 Yoshihiko Shimokawa1,2
Yoshihiko Shimokawa1,2

- 1 Graduate School of Pharmaceutical Sciences, The University of Tokyo, Bunkyo-ku, Tokyo, Japan
- 2 Japan Science and Technology Agency, Core Research for Evolutional Science and Technology, Chiyoda-ku, Tokyo, Japan
- 3 Institute of Natural Medicine, University of Toyama, Toyama, Japan
Benzalacetone synthase, from the medicinal plant Rheum palmatum (RpBAS), is a plant-specific chalcone synthase (CHS) superfamily of type III polyketide synthase (PKS). RpBAS catalyzes the one-step, decarboxylative condensation of 4-coumaroyl-CoA with malonyl-CoA to produce the C6–C4 benzalacetone scaffold. The X-ray crystal structures of RpBAS confirmed that the diketide-forming activity is attributable to the characteristic substitution of the conserved active-site “gatekeeper” Phe with Leu. Furthermore, the crystal structures suggested that RpBAS employs novel catalytic machinery for the thioester bond cleavage of the enzyme-bound diketide intermediate and the final decarboxylation reaction to produce benzalacetone. Finally, by exploiting the remarkable substrate tolerance and catalytic versatility of RpBAS, precursor-directed biosynthesis efficiently generated chemically and structurally divergent, unnatural novel polyketide scaffolds. These findings provided a structural basis for the functional diversity of the type III PKS enzymes.
Introduction
The diketide benzalacetone scaffold is produced by benzalacetone synthase (BAS; EC 2.3.1.-), a member of the plant-specific chalcone synthase (CHS) superfamily of type III polyketide synthases (PKSs), by the one-step decarboxylative condensation of 4-coumaroyl-CoA with one molecule of malonyl-CoA (Figure 1A; Abe et al., 2001). BAS is thought to play a crucial role in the biosynthesis of the C6–C4 moiety of biologically active phenylbutanoids such as raspberry ketone, the characteristic aroma of raspberry fruit, and the anti-inflammatory glucoside lindleyin in rhubarb (Abe et al., 2001). In contrast, chalcone synthase (CHS; EC 2.3.1. 74), a typical type III PKS ubiquitously distributed in plants, catalyzes iterative condensations of 4-coumaroyl-CoA with three C2 units from malonyl-CoA to produce the tetraketide chalcone (Figure 1A), which is the biosynthetic precursor of flavonoids (Austin and Noel, 2003; Abe and Morita, 2010). To the best of our knowledge, only five BAS-encoding cDNAs have been reported, including those from Rheum palmatum (RpBAS; Abe et al., 2001), Wachendorfia thyrsiflora (WtPKS1; Brand et al., 2006), Rubus idaeus (RiPkS4; Zheng and Hrazdina, 2008), and Polygonum cuspidatum (PcPKS1 and PcPKS2; Ma et al., 2009a,b). The type III PKSs are structurally simple, homodimeric proteins, and their functional diversity is principally derived from small modifications in their active-site architectures, which generate the differences in their selection of starter molecules, numbers of malonyl-CoA condensations, and mechanisms of cyclization reactions (Austin and Noel, 2003; Abe, 2007, 2010; Abe and Morita, 2010). Recent crystallographic and site-directed mutagenesis studies have begun to reveal the intimate structural details of the enzyme-catalyzed processes (Austin and Noel, 2003; Abe, 2007, 2010; Abe and Morita, 2010). This mini-review summarizes our recent studies on the BAS from R. palmatum (Polygonaceae; RpBAS), using an approach combining site-directed mutagenesis, X-ray crystallography, and precursor-directed biosynthesis.
