 Alessandro Grottoli1,2
Alessandro Grottoli1,2 Marzia Beccaccioli1,2
Marzia Beccaccioli1,2 Emma Zoppis3
Emma Zoppis3 Rosita Silvana Fratini2
Rosita Silvana Fratini2 Emily Schifano3
Emily Schifano3 Maria Laura Santarelli4
Maria Laura Santarelli4 Daniela Uccelletti3
Daniela Uccelletti3 Massimo Reverberi1,2*
Massimo Reverberi1,2*- 1SARA EnviMob s.r.l., Rome, Italy
- 2Department of Environmental Biology, Sapienza University of Rome, Rome, Italy
- 3Department of Biology and Biotechnology Charles Darwin, Sapienza University of Rome, Rome, Italy
- 4Department of Chemical Engineering Materials Environment, Sapienza University of Rome, Rome, Italy
Microbiological methodologies allow understanding the causes that lead to the development of a certain microbial community colonizing an artistic surface, to characterize its composition and describe its role in the deterioration of the constituent materials. Metagenomics allows identifying microbial communities directly in their natural environments, bypassing the need for isolation and cultivation of individual species, thus providing a more comprehensive picture of the biodiversity present on a surface compared with standard cultivation methods. Furthermore, molecular analyses require small amounts of material, favoring the preservation of the artistic surface during sampling. Here, we verified the suitability of a protocol consisting in DNA extraction with micro-invasive sampling, using adhesive tape, PCR amplification with universal primers [bacteria (16S), fungi (ITS), and Viridiplantae (18S)], and amplicon sequencing by Oxford Nanopore Technologies (ONT) in the hypogeum of Basilica di San Nicola in Carcere Church (Rome, Italy). Sequence data were analyzed with a bioinformatic pipeline customized for pinpointing cultural heritage spoiling organisms, named “AmpLIcon SequencIng Analysis” (ALISIA). These data were integrated with traditional microbiology techniques that allowed the isolation of cultivable bacteria; three species were also characterized through their capability of biofilm formation and antibiotic resistance. Further, Fourier-transform infrared spectroscopy (FTIR) spectroscopy was performed to characterize the main products present on the masonry surface providing indications on the type of decay present. This novel biological workflow represents a powerful opportunity to investigate the microbial colonization of artistic surfaces aimed at implementing preservation strategies of cultural heritage from bio-spoilage.
Introduction
Cultural heritage (CH), such as statues, books, paintings, and frescoes, may be frequently exposed to the effects of physical and chemical deterioration. Moreover, microorganisms may interact among them and with the surface, establishing a real ecosystem with trophic chains and microbiological successions (Caneva and Salvadori, 1989). Biological interaction with the CH may be an important cause of biodeterioration (Sterflinger and Piñar, 2013). Microorganisms, in fact, carry out micro- and macrosurface decoesidation processes causing physical damage and activate chemical degradation processes due to the emission of primary and secondary metabolism products and assimilation processes that can occur with or without the production of extracellular enzymes (Pinna and Salvadori, 2005). The aesthetic damages are among the most obvious and often compromise the readability of the work. They are due to the precipitation of mineral phases or to the presence of biofilms, which may have different thickness, microstructure, and color. The color depends on the type of biocenosis, the stage of development and the growth phase of the microorganisms involved, and the eventual endo-pigments produced (Warscheid and Braams, 2000). Adopting microbiological methodologies in order to understand the causes that lead to the development of a certain microbial community, identifying its composition, and describing its role in the deterioration of the constituent materials are the first important and necessary steps for understanding the effects of microorganisms on CH. The identification of certain microorganisms on the surface of an artistic object does not link them directly as responsible for deterioration. In fact, some microorganisms might derive from secondary colonization, and therefore, their activity is addressed to organic substances released in the environment by death and lysis of pioneer species (Radaelli et al., 2004). Thus, the reconstruction of the succession of populations on the surface is the only way to understand the role of each identified species in the deterioration process (Caneva et al., 2008). Furthermore, a microbiological point of view allows identifying and developing strategies to manage the growth of biodeteriogens and to study fitting prevention strategies through a better knowledge of the growth-limiting nutrients and the dynamics of biological aerosol diffusion for the conservation and protection of monuments and artworks (González and Saiz-Jiménez, 2005; Caneva et al., 2008).
Microbial diversity can be explored mainly through two distinct approaches: isolation and direct cultivation or indirect molecular analysis. Microorganisms’ isolation and cultivation is the traditional method for exploring the microbial diversity, however, it is estimated that only 0.1 up to 3.0% of the environmental bacteria and up to 70% of fungi (with huge variability depending upon the genera) are cultivable using standard cultivation methods (Amann et al., 1995; Hanson et al., 2016).
On the other hand, molecular approaches are not linked to this limit and thus allow obtaining a more comprehensive picture and a realistic view of the biodiversity present on the surface. Furthermore, by repeating molecular analysis at different moments in time, it is also possible to retrieve important information regarding the history of successive colonization. Another advantage of the use of cultural-independent molecular methods is the significant reduction in the quantity of material that these methods require for sampling. This is an important aspect to consider when working on artistic objects in order to preserve their integrity. Metagenomics is the application of modern genomics techniques to the study of communities of microbial organisms directly in their natural environments, bypassing the need for isolation and lab cultivation of individual species (Hugenholtz, 2002; Rappé and Giovannoni, 2003). Within the molecular techniques, next generation sequencing (NGS) metagenomics-based methods represent a minimally invasive and powerful opportunity to investigate the microbial communities, composition, and functional diversity that may be present on artistic surfaces or in different environments from soil to human gut (Gutarowska et al., 2015). Nonetheless, molecular analysis investigation methods should preferably be supported by cultural-dependent methods, which allow the study of the physiology and metabolism of the isolated microorganisms and understanding of their potential role in biodeterioration (Qi-Wang et al., 2011; Otlewska et al., 2014).
This work aims at identifying by molecular methods, namely ONT sequencing, the microbiological composition of the hypogeum of one of the most characteristic Roman monuments for architectural and historical formation: the Basilica di San Nicola in Carcere Church, in Rome. The structure incorporates three temples and a medieval tower transformed into a bell tower. The Church shares the basement and part of the structures with three Roman temples below; its location near the Tiber River has made it vulnerable since its first construction in 1128 AD. Indeed, the high level of humidity present in the masonries made necessary the periodic reconstruction of its frescoes, which in the last version date back to the XIX century.
Micro-invasive sampling of the hypogeum involving the use of adhesive tape was performed; due to the low recovery of materials, we inserted a PCR amplification step of the total DNA extracted, using universal primers for three kingdoms, 16S for bacteria, ITS for fungi and 18S for Viridiplantae. We then used a metagenomics approach for sequencing the amplicons through ONT and analyzing the output data with a customized bioinformatic pipeline. Parallel, from the same area of sampling, microbial and chemical characterizations were performed. Specifically, cultivable bacteria were evaluated, and three isolates were analyzed in terms of antibiotic resistance and biofilm formation ability, in order to highlight the importance of a combined approach to investigate biodegradation phenomena.
Materials and Methods
Sampling for the Molecular Analysis and Bacteria Isolation
In this study, two different approaches were used in order to investigate the presence of microorganisms on CH surfaces: isolation of microorganisms for the nanopore sequencing analysis and isolation of cultivable bacteria for Fourier-transform infrared spectroscopy (FTIR) analysis. For the sequencing analysis, biological material was collected using a non-invasive or micro-invasive sampling technique for CH and using adhesive tape (Urzí and De Leo, 2001). This non-invasive sampling technique allows collecting enough biological material, without affecting the integrity of the CH. The adhesive tape sampling method was used for collecting samples in Basilica di San Nicola in Carcere Church in Rome for metagenomics analysis along the hypogeum wall of the church. Samples collected (n = 6) were pooled in two samples (Samples 1 and 2) and stored in 50 ml sterile tubes at −20°C until the DNA extraction procedures. In the cases of isolation of cultivable bacteria and FTIR analysis, cotton swabs were utilized for sampling very near the area where adhesive tape sampling was performed. Cotton swabs were pressed firmly over the surface of the wall and then placed in sterile tubes containing 2 ml of Nutrient Broth (NB). Successively, samples were incubated in the laboratory at 30°C for 24 h and plated on NB agar plates. Plates, further incubated at 30°C for 24 h, allowed the isolation and purification of morphologically different colonies. Bacterial isolates were then identified at the molecular level by the amplification of 16S rDNA.
