 Magnus Röding
Magnus Röding Piotr Tomaszewski3
Piotr Tomaszewski3 Shun Yu
Shun Yu- 1Agriculture and Food, Bioeconomy and Health, RISE Research Institutes of Sweden, Göteborg, Sweden
- 2Department of Mathematical Sciences, Chalmers University of Technology and University of Gothenburg, Göteborg, Sweden
- 3Mobility and Systems, Digital Systems, RISE Research Institutes of Sweden, Lund, Sweden
- 4Material and Surface Design, Bioeconomy and Health, RISE Research Institutes of Sweden, Stockholm, Sweden
Small-angle X-ray scattering (SAXS) is a useful technique for nanoscale structural characterization of materials. In SAXS, structural and spatial information is indirectly obtained from the scattering intensity in the spectral domain, known as the reciprocal space. Therefore, characterizing the structure requires solving the inverse problem of finding a plausible structure model that corresponds to the measured scattering intensity. Both the choice of structure model and the computational workload of parameter estimation are bottlenecks in this process. In this work, we develop a framework for analysis of SAXS data from disordered materials. The materials are modeled using Gaussian Random Fields (GRFs). We study the case of two phases, pore and solid, and three phases, where a third phase is added at the interface between the two other phases. Further, we develop very fast GPU-accelerated, Fourier transform-based numerical methods for both structure generation and SAXS simulation. We demonstrate that length scales and volume fractions can be predicted with good accuracy using our machine learning-based framework. The parameter prediction executes virtually instantaneously and hence the computational burden of conventional model fitting can be avoided.
1 Introduction
For heterogeneous, disordered materials, the microstructure i.e. the geometry of the different phases substantially influences the performance of a material, e.g., thermal, electric, mechanical, and mass transport properties (Torquato, 2010). Therefore, characterizing the microstructure is a crucial step towards understanding the material and optimizing its design. A vast number of applications of advanced materials rely on precise control of physical and chemical processes within a microstructure with length scales in the nanometer range, like batteries, chemical separation techniques, and chromatography (Gommes, 2018). To characterize detailed random porous structures, high-resolution 3D imaging techniques e.g., micro/nano X-ray computed tomography (X-ray CT), focused ion beam scanning electron microscopy (FIB-SEM), and transmission electron microscopy tomography (TEMT) can provide high quality information on morphological features. Nevertheless, imaging techniques are frequently time-consuming and require advanced sample preparation methods or sample environments. Moreover, the attainable contrast is strongly sample-dependent and can be prohibitively low.
Complementary to imaging approaches at the nanoscale, small-angle X-ray scattering (SAXS) is a powerful technique that characterizes the nanostructure via a scattering intensity measured in the spectral domain (reciprocal space). More precisely, the spatial distribution of electron density is indirectly observed through the elastic scattering behaviour of X-rays passing through the material. The interaction with the sample forces the X-rays to change direction with a certain angle (the scattering angle). The intensity as a function of scattering angle is related to the electron density distribution through a Fourier transform. With a modern 2D pixel-array detector, SAXS can easily provide structural information for sub-micron length scales and all the way down to a few Ångström, by changing the sample-to-detector distance from meters to millimeters accordingly. In addition, owing to the high penetration power of X-rays, the sample can be studied in different states of matter e.g. gas, solution, or solid, and in other conditions coupled with thermal, mechanical, electrical and magnetic fields (Li et al., 2016). With the latest X-ray detection techniques, SAXS measurements become rather fast, in particular at synchrotron facilities offering high photon flux where hundreds of measurements per second can be performed. This enables so-called in situ/in-operando characterization of continuous nanostructure development. SAXS has been applied to numerous types of materials including nanostructures (Li et al., 2016), biomacromolecules (Blanchet and Svergun, 2013), polymers (Chu and Hsiao, 2001), and porous materials (Welborn and Detsi, 2020).
However, a major challenge of SAXS is that the electron density distribution is in general not uniquely determined by the scattering intensity, owing to the ‘phase problem’ (Taylor, 2003): as X-rays are electromagnetic waves having two important parameters, amplitude and phase, the intensity recorded on the detector is the modulus of the wave’s Poynting vector and proportional to the square of the amplitude of the scattered wave, while the phase information is lost in the experiment. In addition, for isotropic systems, the scattering intensity can be presented in a one-dimensional curve that constitutes spherically-averaged spectral magnitudes, further compressing the information. In effect, the SAXS data contains much less information than the corresponding full, three-dimensional Fourier transform. Limited prior knowledge warrants the fitting of multiple SAXS models in a trial-and-error fashion, which can be a daunting task even for an experienced investigator (Archibald et al., 2020; Do et al., 2020). Further, fitting of non-analytical models can be computationally prohibitive.
Because data analysis constitutes a bottleneck in the use of SAXS, numerous approaches based on machine learning have been proposed in recent years, with the aim of accelerating the data analysis and providing decision support for the operator. Most also focus on biomacromolecules and nanoparticles, which are usually characterized in dilute solution by SAXS. For example, Franke et al. (Franke et al., 2018) use k-nearest neighbors to classify SAXS data based on particle shapes, diameters, and molecular mass; Archibald et al. (Archibald et al., 2020) use weighted k-nearest neighbors and Gaussian processes for classifying SAXS data and determine the most probable type of structure; He et al. (He et al., 2020) use a convolutional neural network-based autoencoder combined with a genetic algorithm to search for structures that are consistent with SAXS data; Scherdel et al. (Scherdel et al., 2021) use machine learning to directly predict effective properties such as thermal conductivity of silica aerogels; Tomaszewski et al. (Tomaszewski et al., 2021) evaluate numerous machine learning approaches to classifying SAXS data.