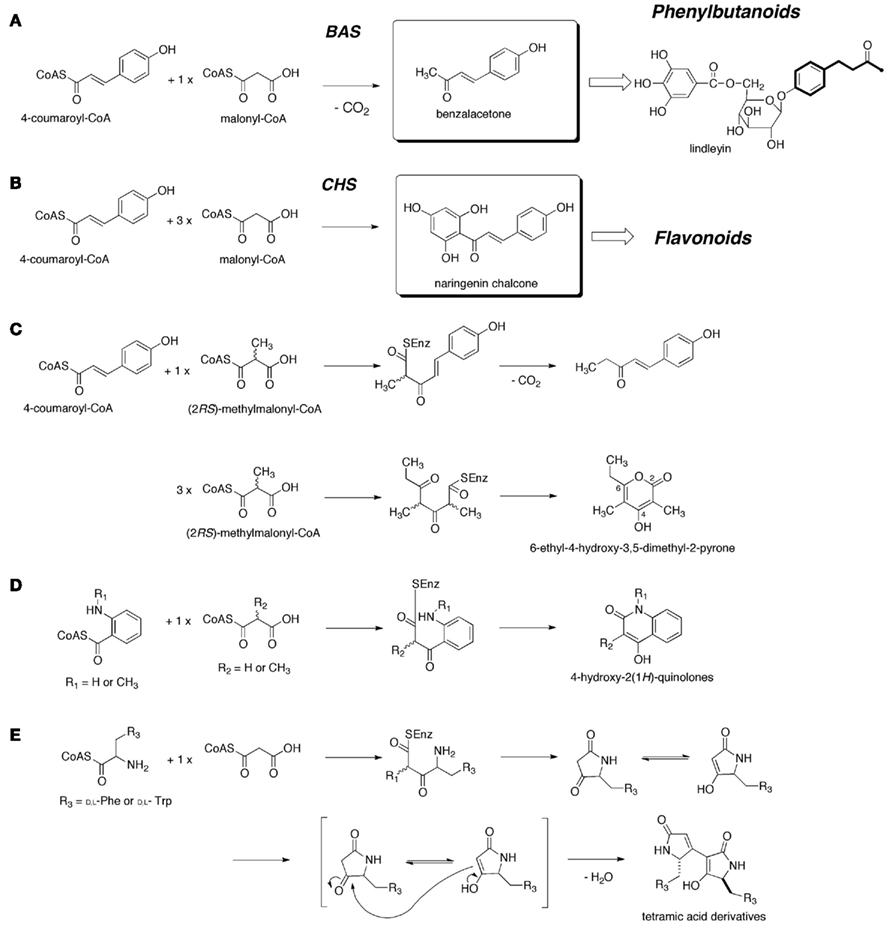
Figure 1. (A) The proposed mechanisms for the formation of benzalacetone by R. palmatum BAS, and naringenin chalcone by CHS. The proposed mechanisms for the formation of (B) the C6–C5 diketide, (C) the methylated C9 triketide pyrone, (D) the diketide quinolones, and (E) the diketide tetramic acid derivatives by R. palmatum BAS.
Cloning, Expression, and Site-Directed Mutagenesis
The cDNA encoding RpBAS was cloned and sequenced from young leaves of R. palmatum by RT-PCR amplification using degenerate primers based on the conserved sequences of the known CHSs (Abe et al., 2001). The deduced amino acid sequence of RpBAS shares 60–75% identity with those of the other members of the CHS-superfamily type III PKSs of plant origin. The recombinant RpBAS was heterologously expressed in Escherichia coli, and afforded the diketide benzalacetone as a single product from 4-coumaroyl-CoA and malonyl-CoA as substrates, within a range of pH 8.0–8.5 (Figure 1; Abe et al., 2001). An enzyme kinetics analysis revealed that RpBAS showed a KM = 10.0 μM and a kcat = 1.79 min−1 for 4-coumaroyl-CoA, and a KM = 23.3 μM and a kcat = 1.78 min−1 for malonyl-CoA, with respect to the benzalacetone-producing activity at pH 8.0. Interestingly, there was a dramatic change in the product profile under acidic pH conditions. Instead of benzalacetone, the triketide pyrone bisnoryangonin (BNY) was obtained as almost the sole product at pH 6.0 (Abe et al., 2003).
