 Simon Evertz1*
Simon Evertz1* Volker Schnabel1Mathias Köhler2
Volker Schnabel1Mathias Köhler2 Ines Kirchlechner2
Ines Kirchlechner2 Paraskevas Kontis2Yen-Ting Chen1Rafael Soler2B. Nagamani Jaya2Christoph Kirchlechner2Denis Music1
Paraskevas Kontis2Yen-Ting Chen1Rafael Soler2B. Nagamani Jaya2Christoph Kirchlechner2Denis Music1 Baptiste Gault2,3Jochen M. Schneider1Dierk Raabe2Gerhard Dehm2
Baptiste Gault2,3Jochen M. Schneider1Dierk Raabe2Gerhard Dehm2- 1Materials Chemistry, RWTH Aachen University, Aachen, Germany
- 2Max-Planck-Institut für Eisenforschung, Düsseldorf, Germany
- 3Department of Materials, Royal School of Mines, Imperial College London, London, United Kingdom
Quantum mechanically guided materials design has been used to predict the mechanical property trends in crystalline materials. Thereby, the identification of composition-structure-property relationships is enabled. However, quantum mechanics based design guidelines and material selection criteria for ultra-strong metallic glasses have been lacking. Hence, based on an ab initio model for metallic glasses in conjunction with an experimental high-throughput methodology geared toward revealing the relationship between chemistry, topology and mechanical properties, we propose principles for the design of tough as well as stiff metallic glasses. The main design notion is that a low fraction of hybridized bonds compared to the overall bonding in a metallic glass can be used as a criterion for the identification of damage-tolerant metallic glass systems. To enhance the stiffness of metallic glasses, the bond energy density must be increased as the bond energy density is the origin of stiffness in metallic glasses. The thermal expansion, which is an important glass-forming identifier, can be predicted based on the Debye-Grüneisen model.
Introduction
Today, 60 years after metallic glasses were first introduced by Klement et al. (1960), a large number of metallic glass compositions have been identified (Li et al., 2017) and their characteristic topological, magnetic, corrosive and mechanical properties have been widely explored (Ashby and Greer, 2006). Metallic glasses are amorphous alloys, lacking long-range order and microstructure (Ashby and Greer, 2006). They are produced by rapid quenching below the glass transition temperature Tg, where kinetic limitations prevent crystallization. Therefore, they are frozen in a local minimum of the energy landscape, exhibiting higher energy than their crystalline counterparts and are hence metastable (Stillinger, 1995; Debenedetti and Stillinger, 2001).
Traditionally, three rules are followed in designing metallic glasses with a high glass forming ability (Inoue, 2000). These rules require for a metallic glass: (a) a multi-component system with more than three constituents; (b) a large atomic size difference between the main constituents, i.e., larger than 12%; (c) negative enthalpies of mixing between the main constituents (Greer, 1993; Inoue, 2000; Greer, 2009). However, deviations from these rules have been found, for instance in the Pd-(Cu)-Ni-P system. Despite not following the empirical rules, a high glass forming ability was reported to be due to the coexistence of two types of short-range ordered clusters in these glasses, namely trigonal prisms and tetragonal dodecahedra in case of the Pd-Cu-Ni-P glass that hinder atomic rearrangements in this material (Inoue, 2000).
Due to their amorphous nature, mechanical properties such as hardness, yield strength, and toughness of metallic glasses are often promising for structural applications (Ashby and Greer, 2006). Metallic glasses can exhibit high resilience together with low damping (Ashby and Greer, 2006), allowing the efficient storage and release of elastic energy. In addition, metallic glasses cover a wide range of fracture toughness together with a high yield strength (Ashby and Greer, 2006; Demetriou et al., 2011). However, tough metallic glasses appear to fail in a brittle manner as shear is concentrated in shear bands leading to work softening (Greer, 2009; Greer et al., 2013). Confinement of shear bands can prohibit catastrophic failure from a single shear band (Greer, 2009; Kumar et al., 2011). Mechanical properties depend on the chemistry but also on sample dimensions and processing history (Greer, 2009; Greer et al., 2013). Processing influences the atomic configuration and especially the free volume in the glass (Li et al., 2007; Cheng and Ma, 2011). The fracture toughness, which is a crucial material parameter for any load-bearing structural material, is enhanced as the free volume is increased (Xu et al., 2010). While intrinsic toughening of metallic glasses focuses on crack initiation, extrinsic toughening strategies focus on crack propagation and utilize finely dispersed phase fractions (Xu et al., 2010) to counteract brittle failure of the material.
Next to their mechanical properties, the soft-magnetic properties of metallic glasses favor their application in transformer sheets and electronic communication (Inoue et al., 2004; Chen et al., 2018). The magnetic moment in metallic glasses is weakened by the introduction of metalloids due to hybridizing bonds (Hostert et al., 2012).
In this review on quantum-mechanically guided design of metallic glasses, we focus on the influence of chemical composition on the electronic structure as well as on the mechanical properties of metallic glasses, especially on stiffness and toughness in monolithic metallic glasses. We investigate composition induced changes in the electronic structure and relate those to property changes to derive design principles that are more precise than the traditional empirical rules. To validate our design approach, we combine ab initio modeling with experimental high-throughput characterization methods to investigate the chemistry-topology relationship and probe the validity of the predicted mechanical properties on the most promising samples thereof.
In the following sections, we review our work on ab initio modeling of metallic glasses as well as on high throughput methods to characterize metallic glasses. Finally, we show that fracture toughness is related to the fraction of hybridized bonds on the overall bonding and stiffness of metallic glasses originates in the bond energy density.
Ab Initio Molecular Dynamics Model of Metallic Glasses
Due to the dense packing and the formation of short-range ordered clusters of atoms in metallic glasses, the modeling of metallic glasses needs to take chemical effects and many-body interactions into account (Greer, 2009). As the selection of reliable interatomic potentials for classical, semi-empirical potential molecular dynamics (MD) is challenging due to the anharmonicity in metallic glasses (Lambson et al., 1986; Crespo et al., 2016; Aitken et al., 2018), density functional theory-based ab initio MD has been used to calculate the properties of a vast number of materials as it does not require the input of interatomic potentials while being able to cover many-body interactions. By ab initio modeling, an in-depth analysis of the structure-property relationship of metallic glasses is possible that is otherwise challenging to probe only by experimental means, including parameters such as the partial pair correlations (Qin et al., 2007; Ganesh and Widom, 2008; Hui et al., 2008b, 2009; Fang et al., 2009; Tian et al., 2011; Zhang et al., 2015; Hunca et al., 2016; Yu et al., 2016), coordination polyhedra (Ganesh and Widom, 2008; Fang et al., 2009; Fujita et al., 2009; Hui et al., 2009; Hirata et al., 2011; Tian et al., 2011; Kumar et al., 2011; Durandurdu, 2012; Wu et al., 2012; Zhang et al., 2015; Huang et al., 2016; Yu et al., 2016) and bonding (Kumar et al., 2011; Hunca et al., 2016). However, modeling the dynamics involved in relaxation and rejuvenation phenomena is out of reach for ab initio molecular dynamics due to the limitations of time steps and cell size as discussed below.
To reveal the structure-property relationship in metallic glasses and predict stiffness and toughness based on the electronic structure, we performed large scale ab initio calculations in the past years. This allows for an in-depth analysis of the electronic structure, which cannot be obtained by classical molecular dynamical calculations. However, the aim of predicting mechanical properties of metallic glasses from ab initio calculations raises two major challenges: First, while the elastic properties and thermal expansion can be directly calculated from ab initio configurations, mechanical properties involving the rearrangement of atoms such as fracture toughness cannot be predicted directly. This requires new predictor functions and their validation. Second, the ab initio configuration has to be representative of the corresponding synthesized materials. Hence, high-throughput synthesis and characterization of topology and microstructure by pair distribution functions and atom probe tomography are employed to compare the synthesized material with the configuration obtained by ab initio calculations. Finally, the predictor functions need to be validated by extracting the mechanical properties by experimental means such as micro-mechanical bending tests. Specifically, we define a cubic supercell with specific chemical composition and obtain an amorphous structure by ab initio heating-quenching cycles. From this glassy cell, mechanical properties are calculated either directly or by the introduction of new predictor functions. These introduced predictor functions are then validated experimentally and revisited to improve their predictive power.
Our ab initio approach starts with a cubic supercell containing in the order of 115 atoms (Hostert et al., 2011), which is in line with the typical size of a supercell for glassy structures found in literature (Hui et al., 2008b; Fang et al., 2009; Fujita et al., 2009; Hirata et al., 2011; Tian et al., 2011; Kumar et al., 2011; Galván-Colín et al., 2015; Zhang et al., 2015; Huang et al., 2016) and represents the lower limit required for physically significant models (Holmström et al., 2010). This cell is then annealed at an extremely high temperature such as 4000 K for 400 fs (Hostert et al., 2011), then quenched to 0 K (Hostert et al., 2011; Schnabel et al., 2015) or room temperature (Zhang et al., 2015) with infinite quenching rate. At the final temperature, the atomic structure is equilibrated (Hostert et al., 2011; Zhang et al., 2015; Yu et al., 2016). The heating – quenching – equilibration cycle is done once (Fang et al., 2009; Fujita et al., 2009; Galván-Colín et al., 2015; Zhang et al., 2015) or repeatedly until the volume is self-consistent (Hostert et al., 2011).
Alternative approaches to obtain an amorphous configuration include: stepwise cooling to simulate a cooling rate (Hui et al., 2008b; Fang et al., 2009; Fujita et al., 2009; Hirata et al., 2011; Tian et al., 2011; Kumar et al., 2011; Durandurdu, 2012; Wu et al., 2012; Hunca et al., 2016; Yu et al., 2016); cooling a supercell from melt, equilibration below the melting point and subsequent cooling to room temperature with an imposed cooling rate (Qin et al., 2007; Jakse and Pasturel, 2008; Hui et al., 2009; Huang et al., 2016); or by conducting several tempering steps (Ganesh and Widom, 2008; Hui et al., 2008a). Thereby, structures consistent with physical samples can be achieved (Galván-Colín et al., 2015). The ab initio models are probed for consistency with physical samples by comparison with experimentally obtained X-ray scattering data (Ganesh and Widom, 2008; Hui et al., 2009; Hirata et al., 2011; Hostert et al., 2011; Wu et al., 2012; Galván-Colín et al., 2015; Huang et al., 2016; Yu et al., 2016), extended X-ray absorption fine structure (EXAFS) (Fujita et al., 2009; Kumar et al., 2011), electron nano-beam diffraction (Hirata et al., 2011), density (Hostert et al., 2011) and coordination numbers (Fang et al., 2009).