DNA Extraction and rDNA Amplification
Genomic DNA extraction was performed using the C-TAB III extraction method as described in Scala et al. (2017), with some modifications. Adhesive tape used for collecting the samples was grounded in liquid nitrogen in the presence of polyvinylpolypyrrolidone. The first step of extraction was performed in a 50 ml tube in presence of 1.5 ml of C-TAB I, 1.5 ml of SOL2A, and 15 μl of Proteinase K (20 mg/ml) and incubated at 55°C at 250 rpm overnight. After this first step, the extraction was performed as protocol. Total DNA extract was resolved by electrophoresis in agarose gel (1%).
Spectrophotometric quantification was performed using a ThermoScientific NanoDrop spectrophotometer, and DNA purity was assessed through the evaluation of 260/280 and 260/230 absorbance ratios. PCR amplification was performed on about 50 ng of DNA of each sample using rDNA kingdom-specific primers; 27 For (5′-GAGATTTGATCCTGGCTCAG-3′) and 1495 Rev (5′-CTACGGCTACCTTGTTACGA-3′) primers were used for the bacteria rDNA 16S region (Lane, 1991); ITS1 (5′-TCCGTAGGTGAACCTGCGG-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) primers were used for the fungi rDNA 18S region (Glass and Donaldson, 1995); and EUK-A (5′-ACCCTGGTTGATCCTGCCA-3′) and EUK-B (5′-TGATCCTTCTGCAGGTTCACCTAC-3′) primers were used for the Viridiplantae rDNA 18S region (Medlin et al., 1988). Amplification was performed by using Taq polymerase (Bioline), and the PCR products were verified through gel electrophoresis in agarose gel (1%).
Amplicon Sequencing
Amplicon sequencing was performed on PCR products deriving from the rDNA amplification belonging to bacteria, fungi, and Viridiplantae present in the total DNA extracted from adhesive tape. PCR products were sequenced through Oxford Nanopore Technologies (ONT) by a MinION sequencer. A total amount of 200 ng using a ratio of 1:1:1 for 16S, ITS, and 18S PCR product was used for sequencing. Rapid barcoding of PCRs was carried out following the protocol released by ONT (SQK-RBK004).
A customized bioinformatic pipeline called “AmpLIcon SequencIng Analysis” (ALISIA) was used in order to pinpoint CH spoiling organisms. ALISIA uses BLASTn version 2.8.1+ (Camacho et al., 2009) following a few steps: (i) Each read is analyzed and associated to a single taxon. (ii) Final output is divided into the three analyzed kingdoms: bacteria, Viridiplantae, and fungi. (iii) Reads are filtered by matching length in order to remove eventual sequencing artifacts following these criteria: 700–1,500 bp for bacteria reads, 250–2,000 bp for Viridiplantae reads, and 500–1,200 bp for fungi reads (Medlin et al., 1988; Lane, 1991; Glass and Donaldson, 1995); moreover, the reads were selected by e-value ≤ 1⋅10–6, length of alignment ≥80%, and percentage of identical matches ≥85% because of the high error rate of the ONT MinION sequencer (Tyler et al., 2018). (iv) Lastly, a virtual gel, representing the density of the sequenced reads, was drawn in R version 3.6.1 (R Core Team, 2018) and using the package ggplot2 version 3.2.0 (Gómez-Rubio, 2017) in order to emulate a DNA electrophoretic run for comparison.
Isolation and Characterization of Bacterial Strains
Different bacteria were isolated from the hypogeum wall in Basilica di San Nicola in Carcere Church, in Rome. Molecular characterization of the isolated bacteria was performed by DNA extraction according to Bergkessel and Guthrie (2013) and the amplification of a region of approximately 1,400 bp from the 16S rRNA gene using the primers F8 (5′-AGAGTTTGATCCTGGCTCAG-3′) and R1492 (5′-GGTTACCTTGTTACGACTT-3′). The PCR reaction was performed utilizing Taq DNA polymerase from Accuzyme DNA Polymerase (Bioline). BMR Genomics (Padova, Italy) sequenced the amplified region, and the obtained sequences were analyzed with the BLAST database. The bacterial species used in this work were Paenibacillus pabuli IP3 (accession number: MN493858), Kocuria rhizophila IP5 (accession number: MN493859), Bacillus licheniformis IP7 (accession number: MN493860), and Stenotrophomonas maltophilia IP9 (accession number: MN493861).
Bacterial strains isolated from hypogeum surfaces were grown in NB at 30°C.
Antibiotic Susceptibility Tests
Antibiotic susceptibility tests were performed according to Schifano et al. (2019). Overnight cultures of the different bacterial cultures (100 μl) were spread onto NB agar plates. Then, antibiotic disks were placed on plates, which were incubated for 24 h at 30°C. The zones of inhibition were measured from the center of the disk and recorded.
Biofilm Formation Assay
The biofilm formation assay on the plastic surface was performed as described in Zanni et al. (2017) with some differences. Briefly, each well was filled with 200 μl of NB broth and a concentration of each bacterial stain of 1⋅107 cell/ml, in triplicate. Wells without bacteria were utilized as controls. Next, plates were incubated at 30°C for 24 h, and Crystal Violet assay was performed. The experiment was repeated three times with three replicates for each treatment. For biofilm formation on glass, 1⋅107 cells/ml of each bacterial strain were separately inoculated in 3 ml of NB, put in a 35 mm Petri plate with a sterile cover slide, and then incubated at 30°C. After 24 h, cultures were gently removed, washed twice with sterile water, and stained using SYTO9 staining (Kit 7012, Invitrogen, Mount Waverley, Australia) for 15 min at room temperature in the dark. Stained cells were observed under a Zeiss Axiovert 25 fluorescence microscope.
Chemical Characterization by FTIR Analysis
The chemical analysis of the efflorescence present on the hypogeum of the church was performed by infrared spectroscopy with a Fourier-transform infrared spectrometer (FTIR, Vertex 70, Bruker Optik GmbH, Ettlingen, Germany) equipped with a single-reflection Diamond ATR cell, a standard MIR source (HeNe), and a room temperature DTGS detector. The ATR-FTIR spectra were recorded with 256 scans in the mid-infrared range (400–4,000 cm–1) at a resolution of 4 cm–1.
