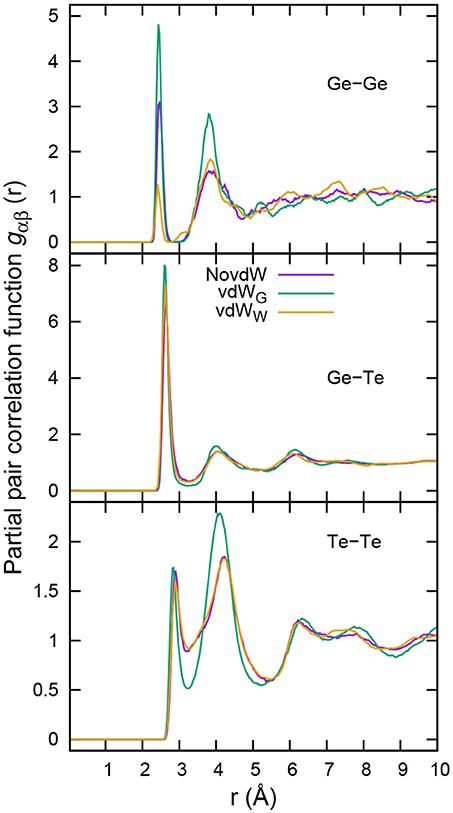
Figure 2. Partial pair correlation functions of g-GeTe4. Results with no inclusion of vdW interactions (purple line) and with inclusion of vdW interactions following the Grimme scheme (vdWG, green line) (as reported by Bouzid et al., 2015b) are compared to those including van der Waals interaction in the vdWW formalism (orange line).
For the Ge-Te pair correlation functions, the results obtained by the three methods are essentially identical, to indicate that the shortest range interactions are not affected by the inclusion of a dispersion scheme. In the Ge-Ge case, the intensities of the first peaks are unequal, indicating small changes in the number of homopolar Ge-Ge bonds. However, vdWG yields by far the most intense first peak. More striking is the difference recorded in the region around r ≃ 4 Å, where the second peak is definitely much higher in the vdWG case, vdWW, and NovdW data being quite close. The consequences of the use of vdWG in comparison with the vdWW and NovdW cases are clearly noticeable in the gTeTe pair correlation function. The pronounced first minima exhibited by gTeTe in the Grimme framework disappear when considering instead the MLWF-based scheme vdWW. Altogether, in the Te-Te case, the effect is quite striking since the profiles of the NovdW and vdWW Te-Te correlation functions are essentially superposed.
4. Discussion and Conclusions
There is a widespread interest in a precise understanding of the impact of dispersion forces on the structural properties of certain classes of disordered materials, such as chalcogenide glasses. When considering networks predominantly based on Te, some pieces of evidence have been produced to prove that such impact cannot be excluded, models containing dispersion forces appearing somewhat closer to experimental counterparts (Micoulaut et al., 2014, 2017; Bouzid et al., 2015b). The present work intends to alert the modeling community on the sensitivity of glassy structural data to different schemes for the dispersion forces. In particular, we show explicitly what happens when one uses a recipe better accounting for the relationship between the van der Waals coefficients and the electronic structure. To this purpose we have produced three models, characterized by the absence of dispersion forces and by the account of dispersion forces in the Grimme scheme (vdWG) (Grimme, 2006) or in the Maximally Localized Wannier scheme (vdWW) (Silvestrelli, 2008). In this approach the van der Waals coefficients adapt to changes undergone by the electronic structure during the motion. We found that the vdWW strategy produces structural data in very good agreement with those obtained in the absence of any account of dispersion forces, thereby differing from the findings previously obtained by adopting vdWG. However, and quite puzzlingly, vdWG features a better agreement with experimental data (pair correlation functions). As a contribution to the debate on the performance of the different van der Waals approaches, it is worth recalling that the general applicability of the Grimme formalism was questioned in the case of glassy GeTe (Raty et al., 2015). This has motivated the use of an alternative scheme proposed by Lee et al. (2010). Conceptual issues related to the proper inclusion of dispersion forces within density functional theory have been extensively reviewed and analyzed in Klimes and Michaelides (2012).