To obtain an amorphous structure in silico, one option to reconstruct the structure from experimental topology data (X-ray scattering, neutron scattering, EXAFS) is the use of Reverse Monte Carlo modeling (RMC) (Jóvári et al., 2007; Mattern et al., 2009; Senkov et al., 2010). In RMC, a starting configuration with N atoms is iteratively fitted to the available experimental data. In every iteration, the difference X 20 of the structure factor of the starting configuration and the experimental structure factor is calculated. Then, one atom is randomly moved. The move is rejected, if it results in an atomic distance smaller than a predefined cut-off distance. After the move, the difference X 2N between the structure factor of the new configuration and the experimental structure is calculated. If X 2N < X 20, the move is accepted. Otherwise, it is accepted with a probability of exp(−(X 2N−X 20)/2). This iteration is repeated until X 2 reaches an equilibrium value (McGreevy, 2001).
We investigated the influence of the quenching rate for the Co-Ta-Fe-B system (Hostert et al., 2011). Comparing supercells quenched with 1014 and 1016 K s–1 and infinite cooling rate, the total energy differences between the two finite and the infinite quenching rates are −0.5% for 1016 K s–1 quenching rate and −1.2% for 1014 K s–1 quenching rate (Hostert et al., 2011). The comparison of Young’s modulus and mass density of the infinitely fast quenched ab initio configuration is consistent with values obtained experimentally from a sample synthesized by magnetron sputtering. To compare the topology of the simulated material with a physical sample, the total pair distribution function (PDF) obtained for the simulated material with an infinite quenching rate is shown in Figure 1 together with the experimental PDF obtained by high-energy X-ray scattering. The calculated PDF shows the same features as the experimental PDF (Hostert et al., 2011). The calculated principal peak indicating the first coordination shell coincides very well with the experimental first coordination shell, while the agreement between theory and experiment of the second and third coordination shell is weaker due to the limited size of the calculated supercell (Hostert et al., 2011).
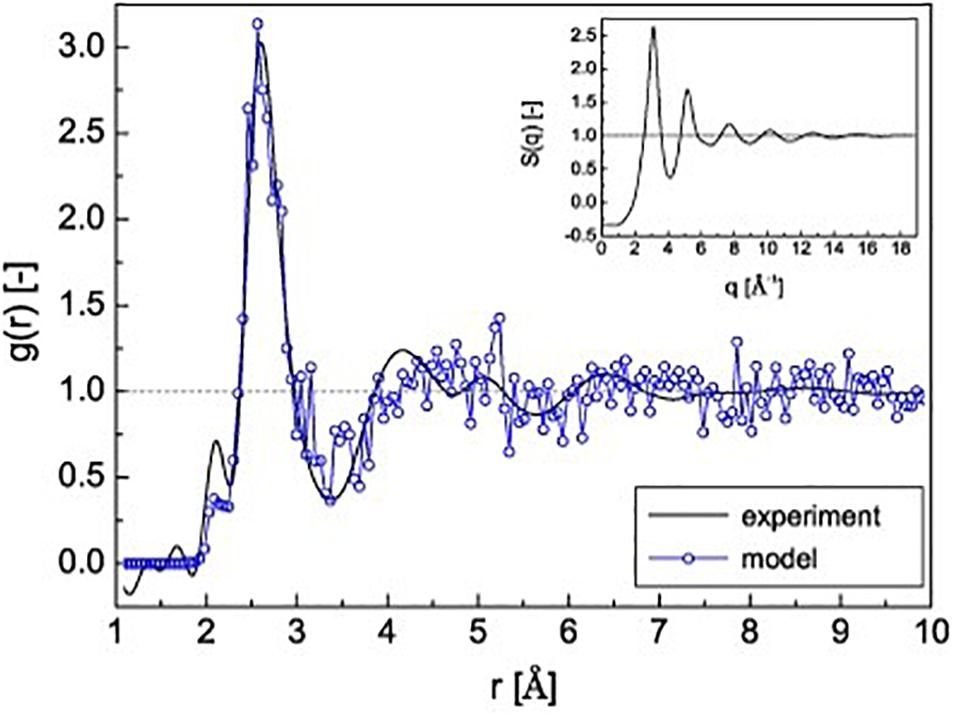
Figure 1. Pair distribution function g(r) of experimentally and ab initio calculated Co45.5Fe24Ta6B24.5. Inset: structure factor S(q) of the experimental sample (Hostert et al., 2011). © IOP Publishing. Reproduced from Hostert et al. (2011) with permission from IOP. All rights reserved.
High-Throughput Characterization
To validate the representativeness of the ab initio configuration and the properties predicted directly by ab initio calculations, a high-throughput synthesis and characterization route is required. To explore a large compositional range, the use of sample libraries with spatially resolved analysis techniques has been proven efficient (Sakurai et al., 2007.; Deng et al., 2007; Li et al., 2008, 2017, 2019; Aono et al., 2010, 2011; Guo et al., 2011; Sakurai et al., 2011; Wang et al., 2011; Ding et al., 2012, 2014; Gregoire et al., 2012; Tsai and Flores, 2014, 2016; Liu et al., 2016; Schnabel et al., 2016b; Bordeenithikasem et al., 2017; Ren et al., 2018; Zhang et al., 2018; Zheng et al., 2019). Examples of synthesis routes for combinatorial samples are physical vapor deposition (Sakurai et al., 2007, 2011; Deng et al., 2007; Li et al., 2008, 2017, 2019; Aono et al., 2011; Guo et al., 2011; Wang et al., 2011; Ding et al., 2012, 2014; Gregoire et al., 2012; Liu et al., 2016; Schnabel et al., 2016b; Bordeenithikasem et al., 2017; Ren et al., 2018; Zhang et al., 2018; Zheng et al., 2019) or laser additive manufacturing (Tsai and Flores, 2014, 2016) of metallic glasses.
Our high-throughput approach to correlate chemistry, topology and mechanical properties is based on magnetron sputtering of combinatorial samples with the same sputtering parameters on silicon and polyimide substrates, enabling to screen samples with in-plane chemical gradients (scheme shown in Figure 2) by energy-dispersive X-ray spectroscopy, high-energy X-ray scattering, atom-probe tomography (APT) and nanoindentation (Schnabel et al., 2016b). The analysis of the Co-Ta-Zr-B system (Figure 2) shows a chemically induced bond weakening of the metal-metal bonds (i.e., increase of bond distances, 1st bond distances correspond predominantly to Co-Co bonds, while 2nd bond distances correspond predominantly to Zr-Zr bonds) with increasing boron content as inferred from a shift in peak positions in the pair distribution function (Schnabel et al., 2016b). However, due to the increased B content, the stiffness is enhanced along the Co-B gradient (Schnabel et al., 2016b). With increasing Zr content, the presence of two Co- and Zr-rich amorphous phases is suggested from the PDF (not shown here, c.f. Schnabel et al., 2016b) and confirmed on selected samples by APT. The periodicity is 20–30 nm independent of composition (Schnabel et al., 2016b). The correlation between composition and mechanical properties can also be revealed by studying thin film metallic glasses on micro-cantilever arrays (Guo et al., 2011).
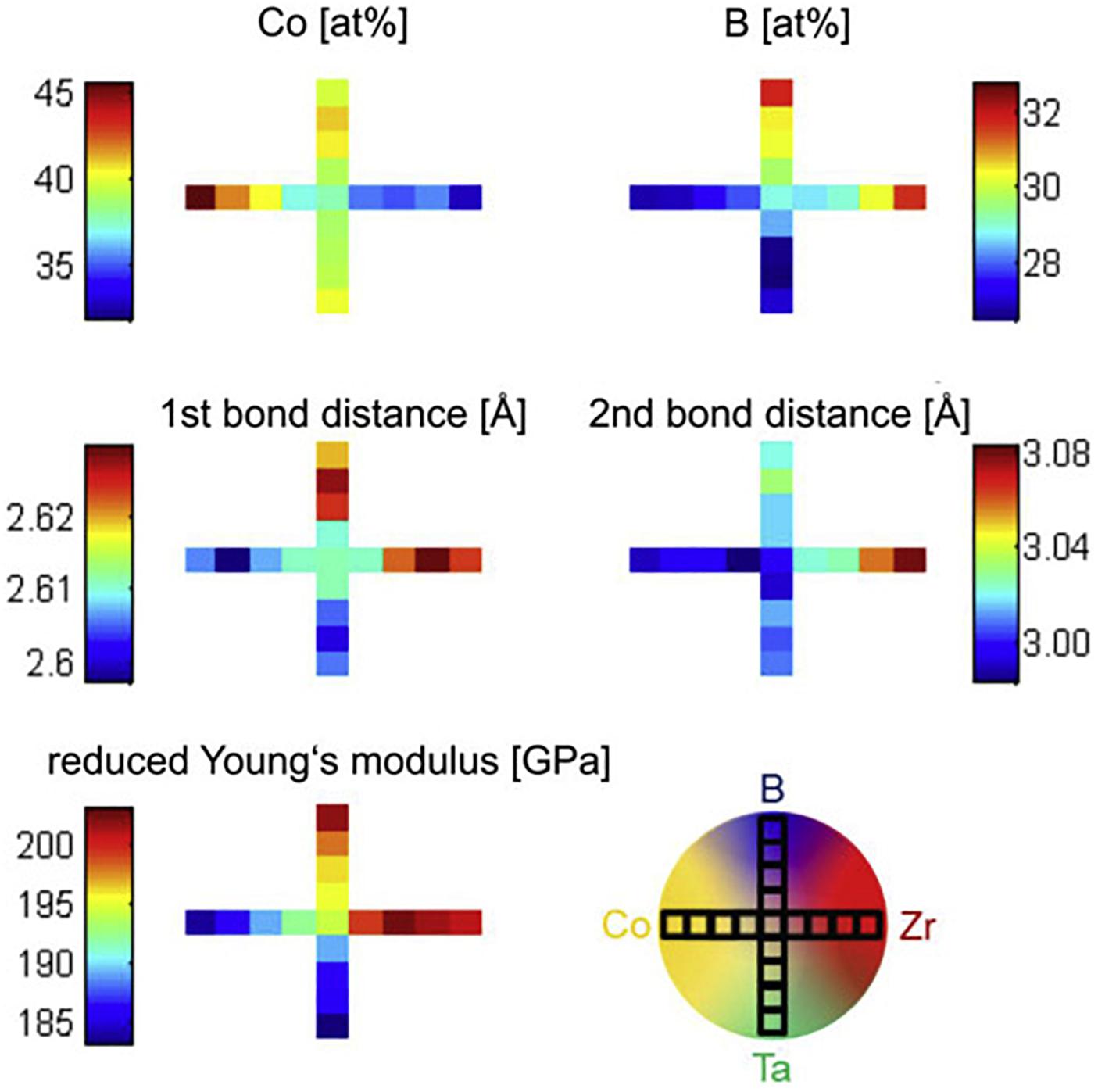
Figure 2. Co and B concentration, 1st and 2nd bond distance and Young’s modulus along the composition gradients of a combinatorial Co-Zr-Ta-B metallic glass as sketched in the lower right scheme of a combinatorial sample. The color scheme indicates the value of the quantity shown along the respective gradients. Each box in the gradient correlates to the measurement location on the investigated sample. Reproduced from Schnabel et al. (2016b) with permission from Elsevier.