A sequence analysis revealed that RpBAS maintains an almost identical CoA binding site and the catalytic triad of Cys164, His303, and Asn336 (numbering in MsCHS; Ferrer et al., 1999). In addition, the active-site residues, including Met137, Gly211, Gly216, Ile254, Gly256, Phe265, Ser338, and Pro375, are well conserved in RpBAS. However, Phe215, which is conserved in all known type III PKSs, is characteristically substituted with Leu (214IL instead of 214LF; Abe et al., 2001). In CHS, the “gatekeeper” Phe215, located at the junction between the active-site cavity and the CoA binding tunnel, is thought to facilitate the decarboxylation of malonyl-CoA and to help orient the substrates and intermediates during the condensation reactions (Ferrer et al., 1999). This led to the proposal that the unique substitution of the “gatekeeper” Phe causes the termination of chain elongation at the diketide stage, which was later supported by the observation that the RpBAS I214L/L215F mutant indeed yielded naringenin chalcone by condensations with three molecules of malonyl-CoA (Abe et al., 2003). The RpBAS I214L/L215F mutant showed a KM = 33.5 μM and a kcat = 0.169 min−1 for 4-coumaroyl-CoA, and a KM = 46.6 μM and a kcat = 0.181 min−1 for malonyl-CoA, with respect to the chalcone-producing activity at pH 6.5 (Abe et al., 2003). The mutant thus exhibited a 36-fold decreases in the kcat/KM for 4-coumaroyl-CoA and a 20-fold decreases in the kcat/KM for malonyl-CoA, as compared with the wild-type RpBAS.
On the other hand, the conserved active-site Thr197 (numbering in MsCHS), which is important for the steric modulation of the active-site cavity in a number of divergent type III PKSs (Austin and Noel, 2003; Abe and Morita, 2010), is uniquely replaced with a reactive Cys. Therefore, it was postulated that RpBAS utilizes this second active-site Cys for the decarboxylation of a diketide intermediate to produce benzalacetone (Austin and Noel, 2003). To test this hypothesis, we constructed a mutant in which the Cys is substituted with Thr; however, the C197T mutant was essentially functionally identical to the wild-type RpBAS, thus excluding the possibility (Abe et al., 2007). In addition, we constructed mutants in which the active-site residues, Cys197, Gly256, and Ser338 (numbering in MsCHS), were respectively replaced with Gly, Leu, and Val (Abe et al., 2007), as in the case of the Aloe arborescens octaketide synthase (AaOKS) that produces the octaketides SEK4/SEK4b by condensations of eight molecules of malonyl-CoA (Abe et al., 2005; Morita et al., 2007). The OKS-like C197G and G256L substitutions are thought to be important for the steric modulation of the active-site cavity, thereby affecting both the substrate and product specificities of the enzyme reaction. In fact, it was previously reported that the S338V mutant of the chalcone-producing CHS yielded trace amounts of SEK4/SEK4b, by the steric modulation of a single residue lining the active-site cavity (Abe et al., 2006b). However, in RpBAS, none of the mutants generated a different product pattern (Abe et al., 2007).
Finally, as described below, crystallographic analyses revealed that the characteristic substitution of the conserved Thr132 (numbering in MsCHS), at the entrance of the “coumaroyl-binding pocket,” with the bulky Leu causes steric contraction of the active-site of RpBAS (Morita et al., 2010), which could also terminate the chain elongation at the diketide stage. Indeed, the L132T substitution also restored the chalcone-producing activity in RpBAS (Shimokawa et al., 2010). The L132T mutant showed a KM = 4.1 μM and a kcat = 1.60 × 10−3 min−1 for 4-coumaroyl-CoA, with respect to the chalcone-producing activity, with a pH optimum of 6.5 (Shimokawa et al., 2010).