Results
ALISIA
PCR amplifications were performed on the two sampling pools (Samples 1 and 2) derived from the hypogeum of Basilica di San Nicola in Carcere Church (see section “Materials and Methods”) in order to investigate the presence of organisms belonging to bacteria, fungi, and Viridiplantae using, respectively, specific rDNA 16S, ITS, and 18S region primers (see section “Materials and Methods”). Electrophoresis agarose gel verified the presence and the length of amplification products for all three regions investigated. Both genomic DNA from Sample 1 and Sample 2 provided amplicons with each kingdom-specific primer: (i) ITS (fungi), one for Sample 1 at ∼600 bp and two separate bands at ∼600 and ∼900 bp in Sample 2; (ii) 18S (Viridiplantae), both samples resulted as positive to a single band ∼1,800 bp; and (iii) 16S (bacteria), single band in both samples at ∼1,500 bp (Figure 1A). However, in every PCR run, a light smear could be identified near the bands of the expected size; this possibly indicated the presence of the DNA of several microorganisms. ONT MinION sequencing was assessed to rapidly classify the PCR amplicon products: the run produced ∼6 Mb of data (∼100 k reads) and ∼11 Mb of data (∼190 k reads) respectively for Samples 1, 2. A customized bioinformatic pipeline called ALISIA was used in order to pinpoint CH spoiling organisms. The sequenced reads were analyzed following four basic steps described in section “Materials and Methods.” In the first two steps, 94,353 reads for Sample 1 and 155,506 reads for Sample 2 were assigned to a taxon (∼80% of the total reads). In the third step, 21,597 reads corresponded to the bacteria kingdom, 47,729 to fungi, and 5,571 to Viridiplantae for Sample 1, while in Sample 2, 34,803 reads corresponded to bacteria, 36,176 to fungi, and 1,055 to Viridiplantae (Figure 2), however, 19,456 reads for Sample 1 and 83,472 reads for Sample 2 corresponded to the Metazoa and Protista kingdoms, or virus or matched with unknown organisms. Interesting, 69,056 reads belonged to the Metazoa kingdom in Sample 2. In fact, the EUK-A and EUK-B primers could have amplified portions of rDNAs belonging to this kingdom (Hugerth et al., 2014). Further analysis will bring to light the convolution of the found species (Table 1). In the last ALISIA steps, a virtual gel was drawn using the density of the reads assigned to a specific kingdom in order to emulate, and in this case compare, a real PCR electrophoretic gel. The virtual gel shows a similar pattern of read length distribution compared to the real agarose PCR gel for both fungi and bacteria lanes, however, in the virtual gel Viridiplantae lanes, the reads were distributed along the Sample 1 lane, while a very low number of reads of Sample 2 were found (respectively 2,026 and 304 reads), showing a different pattern compared to the real agarose PCR gel (Figure 1B). Within these reads, ∼94 and ∼150 k were associated to a single taxon. In bacteria, “uncultured bacteria” was the most abundant association found in the NCBI database for both Samples 1 and 2 (∼1,000 reads, 28.68% of total bacterial reads for Sample 1; ∼2,000 reads, 32.06% of total bacterial reads for Sample 2), however, in both samples, Nesterenkonia was the most abundant genus found (1,666 reads and 812 reads corresponding to 40.80 and 11.30% respectively for Samples 1 and 2) (Table 1). Although also in fungi, in both samples, ∼4,000 reads (respectively 4 and 3% of the total reads for the analyzed samples corresponding to respectively 49.13 and 65.29% of the total fungal species found in the samples) were assigned to an uncultured fungus, in Sample 1, the most abundant fungal taxon found was Capnobotryella spp. (695 reads, 7.82% of total fungal reads), a genus already found on monument surfaces (Sert et al., 2007); while in Sample 2, the most abundant fungal taxon found was Parastagonospora forlicesenica (1,589 reads, 22.66% of total fungal reads) (Table 1). The most abundant plant species found were Parietaria judaica and Platanus occidentalis respectively in Samples 1 and 2 (374 reads and 51 reads corresponding to 18.26 and 15.84% of the total Viridiplantae reads found). However, the second most abundant taxon in Sample 2 was “embryophyte environmental sample” (27 reads, 8.39% of the total Viridiplantae found in the sample) (Table 1).
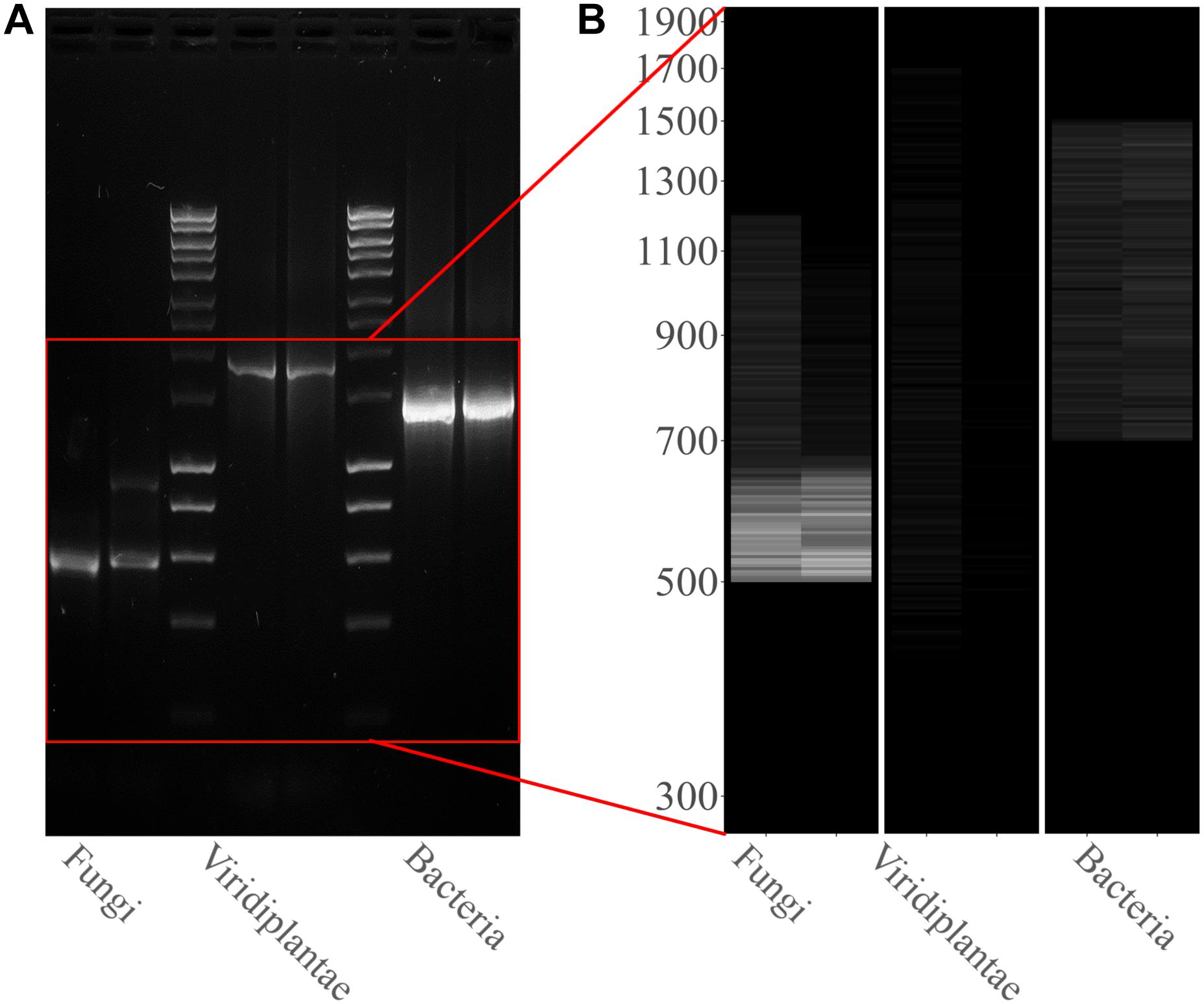
Figure 1. Kingdom rDNA-specific PCR amplicon agarose gel and virtual gel: (A) Electrophoretic gel (agarose 1%) shows three separate and different PCR runs for both samples analyzed: ITS1-4 for fungi, 18S EUK-a/b for Viridiplantae, and 16S for bacteria. Bioline HyperLadderTM 1 kb was used. (B) Virtual gel showing the distribution of the reads per kingdom. Samples in the both images are ordered as follows: Sample 1 and Sample 2.