In addition to mechanical properties, topology, and composition, the glass forming ability can be investigated by high-throughput methods. Ding et al. placed a gas releasing agent below combinatorial magnetron sputtered thin film metallic glasses to investigate thermoplastic formability by blow molding along the concentration gradient (Ding et al., 2014). Sheet resistivity (Li et al., 2019), resistivity (Zhang et al., 2018, Zheng et al., 2019), thermography (Aono et al., 2010, 2011), laser spike annealing (Bordeenithikasem et al., 2017) and image contrast (Ding et al., 2012) have been employed to explore glass forming ability. The use of nanocalorimetry allows the determination of glass transition temperature and glass transition enthalpy (Gregoire et al., 2012). Combinatorial synthesis and characterization are also used to investigate corrosion (Sakurai et al., 2011; Li et al., 2017), antibacterial properties (Liu et al., 2016) and biocompatibility of metallic glasses (Li et al., 2017).
While magnetron sputtering allows for high cooling rates that enable the synthesis of a large range of amorphous compositions, laser deposition allows the high-throughput synthesis via melting which is closer to synthesis conditions of bulk metallic glasses (Tsai and Flores, 2014, 2016). In contrast to magnetron sputtering, this allows the screening of alloys for glass forming ability and mechanical properties as shown by Tsai and Flores for Cu-Zr (Tsai and Flores, 2014) and Cu-Zr-Ti (Tsai and Flores, 2016). However, this method requires more preparation for the investigation of topological changes along the gradients, as samples have to be taken out of the gradient for electron or X-ray scattering experiments.
Despite experimental high-throughput methods, systematic screening by ab initio and experimental means of all possible metallic glass compositions based on the elements in the periodic table would take extremely long. Hence, Ren et al. introduced a machine learning model to predict the glass forming ability (Ren et al., 2018). Starting with a limited amount of data, the predictions of the machine learning model are validated by combinatorial magnetron sputtered samples. The experimental results are fed back into the machine learning model, thereby improving the model (Ren et al., 2018). By the combination of machine learning and combinatorial testing, Ren et al. accelerate the screening of ternary alloys by 100 times compared to traditional methods.
Origin of Stiffness in Metallic Glasses
The stiffness in terms of bulk modulus of metallic glasses is defined by the chemical composition and topology (Davis et al., 1982). Bulk modulus is usually lower in the amorphous structure compared to the crystal structure. This is proposed to originate from strongly bonded metal-metalloid clusters connected by weaker bonds in the amorphous structure (Davis et al., 1982). The lower bulk modulus in the glass compared to the corresponding crystal infers a lower internal bond energy density of the glass compared to the crystal (Gilman, 1975). It has been reported that the bond stiffness and bond strength critically affect the elastic moduli (Zhao et al., 2016). Other work claims variations in bulk modulus are a density effect (Cheng and Ma, 2009). In terms of electronic structure, the origin of stiffness is the valence electron density (Gilman, 2008; Pang et al., 2013), if covalent bonding does not prevail (Pang et al., 2013). The elastic constants can be calculated on the continuum scale from the curvature of the total energy-volume curve of the supercell (Zhang, 2013). As the elastic constants originate from the atomic bonding, they reflect the topology (Davis et al., 1976) and atomic bond strength on a macroscopic scale (Zhang, 2013).
We found, however, that the valence electron density does not scale universally with bulk modulus for metallic glasses (Schnabel et al., 2017). Schnabel et al. (2017) report two different linear relations between B and valence electron density for metal-metal glasses and for metal-metalloid glasses. The comparison of density, electronic structure, and bond energy, obtained by integration of the crystal orbital Hamilton population (Dronskowski and Bloechl, 1993), with bulk moduli of different metal-metal and metal-metalloid metallic glasses indicates the bond energy density as the origin of stiffness in metallic glasses (Schnabel et al., 2017). This underlines the early notion by Gilman that the internal energy density of a glass correlates with the elastic moduli (Gilman, 1975). The fit of bulk modulus as a function of bond energy density (Figure 3) shows the universality of this correlation for metallic glasses. Thereby, the bond energy density is consistent with previous literature as it combines the notions of strong metal-metalloid bonds and the valence electron density.
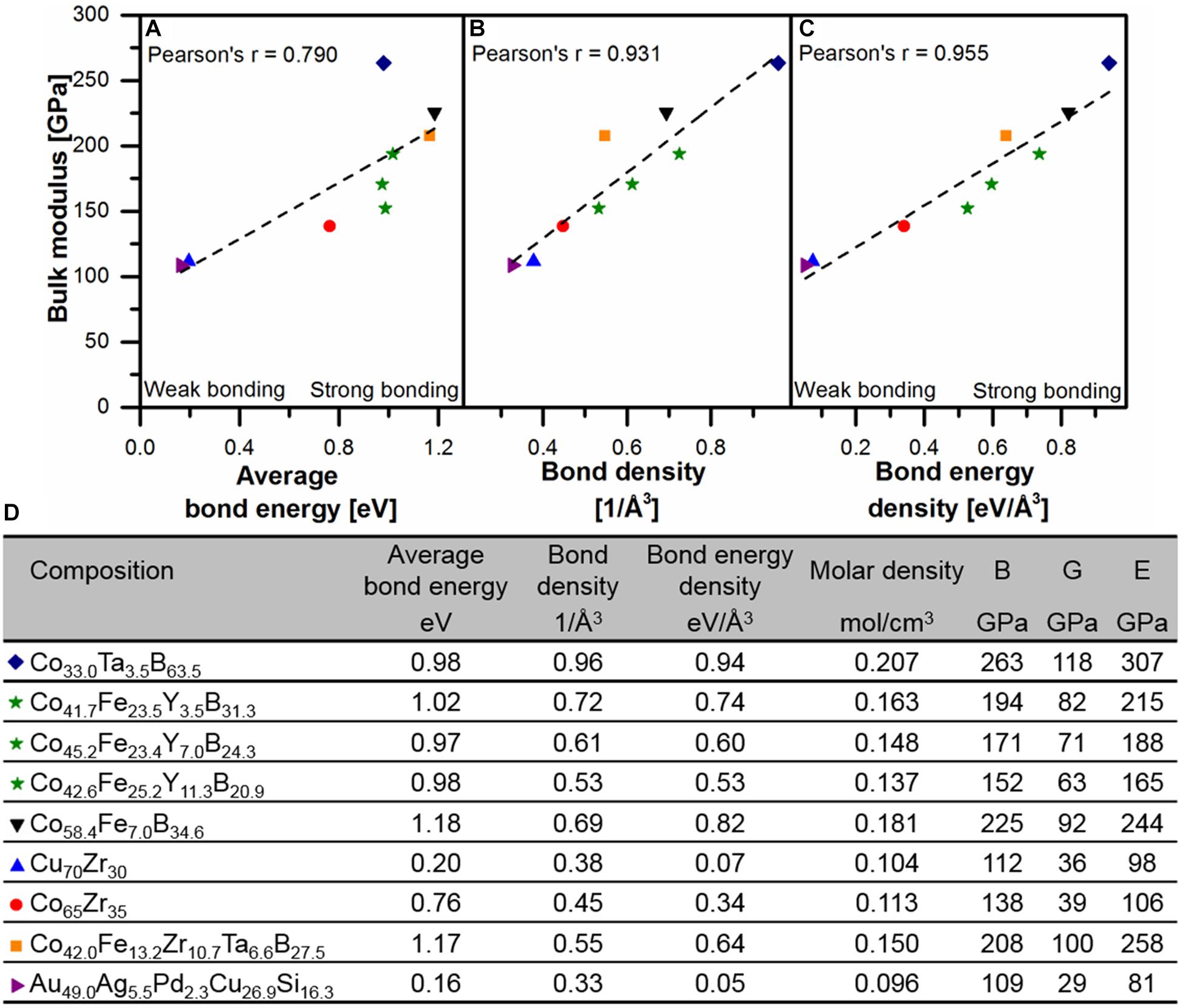
Figure 3. Comparison of bulk modulus as a function of average bond energy (A), bond density (B) and bond energy density (C) based on ab initio calculations. The table (D) summarizes chemical composition, average bond energy, bond density, bond energy density, molar density, bulk, shear and Young’s modulus of data points shown in (A–C). B is the bulk modulus, G shear modulus and E Young’s modulus. © IOP Publishing. Reproduced from Schnabel et al. (2017) with permission from IOP. All rights reserved.
Quantum Mechanical Design Proposal for Fracture Toughness
Next to stiffness, the combination of the mutually exclusive properties strength and toughness (Ritchie, 2011), also referred to as damage tolerance, is of importance for structural applications. While the elastic constants can be directly calculated from the ab initio configuration, for the fracture toughness new predictor functions have to be developed. Therefore, the origin of fracture toughness in metallic glasses needs to be understood. In metallic glasses, a large bulk to shear modulus ratio (B/G) favors damage tolerance, as a low shear modulus facilitates the formation of multiple shear transformation zones, while a high bulk modulus prevents cavity formation inside the glass (Demetriou et al., 2011; Ritchie, 2011). As B/G is proportional to the Poisson’s ratio, a brittle to ductile transition at a Poisson’s ratio of 0.31–0.32 was proposed by Lewandowski et al. (2005). This brittle-to-ductile transition has been proven not to be universal for all metallic glass systems (Raghavan et al., 2009; Zhu et al., 2011; He et al., 2012; Schnabel et al., 2016a). Instead, the shear modulus, the homologous temperature Troom/Tg (Zhu et al., 2011), and the free volume content (Raghavan et al., 2009) are proposed as design guidelines for tough metallic glasses.
Shear transformation zones and free volume are considered as defects in metallic glasses (Spaepen, 2006) and determine the plasticity of metallic glasses (Sun and Wang, 2015). Hence, the toughness of metallic glasses is sensitive to the composition and topology, such as geometrically unfavored structural motifs (Greer et al., 2013; Xu and Ma, 2014). Temperature rises in shear bands are large for brittle and small for tough metallic glasses (Yang et al., 2016). Local fluctuations in the elastic properties are found for metallic glasses with macroscopic plasticity, while they are lacking for brittle metallic glasses (Wang et al., 2009; Stoica et al., 2010). The influence of free volume content on shear band evolution can be observed in nanoindentation (Li et al., 2007). Wang et al. (2010) report a critical free volume content of 2.4% as the onset of yielding for metallic glasses. However, most investigations on the mechanism of plasticity in metallic glasses focus on the topology, whereas the focus should be on the chemical bonding – being the basis of atomic cohesion and hence mechanical properties.