Crystal Structure and Structure-Based Mechanism
The X-ray crystal structures of both the wild-type and chalcone-producing I214L/L215F mutant of RpBAS were solved at 1.8 Å resolution (Morita et al., 2010). Furthermore, the crystal structure of the wild-type RpBAS, with a monoketide coumarate intermediate covalently bound to the catalytic cysteine residue, was solved at 1.6 Å resolution (Morita et al., 2010). This is the first direct evidence that type III PKS utilizes the Cys as both the nucleophile and attachment site for the growing polyketide intermediate.
The overall structures of RpBAS are highly homologous to those of the previously reported plant type III PKSs, including M. sativa CHS (MsCHS; Ferrer et al., 1999) and Pinus sylvestris stilbene synthase (PsSTS; Austin et al., 2004). On the other hand, a comparison of the active-site structure of RpBAS with that of MsCHS revealed that the conformational differences of Leu215 and Ser338 (numbering of MsCHS) cause the loss of the CHS’s “coumaroyl-binding pocket” from the active-site of RpBAS. In addition, the conserved Thr197, at the entrance of the “coumaroyl-binding pocket,” is characteristically substituted with the bulky Leu in RpBAS. As a result, the total cavity volume (350 Å3) of the active-site is much smaller than that of M. sativa CHS (750 Å3; Morita et al., 2010).
The crystal structures revealed that, instead of the “coumaroyl-binding pocket,” RpBAS utilizes an alternative, novel pocket, which protrudes into the “floor” of the conventional CHS active-site, to bind the aromatic moiety of the coumarate (Figure 2; Morita et al., 2010). Furthermore, the F215L substitution causes not only the conformational changes at the active-site, but also a twofold increase in the surface area of the active-site entrance of RpBAS, as compared to that of MsCHS (Morita et al., 2010). This widening of the active-site entrance contributes to the unique substrate and product specificities of RpBAS. In contrast, the crystal structure of the RpBAS I214L/L215F mutant revealed that the substitutions restored the “coumaroyl-binding pocket” within the active-site of the mutant, whereas both the locations and orientations of the residues lining the active-site cavity are almost perfectly conserved with those in the wild-type RpBAS (Figure 2; Morita et al., 2010). Thus, the I214L/L215F substitutions open a gate to the buried “coumaroyl-binding pocket,” thereby increasing the chain elongation up to three condensations with malonyl-CoA, which leads to the recovery of the chalcone-producing activity.
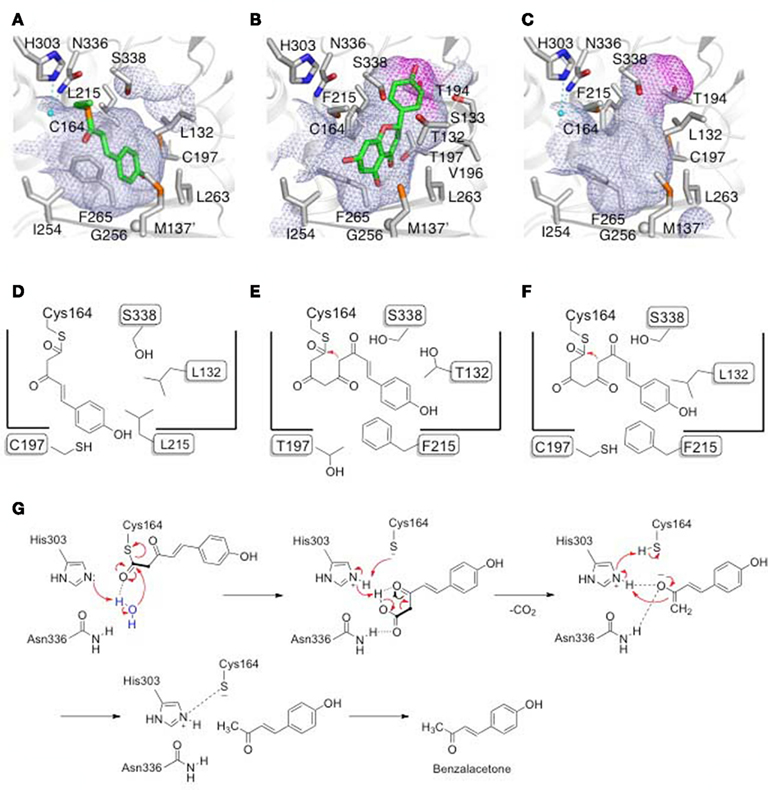
Figure 2. Surface and schematic representations of the active-site cavities of (A,D) wild-type R. palmatum BAS, (B,E) M. sativa CHS, and (C,F) the R. palmatum BAS I207L/L208F mutant (numbering in RpBAS). The bottoms of the “coumaroyl-binding pocket” are highlighted as purple surfaces. The covalently bound coumarate, the water molecules and the hydrogen bonds are highlighted. Proposed mechanism for the BAS enzyme reaction (G).