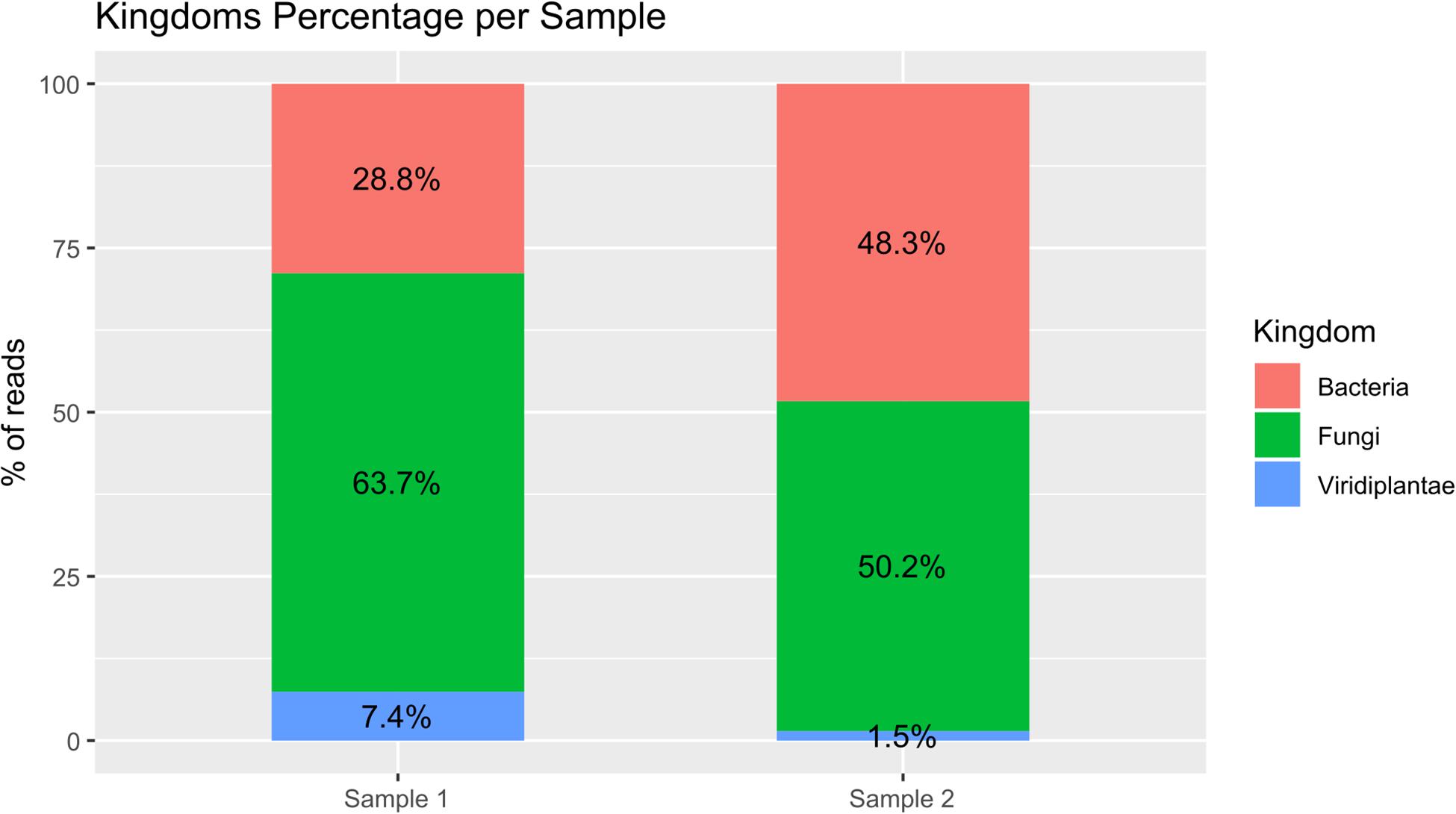
Figure 2. Bar plot of percentage of the three analyzed kingdoms for both samples. The reads assigned to the bacteria kingdom were 28.8 and 48.3% respectively for Samples 1 and 2; the reads assigned to the fungi kingdom were 63.7 and 50.2% respectively for Samples 1 and 2; and the reads assigned to the Viridiplantae kingdom were 7.4 and 1.5% respectively for Samples 1 and 2.
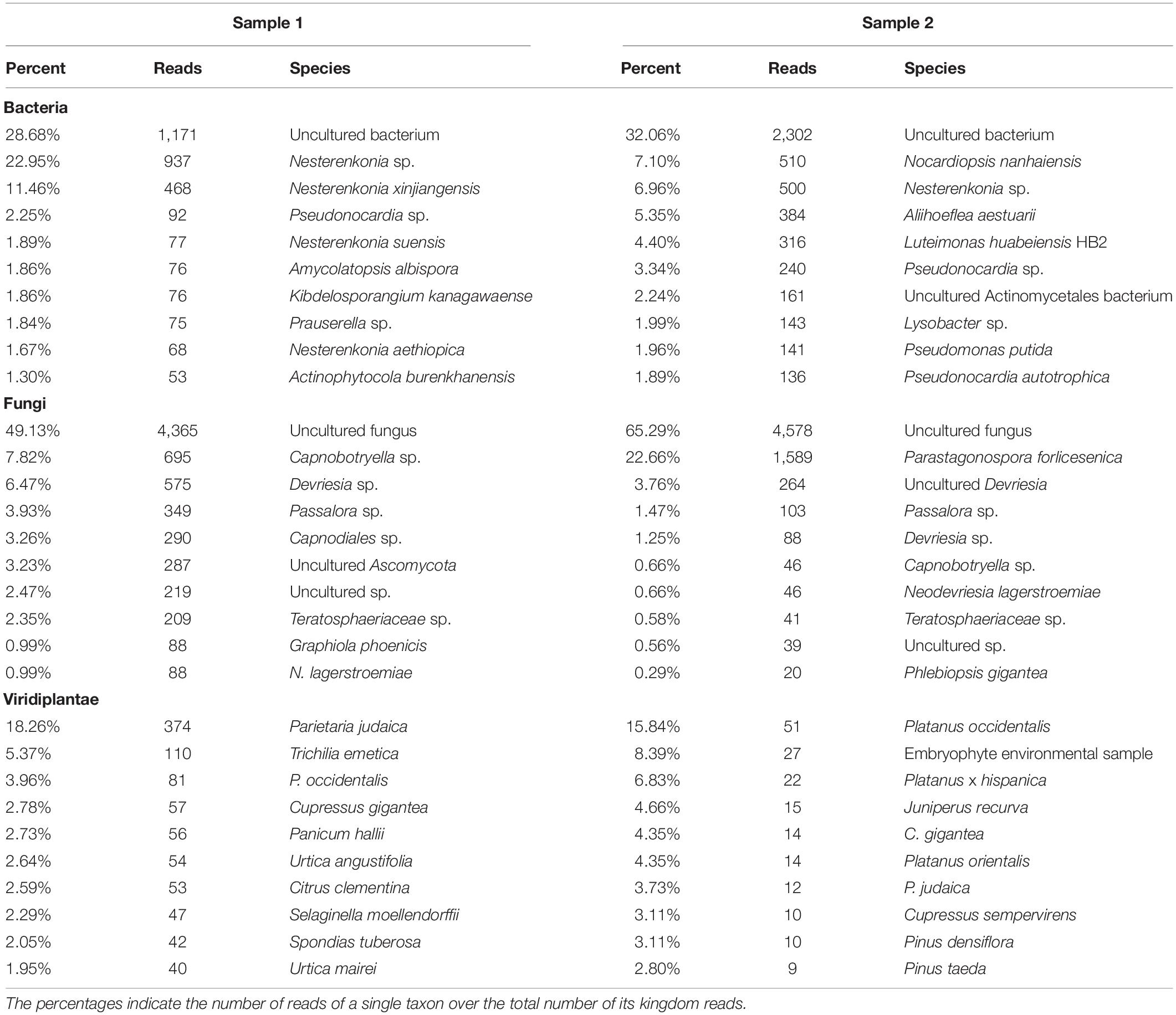
Table 1. The table shows percentage and raw number of reads of the three analyzed kingdoms for Samples 1 and 2.
Identification and Characterization of Culturable Bacterial Strains
Since the most abundant reads found were ascribed to non-cultivable microorganisms, we decided to proceed with standard isolation techniques, taking the case of the culturable bacteria as an example. The amplification of 16S rDNA of bacteria isolated from the hypogeum revealed the presence of different genera: Arthrobacter, Bacillus, Kocuria, Paenibacillus, Pseudomonas, Staphylococcus, Stenotrophomonas, and Streptomyces. Among them, four different isolates, P. pabuli IP3, K. rhizophila IP5, B. licheniformis IP7, and S. maltophilia, IP9 were further characterized. An antibiotic susceptibility test was performed using 19 antibiotics involved in inhibition of cell wall synthesis, protein synthesis, and nucleic acid synthesis. As reported in Table 2, all bacterial strains resulted as resistant to aztreonam. Moreover, S. maltophilia IP9 showed a higher resistance than the other bacterial strains. It was resistant to 12 antibiotics, among which were penicillin, erythromycin, and ampicillin. On the contrary, B. licheniformis IP7 showed a low antibiotic resistance, since inhibition zones were visible only in the case of tetracycline, fosfomycin, and aztreonam. Since the biodeterioration process is mainly caused by microorganisms’ capacity to aggregate and produce an extracellular matrix able to protect them from adverse conditions, biofilm formation of bacterial isolates was evaluated on both plastic and glass surfaces. K. rhizophila IP5, B. licheniformis IP7, and S. maltophilia IP9 showed a high ability of biofilm production on both the two surfaces, while P. pabuli IP3 resulted as being unable to aggregate on plastic and glass (Figure 3).