In this work, we investigate disordered, porous materials where the electron density distribution is modeled using thresholded Gaussian Random Fields (GRFs). This serves as a model system representing continuous multiphase distributions with irregular geometric shapes; this is different from many biomacromolecules and nanoparticles characterized by SAXS which are often treated as isolated systems and can be modelled ab initio. GRFs are frequently used as models for materials microstructures, because they realistically describe phase-separated, heterogeneous materials, originating from a description of spinodal decomposition by Cahn and Hilliard (Cahn and Hilliard, 1958). GRFs have been used in models for scattering data (Berk, 1991; Quintanilla et al., 2007; Gommes, 2013; Gommes and Roberts, 2018) and as material models for a wide variety of materials both for SAXS analysis and otherwise, including microemulsions (Teubner, 1991; Chen et al., 1996), polymer blends (Jinnai et al., 1997; D’hollander et al., 2010; Barman et al., 2019), lithium-ion batteries (Feinauer et al., 2015), porous alloys for energy storage and catalysis (Geslin et al., 2015; Lu et al., 2018), and gels (Roberts, 1997; Gommes and Roberts, 2008). We study the case of two phases, pore (vacuum/air) and solid, and three phases, where the third phase is an intermediate layer residing by the interface between pore and solid. We generate a large number of virtual microstructures for a number of cases. We further develop a Fourier transform-based numerical method for simulating realistic SAXS data. Both the structure generation and SAXS simulation methods are heavily optimized and implemented on GPU with a combined execution time in the order of 1 s. Using the simulated SAXS data as input and the known generation parameters as target output in a machine learning framework, we demonstrate that length scales and volume fractions can be predicted with good accuracy. Our framework is a proof of concept that can be applied to other types of disordered materials as well, in particular other Gaussian random field-based models with different covariance structures.
2 Results and discussion
2.1 Simulation of scattering intensity
In SAXS, the sample is irradiated with collimated or focused X-rays, the incident-radiation wavelength (of the monochromatic X-rays) being λ. The intensity of the elastically scattered X-ray is measured as a function of the magnitude of the scattering vector, q = |q|, where
where
Assume that a virtual electron density ρ is simulated on a periodic cubic domain, i.e. a 3D voxel array, with resolution N3 and voxel size Δx. Then the fast Fourier transform (FFT) can be used to obtain the discrete counterpart to the scattering intensity, which we again denote by I(q). It is also defined on a periodic cubic domain with resolution N3, for all
for i = −N/2, − N/2 + 1, … , N/2–2, N/2–1, and likewise for
where wq is a weight function such that
for qijk = |qijk|, Δq = 2π/(NΔx) (the grid resolution in q space), with
In practice, because of the symmetries in q space, the N3 array I(q) can be folded into a (N/2 + 1)3 array which substantially reduces the computation time (for the steps corresponding to Eqs 3, 4). The simulation is implemented on GPU in Matlab (Mathworks, Natick, MA, US).
2.2 Microstructure model
In the original model by Cahn and Hilliard (Cahn and Hilliard, 1958), GRFs arose as a solution to a spinodal decomposition model described as a superposition of cosine waves,
for M ≫ 1, some wave vectors qm and random phase offsets ηm, 0 ≤ ηm < 2π. The wave vectors follow some probability distribution
Therefore, we instead use a method based on the Fast Fourier Transform (FFT) (Lang and Potthoff, 2011). A GRF can generally be described by a mean value and a covariance function (Liu et al., 2019). The spectral density of this covariance function actually equals
We use the spectral density
a special case of a spectral density used before (Matérn, 1986; Lang and Potthoff, 2011), also in models for materials microstructures (Röding et al., 2020; Prifling et al., 2021) (note that the spectral density is not normalized hence not a probability distribution; this only results in a linear scaling of the GRF, which is of no concern here). Because
We model both two phases, pore (vacuum/air) and solid, and three phases, where the third phase is an intermediate layer residing by the interface between pore and solid. The layer can be regarded as a material condensed at the surface of the solid phase. The microstructures are parameterized by the scaling parameter a, the porosity ϵ, and the fraction ν of the pore space filled up by the intermediate layer; for the two-phase microstructures, ν = 0. The volume fractions are ϵ(1 − ν), ϵν, and 1 − ϵ for pore, layer, and solid. In addition, electron densities need to be specified. We let all electron densities be between 0 (vacuum) and one for convenience; the scattering intensity is proportional to the mean squared fluctuation of the electron density of the material (Welborn and Detsi, 2020) but also to other experimental factors such as X-ray photon flux. Therefore, the scattering intensity can always be rescaled. We choose ρpore = 0, ρlayer = 0.65, and ρsolid = 1. Assuming that the intermediate layer is water, the electron density ratio between the layer and the solid phase is fairly close to that of water to cellulose (density 1.5 g/cm3, ρlayer ≈ 0.69), and water to hard carbon (Nishi and Pistoia, 2014) (density 1.45–1.55 g/cm3, ρlayer ≈ 0.69–0.74), all of which were estimated by using refractive index at around 8 keV (Cu K-alpha X-ray source) from the center of X-ray optics (Henke et al., 1993).
Microstructures are generated from the GRFs in the following manner. First, a binary function is obtained by thresholding,
for T such that p (ψ(x) ≤ T) = ϵ. Second, ψ′ is smoothed with a 3D Gaussian filter (accounting for periodicity; σ = 2 voxels, but this choice is not crucial), yielding ψ″, 0 ≤ ψ″ ≤ 1. Two-phase electron densities can now be defined by
for T such that p (ψ″(x) ≤ T) = ϵ. Three-phase electron densities can be defined similarly by
for T1 such that p (ψ″(x) ≤ T1) = ϵ(1 − ν) and T2 such that p (T1 < ψ″(x) ≤ T2) = ϵν) (and also p (ψ″(x) > T2) = 1 − ϵ)).
Note that the ‘intermediate’ values of ψ″ (not equal to 0 or 1) will be concentrated near what will be the pore-solid interface, which is the reason for defining it this way. It ensures that with high probability, the intermediate layer will be adjacent to both pore and solid. If, on the other hand, ρ(x) would be obtained by thresholding ψ(x) directly, parts of the intermediate layer phase might end up in contact only with pore or only with solid in all directions, which would not be physically plausible. It is also worth pointing out that by using the 3D Gaussian filter also in the two-phase model, the two- and three-phase models are seamlessly integrated into the same framework and the two-phase model is a special case of the three-phase model.
The generation procedure is illustrated with microstructures generated on a grid of size 5123 with voxel size Δx = 0.5 nm (therefore the parameter a also has unit nm). In Figure 1, using a single 2D slice of a three-phase model with a = 4 nm, ϵ = 0.60, and ν = 0.50. In Figure 2, a 3D visualization of the same structure is shown. The simulation is implemented on GPU in Matlab (Mathworks, Natick, MA, US).