Considering the conservation of the overall folding and the catalytic triad, it is likely that RpBAS and CHS utilize similar catalytic mechanisms for the initiation of the enzyme reaction and the condensation of malonyl-CoA (Ferrer et al., 1999). One of the most interesting points is the catalytic mechanism of the thioester bond cleavage of the enzyme-bound diketide intermediate, and the final decarboxylation that produces the C6–C4 scaffold (Morita et al., 2010). Previously, based on a crystallographic comparison of the stilbene-producing PsSTS with the chalcone-producing MsCHS, Noel and Schröder proposed that an “aldol-switch” thioesterase-like hydrogen bond network, including a nucleophilic water molecule and Thr132, neighboring the catalytic Cys164 (numbering in MsCHS), is required for the thioester bond cleavage of the tetraketide intermediate to produce stilbene in PsSTS (Austin et al., 2004). In contrast, the cleavage of the thioester linkage is caused by intramolecular cyclization via a Claisen condensation to produce chalcone in MsCHS (Ferrer et al., 1999). However, a careful examination of the RpBAS crystal structure negated the presence of such hydrogen bond networks, since Thr132 is replaced with the hydrophobic Leu in RpBAS. Instead, the crystal structures clearly demonstrated the presence of a distinct, activated water molecule that forms hydrogen bond networks with the Cys-His-Asn catalytic triad. This finding suggested that RpBAS employs unique catalytic machinery for the thioester bond cleavage of the enzyme-bound diketide intermediate and the final decarboxylation reaction, and that the β-keto acid produced by the nucleophilic attack of the water molecule, presumably activated by His303 (numbering in MsCHS), subsequently undergoes decarboxylation to yield the C6–C4 benzalacetone (Figure 2G; Morita et al., 2010). Thus, the decarboxylation of the β-keto acid proceeds via the proton abstraction by His303, the reactivation by the Cys164 thiolate, and the formation of an enolate anion that is presumably stabilized by the His303–Asn336 oxyanion hole, as in the case of the decarboxylation of malonyl-CoA. Finally, tautomerization to the keto form produces benzalacetone and restores the Cys-His-Asn catalytic triad. This proposal is supported by the detection of the diketide β-keto acid intermediate in the reaction products of RpBAS (Morita et al., 2010).
Substrate Specificity and Precursor-Directed Biosynthesis
As in the cases of the other members of the type III PKSs (Abe et al., 2000; Jez et al., 2002; Austin and Noel, 2003; Abe, 2010, 2012; Abe and Morita, 2010; Morita et al., 2011), RpBAS exhibits unusually broad, promiscuous substrate specificities, and readily accepts diverse non-physiological substrates to produce a variety of chemically and structurally distinct unnatural polyketides. Furthermore, as described, the crystal structures of RpBAS revealed that the characteristic substitution of the “gatekeeper” Phe residue causes not only conformational changes at the active-site, but also a twofold increase in the surface area of the active-site entrance of RpBAS, as compared to that of MsCHS (Morita et al., 2010). This widening of the active-site entrance contributes to the unique substrate specificities of RpBAS, allowing it to accept bulky non-physiological substrates.
