 Mikel Irigoyen1
Mikel Irigoyen1 Jon M. Matxain
Jon M. Matxain Fernando Ruipérez
Fernando Ruipérez- 1Kimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU and Donostia International Physics Center (DIPC), Donostia, Spain
- 2POLYMAT and Physical Chemistry Department, Faculty of Pharmacy, University of the Basque Country UPV/EHU, Vitoria-Gasteiz, Spain
The introduction of dynamic bonds in polymeric materials facilitates the emergence of new functionalities, such as self-healing capacity. Understanding the role of the molecular structure in the efficiency of the self-healing process is fundamental to design new materials with improved features. Computational chemistry has emerged as a valuable tool for the characterization of polymeric materials. In this work, computational chemistry is used to analyze the observed self-healing capacity of a set of disulfide-containing polyurethanes and polymethacrylates, including different hard segments and dynamic bonds. For this purpose, a recently developed theoretical protocol has been used. This protocol is based on three parameters: the probability of generating radicals by cleavage of the disulfide bond, the energetic barrier of the exchange reaction among disulfides and the dynamics of the polymeric chains. This protocol is able to qualitatively explain the experimental self-healing properties of these materials. In particular, it explains both the great performance of two materials and the lack of self-healing capacity of another two. Besides, it can also describe the improvement of the self-healing capacity with increasing temperature. These results demonstrate the robustness and usefulness of this approach for the analysis and prediction of self-healing properties in polymeric materials. Therefore, this protocol allows to predict new materials with improved properties and will help the experimental community in the development of these improved materials.
1 Introduction
The use of dynamic covalent chemistry, introduced by Rowan et al. (2002), has emerged as a powerful tool in the design of new materials in different research fields, ranging from biochemistry to polymer chemistry. It is based on a reversible cleavage and reformation of certain covalent bonds under thermodynamic control, and its use in the design of new materials may introduce new functionalities which could assist creating materials with improved features. This improved features may vary from solid state recycling, property regulation, controllable degradation and reprocessing to self-healing capacity (Zhang et al., 2018). The dynamic character can be introduced by means of non-covalent interactions like hydrogen bonds (Cordier et al., 2008), π−π stacking (Burattini et al., 2009) or metal-ion interactions (Burnworth et al., 2011), but in the last few years, the use of dynamic covalent bonds has increased since, in addition to ensuring dynamic behavior, it also provides robustness to materials (Hager et al., 2010; Billiet et al., 2013; Hillewaere and Du Prez, 2015). Polymeric materials including dynamic bonds as crosslinks in their network have been labelled as covalent adaptive networks (CANs) and the activation of the bond-exchange reactions, which could be triggered by any external stimuli, can induce changes in their topology (Kloxin et al., 2010; Bowman and Kloxin, 2012; Kloxin and Bowman, 2012). Examples of CANs are the Diels-Alder reaction (Chen et al., 2002, 2003; Liu and Chen, 2007), boronic esters (Cash et al., 2015; Cromwell et al., 2015), olefin metathesis (Lu and Guan, 2012), polysiloxanes (Cho et al., 2012; Schmolke et al., 2015) or disulfides (Sarma et al., 2007; Belenguer et al., 2011; Matxain et al., 2016; Nevejans et al., 2016; Rekondo et al., 2014; Martin et al., 2014; Aguirresarobe et al., 2017). The use of disulfides as crosslinks, especially the aromatic ones (Rekondo et al., 2014; Martin et al., 2014; Azcune and Odriozola, 2016; Takahashi et al., 2016), has been revealed as a useful technique to generate responsive materials due to the dynamics and reversibility of this bond, which can be activated without external stimuli (Canadell et al., 2011; Lafont et al., 2012; Amamoto et al., 2012).
One of the most studied polymers in this field are the polyurethanes (PU), because of its great versatility and the ease of functionalization with many chemical moieties (Engels et al., 2013). As a consequence, PUs are very promising class of materials to be used in self-healing applications due to the range of dynamic bonds that can be incorporated (Aguirresarobe et al., 2021). The dynamic character can be introduced in different ways, for example, by urethane exchange (transcarbamoylation) (Fortman et al., 2015; Zheng et al., 2016, 2017), urea exchange (Ying et al., 2014; Zhang et al., 2016; Erice et al., 2018), or by using dichalcogenide bonds as crosslinkers, which can be both aliphatic (Otsuka et al., 2010; Xu et al., 2016; Pan et al., 2017) and aromatic (Rekondo et al., 2014; Martin et al., 2016; de Luzuriaga et al., 2016; An X. et al., 2017; Shin et al., 2020; Eom et al., 2021). The mechanical properties of PUs are greatly affected by supramolecular interactions like hydrogen bonds and by the packaging of the hard segments (Brunette et al., 1982; Mattia and Painter, 2007; Zhang et al., 2014; Erice et al., 2019). The presence of dynamic bonds may also lead to the decrease of the mechanical strength and, therefore, a balance has to be found when designing new self-healing materials (Nevejans et al., 2019).
In this way, Kim et al. studied a set of PUs with different hard segments containing disulfide bonds, in the pursuit of a robust and easily processable polymer that shows self-healing features at room temperature (Kim et al., 2017). The hard segment in PUs is generated by the reaction of isocyanates with short chain diols, and provides strength, hardness and high temperature performance. In particular, these authors synthesized three PUs with different hard segments including aromatic disulfides (Figure 1), providing varying rigidity and labeled after the isocyanate used: IP (isophorone diisocyanate), an asymmetric alicyclic structure, HM [4,4′-methylenebis (cyclohexyl isocyanate)], a symmetric alicyclic structure and M [4,4′-methylenebis (phenyl isocyanate)], an aromatic structure. The IP material was the only one to show self-healing at room temperature with mechanical properties such as robustness, stretchability and durability, surpassing those of room-temperature self-healable materials to date. The HM material showed improved self-healing efficiency with increasing temperature, while the M material showed no self-healing ability even at high temperatures. This lack of self-healing capacity has been explained due to the packaging and the restricted mobility of the chains in this material.
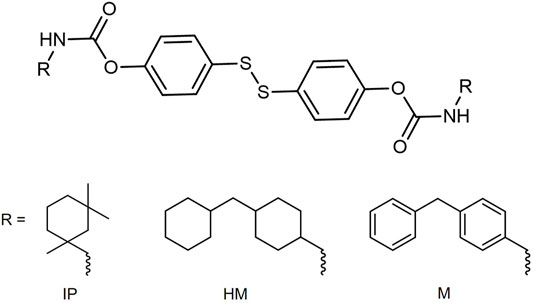
FIGURE 1. Hard segments of self-healing PUs including disulfide bonds proposed in Ref. Kim et al. (2017). The nomenclature is related to the isocyanate used to generate the hard segment: IP = isophorone diisocyanate; HM = 4,4′-methylenebis (cyclohexyl isocyanate) and M = 4,4′-methylenebis (phenyl isocyanate).
Another approach to modify or to provide with self-healing features is changing the dynamic bond. This approach has been used by Takahashi et al. (2017a), whose work focused on polymethacrylate networks including different disulfide bonds (An S. Y. et al., 2017; Liu et al., 2020; Rusayyis and Torkelson, 2020). In particular, these authors synthesized two materials: BTA, including a thermally exchangeable bis(2,2,6,6-tetramethylpiperidin-1-yl)disulfide (BiTEMPS) crosslink (Takahashi et al., 2017b; Tsurumi et al., 2020; Yokochi et al., 2021; Takashima et al., 2020, 2021; Kataoka et al., 2021), and ADSA, including and alkyllic disulfide, Figure 2. The authors observed that BTA, with N atoms linked to the disulfide, presented healing capacity both at room and at high temperature, while ADSA, with aliphatic carbon chains linked to the disulfides, did not show healing capacity even at high temperature, even though the hardener structure was the same. This is an expected result, since it has been previously demonstrated that the presence of nitrogen atoms linked to the sulfur weakens the S-S bond, favoring the disulfide exchange reaction and promoting the self-healing behavior (Ruipérez et al., 2018).