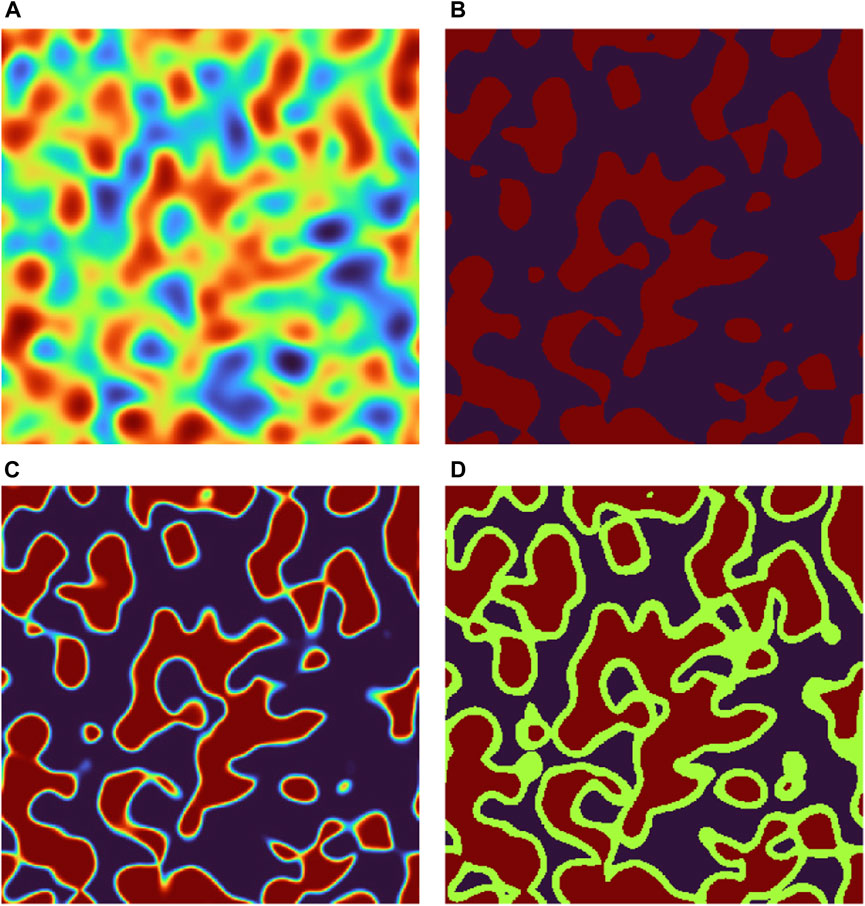
FIGURE 1. Illustration of a single 2D slice of a three-phase model with a =4 nm, ϵ =0.60, and ν =0.50. In (A), the GRF is shown (arbitrary intensity scale). In (B), a binary structure is shown, obtained from (A) by thresholding at the quantile ϵ. In (C), a smooth structure is shown, obtained from (B) by smoothing with a 3D Gaussian filter. In (D), the final electron density is shown, obtained from (C) by thresholding at the quantiles ϵ(1− ν) and ϵ.

FIGURE 2. Illustration of a three-phase model with a =4 nm, ϵ =0.60, and ν =0.50. For an illustration of a single 2D slice from this model, see Figure 1.
2.3 Dataset generation
For developing a machine learning-based model for prediction of microstructural parameters, a large number of microstructures are generated on a grid of size 5123 with voxel size Δx = 0.5 nm (we reiterate that therefore the parameter a also has unit nm, whereas ϵ and ν are dimensionless quantities).
We reiterate that two-phase microstructures are generated using ρpore = 0 and ρsolid = 1; varying ρsolid is unnecessary because SAXS data can always be rescaled. Three-phase microstructures are generated using the same values and additionally ρlayer = 0.65.
For all datasets, the scaling parameter a is uniformly distributed in [0.8, 8] nm. The resulting range of length scales is found to be represented well considering the resolution and simulation box size and to be accessible in the simulated q range. We generate four separate datasets: 1) two-phase, low porosity structures 2) two-phase, high porosity structures, 3) three-phase, low porosity structures, and 4) three-phase, high porosity structures. The porosity ϵ is uniformly distributed in [0.1, 0.5] for the low porosity structures and in [0.5, 0.9] for the high porosity structures. For the three-phase structures, the fraction of the intermediate layer in the pores ν is uniformly distributed in [0.05, 0.5] in both cases. The reason why porosities lower and higher than 0.5 are treated separately is that in the two-phase case, the values ϵ and 1 − ϵ literally cannot be distinguished because they are ‘mirror images’ of each other; therefore, approximate information about the porosity needs to be supplied as an input from e.g. sample contrast. This symmetry is referred to as Babinet’s principle and is reflected in the so-called Porod invariant of a two-phase scattering pattern (Zhang et al., 2012),
where Δρ is the electron density difference between the two phases, which is one in our case. So, whether the structure has low or high porosity is information that has to be supplied by the user. In the three-phase case, the situation is more complex, and unless ρlayer = 1/2 there is no exact mirror image. Nevertheless, we treat two- and three-phase structures consistently in this respect.
For each of the four cases, 219 (524,288) microstructures are generated for the training dataset, 218 (262,144) for the validation dataset, and 217 (131,072) for the test dataset. The scattering intensity is simulated for 500 q values, equidistant between qmin ≈ 0.04 nm−1 and qmax ≈ 3.00 nm−1; these values are taken from an in-house experimental setup using an Anton Paar SAXSpoint 2.0 (Anton Paar, Graz, Austria) and cover a normal SAXS probing range without losing the generality. On an NVIDIA A40 GPU, the average execution time for microstructure generation and simulation of SAXS data combined is approximately 1 s.