For example, RpBAS can accept (2RS)-methylmalonyl-CoA as an extender substrate, and a 4-coumaroyl-CoA starter, to produce an unnatural novel diketide, 1-(4-hydroxyphenyl)pent-1-en-3-one (Figure 1B; Abe et al., 2002). The C6–C5 skeleton was thus produced by the one-step decarboxylative condensation of 4-coumaroyl-CoA with the C2 unit of methylmalonyl-CoA. Notably, since a diketide carboxylic acid was not detected in the reaction mixture, the decarboxylation of the enzyme-bound intermediate should be tightly controlled by the enzyme (Abe et al., 2002). Interestingly, RpBAS also accepts (2RS)-methylmalonyl-CoA as the only substrate to produce a methylated C9 triketide, 6-ethyl-4-hydroxy-3,5-dimethyl-2-pyrone, as a single product from three molecules of (2RS)-methylmalonyl-CoA (Figure 1C; Abe et al., 2006c). It is remarkable that the type III PKS accepts (2RS)-methylmalonyl-CoA as both a starter and an extender substrate, and catalyzes the formation of the C9 triketide pyrone.
Anthranilic acid has been postulated to be a key intermediate in the biosynthesis of quinolone and acridone alkaloids, which occur in their greatest abundance in plants from the family of Rutaceae. Interestingly, RpBAS efficiently catalyzes the condensation of N-methylanthraniloyl-CoA with one molecule of either malonyl-CoA or (2RS)-methylmalonyl-CoA, to produce the corresponding diketide, 4-hydroxy-2(1H)-quinolones (Figure 1D; Abe et al., 2006a). The enzyme reaction with the anthraniloyl starter thus proceeds without the decarboxylation step, and amide formation immediately follows the condensation reactions with malonyl-CoA. Notably, the best yield was obtained with the combination of the non-physiological N-methylanthraniloyl-CoA and the (2RS)-methylmalonyl-CoA. Steady-state enzyme kinetics for the formation of 4-hydroxy-1,3-dimethyl-2(1H)-quinolone revealed a KM = 23.7 μM and a kcat = 1.48 min−1 for N-methylanthraniloyl-CoA, which were comparable to those for the formation of benzalacetone. This result suggests the existence of an as yet unidentified novel type III PKS that produces quinolones from the CoA thioester of anthranilic acid (Abe et al., 2006a).
Finally, RpBAS also accepts a series of aminoacyl-CoA thioesters as starter substrates, and catalyzes their condensation with one molecule of malonyl-CoA to produce the tetramic acid (2,4-pyrrolidinedione) derivatives (Figure 1E; Wakimoto et al., 2011). For example, the enzyme afforded a 1:10 mixture of two products from L-phenylalanoyl-CoA and malonyl-CoA. The minor product was a tetramic acid monomer, which is produced by the condensation of L-phenylalanoyl-CoA with malonyl-CoA, followed by intramolecular lactamization from the enzyme-bound intermediate. On the other hand, the major product was a tetramic acid dimer. Since the active-site of RpBAS is apparently large enough for the monomer, but not the dimer, the dimer is likely to be produced by the non-enzymatic, spontaneous dimerization between each tautomer. Interestingly, RpBAS efficiently accepted both L- and D-phenylalanoyl-CoAs as substrates. Steady-state kinetics analyses revealed a KM = 11.7 μM and a kcat = 27.8 min−1 for L-phenylalanoyl-CoA, and a KM = 3.3 μM and a kcat = 8.1 min−1 for D-phenylalanoyl-CoA. Furthermore, RpBAS also efficiently accepted both L- and D-tryptophanoyl-CoAs as substrates, to produce the corresponding monomeric and dimeric tetramic acid derivatives in nearly equal yields (Wakimoto et al., 2011). The structurally simple RpBAS thus initially accepts the aminoacyl-CoA as a starter, and then recruits malonyl-CoA for a Claisen condensation to generate the γ-amino-β-ketothioester. The free γ-amino group of the enzyme-bound intermediate could cleave the thioester bond, with concomitant intramolecular lactamization. The results suggests the possibility of the further preparation of tetramic acids from various amino acids.