Table 2. Antibiotic susceptibility of the four different bacterial strains.
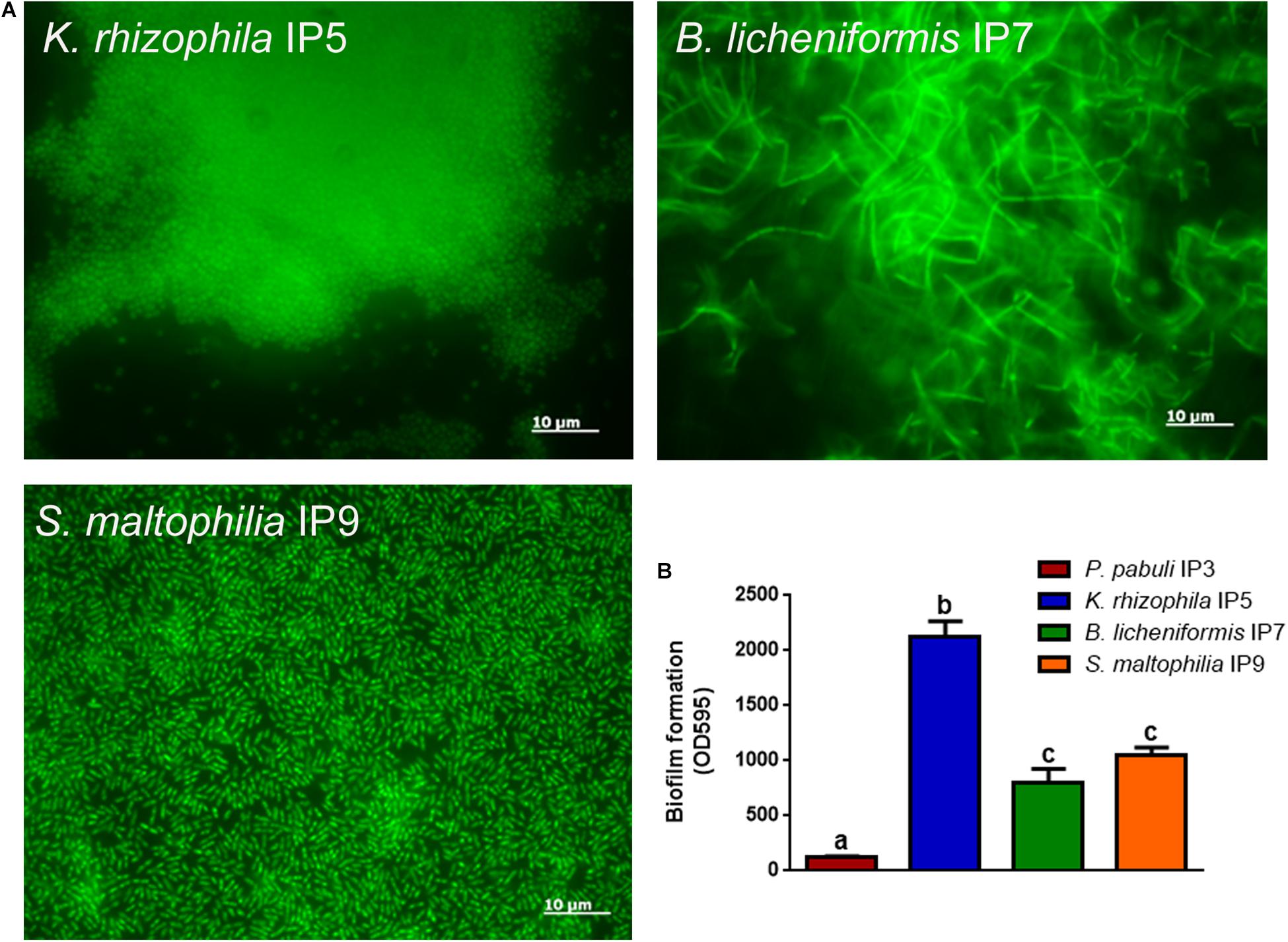
Figure 3. Biofilm formation on glass or plastic surfaces. (A) Fluorescence microscope images of the different bacterial strains forming biofilm on glass. (B) Biofilm formation ability of the bacteria on plastic. To evaluate statistical significance, a one-way ANOVA analysis with the Bonferroni post-test was used (different letters indicate p < 0.05).
FTIR-ATR Characterization of the Efflorescence on the Hypogeum Wall
The formation of efflorescences is due to different phenomena, such as stone weathering. Because of changes in temperature or humidity, salts can precipitate on surfaces, causing a destructive effect (Wilimzig, 1996). Salts deposited are composed of a variety of different chemical compounds, including carbonates, chlorides, nitrates, and sulfates. Salt efflorescences can be observed in environments with high levels of humidity, where they are generated by the action of nitrifying bacteria that convert the organic compounds into nitrate salts (Castrillón Rivera et al., 2019). Thus, studying these efflorescences could provide a means for identifying at least the type of microorganisms that are spoiling that specific CH. The IR spectra of the sample showed the presence of typical vibration bands of calcium sulfate hydrate, known also as gypsum (CaSO4 × 2H2O), centered at 1,109, 669, and 596 cm–1, as well as the stretching and deformation vibrations of the O–H bond of water at 3,525, 3,492, and 3,401 cm–1 and at 1,692 and 1,627 cm–1, respectively. At 1,424 cm–1 is recognized the presence of calcium carbonate; instead, the absorption at 1,343 cm–1 is due to the presence of sodium and/or potassium nitrates. A band of silicates was present at 1,032 and 1,006 cm–1. Gypsum is due to the presence of sulfates in the masonries of the monument for capillarity rise of water from the soil. A strong exchange of cations between the CaCO3 (calcite) of the mortars with the sulfates diluted in the water creates the condition to crystallize gypsum. The presence of silicates is aswell justified by crystallization processes with compounds deriving for capillarity rise of water from the soil.
Discussion
Dealing with CH, the primary concern is that of the preservation of the artistic surface. In this context, sampling must be performed adopting the least invasive method possible and collecting the smallest amount of material required for analysis. The micro-invasive sampling through adhesive tape only causes minimal damage to the superficial layer of the artistic object. However, further investigations could be performed to assess if the micro-invasive character of the method may be maintained also in the case of highly fragile artistic surfaces.
Since this sampling method only allows sampling of the surface of the artistic object, the microbiological components that might have colonized the porous cavities are excluded. It was observed that this sampling method allows the extraction of quantities of DNA enough to carry out genomic analysis without the need of previous amplification. Even so, the overall quantity and quality of the extracted DNA was unsuitable for NGS metagenomics shotgun analysis. To skip this bug, PCR amplification of rDNA kingdom-specific regions allows us to exponentially amplify DNA in order to be suitable for NGS analysis. The use of the ONT MiniION sequencer allows quickly sequencing samples directly in your laboratory for a rapid and real-time analysis. More classic sequencing technologies require a greater amount of time waiting for sequencing data. The high number of reads related to the amplicons belonging to the different organisms sampled by micro-tape in the hypogeum of Basilica di San Nicola in Carcere Church have been decrypted by tailoring a bioinformatic tool for this specific need: ALISIA. ALISIA is a practical bioinformatic tool that allows you to analyze quickly raw amplicon sequencing data from different sequencing technologies, such as Illumina or ONT. The use of ALISIA in CH metagenomics studies provides a clear and immediate view of the microbiome present on the analyzed CH. ALISIA revealed many species present in the microbiome collected by the adhesive tape sampling method in the three kingdoms. Despite the large number of species found, in both the fungi and bacteria kingdoms, the most abundant reads were assigned, by BLAST analysis, to uncultured microorganisms. This can be attributed to the impossibilities and difficulties in isolating microorganisms by traditional cultural methodologies (Amann et al., 1995; Hanson et al., 2016). Thus, it resulted as problematic to have confirmation of the reliability of these taxonomic results.