Thus, based on ab initio calculations, the fraction of hybridized bonds has been proposed as a qualitative fingerprint for fracture toughness in metallic glasses (Schnabel et al., 2016a). Comparing the electronic density of states (DOS) of Cu70Zr30 and Pd57Al24Cu11Y8 metallic glasses exhibiting fracture toughness of 2.7 and 49.0 MPa.m0.5, respectively, a larger band overlap and hence stronger hybridization has been observed between Cu and Zr compared to the major constituents Pd and Al (Figure 4). This is consistent with the analysis of bonding states via crystal orbital Hamilton populations (COHP), where the Pd-based glass shows more anti-bonding states than Cu70Zr30 and hence weaker bonding (Schnabel et al., 2016a). A lower fraction of hybridized bonds hence promotes shear relaxation and enhances toughness by easing the formation of shear transformation zones (Schnabel et al., 2016a). In a sheared metallic glass, the DOS is shifted toward the Fermi level. Therefore, the bonds become weaker and the glass softer. This is consistent with the effects of micro-alloying that affects the hybridization of the constituents (Sun and Wang, 2015).
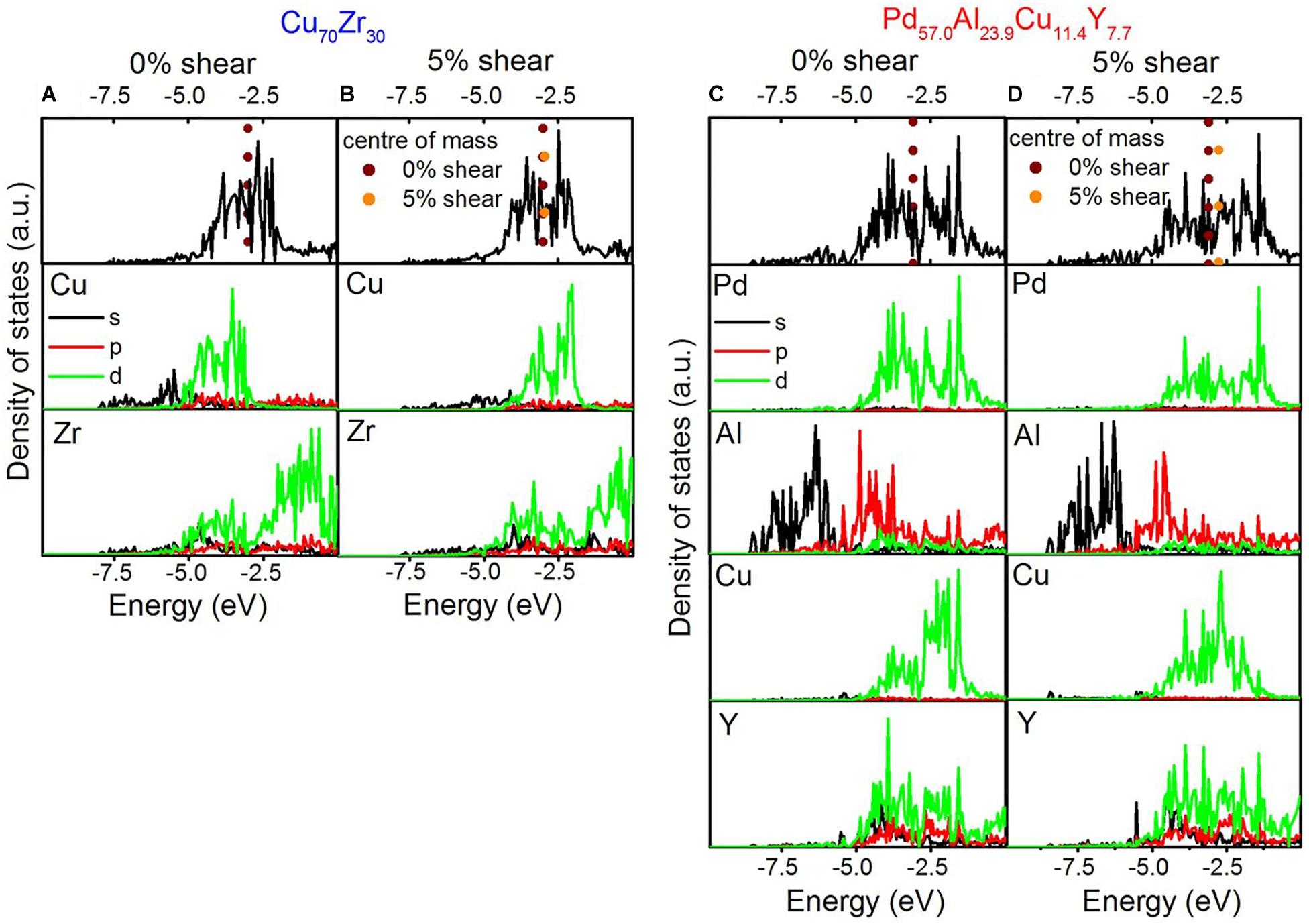
Figure 4. Electronic density of states of Cu70Zr30 (A,B) and Pd57.0Al23.9Cu11.4Y7.7 (C,D). The density of states for the simulation cells without shear is shown in (A,C) and with 5% shear in (B,D). The Fermi level is set to 0 eV. Reproduced from Schnabel et al. (2016a) under the Creative Commons CC BY license.
Based on the notion that the toughness of metallic glasses is defined by the electronic structure (Schnabel et al., 2016a), we predicted quantitatively that the fracture toughness of metallic glasses is maximized as the fraction metallic bonds is maximized (Evertz et al., 2020). However, as it is not possible to quantify the metallic bonds, focusing on the minimization of hybridized bonds enables the quantitative prediction of fracture toughness. Calculating the bond overlap by means of crystal orbital overlap populations (COOP) (Hoffmann, 1987), the hybridized bonds can be analyzed. In a pure metallically bonded material, all electrons at the Fermi level Ef are delocalized and shared in a free electron gas. Hence, the ideal metallic glass in terms of toughness has a COOP(Ef) = 0.
The experimental fracture toughness of metallic glasses scales with COOP(Ef) as shown in (Figure 5A) (Evertz et al., 2020). Considering the COOP(Ef) for a metallic glass with unknown fracture toughness such as Pd57.4Al23.5Y7.8Ni11.3, which has a low fraction of hybridized bonds based on a qualitative analysis of the density of states (Evertz et al., 2020), the fracture toughness can be predicted based on the fit of the data in Figure 5A. This results in a fracture toughness of 95 ± 20 MPa.m0.5 for Pd57.4Al23.5Y7.8Ni11.3. Thereby, not only the fraction of hybridized bonds is crucial, but also the antibonding bonding character at Ef, as antibonding states increase the total energy of the system and promote bond separation (Smith, 2000; Evertz et al., 2020). Comparing the COOPs of the glasses shown in Figure 5A (Figure 5B), a bonding to antibonding transition is observed for all metallic glasses between −5 and −3 eV, while the amount of states at Ef differs significantly. However, the difference in the COOP(Ef) depends mainly on the major constituent of the alloy (Evertz et al., 2020). By this, the design guideline based on the Poisson’s ratio (Lewandowski et al., 2005) may be understood as a useful indicator of the major constituent of tough metallic glasses, while the quantitative prediction of fracture toughness requires the analysis of the electronic structure.
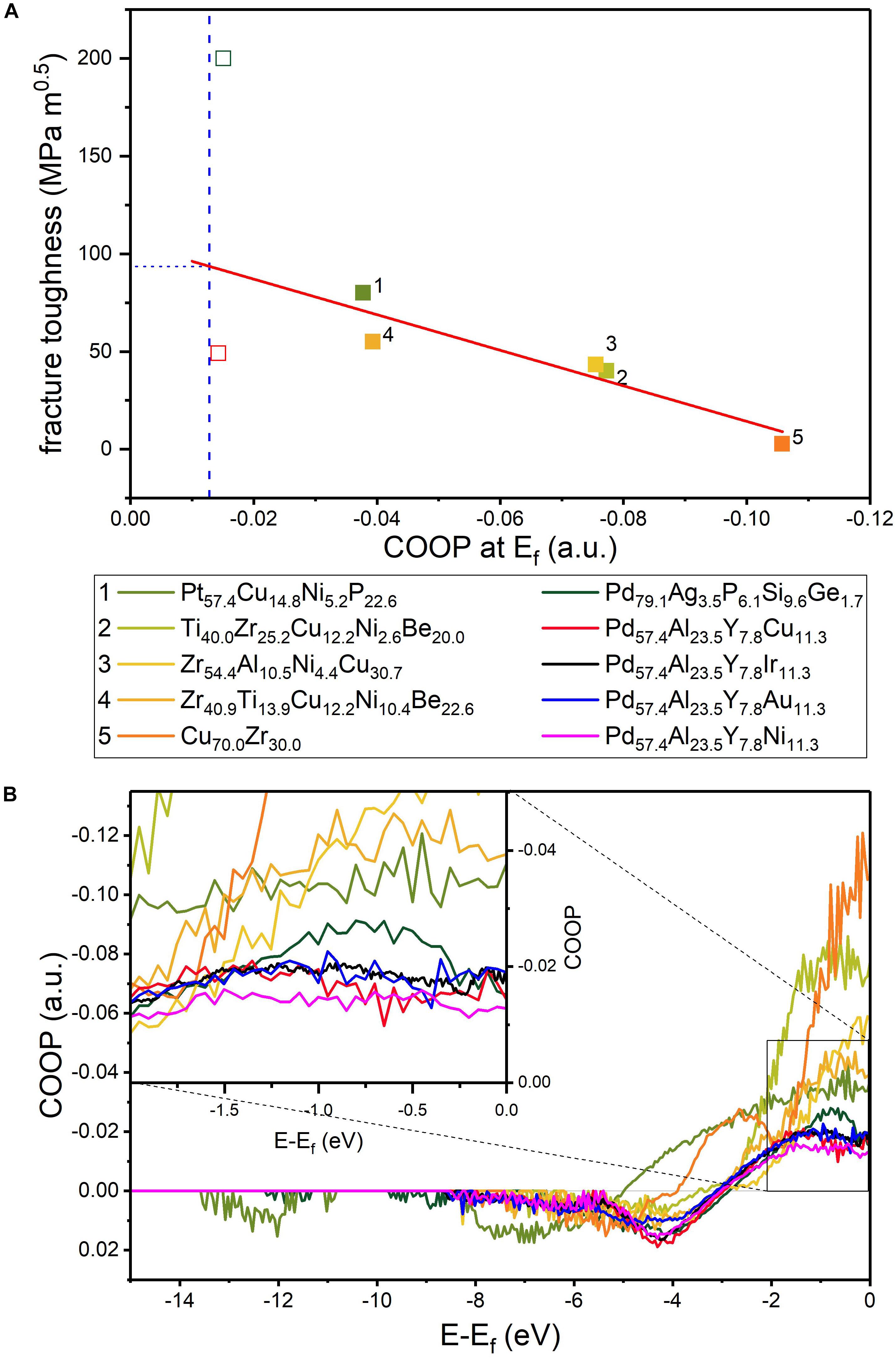
Figure 5. (A) Fracture toughness as a function of COOP at the Fermi level. Open symbols are not included in the fit as they may be sample size dependent (Evertz et al., 2020). The vertical dashed blue line indicates the COOP of Pd57.4Al23.5Y7.8Ni11.3 (B) COOP of the glasses shown in (A). Reproduced from Evertz et al. (2020) under the Creative Commons CC BY license.