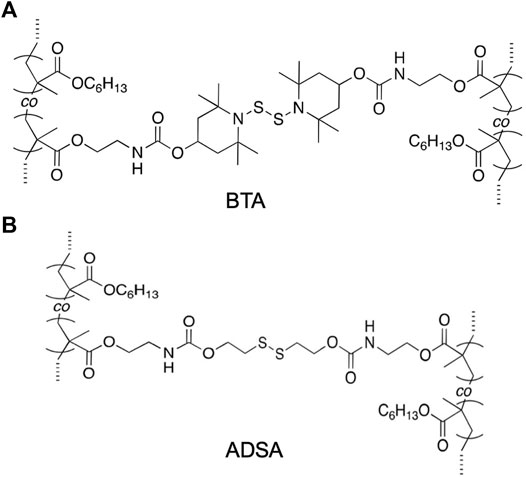
FIGURE 2. Disulfide-containing crosslinks in polymethacrylate networks proposed in Ref. Takahashi et al. (2017a). (A): BiTEMPS-containing diacrylate (BTA); (B): dialkyl disulfide-containing diacrylate (ADSA).
The simultaneous design of dynamic networks combined with reasonable mechanical properties is becoming the landmark of this field. Nevertheless, exploring the multiple polymer/dynamic chemistry combinations to obtain materials with the targeted properties is not trivial and only achievable by trial and error. Thus, computational chemistry appears as a very powerful predictive tool to guide experimental research. It was by means of theoretical chemistry that the good performance of the disulfide bond in the self-healing process was elucidated. Matxain et al. (2016) explored the reaction mechanism involved in aromatic disulfide-based self-healing materials, such as those developed by Rekondo et al. (2014). A (2 + 1) radical-mediated reaction mechanism was found to be responsible for the self-healing reaction, instead of the expected (2 + 2) metathesis mechanism, and it was later verified experimentally in the absence of catalyst (Nevejans et al., 2016). The first step in the (2 + 1) radical-mediated mechanism is the cleavage of the S-S bond, creating sulfenyl radicals, which may further attack neighbouring disulfide bonds. This attack takes place via a three-membered transition state, which eventually leads to a new sulfenyl radical in a single-displacement reaction and another disulfide compound, and thus, to exchanging sulfur atoms in the process. Following this work, the authors were able to establish a theoretical protocol to assess the theoretical self-healing capacity in these kind of materials, consisting on three different parameters (Formoso et al., 2017; Irigoyen et al., 2019b): 1) the probability to generate sulfenyl radicals, which can be controlled by tuning the strength of the S-S bond, 2) the energetic barrier of the exchange process, and 3) the dynamics of the polymeric chains, governing the ability to flow and reshuffle. In previous works (Matxain et al., 2016; Irigoyen et al., 2019a; Ruipérez et al., 2018; Irigoyen et al., 2019b), different aliphatic and aromatic disulfide- and diselenide-based materials have been studied by using this theoretical protocol. These works showed that the proper choice of the dynamic bond, the non-covalent interactions and the packaging are key features to enhance the self-healing capacity of a polymeric material.
Up to now, efforts have been made concerning the understanding of the dynamic character, at the molecular level, of dichalcogenide-containing materials. The main goal of this work is to test the validity of the proposed theoretical protocol to qualitatively predict the self-healing capacity of real systems. In order to do that, we have performed computational simulations of the materials previously mentioned, IP, HM, M, BTA and ADSA, using the theoretical protocol developed in our group (Formoso et al., 2017; Irigoyen et al., 2019b).
2 Computational Methodology
2.1 Quantum Chemical Calculations
All geometry optimizations were carried out in gas phase within density functional theory (DFT) (Hohenberg and Kohn, 1964; Kohn and Sham, 1965), by using the long-range corrected ωB97XD functional (Chai and Head-Gordon, 2008) combined with the 6-31+G(d,p) basis set (Hehre et al., 1972). Harmonic vibrational frequencies were obtained by analytical differentiation of gradients in order to determine whether the structures found were minima or transition states. The frequencies were then used to evaluate the zero-point vibrational energy (ZPVE) and the thermal (T = 298 K) vibrational corrections to the enthalpy (H) and Gibbs free energy (G) in the harmonic oscillator approximation. Single point calculations using the 6-311++G(2df,2p) basis set (Krishnan et al., 1980) were performed on the optimized structures to refine the electronic energy. All DFT calculations were carried out using the Gaussian 16 package (Frisch et al., 2016).
Ab initio Born–Oppenheimer molecular dynamics (QMD) calculations were carried out for the optimized species in order to determine their thermal stability, using the PBE functional (Perdew et al., 1996; 1997) combined with a DZP quality basis set and the RI formalism, with the corresponding auxiliary basis sets (Deglmann et al., 2004; Eichkorn et al., 1995; Sierka et al., 2003). The calculations were carried out at 298 K by means of the Nose–Hoover thermostat. All these simulations were as long as 40,000 a.u. (9.651 ps), with a time step of 40 a.u. (1.93 ps), and were performed using the TURBOMOLE package (TURBOMOLE Gmbh, 2014).
2.2 Molecular Dynamics Simulations
The molecular dynamics (MD) simulations have been performed using GAFF ff14SB (Maier et al., 2015) amber force-field in the AMBER 14 molecular dynamics simulation package (Case et al., 2014). All structures were built via the LEaP module of Ambertools and the charges were computed using the restrained electrostatic potential (RESP) fitting procedure (Bayly et al., 1993). First, the ESP was calculated with the Gaussian 16 package using the 6–31G* basis set, at the Hartree-Fock level of theory, and then the RESP charges were obtained. All the simulations were carried out in vacuum in a canonical ensemble (NVT) with a 2 fs timestep. 100,000 steps of minimization (50,000 steps steep descent minimization plus 50,000 steps of conjugate gradient minimization) were followed by heating from 0 to 300 K over 200 ps. Covalent bond lengths involving hydrogen were constrained using the SHAKE algorithm.
2.3 Theoretical Self-Healing Capacity
The theoretical protocol to estimate the self-healing capacity of disulfide-based materials is based on three parameters (Formoso et al., 2017; Irigoyen et al., 2019b). The first parameter is the probability to generate sulfenyl radicals (ρ), which is governed by the dissociation of the S-S bond. This parameter is calculated from quantum molecular dynamics, and is defined as the number of simulation steps where the S-S bond distance is longer than 2.3 Å (NS−S), a value for which the bond may be considered dissociated, divided by the total number of simulation steps, Ntot.
The second parameter is the energetic barrier of the exchange reaction, which is used to estimate the rate constant (k), according to the Wigner, Eyring, Polanyi and Evans formulation of the Transition State Theory (Evans and Polanyi, 1935; Eyring, 1935):
where R, kB and h are the ideal gas, Boltzmann and Planck constants, respectively, and ΔG is the activation energy corresponding to the attack of a sulfenyl radical to a disulfide bond.
Finally, the mobility of the polymeric chains is estimated by analyzing the non-covalent interactions among chains, mainly hydrogen bonds. In order to do that, the parameter ω is defined, which is based on the distance between disulfides in the material, that will be governed by the interactions among chains:
Using the radial distribution function of the sulfur atoms, three regions may be defined in order to calculate ω: the reacting region (Ii), where disulfides are close enough to undergo the exchange reaction (R ≤ 4.5 Å), the neighboring region (Iii), where disulfides are far to react but with a non-negligible probability to approach the reacting region (4.5
2.4 System Models
Different model structures were used to carry out both quantum and molecular dynamics simulations. In quantum simulations small chain models were used, including the disulfide unit and the surrounding chemical structure. In particular, the model used for the PUs correspond to the hard segments depicted in Figure 1, while the corresponding to the polymethacrylate materials are represented in Figure 3. In the quantum chemical calculations, both one- and two-chain models were used.
FIGURE 3. Molecular models of the BTA and ADSA materials used in the quantum chemical calculations.
The models used for the molecular dynamics simulations are built with two disulfide units including a small polymeric chain between them (Figure 4, where the model corresponding to the IP material is depicted). 16 chains are put randomly in a simulation box, following the procedure previously validated in our group (Irigoyen et al., 2019b), reproducing the experimental density of PUs and polymethacrylates after equilibration.
FIGURE 4. Molecular chain model of the IP material used in the molecular dynamics simulations.
3 Results and Discussion
In this section, the obtained results will be presented and discussed, in order to rationalize the experimental data obtained in the works of Kim et al. (2017) and Takahashi et al. (2017a), by using the theoretical protocol designed in our group. Concretely, this section will be divided into three subsections. First, we will summarize the experimental self-healing properties of these materials. Then, we will make use of quantum chemical calculations to analyze the formation of sulfenyl radicals by cleavage of the disulfide bond. In order to do that, the bond dissociation energy (BDE) and the probability of radicals to be formed (ρ) will be evaluated. Also, in this subsection the exchange reaction barriers (ΔG) will be calculated. In the third subsection, by means of molecular dynamics simulations, the mobility of the polymeric chains will be analyzed by calculating the probability of sulfenyl radicals to find a neighboring disulfide bond (ω), which will be crucial to trigger the exchange reaction.