Interestingly, ALISIA highlighted the presence of different species of genus Nesterenkonia (Table 1). This genus, belonging to the Micrococcaceae family, includes mesophilic moderate haloalkaliphilic species (Stackebrandt et al., 1995). Even if this genus was not found on the bacteria isolation, Kocuria, a closely related genus (Stackebrandt et al., 1995), was isolated and characterized by molecular analysis. Although there is a mismatch between swab isolation bacteria species found and ALISIA analysis, further investigations will be carried out in order to improve ALISIA’s result accuracy in detecting the microbiome present on a CH surface. In fact, it is difficult to compare the high specificity that metagenomic analysis can reach to isolation and cultivation of individual species. A functional metagenomics analysis can be performed in order to find the degradation molecular mechanisms involved in the analyzed CH, completely skipping the identification step, acting directly on the molecular process. Moreover, despite this being bacteria-centric work, a wide number of fungal species, likely involved in CH deterioration, were found. Nevertheless, a plus in the accuracy of ONT and ALISIA could be suggested by results in 18S amplicon sequencing: most of the plant species found are tree plants very common in Rome, such as Platanus, Pinus, or Cupressus, or herbaceous plants typical of cracks in rock faces: P. judaica. Very likely, the DNA of these plants belongs to pollen deposited on sampled wall surfaces. Metagenomic amplicon sequencing by ONT MinION is revealed to be a fast tool to investigate the taxa present on the surface of CH.
To investigate deeply on the bacterial composition of the masonry of the hypogeum, cultivable bacteria were isolated. While many of them were reported to be involved in the formation of salt efflorescences such as the genera of Bacillus, Kocuria, Paenibacillus, and Arthrobacter (Laiz et al., 2000), Pseudomonas spp. resulted as being involved in calcite production (Rivera et al., 2018). Among the isolates, three strains were chosen as representatives to be further characterized, as they belong to the Gram-positive P. pabuli, K. rhizophila, and B. licheniformis and one to the Gram-negative S. maltophilia species. All of them are involved in biodeterioration, as reported in many works (Tarsitani et al., 2014; Saiz-Jimenez, 2017). P. pabuli, besides the microbial corrosion of glass, is also involved in biodeterioration of historic limestone buildings (Marvasi et al., 2009; Skipper and Skipper, 2016); B. licheniformis, instead, is a facultative anaerobe, and it has been identified on deteriorated paintings (Capodicasa et al., 2010). Moreover, Bacillus, Kocuria, and Paenibacillus spp. can grow in presence of nitrate without oxygen on organic substrates and are reported to be involved in salt efflorescence (Laiz et al., 2000). Indeed, nitrifying bacteria oxidize ammonia to nitrite and nitrate ions, which lead to nitric acid formation. This process causes stone dissolution and formation of soluble nitrate salts, appearing as efflorescence; this phenomenon is also triggered by humidity (Kumar and Kumar, 1999). Basilica di San Nicola in Carcere Church, located near the Tiber River, is rich in humidity, favoring the growth of microorganisms. Humidity can react with the matrix of stones, inducing degradation, and especially may cause dissolution of carbonates, solubilization of other elements, and subsequently, erosion of the materials. Microorganisms, together with these abiotic factors, use stone materials as a substrate, contributing to the increase of deterioration and the aesthetic damage (Sterflinger and Piñar, 2013; Dubey and Jain, 2014). Indeed, it is known that the biodeterioration process can be due to the production of inorganic acids like nitrous and nitric acid and salts like nitrite and nitrate by nitrifying bacteria (Mansch and Bock, 1998). Therefore, the results of FTIR analysis, which highlighted the presence of nitrate residues and gypsum on salt efflorescences, strongly correlate with metabolism of Bacillus, Kocuria, and Paenibacillus species isolated from the hypogeum wall (Laiz et al., 2000; Castrillón Rivera et al., 2019) (Figure 4). In agreement, at high humidity, soluble calcium carbonate is transformed into calcium sulfate, namely gypsum, causing visible salt efflorescence (Atlas et al., 1988).
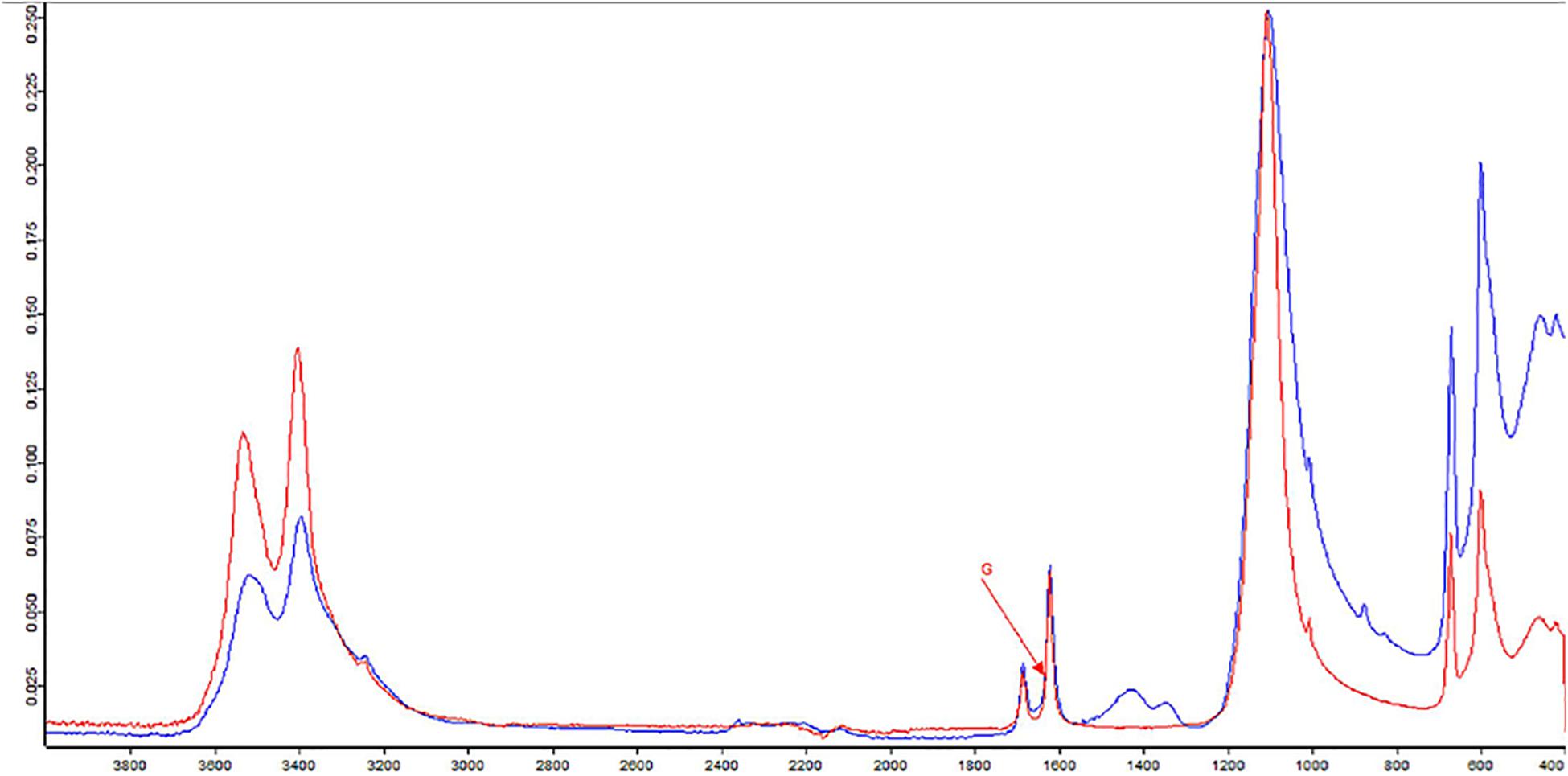
Figure 4. Fourier-transform infrared spectroscopy (FTIR) spectra of sample from hypogeum in blue; in red, the reference (G = gypsum).