To appraise critically the predicted fracture toughness of 95 ± 20 MPa.m0.5 for Pd57.4Al23.5Y7.8Ni11.3 and the new predictor function introduced in Evertz et al. (2020), micro-mechanical beam bending experiments were employed. In these experiments, the Pd57.9Al25.0Y4.9Ni12.2 metallic glass cantilever did not break while showing the formation of multiple shear bands that are confined by the stress gradient in the bent cantilever (Evertz et al., 2020). Moreover, since no crack extension has been observed, the fracture toughness exceeds the boundaries of micro-mechanical fracture experiments (Evertz et al., 2020). This is consistent with the high fracture toughness predicted based on the electronic structure (Evertz et al., 2020).
Prediction of Thermal Expansion
The thermal expansion is important for dimensional accuracy and thermal stresses during near net shape processing of metallic glasses (Telford, 2004; Schroers, 2010). The thermal expansion of metallic glasses is similar to that of crystalline metals such as steel (Telford, 2004). Due to the lack of a first-order phase transition, the volume shrinkage is already reduced to a minimum compared with crystalline materials (Schroers, 2010) but the maximum thermal expansion before Tg is only 25% of the expansion of metals before melting. Thermal expansion is related to the glass transition, as the product of thermal expansion coefficient and Tg is constant (Kato et al., 2008) and can hence be employed to predict the glass forming ability (Li et al., 2009). Experimentally, the thermal expansion of metallic glasses can be measured by high energy X-ray scattering as it is connected to the shift of the principal peak in the structure factor during heating (Mattern et al., 2003, 2004, 2012; Hajlaoui et al., 2004; Yavari et al., 2005; Louzguine-Luzgin et al., 2006; Louzguine-Luzgin and Inoue, 2007; Bednarcik et al., 2011) or by dilatometry (Nishiyama et al., 2000; Gonchukova and Drugov, 2003; Jing, 2003; Jing et al., 2007; Kato et al., 2008; Li et al., 2009).
The coefficient of thermal expansion had been predicted by the volume change of an ab initio configuration as a function of the temperature (Hunca et al., 2016), which is rather expensive in terms of computational resources. Theoretically, the thermal expansion coefficient of metallic glasses can be predicted by the Debye-Grüneisen model. For Pd57.4Al23.5Y7.8Ni11.3 the thermal expansion coefficient has been predicted to be 3.4⋅10−5 K–1 at room temperature, which is in agreement with the experimental value 3.5⋅10−5 K–1 obtained by high energy X-ray scattering (Figure 6; Evertz et al., 2017). However, the prediction of thermal expansion for higher temperatures is only possible as long as no structural changes appear. The deviation between predicted and measured thermal expansion coefficient does indicate structural relaxation or rejuvenation in the physical sample (Evertz et al., 2017). The time scales of the relaxation and rejuvenation processes are too large to cover with ab initio calculations and computational resources which are available today. However, after complete relaxation, the thermal expansion is reversible (Bednarcik et al., 2011) and could hence be covered by ab initio calculations.
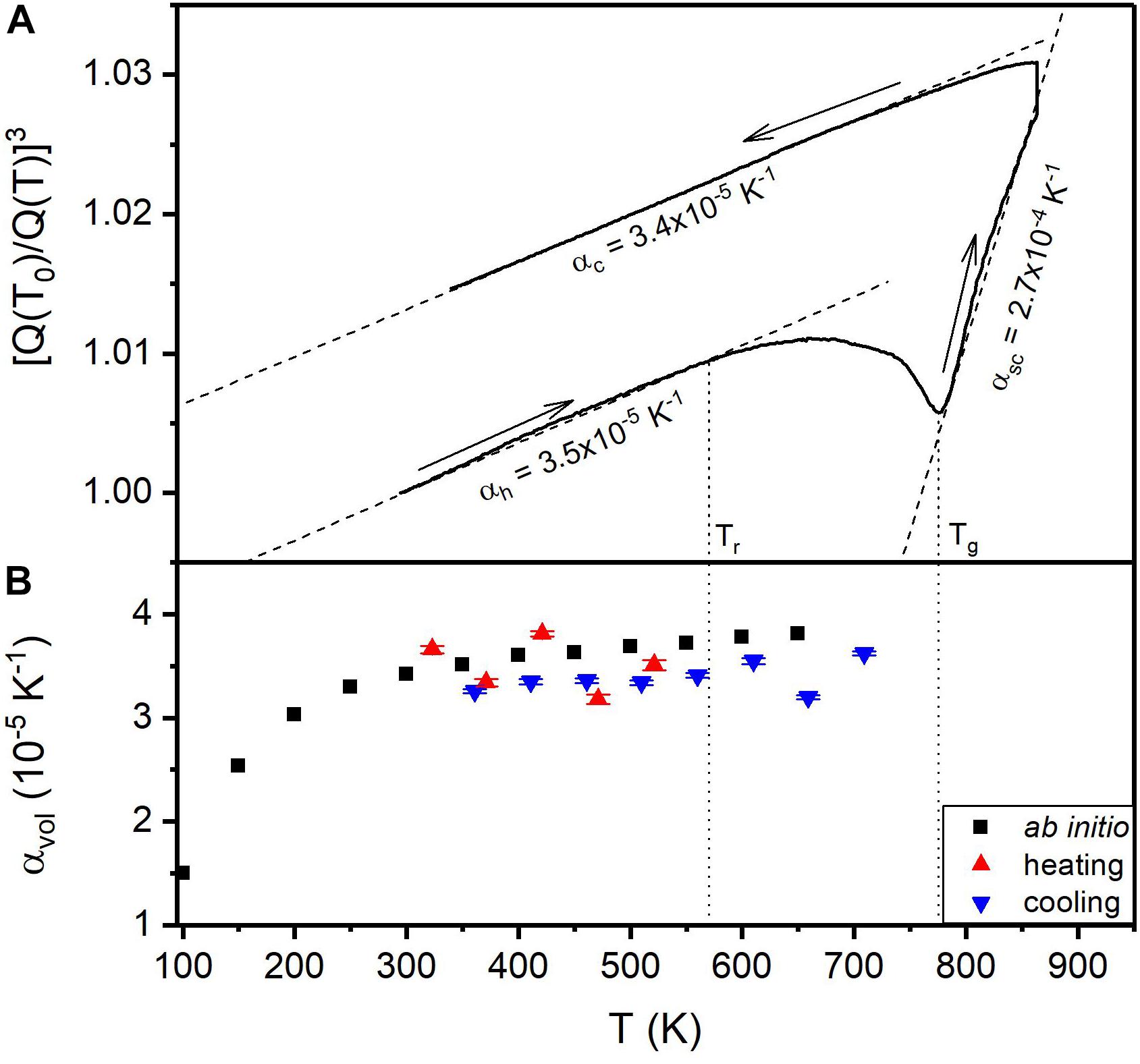
Figure 6. Shift of the principal peak of the structure factor of Pd62.1Al27.3Y4.2Ni6.4 (A), an indicator of volume expansion. (B) Coefficient of thermal expansion calculated ab initio by the Debye-Grüneisen model (black) in comparison with experimental values (red and blue). Reproduced from Evertz et al. (2017) under the Creative Commons CC BY license.
The nature of the thermal expansion for metallic glasses is not associated with an increase of interatomic distances but rather with an increase in free volume content between the short-range ordered clusters (Bar’yakhtar et al., 1989; Qu et al., 2011). This is because the principle peak of the structure factor describes medium to long-range order, as can be observed by comparing pair distribution functions calculated from the principal peak of the structure factor and from the complete structure factor (Bednarcik et al., 2011). However, more recent studies report that the thermal expansion is not equal to changes in free volume, as free volume is annihilated during annealing (structural relaxation) by atomic rearrangements in the glass (Chen, 1978; Nishiyama et al., 2000; Jing, 2003; Hajlaoui et al., 2004; Meng et al., 2006). Especially above Tg atomic rearrangements are no longer negligible, hence the volume change in the supercooled and liquid states cannot be attributed to pure thermal expansion (Mattern et al., 2003, 2012; Georgarakis et al., 2011). The change of thermal expansion, thereby, can serve as an indicator of the fragility of the glass (Bendert et al., 2013). This can be understood in terms of an energy landscape, where at low temperature the glassy state is dominated by local energy minima or basins (Debenedetti and Stillinger, 2001). At high temperatures, the glass can sample the whole potential energy landscape due to enhanced atomic mobility. Hence, in the landscape dominated region, thermal vibrations dominate thermal expansion, while in the high temperature range, structural changes are more important (Bendert et al., 2012) and surpass thermal expansion.
Discussion
Based on the literature discussed above, ab initio calculations are the state of the art to investigate the structure and topology of metallic glasses in depth. The protocols to create a glassy structure representing real physical glasses are thereby similar while differing in the number of cooling steps, heating rates, and heating-quenching cycles. To evaluate the relevance of the ab initio structures, the topology of the simulated samples is compared to the topology of the experimental samples. If the overall topologies are consistent, the structural analysis of the prediction often yields more detail than experimental studies (Qin et al., 2007; Ganesh and Widom, 2008; Hui et al., 2008b; Fang et al., 2009; Fujita et al., 2009; Hui et al., 2009; Hirata et al., 2011; Tian et al., 2011; Kumar et al., 2011; Durandurdu, 2012; Wu et al., 2012; Zhang et al., 2015; Huang et al., 2016; Hunca et al., 2016; Yu et al., 2016). However, the electronic structure of metallic glasses is analyzed in only few studies (Hostert et al., 2011; Kumar et al., 2011; Hunca et al., 2016; Schnabel et al., 2016a, 2017).
High-throughput characterization techniques have been applied for metallic glasses to investigate glass forming ability, thermoplastic formability, composition, topology, and mechanical properties. However, most high-throughput studies are purely experimental, relying on trial and error (Sakurai et al., 2007, 2011; Deng et al., 2007; Aono et al., 2010, 2011; Guo et al., 2011; Wang et al., 2011; Ding et al., 2012, 2014; Gregoire et al., 2012; Tsai and Flores, 2014, 2016; Liu et al., 2016; Bordeenithikasem et al., 2017; Li et al., 2017, 2019; Zhang et al., 2018; Zheng et al., 2019); or empirical, such as machine learning approaches (Ren et al., 2018). In recent years, we combined ab initio calculations with high-throughput characterization techniques. Therefore, we propose quantum-mechanically based design guidelines of crucial properties for structural applications of metallic glasses (Schnabel et al., 2016a, 2017; Evertz et al., 2017, 2020). These quantum mechanical design proposals are: a) the fraction of hybridized bonds is the fingerprint for damage tolerance in metallic glasses (Schnabel et al., 2016a); b) the bond energy density is proposed to be the origin of the stiffness of metallic glasses (Schnabel et al., 2017); and c) the Debye-Grüneisen model is suitable to predict thermal expansion (Evertz et al., 2017).