In view of the consequences implied by our findings, we do find appropriate to stress the preliminary character of the present results, since they need to be confirmed by a full assessment of their statistical accuracy (i.e., through the production of several equilibrium trajectories) and by the use of alternative schemes for the dispersion forces [as the ones given in (Lee et al., 2010) and in (Tkatchenko and Scheffler, 2009)]. However, the mere observation of what we found with one trajectory is already quite compelling in terms of similarities between the total and partial correlation functions for the NovdW and the vdWW cases. The question arises on the capability of dispersion forces recipes to play a role when they cannot be considered negligible and, in turn, vanish when their specific weight is much smaller in comparison to other bonding contributions. Our results indicate that this cannot be always the case, thereby rising an unexpected ambiguity on the actual consistency of such schemes. Should these findings be confirmed, it would be desirable to reconsider any known statement on the expected impact of dispersion forces in the case of disordered chalcogenides, with particular attention devoted to Te-based ones.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^More details on the MLWF formalism are given later in the section devoted to the methodology.
References
Andersson, Y., Langreth, D. C., and Lundqvist, B. I. (1996). van der Waals interactions in Density-Functional Theory. Phys. Rev. Lett. 76:102.
Becke, A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38:3098.
Blöchl, P. E., and Parrinello, M. (1992). Adiabaticity in first-principles molecular dynamics. Phys. Rev. B 45:9413.
Boolchand, P., and Bresser, W. (2001). Mobile silver ions and glass formation in solid electrolytes. Nature 410:1070. doi: 10.1038/35074049
Bouzid, A., Le Roux, S., Ori, G., Boero, M., and Massobrio, C. (2015a). Origin of structural analogies and differences between the atomic structures of GeSe4 and GeS4 glasses: a first principles study. J. Chem. Phys. 143:034504. doi: 10.1063/1.4926830
Bouzid, A., Massobrio, C., Boero, M., Ori, G., Sykina, K., and Furet, E. (2015b). Role of the van der Waals interactions and impact of the exchange-correlation functional in determining the structure of glassy GeTe4. Phys. Rev. B 92:134208. doi: 10.1103/PhysRevB.92.134208
Car, R., and Parrinello, M. (1985). Unified approach for molecular dynamics and density-functional theory. Phys. Rev. Lett. 55:2471.
Chaker, Z., Ori, G., Boero, M., Massobrio, C., Furet, E., and Bouzid, A. (2018a). First-principles study of the atomic structure of glassy Ga10Ge15Te75. J. Non-Cryst. Solids 498:338. doi: 10.1016/j.jnoncrysol.2018.03.039
Chaker, Z., Ori, G., Tugene, C., Le Roux, S., Boero, M., Massobrio, C., et al. (2018b). The role of dispersion forces on the atomic structure of glassy chalcogenides: the case of GeSe4 and GeS4. J. Non-Cryst. Solids 499:167. doi: 10.1016/j.jnoncrysol.2018.07.012
Grimme, S. (2006). Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799. doi: 10.1002/jcc.20495
Hoover, W. G. (1985). Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A 31:1695.
Huang, C.-C., Hewak, D., and Badding, J. (2004). Deposition and characterization of germanium sulphide glass planar waveguides. Opt. Express 12, 2501–2506. doi: 10.1364/OPEX.12.002501
Hutter, J., Parrinello, M., Marx, D., Focher, P., Tuckerman, M., Andreoni, W., et al. (1995). Computer Code CPMD. IBM Zurich Research Laboratory and MPI für Festkörperforschung, 2001.