Moreover, among the isolated bacteria, K. rhizophila IP5, B. licheniformis IP7, and S. maltophilia IP9 were able to form biofilm. This bacterial aggregation is the main cause of biodeterioration, since microorganisms can resist adverse abiotic conditions and many biocides and are also able to deteriorate manufactures (Stewart, 2002; Dakal and Cameotra, 2012; Rivera et al., 2018). This was confirmed by the antibiotic susceptibility test that revealed several resistances for the different strains; indeed, it has been reported that some resistance mechanisms are common to both biocides and antibiotics (Scientific Committee on Emerging and Newly Identified Health Risks [SCENIHR], 2009). In recent years, to solve problems linked to resistance to biocides, new technologies have been developed to combat biodeterioration and to restore stone buildings, using bacterial metabolism. Microbially induced carbonate precipitation was studied as a promising technology for the bioremediation of such structures, thanks to the eco–friendly, self-healing, and highly durable nature of these remedies. Furthermore, nitrate-reducing aerobic and facultative anaerobic bacteria could be chosen for their ability to remove nitrate crusts and efflorescences from stone materials (Dhami et al., 2014; Romano et al., 2019).
The combination of the metagenomic approach with the traditional microbiology integrated with the powerful diagnostic technique of FTIR spectroscopy might reveal new complex structures of microbial communities that will help to establish preservation and restoration strategies.
Conclusion
Preservation of CH is very difficult, and multidisciplinary approaches are highly demanded. Abiotic- and biotic-derived damages in CH mix continuously make the individuation of the cause impossible or extremely difficult, as well as its removal. For instance, efflorescence from a fresco could pave the way for microbial colonization that, in turn, could activate other chemical reactions transforming the materials. Thus, combined strategies for monitoring damages on CH and finding their cause are needed. This study proposes and tries validating a workflow consisting in novel molecular approaches and standard microbiological and chemical analysis. Notably, we here define an intervention protocol for sampling, sequencing, and analysis of the taxonomical composition of the microbiome on the surface of the hypogeum of the Basilica di San Nicola in Carcere Church. The overlap between the two approaches is feeble but present; probably, the huge number of uncultivable microorganisms bias the output and make the comparison with standard isolation procedures difficult. Nevertheless, the new bioinformatic tool here presented, ALISIA, could represent a fast and reliable means for understanding the complex composition of the biotic components of CH such as frescos, paintings, and statues, allowing, where needed, a tailored intervention for restoring the manufactures.
Data Availability Statement
The datasets generated and analyzed for this study can be found in the repositories DDBJ/ENA/GenBank as BioProject PRJNA578894 and BioSample SAMN13086612.
Author Contributions
AG designed ALISIA, performed the sequencing, performed the bioinformatic analysis, and edited the manuscript. MB performed the molecular analysis, performed the metadata analysis, and edited the manuscript. EZ performed the molecular analysis and drafted and edited the manuscript. RF performed the molecular analysis. ES performed the microbiological experiments. MS performed the FTIR analysis. DU conceived the study, edited the manuscript, and provided funding. MR conceived the study, drafted and edited the manuscript, and provided funding.
Funding
This research was partially supported by the project ADAMO B86C18001220002 of the Excellence Centre at the Lazio Technological District for Cultural Heritage (DTC).
Conflict of Interest
AG, MB, and MR are associates of the academic startup of Sapienza, SARA EnviMob s.r.l.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Alessandra Acconci and Dr. Marco Mangano for the possibility of sampling in the case study selected in the framework of the ADAMO project.
References
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169. doi: 10.1128/mmbr.59.1.143-169.1995
Atlas, R. M., Chowdhury, A. N., and Gauri, K. L. (1988). Microbial calcification of gypsum – rock and sulfated marble. Stud. Conserv. 33, 149–153. doi: 10.1179/sic.1988.33.3.149
Bergkessel, M., and Guthrie, C. (2013). Colony PCR. Biolab 529, 299–309. doi: 10.1016/B978-0-12-418687-3.00025-2
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Caneva, G., Nugari, M. P., and Salvadori, O. (2008). “Control of biodeterioration and bioremediation techniques,” in Plant Biology for Cultural Heritage: Biodeterioration and Conservation, eds M. P. Nugari, and O. Salvadori (Los Angeles: Getty Conservation Institute).
Caneva, G., and Salvadori, O. (1989). “Sistematica e sinsistematica delle comunità vegetali nella pianificazione di interventi di restauro TT - Systematics and synsystematics of plant communities in the planning of conservation treatment,” in Il Cantiere Della Conoscenza: il Cantiere del RESTAuro: Atti Del Convegno Di Studi, Bressanone.
Capodicasa, S., Fedi, S., Porcelli, A. M., and Zannoni, D. (2010). The microbial community dwelling on a biodeteriorated 16th century painting. Int. Biodeterior. Biodegrad. 64, 727–733. doi: 10.1016/j.ibiod.2010.08.006
Castrillón Rivera, L., Palma Ramos, A. I., Castañeda Sánchez, J., and Elisa Drago Serrano, M. (2019). “Origin and control strategies of biofilms in the cultural heritage,” in Antimicrobials, Antibiotic Resistance, Antibiofilm Strategies and Activity Methods, ed. S. Kırmusaoğlu (London: IntechOpen), doi: 10.5772/intechopen.79617
Dakal, T. C., and Cameotra, S. S. (2012). Microbially induced deterioration of architectural heritages: routes and mechanisms involved. Environ. Sci. Eur. 24:36. doi: 10.1186/2190-4715-24-36
Dhami, N. K., Reddy, M. S., and Mukherjee, A. (2014). Application of calcifying bacteria for remediation of stones and cultural heritages. Front. Microbiol. 5:304. doi: 10.3389/fmicb.2014.00304
Dubey, S., and Jain, S. K. (2014). Effect of humidity on fungal deteriogens of ancient monuments. Int. Res. J. Biol. Sci. 3, 84–86.
Glass, N. L., and Donaldson, G. C. (1995). Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 61, 1323–1330. doi: 10.1128/aem.61.4.1323-1330.1995
Gómez-Rubio, V. (2017). ggplot2 - elegant graphics for data analysis. J. Stat. Softw. 77, 1–3. doi: 10.18637/jss.v077.b02
González, J. M., and Saiz-Jiménez, C. (2005). Application of molecular nucleic acid-based techniques for the study of microbial communities in monuments and artworks. Int. Microbiol. 8, 189–194.
Gutarowska, B., Celikkol-Aydin, S., Bonifay, V., Otlewska, A., Aydin, E., Oldham, A. L., et al. (2015). Metabolomic and high-throughput sequencing analysis-modern approach for the assessment of biodeterioration of materials from historic buildings. Front. Microbiol. 6:979. doi: 10.3389/fmicb.2015.00979
Hanson, B., Zhou, Y., Bautista, E. J., Urch, B., Speck, M., Silverman, F., et al. (2016). Characterization of the bacterial and fungal microbiome in indoor dust and outdoor air samples: a pilot study. Environ. Sci. Process. Impacts 18, 713–724. doi: 10.1039/C5EM00639B
Hugenholtz, P. (2002). Exploring prokaryotic diversity in the genomic era. Genom. Biol. 3:REVIEWS0003.
Hugerth, L. W., Muller, E. E. L., Hu, Y. O. O., Lebrun, L. A. M., Roume, H., Lundin, D., et al. (2014). Systematic design of 18S rRNA gene primers for determining eukaryotic diversity in microbial consortia. PLoS One 9:e95567. doi: 10.1371/journal.pone.0095567
Kumar, R., and Kumar, A. V. (1999). Biodeterioration of Stone in Tropical Environments: An Overview. Los Angeles, CA: Getty Publications.