However, ab initio calculations still have some drawbacks due to their high computational cost, leading to the analysis of some hundred atoms only, which means that only the short-range order, but not the medium-range order in metallic glasses can be studied. Moreover, ab initio produced metallic glasses are exposed to very high cooling rates (Hostert et al., 2011), originating in the small time scales accessible by ab initio methods. Hence, the simulation of structural relaxation and rejuvenation in metallic glasses is not achievable with conventional DFT codes and available resources today, which might result in different free volume contents compared to physical samples, affecting the mechanical properties (Li et al., 2007; Wang et al., 2010; Sun and Wang, 2015), especially plastic deformations. No systematic ab initio studies on the correlation between free volume and electronic structure are known to the authors so far. Hence, the effects of free volume on the electronic structure remain an open question for future studies.
Conclusion
Ab initio calculations have been used to explore composition induced changes of the topology and properties of metallic glasses. In recent years, we correlated theoretical and experimental approaches to propose design guidelines for fracture toughness and stiffness of metallic glasses. These predictions have been validated experimentally by micro-mechanical experiments. One grand future challenge is that the effect of free volume has to be included in the ab initio models to enhance the quantum-mechanical design for metallic glasses. Further, the formation of shear bands has not been understood and the role of the electronic structure for the onset of shear bands needs to be investigated.
Author Contributions
All authors contributed to the conception of the review, manuscript revision, read, and approved the submitted version. SE wrote the first draft of the manuscript.
Funding
Financial support of the German Research Foundation within the SPP 1594 “Topological Engineering of Ultrastrong Glasses” is acknowledged (DE 796/9-2, SCHN 735/22-2, and RA 659/18-2).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aitken, Z. H., Jafary-Zadeh, M., Lewandowski, J. J., and Zhang, Y.-W. (2018). Anharmonic model for the elastic constants of bulk metallic glass across the glass transition. Phys. Rev. B 97:014101. doi: 10.1103/PhysRevB.97.014101
Aono, Y., Sakurai, J., Ishida, T., Shimokohbe, A., and Hata, S. (2010). High-throughput measurement method for time–temperature-transformation diagram of thin film amorphous alloys. Appl. Phys. Express 3:125601. doi: 10.1143/APEX.3.125601
Aono, Y., Sakurai, J., Shimokohbe, A., and Hata, S. (2011). High-throughput characterization method for crystallization temperature of integrated thin film amorphous alloys using thermography. Jpn. J. Appl. Phys. 50:55601. doi: 10.1143/JJAP.50.055601
Ashby, M., and Greer, A. L. (2006). Metallic glasses as structural materials. Scr. Mater. 54, 321–326. doi: 10.1016/j.scriptamat.2005.09.051
Bar’yakhtar, V. G., Mikhailova, L. E., Il’inskii, A. G., Romanova, A. V., and Khristenko, T. M. (1989). Thermal expansion of liquid metals. Zh. Eksp. Teor. Fiz. 95, 1404–1411.
Bednarcik, J., Michalik, S., Sikorski, M., Curfs, C., Wang, X. D., Jiang, J. Z., et al. (2011). Thermal expansion of a La-based bulk metallic glass: insight from in situ high-energy x-ray diffraction. J. Phys. Condens. Matter 23:254204. doi: 10.1088/0953-8984/23/25/254204
Bendert, J. C., Blodgett, M. E., Gangopadhyay, A. K., and Kelton, K. F. (2013). Measurements of volume, thermal expansion, and specific heat in Zr 57 Cu 15.4 Ni 12.6 Al 10 Nb 5 and Zr 58.5 Cu 15.6 Ni 12.8 Al 10.3 Nb 2.8 liquids and glasses. Appl. Phys. Lett. 102:211913. doi: 10.1063/1.4808030
Bendert, J. C., Gangopadhyay, A. K., Mauro, N. A., and Kelton, K. F. (2012). Volume expansion measurements in metallic liquids and their relation to fragility and glass forming ability: an energy landscape interpretation. Phys. Rev. Lett. 109:185901. doi: 10.1103/PhysRevLett.109.185901
Bordeenithikasem, P., Liu, J., Kube, S. A., Li, Y., Ma, T., Scanley, B. E., et al. (2017). Determination of critical cooling rates in metallic glass forming alloy libraries through laser spike annealing. Sci. Rep. 7:7155. doi: 10.1038/s41598-017-07719-2
Chen, H., Dong, B., Zhou, S., Li, X., and Qin, J. (2018). Structural, magnetic, and electronic properties of Fe82Si4B10P4 metallic glass. Sci Rep 8:1331. doi: 10.1038/s41598-018-23952-9
Chen, H. S. (1978). The influence of structural relaxation on the density and Young’s modulus of metallic glasses. J. Appl. Phys. 49, 3289–3291. doi: 10.1063/1.325279
Cheng, Y. Q., and Ma, E. (2009). Configurational dependence of elastic modulus of metallic glass. Phys. Rev. B 80:611. doi: 10.1103/PhysRevB.80.064104
Cheng, Y. Q., and Ma, E. (2011). Atomic-level structure and structure–property relationship in metallic glasses. Prog. Mater. Sci. 56, 379–473. doi: 10.1016/j.pmatsci.2010.12.002
Crespo, D., Bruna, P., Valles, A., and Pineda, E. (2016). Phonon dispersion relation of metallic glasses. Phys. Rev. B 94:144205. doi: 10.1103/PhysRevB.94.144205
Davis, L. A., Chou, C.-P., Tanner, L. E., and Ray, R. (1976). Strenghts and stiffnesses of metallic glasses. Scr. Metallurg. 10, 937–940. doi: 10.1016/0036-9748(76)90217-9
Davis, L. A., Yeow, Y. T., and Anderson, P. M. (1982). Bulk stiffnesses of metallic glasses. J. Appl. Phys. 53, 4834–4837. doi: 10.1063/1.331313
Debenedetti, P. G., and Stillinger, F. H. (2001). Supercooled liquids and the glass transition. Nature 410, 259–267. doi: 10.1038/35065704
Demetriou, M. D., Launey, M. E., Garrett, G., Schramm, J. P., Hofmann, D. C., Johnson, W. L., et al. (2011). A damage-tolerant glass. Nat. Mater. 10, 123–128. doi: 10.1038/NMAT2930
Deng, Y. P., Guan, Y. F., Fowlkes, J. D., Wen, S. Q., Liu, F. X., Pharr, G. M., et al. (2007). A combinatorial thin film sputtering approach for synthesizing and characterizing ternary ZrCuAl metallic glasses. Intermetallics 15, 1208–1216. doi: 10.1016/j.intermet.2007.02.011
Ding, S., Gregoire, J., Vlassak, J. J., and Schroers, J. (2012). Solidification of Au-Cu-Si alloys investigated by a combinatorial approach. J. Appl. Phys. 111:114901. doi: 10.1063/1.4722996
Ding, S., Liu, Y., Li, Y., Liu, Z., Sohn, S., Walker, F. J., et al. (2014). Combinatorial development of bulk metallic glasses. Nat. Mater. 13, 494–500. doi: 10.1038/nmat3939
Dronskowski, R., and Bloechl, P. E. (1993). Crystal orbital hamilton populations (COHP): energy-resolved visualization of chemical bonding in solids based on density-functional calculations. J. Phys. Chem. 97, 8617–8624. doi: 10.1021/j100135a014
Durandurdu, M. (2012). Ab initio modeling of metallic Pd80Si20 glass. Comp. Mater. Sci. 65, 44–47. doi: 10.1016/j.commatsci.2012.06.040
Evertz, S., Kirchlechner, I., Soler, R., Kirchlechner, C., Kontis, P., Bednarcik, J., et al. (2020). Electronic structure based design of thin film metallic glasses with superior fracture toughness. Mater. Des. 186:108327. doi: 10.1016/j.matdes.2019.108327
Evertz, S., Music, D., Schnabel, V., Bednarcik, J., and Schneider, J. M. (2017). Thermal expansion of Pd-based metallic glasses by ab initio methods and high energy X-ray diffraction. Sci. Rep. 7:15744. doi: 10.1038/s41598-017-16117-7
Fang, H. Z., Hui, X., Chen, G. L., and Liu, Z. K. (2009). Al-centered icosahedral ordering in Cu46Zr46Al8 bulk metallic glass. Appl. Phys. Lett. 94:91904. doi: 10.1063/1.3086885
Fujita, T., Konno, K., Zhang, W., Kumar, V., Matsuura, M., Inoue, A., et al. (2009). Atomic-scale heterogeneity of a multicomponent bulk metallic glass with excellent glass forming ability. Phys. Rev. Lett. 103:75502. doi: 10.1103/PhysRevLett.103.075502
Galván-Colín, J., Valladares, A. A., Valladares, R. M., and Valladares, A. (2015). Short-range order in ab initio computer generated amorphous and liquid Cu–Zr alloys: a new approach. Phys. B Condens. Matter 475, 140–147. doi: 10.1016/j.physb.2015.07.027
Ganesh, P., and Widom, M. (2008). Ab initio simulations of geometrical frustration in supercooled liquid Fe and Fe-based metallic glass. Phys. Rev. B 77:467. doi: 10.1103/PhysRevB.77.014205
Georgarakis, K., Louzguine-Luzgin, D. V., Antonowicz, J., Vaughan, G., Yavari, A. R., Egami, T., et al. (2011). Variations in atomic structural features of a supercooled Pd–Ni–Cu–P glass forming liquid during in situ vitrification. Acta Mater. 59, 708–716. doi: 10.1016/j.actamat.2010.10.009
Gilman, J. J. (1975). Mechanical behavior of metallic glasses. J. Appl. Phys. 46, 1625–1633. doi: 10.1063/1.321764
Gilman, J. J. (2008). Electronic Basis of the Strength of Materials. Cambridge: Cambridge University Press.