Ikeda, T., and Boero, M. (2015). Role of van der Waals corrections in first principles simulations of alkali metal ions in aqueous solutions. J. Chem. Phys. 143:194510. doi: 10.1063/1.4935932
Kaban, I., Halm, T., Hoyer, W., Jovari, P., and Neuefeind, J. (2003). Short-range order in amorphous germanium-tellurium alloys. J. Non-Cryst. Solids 326:120. doi: 10.1016/S0022-3093(03)00389-2
Kato, N., Fukano, T., Takeda, Y., Takeichi, A., Motohiro, T., and Kawai, S. (2006). GeS2/metal thin film bilayered structures as write-once-type optical recording materials. J. Appl. Phys. 100:113115. doi: 10.1063/1.2398556
Klimes, J., and Michaelides, A. (2012). Perspective: advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 137:120901. doi: 10.1063/1.4754130
Lampin, E., Bouzid, A., Ori, G., Boero, M., and Massobrio, C. (2017). Impact of dispersion forces on the atomic structure of a prototypical network-forming disordered system: the case of liquid GeSe2. J. Chem. Phys. 147:044504. doi: 10.1063/1.4986166
Le Roux, S., Bouzid, A., Boero, M., and Massobrio, C. (2012). Structural properties of glassy Ge2Se3 from first-principles molecular dynamics. Phys. Rev. B 86:224201. doi: 10.1103/PhysRevB.86.224201
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37:785.
Lee, K., Murray, E. D., Kong, L., Lundqvist, B. I., and Langreth, D. C. (2010). Higher-accuracy van der Waals density functional. Phys. Rev. B 82:081101. doi: 10.1103/PhysRevB.82.081101
Micoulaut, M. (2013). Van der waal corrections for an improved structural description of telluride based materials. J. Chem. Phys. 138:061103. doi: 10.1063/1.4792195
Micoulaut, M., Coulet, M.-V., Piarristeguy, A., Johnson, M. R., Cuello, G. J., Bichara, C., et al. (2014). Effect of concentration in Ge-Te liquids: a combined density functional and neutron scattering study. Phys. Rev. B 89:174205. doi: 10.1103/PhysRevB.89.174205
Micoulaut, M., Piarristeguy, A., Flores-Ruiz, H., and Pradel, A. (2017). Towards accurate models for amorphous GeTe: Crucial effect of dispersive van der Waals corrections on the structural properties involved in the phase-change mechanism. Phys. Rev. B 96:184204. doi: 10.1103/PhysRevB.96.184204
Nosé, S. (1984). A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81:511–519.
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77:3865.
Perdew, J. P., Burke, K., and Ernzerhof, M. (1997). Generalized gradient approximation made simple. Erratum Phys. Rev. Lett. 78:1396.
Raty, J. Y., Zhang, W., Luckas, J., Mazzarello, R., Bichara, C., and Wuttig, M. (2015). Aging mechanisms in amorphous phase-change materials. Nat. Comm. 6:7467. doi: 10.1038/ncomms8467
Silvestrelli, P. L. (2008). Van der Waals interactions in DFT made easy by Wannier functions. Phys. Rev. Lett. 100:053002. doi: 10.1103/PhysRevLett.100.053002
Silvestrelli, P. L., Marzari, N., Vanderbilt, D., and Parrinello, M. (1998). Maximally-localized Wannier functions for disordered systems: Application to amorphous silicon. Solid State Commun. 107, 7–11.
Tkatchenko, A., and Scheffler, M. (2009). Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102:073005. doi: 10.1103/PhysRevLett.102.073005
Troullier, N., and Martins, J. L. (1991). Efficient pseudopotentials for plane-wave calculations. II. operators for fast iterative diagonalization. Phys. Rev. B 43:8861.
Keywords: Car-Parrinello molecular dynamics (CPMD), atomic structure, di-chalcogenides, van der Waals, amorphous materials
Citation: Massobrio C, Martin E, Chaker Z, Boero M, Bouzid A, Le Roux S and Ori G (2018) Sensitivity to Dispersion Forces in First-Principles Modeling of Disordered Chalcogenides. Front. Mater. 5:78. doi: 10.3389/fmats.2018.00078
Received: 03 October 2018; Accepted: 12 December 2018;
Published: 21 December 2018.
Edited by:
Jincheng Du, University of North Texas, United StatesReviewed by:
Alfonso Pedone, Università degli Studi di Modena e Reggio Emilia, ItalyGuillaume Pierre Jean Ferlat, Université Pierre et Marie Curie, France
Copyright © 2018 Massobrio, Martin, Chaker, Boero, Bouzid, Le Roux and Ori. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Massobrio, Y2FybG8ubWFzc29icmlvQGlwY21zLnVuaXN0cmEuZnI=