3.1 Self-Healing Properties of the Studied Materials
The polyurethanes synthesized by Kim et al. showed healing efficiency in the following order: IP
The self-healing properties of the polymethacrylate networks studied by Takahashi et al. were tested by using strength and strain at break. First, a sample of BTA network was cut and the surfaces were put together at 120°C under mild pressure. The scar became invisible after 8 h and healing ratios of up to 85 and 92% for the strength and strain at break were recorded, respectively. Inferior recovery performance was observed for ADSA, and visible scars appeared. The difference in self-healing properties was also supported from fracture behavior in tensile tests. BTA fractured at different positions, similar to the pristine material, while ADSA fractured in the cut line.
3.2 Radical Formation and Disulfide Exchange
Several energetic and structural parameters have been considered in order to study the sulfenyl radicals formation and their influence in the disulfide exchange. The disulfide bond strength has been analyzed by calculating the BDE considering two different chain conformations, labeled as open and closed, in a single-chain model (Figure 5A). In addition to this, the interaction between two chains has been also calculated, to take into account the interchain non-covalent interactions, such as hydrogen bonding and π − π stacking among phenyl rings (Figure 5B).
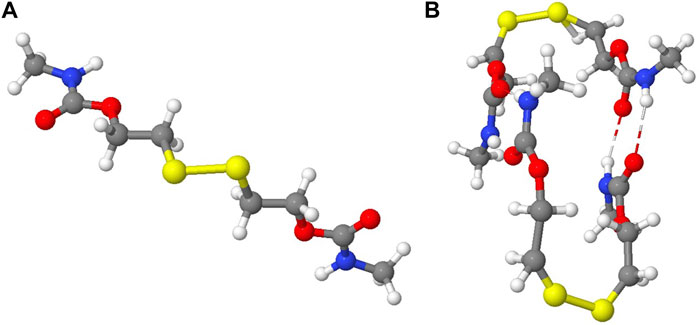
FIGURE 5. (A) ADSA single-chain model in an open conformation. (B) Interaction between two ADSA chains.
3.2.1 Bond Dissociation Energies
In Table 1 are collected the geometrical parameters obtained for the open and closed conformations, together with the BDEs. In the open conformation, the R-SS-R dihedral angles are large, more than 100°, which means that both chains linked to the sulfur atoms are rather far from each other, hindering the interaction among them, as depicted in Figure 5A. In the closed conformation, otherwise, the dihedral angle is small, of around 60° or less, which allows the intrachain interactions. See Figure 5B, where two chains interacting in a closed conformation are represented. In principle, a combination of different conformations would be present in the polymeric material due to the presence of multiple chains. However, in those materials including diphenyl disulfides (IP, HM, and M), the closed conformation seems to be the most favored one, since the interaction between the phenyl rings adjacent to the sulfur atoms helps reducing the steric repulsion with the rest of the chain. In the M and ADSA models both conformations can be obtained, while the steric repulsion in BTA prevents the formation of the closed conformation.
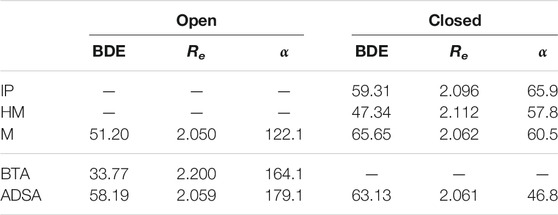
TABLE 1. Bond dissociation energy (BDE), in kcal/mol, S-S bond length (Re), in Å, and R-SS-R dihedral angle (α), in degrees, for the open and closed conformations of the one-chain molecular models.
In previous works (Matxain et al., 2016; Ruipérez et al., 2018), the BDE of different disulfides was calculated to be around 48 kcal/mol for diphenyl disulfides, 33 kcal/mol for sulfenamides (molecules containing S-N bonds) and 60 kcal/mol for aliphatic disulfides. The calculated values in this work for BTA, a sulfenamide (33.77 kcal/mol), ADSA, an aliphatic disulfide (58.19 kcal/mol, open conformation, and 63.13 kcal/mol, closed conformation), and HM, an aromatic disulfide (47.34 kcal/mol), are in agreement with those values. Nevertheless, the BDEs calculated for IP and M (closed conformation) are notably larger, 59.31 and 65.65 kcal/mol, respectively. This suggests the presence of stronger intrachain interactions in this conformation (hydrogen bonding and π−π stacking). Note that the BDE is calculated as the energy difference between the whole molecule (disulfide) and the two separated sulfenyl fragments. This means that these non-covalent interactions are also included in the calculation of the BDE. Therefore, in the open conformation, where the non-covalent interactions are absent, the BDE values are expected to be lower and closer to the calculated in previous works. This effect is observed in the M model, for which the BDE in the open conformation is smaller (51.20 kcal/mol) than in the closed one (65.65 kcal/mol). In Table 1 is also observed that lower BDEs correspond to longer S-S bond distances.
Since intermolecular interactions have been found to be important in the self-healing capacity of materials, in this work the interchain interactions have been studied using a two-chain model (Figure 5B). A deeper analysis will be carried out later by means of molecular dynamics simulations, including more and larger chains. In Table 2 are collected the geometrical parameters of each chain and the interaction energy between them. Inspecting the calculated interaction energies (ΔH), it is observed that in all cases interchain interactions are rather strong, around 30 kcal/mol. This suggests that the mobility of the chains may be limited.
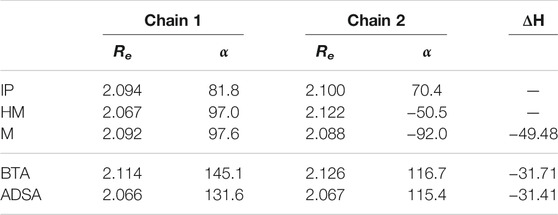
TABLE 2. S-S bond length (Re), in Å, R-SS-R dihedral angle (α), in degrees, and interaction energy (ΔH), in kcal/mol, between two polymeric chains.
3.2.2 Radical Formation
The BDE gives a measure of the strength of the bond and is relevant to estimate the probability to generate sulfenyl radicals (ρ, Eq. 1). Since the BDE is affected by chain conformation and interchain interactions, in order to estimate this probability, it is necessary to take into account the dynamics of many chains interacting at the same time in the material. Thus, ρ has been calculated for the five model systems by means of quantum molecular dynamics (QMD) simulations at different temperatures, those in which the experiments have been carried out, to see the influence of temperature in the formation of radicals. So that, four different temperatures have been used in the simulations (25, 40, 60, and 80°C) for the IP, HM and M systems, while two temperatures (25 and 120°C) have been used for the BTA and ADSA systems. In Table 3 are collected the results of these simulations.
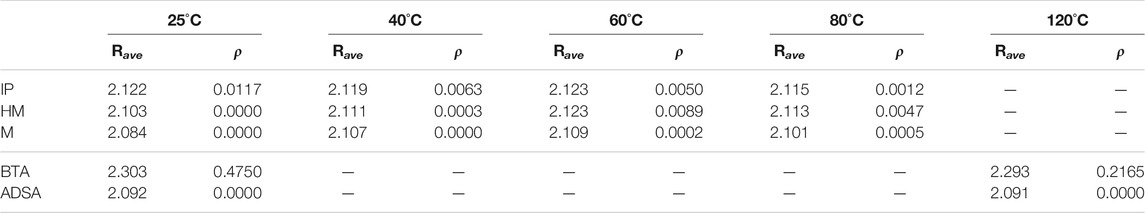
TABLE 3. Average S-S bond distances (Rave), in Å, obtained in QMD simulations, and probability to generate sulfenyl radicals (ρ) as a function of temperature. The calculated temperatures are those in which experiments were performed, 25, 40, 60, and 80°C for IP, HM and M, and 25°C and 120°C for BTA and ADSA.