Gonchukova, N. O., and Drugov, A. N. (2003). Thermal expansion of amorphous alloys. Glass Phys. Chem. 29, 184–187. doi: 10.1023/A:1023463210498
Greer, A. L. (2009). Metallic glasses on the threshold. Mater. Today 12, 14–22. doi: 10.1016/s1369-7021(09)70037-9
Greer, A. L., Cheng, Y. Q., and Ma, E. (2013). Shear bands in metallic glasses. Mater. Sci. Eng. R 74, 71–132. doi: 10.1016/j.mser.2013.04.001
Gregoire, J. M., McCluskey, P. J., Dale, D., Ding, S., Schroers, J., and Vlassak, J. J. (2012). Combining combinatorial nanocalorimetry and X-ray diffraction techniques to study the effects of composition and quench rate on Au–Cu–Si metallic glasses. Scr. Mater. 66, 178–181. doi: 10.1016/j.scriptamat.2011.10.034
Guo, Q., Zhang, L., Zeiger, A. S., Li, Y., van Vliet, K. J., and Thompson, C. V. (2011). Compositional dependence of Young’s moduli for amorphous Cu–Zr films measured using combinatorial deposition on microscale cantilever arrays. Scr. Mater. 64, 41–44. doi: 10.1016/j.scriptamat.2010.08.061
Hajlaoui, K., Benameur, T., Vaughan, G., and Yavari, A. R. (2004). Thermal expansion and indentation-induced free volume in Zr-based metallic glasses measured by real-time diffraction using synchrotron radiation. Scr. Mater. 51, 843–848. doi: 10.1016/j.scriptamat.2004.07.008
He, Q., Shang, J. K., Ma, E., and Xu, J. (2012). Crack-resistance curve of a Zr–Ti–Cu–Al bulk metallic glass with extraordinary fracture toughness. Acta Mater. 60, 4940–4949. doi: 10.1016/j.actamat.2012.05.028
Hirata, A., Guan, P., Fujita, T., Hirotsu, Y., Inoue, A., Yavari, A. R., et al. (2011). Direct observation of local atomic order in a metallic glass. Nat. Mater. 10, 28–33. doi: 10.1038/NMAT2897
Hoffmann, R. (1987). How chemistry and physics meet in the solid state. Angew. Chem. Int. Ed. Engl. 26, 846–878. doi: 10.1002/anie.198708461
Holmström, E., Bock, N., Peery, T., Chisolm, E., Lizárraga, R., de Lorenzi-Venneri, G., et al. (2010). Structure discovery for metallic glasses using stochastic quenching. Phys. Rev. B 82:024203. doi: 10.1103/PhysRevB.82.024203
Hostert, C., Music, D., Bednarcik, J., Keckes, J., Kapaklis, V., Hjorvarsson, B., et al. (2011). Ab initio molecular dynamics model for density, elastic properties and short range order of Co-Fe-Ta-B metallic glass thin films. J. Phys. Condens. Matter 23:475401. doi: 10.1088/0953-8984/23/47/475401
Hostert, C., Music, D., Bednarcik, J., Keckes, J., and Schneider, J. M. (2012). Quantum mechanically guided design of Co43Fe20Ta(5.5)X(31.5) (X=B, Si, P, S) metallic glasses. J. Phys. Condens. Matter 24:175402. doi: 10.1088/0953-8984/24/17/175402
Huang, Y., Huang, L., Wang, C. Z., Kramer, M. J., and Ho, K. M. (2016). Ab initio molecular dynamics simulations of short-range order in Zr50Cu45Al5 and Cu50Zr45Al5 metallic glasses. J. Phys. Condens. Matter 28:85102. doi: 10.1088/0953-8984/28/8/085102
Hui, X., Fang, H. Z., Chen, G. L., Shang, S. L., Wang, Y., and Liu, Z. K. (2008a). Icosahedral ordering in Zr41Ti14Cu12.5Ni10Be22.5 bulk metallic glass. Appl. Phys. Lett. 92:201913. doi: 10.1063/1.2931702
Hui, X., Fang, H. Z., Chen, G. L., Shang, S. L., Wang, Y., Qin, J. Y., et al. (2009). Atomic structure of Zr41.2Ti13.8Cu12.5Ni10Be22.5 bulk metallic glass alloy. Acta Mater. 57, 376–391. doi: 10.1016/j.actamat.2008.09.022
Hui, X., Gao, R., Chen, G. L., Shang, S. L., Wang, Y., and Liu, Z. K. (2008b). Short-to-medium-range order in Mg65Cu25Y10 metallic glass. Phys. Lett. A 372, 3078–3084. doi: 10.1016/j.physleta.2008.01.031
Hunca, B., Dharmawardhana, C., Sakidja, R., and Ching, W.-Y. (2016). Ab initio calculations of thermomechanical properties and electronic structure of vitreloy Zr41.2Ti13.8Cu12.5Ni10Be22.5. Phys. Rev. B 94:144207. doi: 10.1103/PhysRevB.94.144207
Inoue, A. (2000). Stabilization of metallic supercooled liquid and bulk amorphous alloys. Acta Mater. 48, 279–306. doi: 10.1016/S1359-6454(99)00300-6
Inoue, A., Shen, B. L., Koshiba, H., Kato, H., and Yavari, A. R. (2004). Ultra-high strength above 5000 MPa and soft magnetic properties of Co? Fe?Ta?B bulk glassy alloys. Acta Mater. 52, 1631–1637. doi: 10.1016/j.actamat.2003.12.008
Jakse, N., and Pasturel, A. (2008). Local order and dynamic properties of liquid and undercooled CuxZr1-x alloys by ab initio molecular dynamics. Phys. Rev. B 78:425. doi: 10.1103/PhysRevB.78.214204
Jing, G., Xiufang, B., Tao, L., Yan, Z., Li, T., Bo, Z., et al. (2007). Formation and interesting thermal expansion behavior of novel Sm-based bulk metallic glasses. Intermetallics 15, 929–933. doi: 10.1016/j.intermet.2006.11.003
Jing, Q. (2003). Thermal expansion behavior and structure relaxation of ZrTiCuNiBe bulk amorphous alloy. Scr. Mater. 49, 111–115. doi: 10.1016/S1359-6462(03)00240-9
Jóvári, P., Saksl, K., Pryds, N., Lebech, B., Bailey, N. P., Mellergård, A., et al. (2007). Atomic structure of glassy Mg60Cu30Y10 investigated with EXAFS, x-ray and neutron diffraction, and reverse Monte Carlo simulations. Phys. Rev. B 76:42. doi: 10.1103/PhysRevB.76.054208
Kato, H., Chen, H.-S., and Inoue, A. (2008). Relationship between thermal expansion coefficient and glass transition temperature in metallic glasses. Scr. Mater. 58, 1106–1109. doi: 10.1016/j.scriptamat.2008.02.006
Klement, W., Willens, R. H., and Duwez, P. O. L. (1960). Non-crystalline structure in solidified Gold–Silicon alloys. Nature 187, 869–870. doi: 10.1038/187869b0
Kumar, G., Desai, A., and Schroers, J. (2011). Bulk metallic glass: the smaller the better. Adv. Mater. 23, 461–476. doi: 10.1002/adma.201002148
Kumar, V., Fujita, T., Konno, K., Matsuura, M., Chen, M. W., Inoue, A., et al. (2011). Atomic and electronic structure of Pd 40 Ni 40 P 20 bulk metallic glass from ab initio simulations. Phys. Rev. B 84:134204. doi: 10.1103/PhysRevB.84.134204
Lambson, E. F., Lambson, W. A., Macdonald, J. E., Gibbs, M. R., Saunders, G. A., and Turnbull, D. (1986). Elastic behavior and vibrational anharmonicity of a bulk Pd40Ni40P20 metallic glass. Phys. Rev. B 33, 2380–2385. doi: 10.1103/physrevb.33.2380
Lewandowski, J. J., Wang, W. H., and Greer, A. L. (2005). Intrinsic plasticity or brittleness of metallic glasses. Phil. Mag. Lett. 85, 77–87. doi: 10.1080/09500830500080474
Li, G. H., Wang, W. M., Bian, X. F., Zhang, J. T., Li, R., and Qin, J. Y. (2009). Correlation between thermal expansion coefficient and glass formability in amorphous alloys. Mater. Chem. Phys. 116, 72–75. doi: 10.1016/j.matchemphys.2009.02.041
Li, J., Gittleson, F. S., Liu, Y., Liu, J., Loye, A. M., McMillon-Brown, L., et al. (2017). Exploring a wider range of Mg-Ca-Zn metallic glass as biocompatible alloys using combinatorial sputtering. Chem. Commun. (Cambridge, England) 53, 8288–8291. doi: 10.1039/c7cc02733h
Li, M.-X., Zhao, S.-F., Lu, Z., Hirata, A., Wen, P., Bai, H.-Y., et al. (2019). High-temperature bulk metallic glasses developed by combinatorial methods. Nature 569, 99–103. doi: 10.1038/s41586-019-1145-z
Li, N., Liu, L., Chen, Q., Pan, J., and Chan, K. C. (2007). The effect of free volume on the deformation behaviour of a Zr-based metallic glass under nanoindentation. J. Phys. D Appl. Phys. 40, 6055–6059. doi: 10.1088/0022-3727/40/19/043
Li, Y., Guo, Q., Kalb, J. A., and Thompson, C. V. (2008). Matching glass-forming ability with the density of the amorphous phase. Science (New York, N.Y.) 322, 1816–1819. doi: 10.1126/science.1163062
Li, Y., Zhao, S., Liu, Y., Gong, P., and Schroers, J. (2017). How many bulk metallic glasses are there? ACS Combinat. Sci. 19, 687–693. doi: 10.1021/acscombsci.7b00048
Liu, Y., Padmanabhan, J., Cheung, B., Liu, J., Chen, Z., Scanley, B. E., et al. (2016). Combinatorial development of antibacterial Zr-Cu-Al-Ag thin film metallic glasses. Sci. Rep. 6:26950. doi: 10.1038/srep26950
Louzguine-Luzgin, D. V., and Inoue, A. (2007). Thermal expansion of an amorphous alloy. Reciprocal-space versus real-space distribution functions. Physica B Condens. Matter 388, 290–293. doi: 10.1016/j.physb.2006.06.143
Louzguine-Luzgin, D. V., Inoue, A., Yavari, A. R., and Vaughan, G. (2006). Thermal expansion of a glassy alloy studied using a real-space pair distribution function. Appl. Phys. Lett. 88:121926. doi: 10.1063/1.2187955
Mattern, N., Hermann, H., Roth, S., Sakowski, J., Macht, M.-P., Jovari, P., et al. (2003). Structural behavior of Pd40Cu30Ni10P20 bulk metallic glass below and above the glass transition. Appl. Phys. Lett. 82, 2589–2591. doi: 10.1063/1.1567457
Mattern, N., Jóvári, P., Kaban, I., Gruner, S., Elsner, A., Kokotin, V., et al. (2009). Short-range order of Cu–Zr metallic glasses. J. Alloys Compd. 485, 163–169. doi: 10.1016/j.jallcom.2009.05.111
Mattern, N., Kühn, U., Hermann, H., Roth, S., Vinzelberg, H., and Eckert, J. (2004). Thermal behavior and glass transition of Zr-based bulk metallic glasses. Mater. Sci. Eng. 375–377, 351–354. doi: 10.1016/j.msea.2003.10.125