Laiz, L., Recio, D., Hermosin, B., and Saiz-Jimenez, C. (2000). “Microbial communities in salt efflorescences,” in Of Microbes and Art, eds O. Ciferri, P. Tiano, and G. Mastromei (Boston, MA: Springer), 77–88. doi: 10.1007/978-1-4615-4239-1-6
Lane, D. J. (1991). “6S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematic, eds E. Stackebrandt and M. Goodfellow (New York: John Wiley and Sons), 115–175.
Mansch, R., and Bock, E. (1998). Biodeterioration of natural stone with special reference to nitrifying bacteria. Biodegradation 9, 47–64. doi: 10.1023/A:1008381525192
Marvasi, M., Vedovato, E., Balsamo, C., Macherelli, A., Dei, L., Mastromei, G., et al. (2009). Bacterial community analysis on the mediaeval stained glass window “Natività” in the florence cathedral. J. Cult. Herit. 10, 124–133. doi: 10.1016/j.culher.2008.08.010
Medlin, L., Elwood, H. J., Stickel, S., and Sogin, M. L. (1988). The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71, 491–499. doi: 10.1016/0378-1119(88)90066-2
Otlewska, A., Adamiak, J., and Gutarowska, B. (2014). Application of molecular techniques for the assessment of microorganism diversity on cultural heritage objects. Acta Biochim. Pol. 61, 217–225.
Pinna, D., and Salvadori, O. (2005). “Meccanismi generali dei processi di biodeterioramento,” in La Biologia Vegetale Per i Beni Culturali, Biodeterioramento e Conservazione, eds G. Caneva, M. P. Nugari, and O. Salvadori (Firenze: Nardini Editore), 15–34.
Qi-Wang, Ma, G. Y., He, L. Y., and Sheng, X. F. (2011). Characterization of bacterial community inhabiting the surfaces of weathered bricks of Nanjing Ming city walls. Sci. Total Environ. 409, 756–762. doi: 10.1016/j.scitotenv.2010.11.001
Radaelli, A., Paganini, M., Basavecchia, V., Elli, V., Neri, M., Zanotto, C., et al. (2004). Identification, molecular biotyping and ultrastructural studies of bacterial communities isolated from two damaged frescoes of St Damian’s Monastery in Assisi. Lett. Appl. Microbiol. 38, 447–453. doi: 10.1111/j.1472-765X.2004.01514.x
Rappé, M. S., and Giovannoni, S. J. (2003). The uncultured microbial majority. Annu. Rev. Microbiol. 57, 369–394. doi: 10.1146/annurev.micro.57.030502.090759
Rivera, L. E. C., Ramos, A. P., Sánchez, J. I. C., and Serrano, M. E. D. (2018). “Origin and control strategies of biofilms in the cultural heritage,” in Antimicrobials, Antibiotic Resistance, Antibiofilm Strategies and Activity Methods, ed. S. Kırmusaoğlu (London: IntechOpen), doi: 10.5772/intechopen.79617
Romano, I., Abbate, M., Poli, A., and D’Orazio, L. (2019). Bio-cleaning of nitrate salt efflorescence on stone samples using extremophilic bacteria. Sci. Rep. 9:1668. doi: 10.1038/s41598-018-38187-x
Saiz-Jimenez, C. (2017). Molecular Biology and Cultural Heritage. Milton Park: Taylor & Francis Company, doi: 10.1201/9780203746578
Scala, V., Grottoli, A., Cigliano, R. A., Anzar, I., Beccaccioli, M., Fanelli, C., et al. (2017). Careful with that axe, gene, genome perturbation after a PEG-mediated protoplast transformation in Fusarium verticillioides. Toxins 9:E183. doi: 10.3390/toxins9060183
Schifano, E., Zinno, P., Guantario, B., Roselli, M., Marcoccia, S., Devirgiliis, C., et al. (2019). The Foodborne strain Lactobacillus fermentum MBC2 triggers pept-1-dependent pro-longevity effects in Caenorhabditis elegans. Microorganisms 7:45. doi: 10.3390/microorganisms7020045
Scientific Committee on Emerging and Newly Identified Health Risks [SCENIHR] (2009). Assessment of the antibiotic resistance effects of biocides. Eur. Comm. 6, 25–64.
Sert, H., Sümbül, H., and Sterflinger, K. (2007). A new species of Capnobotryella from monument surfaces. Mycol. Res. 111, 1235–1241. doi: 10.1016/j.mycres.2007.06.011
Skipper, P., and Skipper, L. (2016). “A survey of bacterial colonisation of historic limestone buildings: lincoln cathedral and St. Peter at Gowts, United Kingdom,” in Rehab 2014 – Proceedings of the International Conference on Preservation, Maintenance and Rehabilitation of Historic Buildings and Structures (Hoboken, NJ: Green Lines Institut).
Stackebrandt, E., Koch, C., Gvozdiak, O., and Schumann, P. (1995). Taxonomic dissection of the genus Micrococcus: Kocuria gen. nov., Nesterenkonia gen. nov., Kytococcus gen. nov., Dermacoccus gen. nov., and micrococcus cohn 1872 gen. emend. Int. J. Syst. Bacteriol. 45, 682–692. doi: 10.1099/00207713-45-4-682
Sterflinger, K., and Piñar, G. (2013). Microbial deterioration of cultural heritage and works of art — tilting at windmills? Appl. Microbiol. Biotechnol. 97, 9637–9646. doi: 10.1007/s00253-013-5283-1
Stewart, P. S. (2002). Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 292, 107–113. doi: 10.1078/1438-4221-00196
Tarsitani, G., Moroni, C., Cappitelli, F., Pasquariello, G., and Maggi, O. (2014). Microbiological analysis of surfaces of leonardo da vinci’s atlantic codex: biodeterioration risk. Int. J. Microbiol. 2014:214364. doi: 10.1155/2014/214364
Tyler, A. D., Mataseje, L., Urfano, C. J., Schmidt, L., Antonation, K. S., Mulvey, M. R., et al. (2018). Evaluation of oxford nanopore’s minion sequencing device for microbial whole genome sequencing applications. Sci. Rep. 8:10931. doi: 10.1038/s41598-018-29334-5
Urzí, C., and De Leo, F. (2001). Sampling with adhesive tape strips: an easy and rapid method to monitor microbial colonization on monument surfaces. J. Microbiol. Methods 44, 1–11. doi: 10.1016/S0167-7012(00)00227-X
Warscheid, T., and Braams, J. (2000). Biodeterioration of stone: a review. Int. Biodeterior. Biodegrad. 46, 343–368. doi: 10.1016/s0964-8305(00)00109-8
Wilimzig, M. (1996). “Deterioration and conservation of stone,” in Proceedings of the 8th International Congress on Deterioration and Conservation of Stone (Dixon, CA), 579–584.
Zanni, E., Schifano, E., Motta, S., Sciubba, F., Palleschi, C., Mauri, P., et al. (2017). Combination of metabolomic and proteomic analysis revealed different features among Lactobacillus delbrueckii subspecies bulgaricus and lactis strains while in vivo testing in the model organism Caenorhabditis elegans highlighted probiotic properties. Front. Microbiol. 8:1206. doi: 10.3389/fmicb.2017.01206
Keywords: nanopore, bioinformatic, microbial community, artistic surfaces, non invasive approach
Citation: Grottoli A, Beccaccioli M, Zoppis E, Fratini RS, Schifano E, Santarelli ML, Uccelletti D and Reverberi M (2020) Nanopore Sequencing and Bioinformatics for Rapidly Identifying Cultural Heritage Spoilage Microorganisms. Front. Mater. 7:14. doi: 10.3389/fmats.2020.00014
Received: 21 October 2019; Accepted: 14 January 2020;
Published: 21 February 2020.
Edited by:
Gabriel Maria Ingo, Italian National Research Council, ItalyReviewed by:
David Chelazzi, University of Florence, ItalyMonica Albini, Italian National Research Council, Italy
Copyright © 2020 Grottoli, Beccaccioli, Zoppis, Fratini, Schifano, Santarelli, Uccelletti and Reverberi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Massimo Reverberi, bWFzc2ltby5yZXZlcmJlcmlAdW5pcm9tYTEuaXQ=