Note that the simulation of the scattering intensity I(q) described above does not account for measurement noise. The specifics of the noise are dependent on the experimental conditions (such as photon flux and quantum efficiency of the detector) and thereby on the intensity scale determined by the unknown prefactor I0, as aforementioned. We use a noise model inspired by the Poisson distributed photon counting nature of the data acquisition (Sedlak et al., 2017). A Poisson model would imply that the variance of the noise is σ2(q) = I(q); however, considering the intensity scales resulting from using I0 = 1, this model assumption does not produce realistic noise levels. Instead, we use a lognormal noise model with mean I(q) and σ2(q) = αI(q), where a value of α is sampled from a log-uniform distribution in [102, 105.5] for each SAXS curve. The lognormal distribution only produces positive values, whereas the commonly suggested normal approximation can produce physically implausible, negative values (which it did, in this case). Note that whereas we account for measurement noise, we do not account for the finite instrument resolution which would yield a slight blurring of the SAXS curves.
Further, analogously to Gommes (2018) (Gommes, 2018), each simulated SAXS curve is normalized, dividing by a total intensity approximated by
where
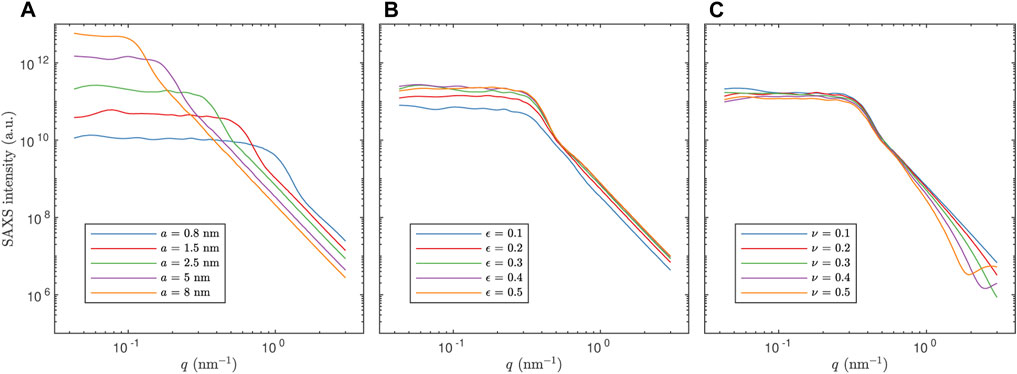
FIGURE 3. Examples of simulated SAXS curves. In (A), a is varied for ϵ =0.3 and ν =0 (two-phase model). In (B), ϵ is varied for a =2.5 nm and ν =0 (two-phase model). In (C), ν is varied for a =2.5 nm and ϵ =0.3 (three-phase model). For clarity, measurement noise has not been added to these curves.
2.4 Prediction of model parameters
The classical approach to estimating parameters in a model would be some variety of curve fitting, using e.g. least squares to find a scattering intensity curve that deviates minimally from the data and extract its parameters. However, predicting the model parameters can alternatively be considered a nonlinear regression and supervised learning problem: given a set of inputs i.e. the scattering intensities and the corresponding outputs i.e. the model parameters, find a function that approximates the mapping from input to output. Virtually any machine learning method for regression can be used in this setting. We rely on XGBoost (Chen and XGBoost, 2016), a specific implementation of gradient boosted trees. XGBoost has proven useful and accurate in another recent SAXS study where it was found to be superior to several other methods (Tomaszewski et al., 2021). Conveniently, and contrarily to some other candidate methods, XGBoost provides support for GPU acceleration. The orders-of-magnitude speedup delivered by the GPU acceleration makes XGBoost a particularly pragmatic choice for experiments involving large amounts of data, as it significantly shortens the experimentation time. The prediction model is a tree ensemble that combines a large number of weak, decision tree-based prediction models to produce a single, stronger prediction model. Each regression tree is constructed by recursively splitting the input data space into partitions or “branches”. After a sufficient amount of splitting, the space is divided into “leafs”, where each leaf corresponds to a single, scalar value which is the prediction of the output. Whereas a single regression tree is a poor approximation to the function mapping input to output, combining a large number of regression trees is a very powerful approach with performance on par with artificial neural networks. While deep neural networks dominate machine learning for computer vision and natural language processing, tree ensemble methods are generally the recommended approach for tabular data (Shwartz-Ziv and Armon, 2022).
The inputs of the training set are first preprocessed by transforming to logarithmic scale. Then, they are standardized by computing the mean and variance for each dimension separately on the training set, and then rescaling to zero mean and unit variance. Finally, the same rescaling (using the mean and variance of the training set) is applied to the validation and test sets (and also to new data once the prediction model is finalized and used). The outputs are not preprocessed. During training, the trees of the XGBoost model are optimized with respect to mean squared error (MSE) loss,
where y is a target value and

TABLE 1. List of hyperparameters investigated, with a brief explanation of their meaning and range of their values.
The results for the final selected XGBoost models is shown in Table 2. In addition to MSE, we also use the more intuitive mean absolute percentage error (MAPE) loss,
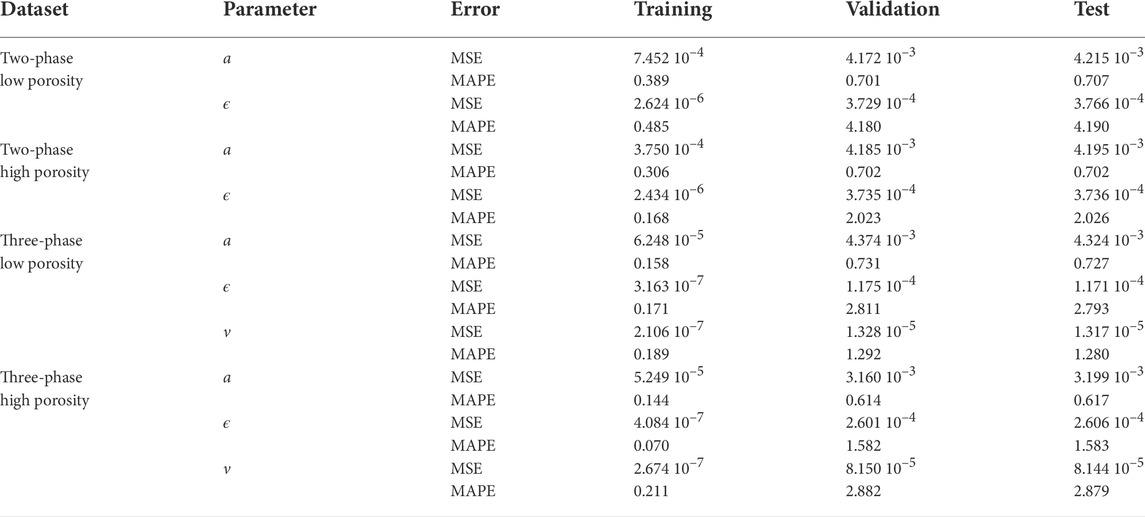
TABLE 2. Error measures for the prediction of the parameters, where MSE and MAPE (in %) is given for the training, validation and test sets.