Summary
Crystallographic and site-directed mutagenesis studies on the functionally diverse type III PKSs have begun to reveal the intimate structural details of the enzyme-catalyzed processes. It is remarkable that the catalytic plasticity of the structurally and mechanistically conventional type III PKSs evolved from simple modifications of the substrate structures and the active-sites of the enzymes. By exploiting the remarkable substrate tolerance and catalytic versatility of the enzymes, the precursor-directed biosynthesis will contribute to the further production of chemically and structurally divergent, unnatural novel polyketides.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to express their sincere appreciation to the excellent group of coworkers whose contributions are cited in the text. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by the CREST program from Japan Science and Technology Agency, Japan. Yoshihiko Shimokawa is a recipient of the JSPS Fellowship for Young Scientist.
References
Abe, I. (2010). Engineered biosynthesis of plant polyketides: structure-based and precursor-directed approach. Top. Curr. Chem. 297, 45–66.
Abe, I. (2012). Novel applications of plant polyketide synthases. Curr. Opin. Chem. Biol. 16, doi: 10.1016/j.cbpa.2011.12.016.. (in press).
Abe, I., Abe, T., Wanibuchi, K., and Noguchi, H. (2006a). Enzymatic formation of quinolone alkaloids by a plant type III polyketide synthase. Org. Lett. 8, 6063–6065.
Abe, I., Watanabe, T., Morita, H., Kohno, T., and Noguchi, H. (2006b). Engineered biosynthesis of plant polyketides: manipulation of chalcone synthase. Org. Lett. 8, 499–502.
Abe, T., Noma, H., Noguchi, H., and Abe, I. (2006c). Enzymatic formation of an unnatural methylated triketide by plant type III polyketide synthases. Tetrahedron Lett. 47, 8727–8730.
Abe, I., and Morita, H. (2010). Structure and function of the chalcone synthase superfamily of plant type III polyketide synthases. Nat. Prod. Rep. 27, 809–838.
Abe, I., Morita, H., Nomura, A., and Noguchi, H. (2000). Substrate specificity of chalcone synthase: Enzymatic formation of unnatural polyketides from synthetic cinnamoyl-CoA analogs. J. Am. Chem. Soc. 122, 11242–11243.
Abe, I., Oguro, S., Utsumi, Y., Sano, Y., and Noguchi, H. (2005). Engineered biosynthesis of plant polyketides: chain length control in an octaketide-producing plant type III polyketide synthase. J. Am. Chem. Soc. 127, 12709–12716.
Abe, I., Sano, Y., Takahashi, Y., and Noguchi, H. (2003). Site-directed mutagenesis of benzalacetone synthase: the role of Phe215 in plant type III polyketide synthases. J. Biol. Chem. 278, 25218–25226.
Abe, I., Takahashi, Y., Morita, H., and Noguchi, H. (2001). Benzalacetone synthase: a novel polyketide synthase that plays a crucial role in the biosynthesis of phenylbutanones in Rheum palmatum. Eur. J. Biochem. 268, 3354–3359.
Abe, I., Takahashi, Y., and Noguchi, H. (2002). Enzymatic formation of an unnatural C6-C5 aromatic polyketide by plant type III polyketide synthases. Org. Lett. 4, 3623–3626.
Abe, T., Morita, H., Noma, H., Kohno, T., Noguchi, H., and Abe, I. (2007). Structure function analysis of benzalacetone synthase from Rheum palmatum. Bioorg. Med. Chem. Lett. 17, 3161–3166.