Mattern, N., Stoica, M., Vaughan, G., and Eckert, J. (2012). Thermal behaviour of Pd40Cu30Ni10P20 bulk metallic glass. Acta Mater. 60, 517–524. doi: 10.1016/j.actamat.2011.10.032
McGreevy, R. L. (2001). Reverse monte carlo modelling. J. Phys. Condens. Matter 13, R877–R913. doi: 10.1088/0953-8984/13/46/201
Meng, Q. G., Zhang, S. G., Li, J. G., and Bian, X. F. (2006). Dilatometric measurements and glass-forming ability in Pr-based bulk metallic glasses. Scr. Mater. 55, 517–520. doi: 10.1016/j.scriptamat.2006.05.036
Nishiyama, N., Horino, M., and Inoue, A. (2000). Thermal expansion and specific volume of Pd40Cu30Ni10P20 alloy in various states. Mater. Trans. JIM 41, 1432–1434. doi: 10.2320/matertrans1989.41.1432
Pang, J. J., Tan, M. J., and Liew, K. M. (2013). On valence electron density, energy dissipation and plasticity of bulk metallic glasses. J. Alloys Compd. 577, S56–S65. doi: 10.1016/j.jallcom.2012.03.036
Qin, J., Gu, T., Yang, L., and Bian, X. (2007). Study on the structural relationship between the liquid and amorphous Fe78Si9B13 alloys by ab initio molecular dynamics simulation. Appl. Phys. Lett. 90:201909. doi: 10.1063/1.2737937
Qu, D., Liss, K.-D., Yan, K., Reid, M., Almer, J. D., Wang, Y., et al. (2011). On the atomic anisotropy of thermal expansion in bulk metallic glass. Adv. Eng. Mater. 13, 861–864. doi: 10.1002/adem.201000349
Raghavan, R., Murali, P., and Ramamurty, U. (2009). On factors influencing the ductile-to-brittle transition in a bulk metallic glass. Acta Mater. 57, 3332–3340. doi: 10.1016/j.actamat.2009.03.047
Ren, F., Ward, L., Williams, T., Laws, K. J., Wolverton, C., Hattrick-Simpers, J., et al. (2018). Accelerated discovery of metallic glasses through iteration of machine learning and high-throughput experiments. Sci. Adv. 4:eaaq1566. doi: 10.1126/sciadv.aaq1566
Ritchie, R. O. (2011). The conflicts between strength and toughness. Nat. Mater. 10, 817–822. doi: 10.1038/NMAT3115
Sakurai, J., Hata, S., Yamauchi, R., and Shimokohbe, A. (2007). Searching for novel Ru-based thin film metallic glass by combinatorial Arc plasma deposition. Jpn. J. Appl. Phys. 46, 1590–1595. doi: 10.1143/jjap.46.1590
Sakurai, J., Kozako, H., Mukai, N., Ohnuma, Y., Takahashi, T., and Hata, S. (2011). Combinatorial search for Ni–Nb–Ti thin film amorphous alloys with high corrosion resistances. Jpn. J. Appl. Phys. 50:87201. doi: 10.1143/JJAP.50.087201
Schnabel, V., Evertz, S., Ruess, H., Music, D., and Schneider, J. M. (2015). Stiffness and toughness prediction of Co-Fe-Ta-B metallic glasses, alloyed with Y, Zr, Nb, Mo, Hf, W, C, N and O by ab initio molecular dynamics. J. Phys. Condens. Matter 27:105502. doi: 10.1088/0953-8984/27/10/105502
Schnabel, V., Jaya, B. N., Kohler, M., Music, D., Kirchlechner, C., Dehm, G., et al. (2016a). Electronic hybridisation implications for the damage-tolerance of thin film metallic glasses. Sci. Rep. 6:36556. doi: 10.1038/srep36556
Schnabel, V., Köhler, M., Evertz, S., Gamcova, J., Bednarcik, J., Music, D., et al. (2016b). Revealing the relationships between chemistry, topology and stiffness of ultrastrong Co-based metallic glass thin films: a combinatorial approach. Acta Mater. 107, 213–219. doi: 10.1016/j.actamat.2016.01.060
Schnabel, V., Köhler, M., Music, D., Bednarcik, J., Clegg, W. J., Raabe, D., et al. (2017). Ultra-stiff metallic glasses through bond energy density design. J. Phys. Condens. Matter 29:265502. doi: 10.1088/1361-648X/aa72cb
Schroers, J. (2010). Processing of bulk metallic glass. Adv. Mater. 22, 1566–1597. doi: 10.1002/adma.200902776
Senkov, O. N., Miracle, D. B., Barney, E. R., Hannon, A. C., Cheng, Y. Q., and Ma, E. (2010). Local atomic structure of Ca-Mg-Zn metallic glasses. Phys. Rev. B 82:193. doi: 10.1103/PhysRevB.82.104206
Spaepen, F. (2006). Homogeneous flow of metallic glasses: a free volume perspective. Scr. Mater. 54, 363–367. doi: 10.1016/j.scriptamat.2005.09.046
Stillinger, F. H. (1995). A topographic view of supercooled liquids and glass formation. Science (New York, N.Y.) 267, 1935–1939. doi: 10.1126/science.267.5206.1935
Stoica, M., Das, J., Bednarèik, J., Wang, G., Vaughan, G., Wang, W. H., et al. (2010). Mechanical response of metallic glasses: insights from in-situ high energy X-ray diffraction. JOM 62, 76–82. doi: 10.1007/s11837-010-0037-3
Sun, B. A., and Wang, W. H. (2015). The fracture of bulk metallic glasses. Prog. Mater. Sci. 74, 211–307. doi: 10.1016/j.pmatsci.2015.05.002
Telford, M. (2004). The case for bulk metallic glass. Mater. Today 7, 36–43. doi: 10.1016/S1369-7021(04)00124-5
Tian, H., Zhang, C., Wang, L., Zhao, J., Dong, C., Wen, B., et al. (2011). Ab initio molecular dynamics simulation of binary Cu64Zr36 bulk metallic glass: validation of the cluster-plus-glue-atom model. J. Appl. Phys. 109:123520. doi: 10.1063/1.3599882
Tsai, P., and Flores, K. M. (2014). A combinatorial strategy for metallic glass design via laser deposition. Intermetallics 55, 162–166. doi: 10.1016/j.intermet.2014.07.017
Tsai, P., and Flores, K. M. (2016). High-throughput discovery and characterization of multicomponent bulk metallic glass alloys. Acta Mater. 120, 426–434. doi: 10.1016/j.actamat.2016.08.068
Wang, J. G., Zhao, D. Q., Pan, M. X., Shek, C. H., and Wang, W. H. (2009). Mechanical heterogeneity and mechanism of plasticity in metallic glasses. Appl. Phys. Lett. 94:31904. doi: 10.1063/1.3073985
Wang, J. G., Zhao, D. Q., Pan, M. X., Wang, W. H., Song, S. X., and Nieh, T. G. (2010). Correlation between onset of yielding and free volume in metallic glasses. Scr. Mater. 62, 477–480. doi: 10.1016/j.scriptamat.2009.12.015
Wang, Z. T., Zeng, K. Y., and Li, Y. (2011). The correlation between glass formation and hardness of the amorphous phase. Scr. Mater. 65, 747–750. doi: 10.1016/j.scriptamat.2011.06.043
Wu, S., Kramer, M. J., Fang, X. W., Wang, S. Y., Wang, C. Z., Ho, K. M., et al. (2012). Icosahedral short-range order in amorphous Cu80Si20 by ab initio molecular dynamics simulation study. Intermetallics 30, 122–126. doi: 10.1016/j.intermet.2012.03.018
Xu, J., and Ma, E. (2014). Damage-tolerant Zr–Cu–Al-based bulk metallic glasses with record-breaking fracture toughness. J. Mater. Res. 29, 1489–1499. doi: 10.1557/jmr.2014.160
Xu, J., Ramamurty, U., and Ma, E. (2010). The fracture toughness of bulk metallic glasses. JOM 62, 10–18. doi: 10.1007/s11837-010-0052-4
Yang, G. N., Shao, Y., and Yao, K. F. (2016). The material-dependence of plasticity in metallic glasses: an origin from shear band thermology. Mater. Des. 96, 189–194. doi: 10.1016/j.matdes.2016.02.007
Yavari, A. R., Le Moulec, A., Inoue, A., Nishiyama, N., Lupu, N., Matsubara, E., et al. (2005). Excess free volume in metallic glasses measured by X-ray diffraction. Acta Mater. 53, 1611–1619. doi: 10.1016/j.actamat.2004.12.011
Yu, Q., Wang, X. D., Lou, H. B., Cao, Q. P., and Jiang, J. Z. (2016). Atomic packing in Fe-based metallic glasses. Acta Mater. 102, 116–124. doi: 10.1016/j.actamat.2015.09.001
Zhang, H., Lee, D., Shen, Y., Miao, Y., Bae, J., Liu, Y., et al. (2018). Combinatorial temperature resistance sensors for the analysis of phase transformations demonstrated for metallic glasses. Acta Mater. 156, 486–495. doi: 10.1016/j.actamat.2018.07.012
Zhang, S. G. (2013). Signature of properties in elastic constants of no-metalloid bulk metallic glasses. Intermetallics 35, 1–8. doi: 10.1016/j.intermet.2012.11.017
Zhang, X., Li, R., and Zhang, T. (2015). Ab initio molecular dynamics simulation of the surface composition of Co 54 Ta 11 B 35 metallic glasses. J. Non Cryst. Solids 425, 199–206. doi: 10.1016/j.jnoncrysol.2015.04.013
Zhao, W., Cheng, J. L., Feng, S. D., Li, G., and Liu, R. P. (2016). Intrinsic correlation between elastic modulus and atomic bond stiffness in metallic glasses. Mater. Lett. 175, 227–230. doi: 10.1016/j.matlet.2016.03.037
Zheng, J., Zhang, H., Miao, Y., Chen, S., and Vlassak, J. J. (2019). Temperature-resistance sensor arrays for combinatorial study of phase transitions in shape memory alloys and metallic glasses. Scr. Mater. 168, 144–148. doi: 10.1016/j.scriptamat.2019.04.027
Keywords: metallic glass, ab initio, quantum mechanical materials design, toughness, stiffness, micro-mechanics
Citation: Evertz S, Schnabel V, Köhler M, Kirchlechner I, Kontis P, Chen Y-T, Soler R, Jaya BN, Kirchlechner C, Music D, Gault B, Schneider JM, Raabe D and Dehm G (2020) Review on Quantum Mechanically Guided Design of Ultra-Strong Metallic Glasses. Front. Mater. 7:89. doi: 10.3389/fmats.2020.00089
Received: 06 February 2020; Accepted: 25 March 2020;
Published: 17 April 2020.
Edited by:
Jürgen Horbach, Heinrich Heine University of Düsseldorf, GermanyReviewed by:
Roger Jay Loucks, Alfred University, United StatesHaizheng Tao, Wuhan University of Technology, China
Copyright © 2020 Evertz, Schnabel, Köhler, Kirchlechner, Kontis, Chen, Soler, Jaya, Kirchlechner, Music, Gault, Schneider, Raabe and Dehm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon Evertz, ZXZlcnR6QG1jaC5yd3RoLWFhY2hlbi5kZQ==