Further, in Figures 4, 5, we show scatter plots of the predictions of all parameters for the test sets. We note that the scaling parameter a is consistently the easiest to predict in terms of MAPE (considering the substantial impact of the value of a on both the length and magnitude of the low-q plateau as seen in Figure 3, this is no surprise), but evidently a bit more difficult for increasing values of a because the relative error increases (not shown). This might be partly because of resolution limitations for low values of q (the simulated values of I(q) for low q is based on a very small number of grid points in q space; and large length scales i.e. large a correspond to low q). Also, the porosity ϵ is consistently the most difficult to predict. Finally, the fraction ν of the pore spaced filled up by intermediate layer is predicted a bit better than ϵ, but has more pronounced outliers in the low porosity case, in particular for low ν. This is likely because the fraction of the third phase is very low for low values of both ϵ and ν, and therefore the simulated SAXS data contains very limited information about that phase. For all parameters, the predictions have a positive bias near the lower bound of the range of true values. Likewise, the predictions have a negative bias near the upper bound of the range of true values. This is simply because the models are not trained to predict values outside the range and hence are unlikely to make such predictions. This fact also illustrates very clearly that the models cannot be expected to extrapolate well, but will rather provide reasonable predictions only within the domain of applicability (Sutton et al., 2020), which is determined by the distribution of inputs and outputs in the training set and the prediction model itself. It is also worth pointing out that if the prediction model would have been trained to predict the porosity on the low-porosity and the high-porosity data jointly, the predicted values would be nonsensical, and accordingly, the reported accuracy would be substantially lower.
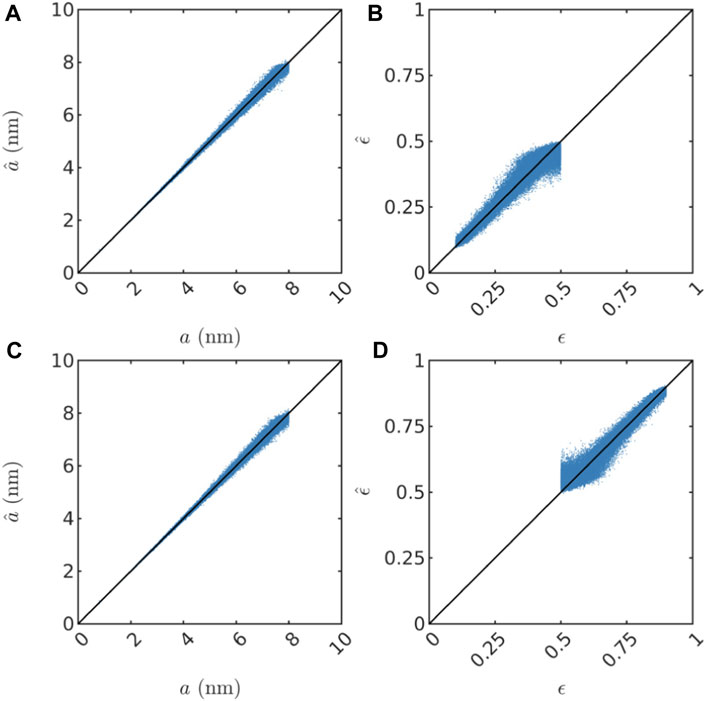
FIGURE 4. Scatter plots showing prediction results on the test set for both two-phase datasets. In (A,B), predictions of a and ϵ are shown for the two-phase low porosity structures. In (C,D), predictions of a and ϵ are shown for the two-phase high porosity structures.
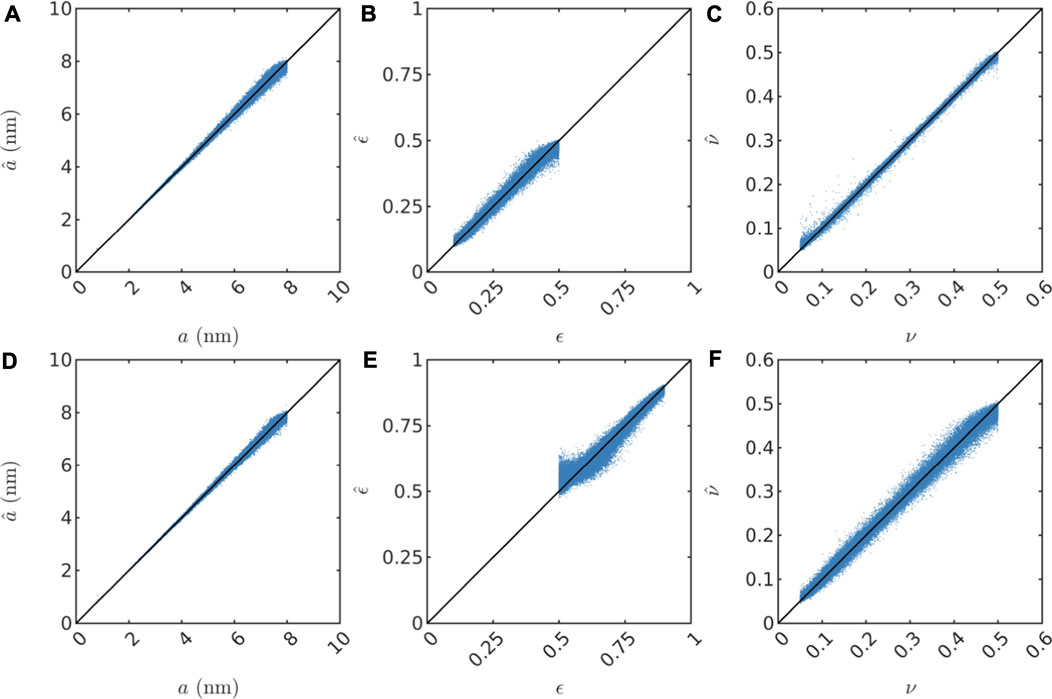
FIGURE 5. Scatter plots showing prediction results on the test set for both three-phase datasets. In (A–C), predictions of a, ϵ, and ν are shown for the three-phase low porosity structures. In (D–F), predictions of a, ϵ, and ν are shown for the three-phase high porosity structures.