Starting with the PUs, it can be seen that, at room temperature, IP is the only material presenting a value of ρ different from 0, which means that, theoretically, this would be the only one that could trigger the exchange reaction at that temperature due to the presence of radicals, in agreement with the experimental findings. At increasing temperatures, non-zero probabilities are calculated for the three PUs, but it is remarkable the almost negligible values calculated for the M material at any temperature, again consistent with the experimental results. This supports the assumption that the probability to break the S-S bond is not only dependent on the electronic features of the bond and its surroundings, but is also largely determined by the mobility of the chains, which in the case of the M material is notably reduced due to the packaging developed by the π–π interactions among phenyl rings. This has as a consequence a lower or even absent self-healing efficiency.
Inspecting the polymethacrylates, it can be seen that ρ for ADSA is zero at both room temperature and 120°C, meanwhile BTA presents high values at both temperatures, meaning that sulfenyl radicals are easily created and the exchange reaction promoted, in agreement with the self-healing efficiency observed experimentally in this material.
In Table 3, it can also be observed a non-regular trend for the variation of ρ with temperature. One could expect that increasing temperature would favor the dissociation of the bond. However, in some cases the opposite trend is observed. In Figure 6, the evolution of the disulfide bond distances during the simulation, used for the calculation of ρ, are depicted for both chains in a two-chain model of IP material, for which ρ shows a decreasing trend with increasing temperature. It is clearly seen that the disulfide of one of the chains shows a much larger variation of the S-S bond distance along the simulation (green line), while the other one (red line) remains with bond distances very similar to those calculated for the single-chain system. This can be related to the conformation adopted by the chain (open and closed), which, as it has been analyzed before, affects the bond distance (and the BDE). Thus, the red line would correspond to a chain with a closed conformation, where the π-π interactions between phenyl rings keep the sulfur atoms closer during the simulation, while the green line would correspond to a chain with an open conformation, providing more mobility to the sulfur atoms. This suggests that the bond distance and, therefore, ρ, may be affected by the initial conformation of the chains, that would be basically determined by both the starting conformation and the simulation length. In Table 3, ρ is extracted only from the closed conformation in order to relieve the computational effort. Nonetheless, several calculations were performed using both conformations as starting point, and it was observed that the value obtained for the closed one always corresponded to the smallest, therefore, we use it as the lower limit of this parameter. This feature hinders the observation of a regular trend in ρ, and could be partially solved by exploring a large number of conformations and using longer simulation times, which would be very costly computationally using quantum molecular dynamics simulations.
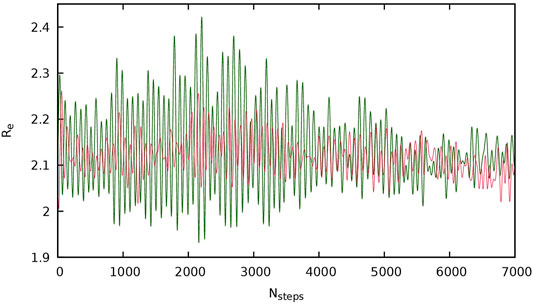
FIGURE 6. Variation of the S-S bond length (Re), in Å, in the QMD simulations for each chain in the two-chain model of IP material (red and green lines). Nsteps states for number of steps in the simulation.
3.2.3 Barrier of the Exchange Reaction
Finally, the other parameter obtained by means of quantum chemical calculations is the exchange reaction barrier, calculated as the energy of the transition state, ΔG, which gives information about the kinetics of the exchange reaction (Eq. 2). In previous works (Matxain et al., 2016; Ruipérez et al., 2018; Irigoyen et al., 2019a), it was concluded that the chemical structure around the dynamic bond was a key feature in the calculation of the barrier. Since the calculation of the transition states is very demanding computationally and the systems studied in this work are rather large, we decided to compute only the reaction barrier for the IP material, obtaining a value of 12.02 kcal/mol, close to those previously calculated with a similar chemical structure around the disulfide bond, that is, an aromatic disulfide with a urethane unit attached to the ring in para position (12.21 kcal/mol) (Irigoyen et al., 2019a). So that, we can assume that the reaction barrier for all of IP, M, and HM will be similar, since all of them have the same chemical structure around the dynamic bond. Similarly, we can use our previous values calculated for sulfenamides (16–18 kcal/mol) (Ruipérez et al., 2018) to estimate the approximate reaction barrier of BTA material.
These results suggest that the exchange reaction barrier is not a relevant factor in the different self-healing performances observed in these materials.
3.3 Influence of the Hard Segment and Crosslinks in the Chain Mobility
The mobility of the chains is a key factor in the self-healing process, as it determines the ability of chains, and sulfur atoms in particular, to move and rearrange, which is essential for sulfur radicals to attack disulfide bonds and trigger the exchange reaction. This mobility is determined by the non-covalent interactions among chains which, as it has been demonstrated in the previous section, also affects the S-S bond dissociation process, the first step in the exchange mechanism.
Therefore, in this section, by means of classical molecular dynamics simulations, we will not only analyze the chain mobility by computing the ω parameter (Eq. 3), but also ρ will be reevaluated and compared with that obtained by QMD simulations. In classical simulations, many more and longer chains are included and, since the starting point is a random configuration, lots of different conformations are taken into account, removing the initial bias observed in the quantum simulations.
Inspecting the radial distribution functions of the sulfur atoms obtained from the molecular dynamics simulations (Figure 7), two different regions can be observed: one corresponding to the sulfur atoms within a disulfide bond, with a high, narrow peak at around the S-S equilibrium bond distance (∼2.10 Å) and a large region, ranging from 4 to 25 Å, approximately (enlarged in the small box of Figure 7), which corresponds to the distances between sulfur atoms of different disulfide bonds. The parameter ω is obtained by integration of different regions of the radial distribution functions to estimate the number of disulfides in each region. In the same manner, a value for ρ can be defined by integrating the radial distribution function in the region of the S-S bond equilibrium distance (Figure 8), with the aim of obtaining a ratio of the sulfur atoms located away from the equilibrium distance, which could be considered as dissociated.
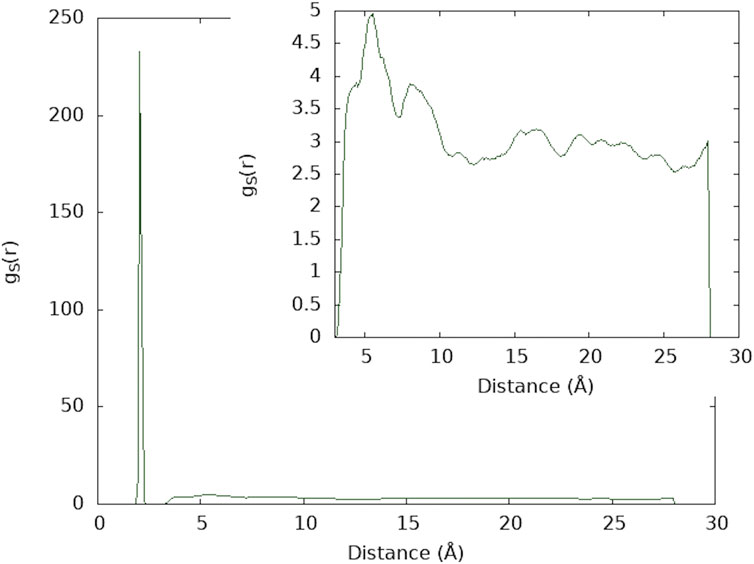
FIGURE 7. Radial distribution function of sulfur atoms for IP material. The smaller box shows an enlargement of the region between 4 and 25 Å.
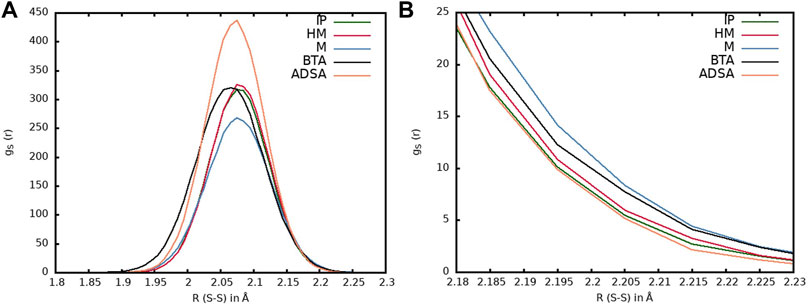
FIGURE 8. (A) Radial distribution functions corresponding to the sulfur atoms within the same disulfide bond for the five models: IP (green line), HM (red line), M (blue line), BTA (black line) and ADSA (orange line). (B) Enlargement of the region between 2.18 and 2.23 Å.