Austin, M. B., Bowman, M. E., Ferrer, J.-L., Schröder, J., and Noel, J. P. (2004). An aldol switch discovered in stilbene synthases mediates cyclization specificity of type III polyketide synthases. Chem. Biol. 11, 1179–1194.
Austin, M. B., and Noel, J. P. (2003). The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110.
Brand, S., Hölscher, D., Schierhorn, A., Svatos, A., Schröder, J., and Schneider, B. (2006). A type III polyketide synthase from Wachendorfia thyrsiflora and its role in diarylheptanoid and phenylphenalenone biosynthesis. Planta 224, 413–428.
Ferrer, J. L., Jez, J. M., Bowman, M. E., Dixon, R. A., and Noel, J. P. (1999). Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nat. Struct. Biol. 6, 775–784.
Jez, J. M., Bowman, M. E., and Noel, J. P. (2002). Expanding the biosynthetic repertoire of plant type III polyketide synthases by altering starter molecule specificity. Proc. Natl. Acad. Sci. U.S.A. 99, 5319–5324.
Ma, L. Q., Guo, Y. W., Gao, D. Y., Ma, D. M., Wang, Y. N., Li, G. F., Liu, B. Y., Wang, H., and Ye, H. C. (2009a). Identification of a Polygonum cuspidatum three-intron gene encoding a type III polyketide synthase producing both naringenin and p-hydroxybenzalacetone. Planta 229, 1077–1086.
Ma, L. Q., Pang, X. B., Shen, H. Y., Pu, G. B., Wang, H. H., Lei, C. Y., Wang, H., Li, G. F., Liu, B. Y., and Ye, H. C. (2009b). A novel type III polyketide synthase encoded by a three-intron gene from Polygonum cuspidatum. Planta 229, 457–469.
Morita, H., Kondo, S., Oguro, S., Noguchi, H., Sugio, S., Abe, I., and Kohno, T. (2007). Structural insight into chain-length control and product specificity of pentaketide chromone synthase from Aloe arborescens. Chem. Biol. 14, 359–369.
Morita, H., Shimokawa, Y., Tanio, M., Kato, R., Noguchi, H., Sugio, S., Kohno, T., and Abe, I. (2010). A structure-based mechanism for benzalacetone synthase from Rheum palmatum. Proc. Natl. Acad. Sci. U.S.A. 107, 669–673.
Morita, H., Yamashita, M., Shi, S.-P., Wakimoto, T., Kondo, S., Kato, R., Sugio, S., Kohno, T., and Abe, I. (2011). Synthesis of unnatural alkaloid scaffolds by exploiting plant polyketide synthase. Proc. Natl. Acad. Sci. U.S.A. 108, 13504–13509.
Shimokawa, Y., Morita, H., and Abe, I. (2010). Structure-based engineering of benzalacetone synthase. Bioorg. Med. Chem. Lett. 20, 5099–5103.
Wakimoto, T., Mori, T., Morita, H., and Abe, I. (2011). Cytotoxic tetramic acid derivative produced by a plant type-III polyketide synthase. J. Am. Chem. Soc. 133, 4746–4749.
Keywords: benzalacetone synthase, polyphenol, polyketide synthase, enzyme, biosynthesis
Citation: Shimokawa Y Morita H and Abe I (2012) Benzalacetone synthase. Front. Plant Sci. 3:57. doi: 10.3389/fpls.2012.00057
Received: 14 February 2012; Paper pending published: 26 February 2012;
Accepted: 06 March 2012; Published online: 21 March 2012.
Edited by:
Jorge M. Vivanco, Colorado State University, USAReviewed by:
Ute Roessner, The University of Melbourne, AustraliaJorge M. Vivanco, Colorado State University, USA
Copyright: © 2012 Shimokawa, Morita and Abe. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ikuro Abe, Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. e-mail:YWJlaUBtb2wuZi51LXRva3lvLmFjLmpw