In this context, it is important to note that the structures are random and not uniquely defined by the set of parameter values used to generate them; each set of parameter values can yield a very large number of different structures that in turn yield an equal number of different SAXS curves. Therefore, a SAXS curve cannot be uniquely mapped to a set of parameter values, even in the absence of measurement noise; their relationship is inherently random. It follows that the prediction loss is due to a combination of the randomness of the structures and the randomness induced by the added measurement noise. Therefore, there is in practice a lower bound on the attainable accuracy. This effect is essentially a result of the limited resolution and field of view of the simulated data and not as such a fundamental limitation of SAXS.
It is worth noting that we investigate two other techniques for regression. The first is also based on XGBoost but utilizing chained regression. This means that the different outputs are predicted sequentially such that the predictions of the first are used as input for prediction of the second, and the predictions of the first and second are used as inputs for prediction of the third. Also we investigate fully-connected artificial neural networks. An initial investigation suggests that neither of these two attempts yield better results than the ‘plain’ XGBoost approach presented, and are therefore not shown herein.
2.5 Simulated case study
To illustrate the performance of the method more clearly, we do a case study on simulated data using the three-phase model. Because the prediction model does not capture variability, the uncertainty of the predictions cannot be assessed using a single SAXS measurement. Therefore, we simulate a large number of measurements using the same parameter values, akin to performing replicate real measurements. Indeed, for a = 4 nm, ϵ = 0.70, and ν = 0.20, we generate 500 SAXS curves with the same noise model as before and use the three-phase high porosity model for prediction. The results are shown in Figure 6. The combined results are
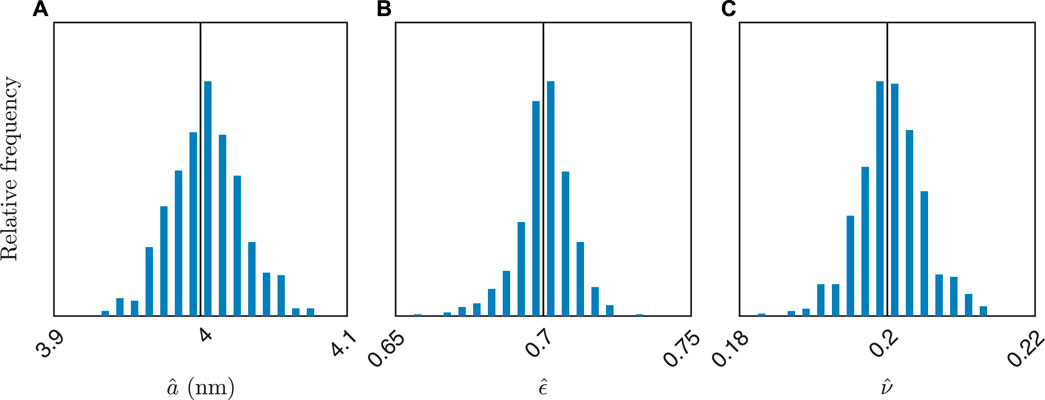
FIGURE 6. Histogram of estimated values for 500 simulated SAXS curves for a = 4 nm, ϵ = 0.70, and ν = 0.20. In (A–C), the distribution of estimated values of a, ϵ, and ν are shown. The true values are also indicated (vertical black lines).
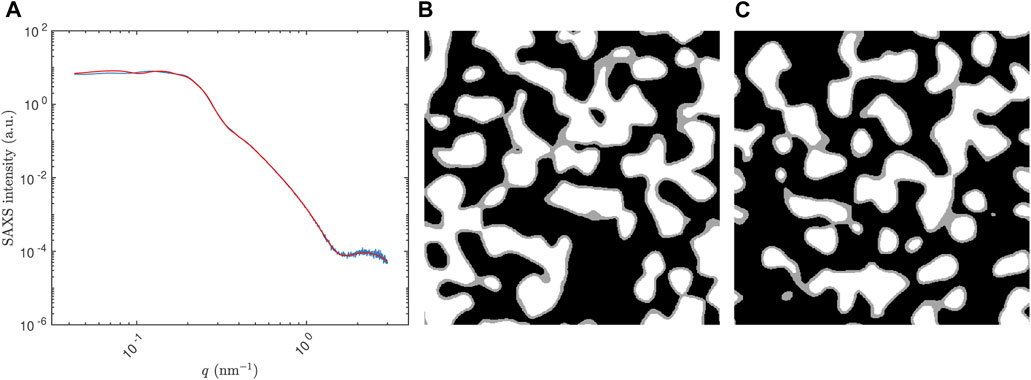
FIGURE 7. Results for a single structure in the case study. In (A), the simulated SAXS curve for a =4 nm, ϵ =0.70, and ν =0.20 (blue), and a (noiseless) SAXS curve of a structure generated using the predicted parameters
3 Conclusion
We have implemented a machine learning-based approach to fast estimation of microstructural parameters from SAXS data. The microstructure model is based on a periodic Gaussian random field with variable length scale, which is processed and thresholded to yield two-phase (pore and solid) and three-phase (pore, intermediate layer, and solid) structures, with all phases having different electron densities. We also develop a Fourier transform-based method to simulate SAXS data. Both microstructure generation and SAXS simulation are implemented on the GPU and very fast. We generate four very large, separate datasets: 1) two-phase, low porosity structures 2) two-phase, high porosity structures, 3) three-phase, low porosity structures, and 4) three-phase, high porosity structures. We demonstrate that by performing regression using XGBoost, a decision tree-based machine learning framework, the parameters of the models can be predicted with good accuracy. Given that artificial neural networks did not perform better than XGBoost, and given that there is no time dependence or translational invariance in the data to further exploit, it is unlikely that more advanced architectures such as recurrent or convolutional neural networks would perform better. Further, the parameter prediction executes virtually instantaneously. Hence the computational burden of conventional model fitting can be avoided, enabling for the SAXS practitioner to efficiently analyze many measurements.