3.3.1 Reevaluation of ρ by Classical Molecular Dynamics
First, we will focus on the region of the radial distribution function corresponding to the sulfur atoms within each disulfide bond. In Figure 8A, the radial distribution functions of all the studied systems at 25°C are represented, while the region corresponding to larger bond distances are magnified in the right panel. It can be seen that both M (blue line) and BTA (black line) materials show the highest population of sulfur atoms at larger distances, while ADSA (orange line) shows the lowest one, with IP (green line) and HM (red line) laying in between with a very similar profile (Figure 8B). These results are a consequence of the chemical structure around the disulfide bond. BTA is a sulfenamide and, as it has been shown in the previous section, the N atoms linked to the sulfur produce weaker S-S bonds and larger bond distances (Table 1). The ADSA system consists on an aliphatic disulfide with stronger and, thus, shorter bonds. IP and HM have the same structure around the disulfide, that is, an aromatic ring attached to each sulfur atom, producing intermediate bond strengths and similar profiles. The exception to this trend corresponds to M, also an aromatic disulfide, but it presents the largest bond distances. In Table 1 can be observed that the M system, unlike IP and HM, may be found in both the open and closed conformation. The absence of the intrachain interactions in the open conformation makes the S-S bond dissociation easier and, thus, longer bond distances are observed.
In Figure 9A, the radial distribution functions for the IP system at different temperatures are represented. It can be observed that the higher the temperature, the lower the peak of the curve, as expected, since increasing the temperature leads to longer S-S bonds and the distribution is broader. In Figure 9B, an enlargement of the region corresponding to longer bond distances is represented and the same behavior can be observed, where higher temperatures show a larger amount of sulfur atoms at longer distances. This would correspond to a large probability for the disulfide bonds to be dissociated.
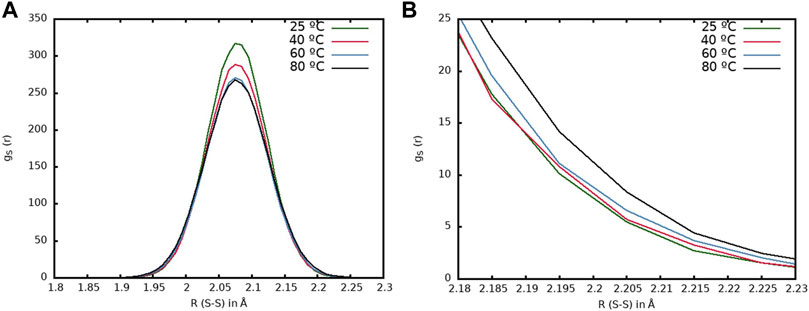
FIGURE 9. (A) Radial distribution function corresponding to the sulfur atoms of the disulfide bonds for the IP model at different temperatures: 25°C (green line), 40°C (red line), 60°C (blue line) and 80°C (black line). (B) Enlargement of the region between 2.16 and 2.23 Å.
This is the region of the radial distribution function that will be integrated in order to estimate the ratio of sulfur atoms laying at longer distances and, thus, with a higher probability to be dissociated. For that, a similar expression to that of ω parameter has been used, leading to the so-called ρMD parameter (Eq. 4).
The regions selected for the integration are the following: II ranges from 2.20 to 3.00 Å, where the sulfur atoms with higher probability to be dissociated are included, and III, ranging from 1.80 to 3.00 Å. In this way, we obtain a proportion of the sulfur atoms that could be dissociated, similar to that obtained by using QMD simulations. The results are collected in Table 4. Now, it can be observed a more regular trend, where increasing temperatures corresponds to larger probabilities of the bond to be dissociated. This confirms that classical molecular dynamics simulations remove almost completely the limitations found in the quantum molecular dynamics calculations. Comparing Table 3 and Table 4, differences arise because ρ is calculated differently. In both cases, ρ is defined similarly in QMD and MD calculations, i.e., as the probability of S-S bond distance to be larger than a given threshold. In this way, using QMD we are able to study the formation and breaking of bonds, while using MD it is not possible. Thus, in the MD calculations, the S-S distance is defined by the contributions included in the force field, while in the QMD calculations, the S-S distances change according to quantum effects. Hence, for considering the formation of radicals, QMD methods are necessary. According to these results, the values for M and ADSA are 0 since in the QMD simulations no S-S distances are found to be longer than the threshold, due to electronic quantum effects. In both cases, the 0 value arise directly from the calculation. MD results are used to study the temperature effect, but it is not possible to perform a direct comparison with ρ from QMD calculations, since MD is not able to describe formation and breaking of the bond.
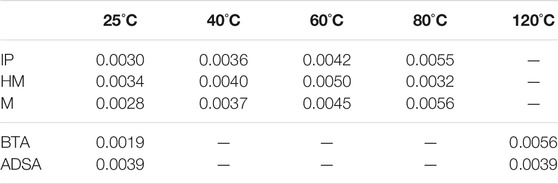
TABLE 4. Probability to generate sulfenyl radicals obtained by classical molecular dynamic simulations (ρMD) for all the studied systems at different temperatures.
3.3.2 Mobility of the Polymeric Chains
Next, an analysis of the region of the radial distribution functions corresponding to longer distances, those between different disulfide units, will be performed (see the enlarged region in Figure 7). This will allow us to understand the mobility of the sulfur atoms and their ability to reach neighboring disulfides in order to trigger the exchange reaction. To do that, the following regions are defined and integrated to evaluate the ω parameter (Eq. 3): II, the reacting region, ranging from 3 Å to 4.5Å, where sulfurs are close enough to react, and III, the neighboring region, from 4.5 Å to 20 Å, where the disulfides are further away, but with a non-zero probability to enter in the reacting region.
The results, at different temperatures, are collected in Table 5. It can be observed that the temperature has small influence in ω, as it fluctuates around a certain value. Since the materials are solids, the variations in the molecular structure caused by temperature are small in this range of temperatures, as they are well below the Tg of polyurethanes (120–130°C). So that, it makes sense to define an average value (ωave), which should be characteristic of each material at temperatures below the Tg.
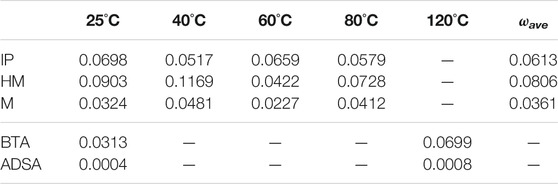
TABLE 5. Ratio of disulfides in the reacting region (ω) at different temperatures, together with the average value (ωave).
The differences observed in the PUs can be explained by inspecting their chemical structures. The highest ωave is obtained for HM, which includes aliphatic cyclohexanes, while the lowest one is obtained for M, which contains phenyl groups (Figure 1). The aromatic groups favor more rigid structures due to their planarity and the π-π stacking interactions, while the cyclohexanes possess more degrees of freedom and, therefore, more mobility. The intermediate value for the IP system can be explained by a more limited mobility of the substituted cyclohexanes, still larger than that of the phenyl rings.
Regarding the polymethacrylates, a large difference is observed between BTA and ADSA, which can also be ascribed to their molecular structure. ADSA contains aliphatic hydrocarbon chains attached to the disulfide bond, which can be effectively packed and favoring the hydrogen bonding between urethane moieties. This hinders the mobility of the chains and a very low value of ωave is found (0.0006). In BTA, instead, the presence of a cyclohexane next to the disulfide leads to a much less packed molecular structure and prevents the formation of hydrogen bonds between urethanes. Thus, a much larger value of ωave is calculated (0.0506).
4 Conclusion
In this work, we have analyzed the theoretical self-healing capacity of several materials and compared it to the experimental results. In general, a good agreement is found and the theoretical calculations are able to explain qualitatively the experimental self-healing capacities.
In particular, it explains the great performance of IP and BTA, as well as the lack of self-healing capacity of M and ADSA. More interestingly, it can also describe the improvement of the self-healing capacity of HM with increasing temperatures. However, an accurate representation of the temperature effects in the dissociation of the disulfide bond is problematic with the present protocol, since the initial conformation in the quantum molecular dynamics used to calculate ρ may introduce a conformational bias. This issue may be overcome by using classical molecular dynamics, that allow including many more conformations, reducing at a great extent the initial bias. Nevertheless, it is not possible to describe the cleavage of the bond and, therefore, the formation of radicals, by classical dynamics. This means that this technique can only be used to estimate the influence of the temperature by inspecting the variation of the bond distance during the simulation.