We observed positive and negative bias in the predictions observed near the lower and upper bound of the simulated parameter ranges. This bias could be reduced by using a wider range of parameters (where possible) for the training set while maintaining the ranges for the validation and test sets. In this manner, the performance of the prediction will be assessed in a smaller parameter space, which should then be considered the domain of applicability.
Although the microstructure models herein are aimed at mimicking a certain type of morphology and certain ranges of the parameters, similar models can be expected to perform well for other types of microstructures (i.e., fibers, foams, granules) and other parameter ranges. The only requirement is that the microstructure model is efficiently implemented so that a large, representative dataset can be generated, and that the corresponding SAXS curves are sufficiently informative regarding the parameters to be predicted. Although the approach is evaluated on a specific type of morphology, it is a proof of concept that can be used for other types of materials, both with regard to spatial structure and electron density values, and also for other experimental parameters such as other q value ranges, and non-equidistant q values. Indeed, generalizing this investigation to multiple classes of Gaussian random field-based models would be an interesting prospect for further work.
In conclusion, this proof of concept illustrates the usefulness not only of the machine learning-based approached but also of the efficient GPU-accelerated scheme for simulating the materials structures and the corresponding SAXS data and the new three-phase model. Finally, all the data and codes used herein are publicly available to facilitate further development in this field.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: DOI:10.5281/zenodo.5948941.
Author contributions
SY and JR conceived the idea. MR developed the microstructure generation and SAXS simulation methods together with SY. MR, PT, and MB developed the machine learning methods. MR and JR coordinated the work. All authors contributed to designing the study and to writing the manuscript.
Funding
MR acknowledges the financial support of the Swedish Research Council for Sustainable Development (grant number 2019-01295). SY acknowledges the financial support of the Swedish Research Council (grant number 2018-06378).
Acknowledgments
The computations were in part performed on resources at Chalmers Centre for Computational Science and Engineering (C3SE) provided by the Swedish National Infrastructure for Computing (SNIC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Archibald, R. K., Doucet, M., Johnston, T., Young, S. R., Yang, E., and Heller, W. T. (2020). Classifying and analyzing small-angle scattering data using weighted k nearest neighbors machine learning techniques. J. Appl. Crystallogr. 53, 326–334. doi:10.1107/s1600576720000552
Barman, S., Rootzén, H., and Bolin, D. (2019). Prediction of diffusive transport through polymer films from characteristics of the pore geometry. AIChE J. 65, 446–457. doi:10.1002/aic.16391
Bergstra, J., and Bengio, Y. (2012). Random search for hyper-parameter optimization. J. Mach. Learn. Res. 13, 281.
Berk, N. F. (1991). Scattering properties of the leveled-wave model of random morphologies. Phys. Rev. A . Coll. Park. 44, 5069–5079. doi:10.1103/physreva.44.5069
Blanchet, C. E., and Svergun, D. I. (2013). Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution. Annu. Rev. Phys. Chem. 64, 37–54. doi:10.1146/annurev-physchem-040412-110132
Cahn, J. W., and Hilliard, J. E. (1958). Free energy of a nonuniform system. I. Interfacial free energy. J. Chem. Phys. 28, 258–267. doi:10.1063/1.1744102
Chen, S. H., Lee, D. D., Kimishima, K., Jinnai, H., and Hashimoto, T. (1996). Measurement of the Gaussian curvature of the surfactant film in an isometric bicontinuous one-phase microemulsion. Phys. Rev. E 54, 6526–6531. doi:10.1103/physreve.54.6526
Chen, T., and XGBoost, C. G. (2016). “A scalable tree boosting system,” in Proceedings of the 22nd ACM SIGKDD Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, August 13-17, 2016, 785–794.
Chu, B., and Hsiao, B. S. (2001). Small-angle X-ray scattering of polymers. Chem. Rev. 101, 1727–1762. doi:10.1021/cr9900376
D’hollander, S., Gommes, C. J., Mens, R., Adriaensens, P., Goderis, B., and Du Prez, F. (2010). Modeling the morphology and mechanical behavior of shape memory polyurethanes based on solid-state NMR and synchrotron SAXS/WAXD. J. Mat. Chem. 20, 3475–3486. doi:10.1039/b923734h
Do, C., Chen, W.-R., and Lee, S. (2020). Small angle scattering data analysis assisted by machine learning methods. MRS Adv. 5, 1577–1584. doi:10.1557/adv.2020.130
Feinauer, J., Brereton, T., Spettl, A., Weber, M., Manke, I., and Schmidt, V. (2015). Stochastic 3D modeling of the microstructure of lithium-ion battery anodes via Gaussian random fields on the sphere. Comput. Mater. Sci. 109, 137–146. doi:10.1016/j.commatsci.2015.06.025
Franke, D., Jeffries, C. M., and Svergun, D. I. (2018). Machine learning methods for X-ray scattering data analysis from biomacromolecular solutions. Biophysical J. 114, 2485–2492. doi:10.1016/j.bpj.2018.04.018
Geslin, P.-A., McCue, I., Gaskey, B., Erlebacher, J., and Karma, A. (2015). Topology-generating interfacial pattern formation during liquid metal dealloying. Nat. Commun. 6 (1–8), 8887. doi:10.1038/ncomms9887
Gommes, C. J., and Roberts, A. P. (2018). Stochastic analysis of capillary condensation in disordered mesopores. Phys. Chem. Chem. Phys. 20, 13646–13659. doi:10.1039/c8cp01628c
Gommes, C. J., and Roberts, A. P. (2008). Structure development of resorcinol-formaldehyde gels: Microphase separation or colloid aggregation. Phys. Rev. E 77, 041409. doi:10.1103/physreve.77.041409
Gommes, C. J. (2018). Stochastic models of disordered mesoporous materials for small-angle scattering analysis and more. Microporous Mesoporous Mater. 257, 62–78. doi:10.1016/j.micromeso.2017.08.009
Gommes, C. J. (2013). Three-dimensional reconstruction of liquid phases in disordered mesopores using in situ small-angle scattering. J. Appl. Crystallogr. 46, 493–504. doi:10.1107/s0021889813003816
He, H., Liu, C., and Liu, H. (2020). Model reconstruction from small-angle X-ray scattering data using deep learning methods. iScience 23, 100906. doi:10.1016/j.isci.2020.100906
Henke, B. L., Gullikson, E. M., and Davis, J. C. (1993). X-Ray interactions: Photoabsorption, scattering, transmission, and reflection at E = 50-30, 000 eV, Z = 1-92. Atomic Data Nucl. Data Tables 54, 181–342. doi:10.1006/adnd.1993.1013
Jinnai, H., Hashimoto, T., Lee, D., and Chen, S.-H. (1997). Morphological characterization of bicontinuous phase-separated polymer blends and one-phase microemulsions. Macromolecules 30, 130–136. doi:10.1021/ma960486x
Lang, A., and Potthoff, J. (2011). Fast simulation of Gaussian random fields. Monte Carlo Methods Appl. 17, 195–214. doi:10.1515/mcma.2011.009
Li, T., Senesi, A. J., and Lee, B. B. (2016). Small angle X-ray scattering for nanoparticle research. Chem. Rev. 116, 11128–11180. doi:10.1021/acs.chemrev.5b00690
Liu, Y., Li, J., Sun, S., and Yu, B. (2019). Advances in Gaussian random field generation: A review. Comput. Geosci. 23, 1011–1047. doi:10.1007/s10596-019-09867-y
Lu, Z., Li, C., Han, J., Zhang, F., Liu, P., Wang, H., et al. (2018). Three-dimensional bicontinuous nanoporous materials by vapor phase dealloying. Nat. Commun. 9 (1–7), 276. doi:10.1038/s41467-017-02167-y
Nishi, Y. (2014). “2 - past, present and future of lithium-ion batteries: Can new technologies open up new horizons?,” in Lithium-ion batteries. Editor G. Pistoia (Amsterdam: Elsevier), 21–39.
Prifling, B., Röding, M., Townsend, P., Neumann, M., and Schmidt, V. (2021). Large-scale statistical learning for mass transport prediction in porous materials using 90, 000 artificially generated microstructures. Front. Mat. 8, 786502. doi:10.3389/fmats.2021.786502
Quintanilla, J. A., Chen, J. T., Reidy, R. F., and Allen, A. J. (2007). Versatility and robustness of Gaussian random fields for modelling random media. Model. Simul. Mat. Sci. Eng. 15, S337–S351. doi:10.1088/0965-0393/15/4/s02
Roberts, A. P. (1997). Morphology and thermal conductivity of model organic aerogels. Phys. Rev. E 55, R1286–R1289. doi:10.1103/physreve.55.r1286
Röding, M., Ma, Z., and Torquato, S. (2020). Predicting permeability via statistical learning on higher-order microstructural information. Sci. Rep. 10, 15239. doi:10.1038/s41598-020-72085-5
Scherdel, C., Miller, E., Reichenauer, G., and Schmitt, J. (2021). Advances in the development of sol-gel materials combining small-angle X-ray scattering (SAXS) and machine learning (ML). Processes 9, 672. doi:10.3390/pr9040672
Schmidt-Rohr, K. (2007). Simulation of small-angle scattering curves by numerical Fourier transformation. J. Appl. Crystallogr. 40, 16–25. doi:10.1107/s002188980604550x
Sedlak, S. M., Bruetzel, L. K., and Lipfert, J. (2017). Quantitative evaluation of statistical errors in small-angle X-ray scattering measurements. J. Appl. Crystallogr. 50, 621–630. doi:10.1107/s1600576717003077
Shwartz-Ziv, R., and Armon, A. (2022). Tabular data: Deep learning is not all you need. Inf. Fusion 81, 84–90. doi:10.1016/j.inffus.2021.11.011
Sorbier, L., Moreaud, M., and Humbert, S. (2019). Small-angle X-ray scattering intensity of multiscale models of spheres. J. Appl. Crystallogr. 52, 1348–1357. doi:10.1107/s1600576719013839
Sutton, C., Boley, M., Ghiringhelli, L. M., Rupp, M., Vreeken, J., and Scheffler, M. (2020). Identifying domains of applicability of machine learning models for materials science. Nat. Commun. 11 (1–9), 4428. doi:10.1038/s41467-020-17112-9
Taylor, G. (2003). The phase problem. Acta Crystallogr. D. Biol. Crystallogr. 59, 1881–1890. doi:10.1107/s0907444903017815
Teubner, M. (1991). Level surfaces of Gaussian random fields and microemulsions. Europhys. Lett. 14, 403–408. doi:10.1209/0295-5075/14/5/003
Tomaszewski, P., Yu, S., Borg, M., and Rönnols, Jerk (2021). “Machine learning-assisted analysis of small angle X-ray scattering,” in Proceedings of the 2021 Swedish Workshop on Data Science, Växjö, Sweden, December 2-3, 2021.1–6
Torquato, S. (2010). Optimal design of heterogeneous materials. Annu. Rev. Mat. Res. 40, 101–129. doi:10.1146/annurev-matsci-070909-104517
Welborn, S. S., and Detsi, E. (2020). Small-angle X-ray scattering of nanoporous materials. Nanoscale Horiz. 5 (12–24), 12–24. doi:10.1039/c9nh00347a
Keywords: machine learning, Gaussian random field, regression, porous material, disordered material, small angle X-ray scattering, boosted trees
Citation: Röding M, Tomaszewski P, Yu S, Borg M and Rönnols J (2022) Machine learning-accelerated small-angle X-ray scattering analysis of disordered two- and three-phase materials. Front. Mater. 9:956839. doi: 10.3389/fmats.2022.956839
Received: 30 May 2022; Accepted: 30 August 2022;
Published: 27 September 2022.
Edited by:
M. K. Samal, Bhabha Atomic Research Centre (BARC), IndiaReviewed by:
Sagar Chandra, Homi Bhabha National Institute, IndiaAvik Das, Bhabha Atomic Research Centre (BARC), India
Suresh Koppoju, International Advanced Research Centre for Powder Metallurgy and New Materials, India
Copyright © 2022 Röding, Tomaszewski, Yu, Borg and Rönnols. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magnus Röding, bWFnbnVzLnJvZGluZ0ByaS5zZQ==