The mobility of the polymeric chains has been estimated using the ω parameter. Large values of ω correspond to greater mobility of the chains. In PUs, the following trend has been calculated for the three hardeners: ωHM > ωIP > ωM. This suggests that, for HM and IP, the sulfenyl radicals will have more mobility to approach another disulfide and favoring the self-healing process, while for M the opposite will take place, in agreement with the experimental findings. For the polymethacrylates, BTA shows a large ω value, while that of ADSA is almost zero, again in agreement with the differences observed in the self-healing behaviour of the two materials. Besides, we have observed that the increment of the temperature does not really have an impact in ω, as the properties of the bulky materials are not affected that much when working at temperatures well below their Tg. We can conclude, then, that the effect of temperature in the self-healing capacity is mainly related to a greater generation of radicals in the material. Finally, the estimation of the kinetic barriers of the exchange reaction suggests that this is not a key factor in the self-healing capacity of these materials.
As a summary, we believe that this theoretical protocol will assist in the development of new materials with improved self-healing properties.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
Conceptualization, JMM and FR; methodology, JMM and FR; formal analysis, MI, JMM, and FR; resources, JMM and FR; writing, MI, JMM, and FR; supervision, JMM and FR.
Funding
This research was funded by Eusko Jaurlaritza grant number IT1254-19.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Technical and human support provided by IZO-SGI, SGIker (UPV/EHU, MICINN, GV/EJ, ERDF and ESF) is gratefully acknowledged for assistance and generous allocation of computational resources.
References
Aguirresarobe, R. H., Martin, L., Fernandez-Berridi, M. J., and Irusta, L. (2017). Autonomic Healable Waterborne Organic-Inorganic Polyurethane Hybrids Based on Aromatic Disulfide Moieties. Express Polym. Lett. 11, 266–277. doi:10.3144/expresspolymlett.2017.27
Aguirresarobe, R. H., Nevejans, S., Reck, B., Irusta, L., Sardon, H., Asua, J. M., et al. (2021). Healable and Self-Healing Polyurethanes Using Dynamic Chemistry. Prog. Polym. Sci. 114, 101362. doi:10.1016/j.progpolymsci.2021.101362
Amamoto, Y., Otsuka, H., Takahara, A., and Matyjaszewski, K. (2012). Self-healing of Covalently Cross-Linked Polymers by Reshuffling Thiuram Disulfide Moieties in Air under Visible Light. Adv. Mater. 24, 3975–3980. doi:10.1002/adma.201201928
An, S. Y., Noh, S. M., and Oh, J. K. (2017a). Multiblock Copolymer-Based Dual Dynamic Disulfide and Supramolecular Crosslinked Self-Healing Networks. Macromol. Rapid Commun. 38, 1600777. doi:10.1002/marc.201600777
An, X., Aguirresarobe, R. H., Irusta, L., Ruipérez, F., Matxain, J. M., Pan, X., et al. (2017b). Aromatic Diselenide Crosslinkers to Enhance the Reprocessability and Self-Healing of Polyurethane Thermosets. Polym. Chem. 8, 3641–3646. doi:10.1039/c7py00448f
Azcune, I., and Odriozola, I. (2016). Aromatic Disulfide Crosslinks in Polymer Systems: Self-Healing, Reprocessability, Recyclability and More. Eur. Polym. J. 84, 147–160. doi:10.1016/j.eurpolymj.2016.09.023
Bayly, C. I., Cieplak, P., Cornell, W., and Kollman, P. A. (1993). A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: the Resp Model. J. Phys. Chem. 97, 10269–10280. doi:10.1021/j100142a004
Belenguer, A. M., Friščić, T., Day, G. M., and Sanders, J. K. M. (2011). Solid-state Dynamic Combinatorial Chemistry: Reversibility and Thermodynamic Product Selection in Covalent Mechanosynthesis. Chem. Sci. 2, 696. doi:10.1039/c0sc00533a
Billiet, S., Hillewaere, X. K. D., Teixeira, R. F. A., and Du Prez, F. E. (2013). Chemistry of Crosslinking Processes for Self-Healing Polymers. Macromol. Rapid Commun. 34, 290–309. doi:10.1002/marc.201200689
Bin Rusayyis, M., and Torkelson, J. M. (2020). Recyclable Polymethacrylate Networks Containing Dynamic Dialkylamino Disulfide Linkages and Exhibiting Full Property Recovery. Macromolecules 53, 8367–8373. doi:10.1021/acs.macromol.0c01539
Bowman, C. N., and Kloxin, C. J. (2012). Covalent Adaptable Networks: Reversible Bond Structures Incorporated in Polymer Networks. Angew. Chem. Int. Ed. 51, 4272–4274. doi:10.1002/anie.201200708
Brunette, C. M., Hsu, S. L., and MacKnight, W. J. (1982). Hydrogen-bonding Properties of Hard-Segment Model Compounds in Polyurethane Block Copolymers. Macromolecules 15, 71–77. doi:10.1021/ma00229a014
Burattini, S., Colquhoun, H. M., Fox, J. D., Friedmann, D., Greenland, B. W., Harris, P. J. F., et al. (2009). A Self-Repairing, Supramolecular Polymer System: Healability as a Consequence of Donor-Acceptor π-π Stacking Interactions. Chem. Commun. 2009, 6717. doi:10.1039/b910648k
Burnworth, M., Tang, L., Kumpfer, J. R., Duncan, A. J., Beyer, F. L., Fiore, G. L., et al. (2011). Optically Healable Supramolecular Polymers. Nature 472, 334–337. doi:10.1038/nature09963
Canadell, J., Goossens, H., and Klumperman, B. (2011). Self-healing Materials Based on Disulfide Links. Macromolecules 44, 2536–2541. doi:10.1021/ma2001492
Case, D., Babin, V., Berryman, J., Betz, R., Cai, Q., Cerutti, D., et al. (2014). AMBER 14. San Francisco: University of California.
Cash, J. J., Kubo, T., Bapat, A. P., and Sumerlin, B. S. (2015). Room-temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters. Macromolecules 48, 2098–2106. doi:10.1021/acs.macromol.5b00210
Chai, J.-D., and Head-Gordon, M. (2008). Long-range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 10, 6615. doi:10.1039/b810189b
Chen, X., Dam, M. A., Ono, K., Mal, A., Shen, H., Nutt, S. R., et al. (2002). A Thermally Re-mendable Cross-Linked Polymeric Material. Science 295, 1698–1702. doi:10.1126/science.1065879
Chen, X., Wudl, F., Mal, A. K., Shen, H., and Nutt, S. R. (2003). New Thermally Remendable Highly Cross-Linked Polymeric Materials. Macromolecules 36, 1802–1807. doi:10.1021/ma0210675
Cho, S. H., White, S. R., and Braun, P. V. (2012). Room-temperature Polydimethylsiloxane-Based Self-Healing Polymers. Chem. Mater. 24, 4209–4214. doi:10.1021/cm302501b
Cordier, P., Tournilhac, F., Soulié-Ziakovic, C., and Leibler, L. (2008). Self-healing and Thermoreversible Rubber from Supramolecular Assembly. Nature 451, 977–980. doi:10.1038/nature06669
Cromwell, O. R., Chung, J., and Guan, Z. (2015). Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc. 137, 6492–6495. doi:10.1021/jacs.5b03551
Deglmann, P., May, K., Furche, F., and Ahlrichs, R. (2004). Nuclear Second Analytical Derivative Calculations Using Auxiliary Basis Set Expansions. Chem. Phys. Lett. 384, 103–107. doi:10.1016/j.cplett.2003.11.080
Eichkorn, K., Treutler, O., Öhm, H., Häser, M., and Ahlrichs, R. (1995). Auxiliary Basis Sets to Approximate Coulomb Potentials. Chem. Phys. Lett. 240, 283–290. doi:10.1016/0009-2614(95)00621-a
Engels, H.-W., Pirkl, H.-G., Albers, R., Albach, R. W., Krause, J., Hoffmann, A., et al. (2013). Polyurethanes: Versatile Materials and Sustainable Problem Solvers for Today's Challenges. Angew. Chem. Int. Ed. 52, 9422–9441. doi:10.1002/anie.201302766
Eom, Y., Kim, S.-M., Lee, M., Jeon, H., Park, J., Lee, E. S., et al. (2021). Mechano-responsive Hydrogen-Bonding Array of Thermoplastic Polyurethane Elastomer Captures Both Strength and Self-Healing. Nat. Commun. 12, 621. doi:10.1038/s41467-021-20931-z
Erice, A., Azcune, I., Ruiz de Luzuriaga, A., Ruipérez, F., Irigoyen, M., Matxain, J. M., et al. (2019). Effect of Regioisomerism on Processability and Mechanical Properties of Amine/urea Exchange Based Poly(urea-Urethane) Vitrimers. ACS Appl. Polym. Mater. 1, 2472–2481. doi:10.1021/acsapm.9b00589
Erice, A., Ruiz de Luzuriaga, A., Matxain, J. M., Ruipérez, F., Asua, J. M., Grande, H.-J., et al. (2018). Reprocessable and Recyclable Crosslinked Poly(urea-Urethane)s Based on Dynamic Amine/urea Exchange. Polymer 145, 127–136. doi:10.1016/j.polymer.2018.04.076
Evans, M. G., and Polanyi, M. (1935). Some Applications of the Transition State Method to the Calculation of Reaction Velocities, Especially in Solution. Trans. Faraday Soc. 31, 875. doi:10.1039/tf9353100875
Eyring, H. (1935). The Activated Complex in Chemical Reactions. J. Chem. Phys. 3, 107–115. doi:10.1063/1.1749604
Formoso, E., Asua, J. M., Matxain, J. M., and Ruipérez, F. (2017). The Role of Non-covalent Interactions in the Self-Healing Mechanism of Disulfide-Based Polymers. Phys. Chem. Chem. Phys. 19, 18461–18470. doi:10.1039/c7cp03570e
Fortman, D. J., Brutman, J. P., Cramer, C. J., Hillmyer, M. A., and Dichtel, W. R. (2015). Mechanically Activated, Catalyst-free Polyhydroxyurethane Vitrimers. J. Am. Chem. Soc. 137, 14019–14022. doi:10.1021/jacs.5b08084
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 Revision C.01. Wallingford CT: Gaussian Inc.
Hager, M. D., Greil, P., Leyens, C., van der Zwaag, S., and Schubert, U. S. (2010). Self-healing Materials. Adv. Mater. 22, 5424–5430. doi:10.1002/adma.201003036
Hehre, W. J., Ditchfield, R., and Pople, J. A. (1972). Self-consistent Molecular Orbital Methods. Xii. Further Extensions of Gaussian-type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 56, 2257–2261. doi:10.1063/1.1677527
Hillewaere, X. K. D., and Du Prez, F. E. (2015). Fifteen Chemistries for Autonomous External Self-Healing Polymers and Composites. Prog. Polym. Sci. 49-50, 121–153. doi:10.1016/j.progpolymsci.2015.04.004
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous Electron Gas. Phys. Rev. 136, B864–B871. doi:10.1103/physrev.136.b864
Irigoyen, M., Fernández, A., Ruiz, A., Ruipérez, F., and Matxain, J. M. (2019a). Diselenide Bonds as an Alternative to Outperform the Efficiency of Disulfides in Self-Healing Materials. J. Org. Chem. 84, 4200–4210. doi:10.1021/acs.joc.9b00014
Irigoyen, M., Matxain, J. M., and Ruipérez, F. (2019b). Effect of Molecular Structure in the Chain Mobility of Dichalcogenide-Based Polymers with Self-Healing Capacity. Polymers 11, 1960. doi:10.3390/polym11121960
Kataoka, S., Tsuruoka, A., Aoki, D., and Otsuka, H. (2021). Fast and Reversible Cross-Linking Reactions of Thermoresponsive Polymers Based on Dynamic Dialkylaminodisulfide Exchange. ACS Appl. Polym. Mater. 3, 888–895. doi:10.1021/acsapm.0c01205
Kim, S.-M., Jeon, H., Shin, S.-H., Park, S.-A., Jegal, J., Hwang, S. Y., et al. (2017). Superior Toughness and Fast Self-Healing at Room Temperature Engineered by Transparent Elastomers. Adv. Mater. 30, 1705145. doi:10.1002/adma.201705145
Kloxin, C. J., and Bowman, C. N. (2012). Covalent Adaptable Networks: Smart, Reconfigurable and Responsive Network Systems. Chem. Soc. Rev. 42, 7161–7173. doi:10.1039/c3cs60046g
Kloxin, C. J., Scott, T. F., Adzima, B. J., and Bowman, C. N. (2010). Covalent Adaptable Networks (Cans): A Unique Paradigm in Cross-Linked Polymers. Macromolecules 43, 2643–2653. doi:10.1021/ma902596s
Kohn, W., and Sham, L. J. (1965). Self-consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 140, A1133–A1138. doi:10.1103/physrev.140.a1133
Krishnan, R., Binkley, J. S., Seeger, R., and Pople, J. A. (1980). Self‐consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 72, 650–654. doi:10.1063/1.438955
Lafont, U., van Zeijl, H., and van der Zwaag, S. (2012). Influence of Cross-Linkers on the Cohesive and Adhesive Self-Healing Ability of Polysulfide-Based Thermosets. ACS Appl. Mater. Inter. 4, 6280–6288. doi:10.1021/am301879z
Liu, L., Chu, Z., Liao, Y., Ma, Z., and Li, Y. (2020). Flow-induced Crystallization in Butene-1/1,5-Hexadiene Copolymers: Mutual Effects of Molecular Factor and Flow Stimuli. Macromolecules 53, 8476–8486. doi:10.1021/acs.macromol.0c01318
Liu, Y.-L., and Chen, Y.-W. (2007). Thermally Reversible Cross-Linked Polyamides with High Toughness and Self-Repairing Ability from Maleimide- and Furan-Functionalized Aromatic Polyamides. Macromol. Chem. Phys. 208, 224–232. doi:10.1002/macp.200600445
Lu, Y.-X., and Guan, Z. (2012). Olefin Metathesis for Effective Polymer Healing via Dynamic Exchange of strong Carbon-Carbon Double Bonds. J. Am. Chem. Soc. 134, 14226–14231. doi:10.1021/ja306287s
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14sb: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99sb. J. Chem. Theor. Comput. 11, 3696–3713. doi:10.1021/acs.jctc.5b00255
Martin, R., Rekondo, A., de Luzuriaga, A. R., Casuso, P., Dupin, D., Cabañero, G., et al. (2016). Dynamic Sulfur Chemistry as a Key Tool in the Design of Self-Healing Polymers. Smart Mater. Struct. 25, 084017. doi:10.1088/0964-1726/25/8/084017
Martin, R., Rekondo, A., Ruiz de Luzuriaga, A., Cabañero, G., Grande, H. J., and Odriozola, I. (2014). The Processability of a Poly(urea-Urethane) Elastomer Reversibly Crosslinked with Aromatic Disulfide Bridges. J. Mater. Chem. A. 2, 5710. doi:10.1039/c3ta14927g
Mattia, J., and Painter, P. (2007). A Comparison of Hydrogen Bonding and Order in a Polyurethane and Poly(urethane−urea) and Their Blends with Poly(ethylene Glycol). Macromolecules 40, 1546–1554. doi:10.1021/ma0626362
Matxain, J. M., Asua, J. M., and Ruipérez, F. (2016). Design of New Disulfide-Based Organic Compounds for the Improvement of Self-Healing Materials. Phys. Chem. Chem. Phys. 18, 1758–1770. doi:10.1039/c5cp06660c
Nevejans, S., Ballard, N., Fernández, M., Reck, B., García, S. J., and Asua, J. M. (2019). The Challenges of Obtaining Mechanical Strength in Self-Healing Polymers Containing Dynamic Covalent Bonds. Polymer 179, 121670. doi:10.1016/j.polymer.2019.121670
Nevejans, S., Ballard, N., Miranda, J. I., Reck, B., and Asua, J. M. (2016). The Underlying Mechanisms for Self-Healing of Poly(disulfide)s. Phys. Chem. Chem. Phys. 18, 27577–27583. doi:10.1039/c6cp04028d
Otsuka, H., Nagano, S., Kobashi, Y., Maeda, T., and Takahara, A. (2010). A Dynamic Covalent Polymer Driven by Disulfidemetathesis under Photoirradiation. Chem. Commun. 46, 1150–1152. doi:10.1039/b916128g
Pan, X., Driessen, F., Zhu, X., and Du Prez, F. E. (2017). Selenolactone as a Building Block toward Dynamic Diselenide-Containing Polymer Architectures with Controllable Topology. ACS Macro Lett. 6, 89–92. doi:10.1021/acsmacrolett.6b00944
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/physrevlett.77.3865
Perdew, J. P., Burke, K., and Ernzerhof, M. (1997). Generalized Gradient Approximation Made Simple [Phys. Rev. Lett. 77, 3865 (1996)]. Phys. Rev. Lett. 78, 1396. doi:10.1103/physrevlett.78.1396
Rekondo, A., Martin, R., Ruiz de Luzuriaga, A., Cabañero, G., Grande, H. J., and Odriozola, I. (2014). Catalyst-free Room-Temperature Self-Healing Elastomers Based on Aromatic Disulfide Metathesis. Mater. Horiz. 1, 237–240. doi:10.1039/c3mh00061c
Rowan, S. J., Cantrill, S. J., Cousins, G. R. L., Sanders, J. K. M., and Stoddart, J. F. (2002). Dynamic Covalent Chemistry. Angew. Chem. Int. Ed. 41, 898–952. doi:10.1002/1521-3773(20020315)41:6<898::aid-anie898>3.0.co;2-e
Ruipérez, F., Galdeano, M., Gimenez, E., and Matxain, J. M. (2018). Sulfenamides as Building Blocks for Efficient Disulfide-Based Self-Healing Materials. A Quantum Chemical Study. ChemistryOpen 7, 248–255. doi:10.1002/open.201800003
Ruiz de Luzuriaga, A., Martin, R., Markaide, N., Rekondo, A., Cabañero, G., Rodríguez, J., et al. (2016). Epoxy Resin with Exchangeable Disulfide Crosslinks to Obtain Reprocessable, Repairable and Recyclable Fiber-Reinforced Thermoset Composites. Mater. Horiz. 3, 241–247. doi:10.1039/c6mh00029k
Sarma, R. J., Otto, S., and Nitschke, J. R. (2007). Disulfides, Imines, and Metal Coordination within a Single System: Interplay between Three Dynamic Equilibria. Chem. Eur. J. 13, 9542–9546. doi:10.1002/chem.200701228
Schmolke, W., Perner, N., and Seiffert, S. (2015). Dynamically Cross-Linked Polydimethylsiloxane Networks with Ambient-Temperature Self-Healing. Macromolecules 48, 8781–8788. doi:10.1021/acs.macromol.5b01666
Shin, S.-H., Kim, S.-M., Jeon, H., Hwang, S. Y., Oh, D. X., and Park, J. (2020). Skin-inspired Hydrogel-Elastomer Hybrid Forms a Seamless Interface by Autonomous Hetero-Self-Healing. ACS Appl. Polym. Mater. 2, 5352–5357. doi:10.1021/acsapm.0c00925
Sierka, M., Hogekamp, A., and Ahlrichs, R. (2003). Fast Evaluation of the Coulomb Potential for Electron Densities Using Multipole Accelerated Resolution of Identity Approximation. J. Chem. Phys. 118, 9136–9148. doi:10.1063/1.1567253
Takahashi, A., Goseki, R., Ito, K., and Otsuka, H. (2017a). Thermally Healable and Reprocessable Bis(hindered Amino)disulfide-Cross-Linked Polymethacrylate Networks. ACS Macro Lett. 6, 1280–1284. doi:10.1021/acsmacrolett.7b00762
Takahashi, A., Goseki, R., and Otsuka, H. (2017b). Thermally Adjustable Dynamic Disulfide Linkages Mediated by Highly Air-Stable 2,2,6,6-Tetramethylpiperidine-1-Sulfanyl (Temps) Radicals. Angew. Chem. Int. Ed. 56, 2016–2021. doi:10.1002/anie.201611049
Takahashi, A., Ohishi, T., Goseki, R., and Otsuka, H. (2016). Degradable Epoxy Resins Prepared from Diepoxide Monomer with Dynamic Covalent Disulfide Linkage. Polymer 82, 319–326. doi:10.1016/j.polymer.2015.11.057
Takashima, R., Aoki, D., and Otsuka, H. (2020). Rational Entry to Cyclic Polymers via Thermally Induced Radical Ring-Expansion Polymerization of Macrocycles with One Bis(hindered Amino)disulfide Linkage. Macromolecules 53, 4670–4677. doi:10.1021/acs.macromol.0c00798
Takashima, R., Aoki, D., and Otsuka, H. (2021). Synthetic Strategy for Mechanically Interlocked Cyclic Polymers via the Ring-Expansion Polymerization of Macrocycles with a Bis(hindered Amino)disulfide Linker. Macromolecules 54, 8154–8163. doi:10.1021/acs.macromol.1c01067
Tsurumi, N., Takashima, R., Aoki, D., Kuwata, S., and Otsuka, H. (2020). A Strategy toward Cyclic Topologies Based on the Dynamic Behavior of a Bis(hindered Amino)disulfide Linker. Angew. Chem. Int. Ed. 59, 4269–4273. doi:10.1002/anie.201910722
TURBOMOLE Gmbh (2014). TURBOMOLE Version 6.6, a Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007. Available at: http://www.turbomole.com.
Xu, W. M., Rong, M. Z., and Zhang, M. Q. (2016). Sunlight Driven Self-Healing, Reshaping and Recycling of a Robust, Transparent and Yellowing-Resistant Polymer. J. Mater. Chem. A. 4, 10683–10690. doi:10.1039/c6ta02662a
Ying, H., Zhang, Y., and Cheng, J. (2014). Dynamic Urea Bond for the Design of Reversible and Self-Healing Polymers. Nat. Commun. 5, 3218. doi:10.1038/ncomms4218
Yokochi, H., Ohira, M., Oka, M., Honda, S., Li, X., Aoki, D., et al. (2021). Topology Transformation toward Cyclic, Figure-Eight-Shaped, and Cross-Linked Polymers Based on the Dynamic Behavior of a Bis(hindered Amino)disulfide Linker. Macromolecules 54, 9992–10000. doi:10.1021/acs.macromol.1c01437
Zhang, C., Hu, J., Li, X., Wu, Y., and Han, J. (2014). Hydrogen-bonding Interactions in Hard Segments of Shape Memory Polyurethane: Toluene Diisocyanates and 1,6-hexamethylene Diisocyanate. A Theoretical and Comparative Study. J. Phys. Chem. A. 118, 12241–12255. doi:10.1021/jp508817v
Zhang, Y., Ying, H., Hart, K. R., Wu, Y., Hsu, A. J., Coppola, A. M., et al. (2016). Malleable and Recyclable Poly(urea-Urethane) Thermosets Bearing Hindered Urea Bonds. Adv. Mater. 28, 7646–7651. doi:10.1002/adma.201601242
Zhang, Z. P., Rong, M. Z., and Zhang, M. Q. (2018). Polymer Engineering Based on Reversible Covalent Chemistry: A Promising Innovative Pathway towards New Materials and New Functionalities. Prog. Polym. Sci. 80, 39–93. doi:10.1016/j.progpolymsci.2018.03.002
Zheng, N., Fang, Z., Zou, W., Zhao, Q., and Xie, T. (2016). Thermoset Shape-Memory Polyurethane with Intrinsic Plasticity Enabled by Transcarbamoylation. Angew. Chem. Int. Ed. 55, 11421–11425. doi:10.1002/anie.201602847
Keywords: self-healing, dynamic chemistry, disulfides, polymers, computational chemistry
Citation: Irigoyen M, Matxain JM and Ruipérez F (2022) Combined DFT and MD Simulation Protocol to Characterize Self-Healing Properties in Disulfide-Containing Materials: Polyurethanes and Polymethacrylates as Case Studies. Front. Mater. 9:859482. doi: 10.3389/fmats.2022.859482
Received: 21 January 2022; Accepted: 28 February 2022;
Published: 21 March 2022.
Edited by:
Marianella Hernández Santana, Institute of Polymer Science and Technology (CSIC), SpainReviewed by:
Junheng Zhang, South-Central University for Nationalities, ChinaYu Wang, University of Science and Technology of China, China
Copyright © 2022 Irigoyen, Matxain and Ruipérez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernando Ruipérez, ZmVybmFuZG8ucnVpcGVyZXpAZWh1LmV1cw==; Jon M. Matxain, am9ubWF0dGluLm1hdHhhaW5AZWh1LmV1cw==