 Bin Dong1,2
Bin Dong1,2 Haibin Wang1,2
Haibin Wang1,2 Aiping Song1,2Tao Liu1Yun Chen1Weimin Fang1Sumei Chen1Fadi Chen1Zhiyong Guan1*
Aiping Song1,2Tao Liu1Yun Chen1Weimin Fang1Sumei Chen1Fadi Chen1Zhiyong Guan1* Jiafu Jiang1,2*
Jiafu Jiang1,2*- 1Lab of Chrysanthemum Genetics, Breeding and Molecular Biology, College of Horticulture, Nanjing Agricultural University, Nanjing, China
- 2Jiangsu Province Engineering Lab for Modern Facility Agriculture Technology and Equipment, Nanjing, China
Many plant species are autopolyploid, a condition frequently associated with improvements in both vegetative and reproductive vigor. The possible contribution of miRNAs to this improvement was investigated by characterizing the miRNA content of a diploid and an autotetraploid form of Chrysanthemum nankingense. 162 and 161 known miRNA sequences were identified in 2x and 4x library. The length of 22 and 25 nt was predominant in diploid. However, 21 and 24 nt showed dominance in autotetraploid. It seems likely that autopolyploidization have had an immediate effect the distribution of miRNAs. In addition, the abundance of the miRNAs differed markedly between the two ploidy levels and contributed to their targets diversity. A number of target genes associated with miRNAs play important roles in growth and development. The conclusion was that some miRNAs likely make a contribution to the vigor displayed by autotetraploid C. nankingense.
Introduction
Polyploidization is an important evolutionary driver in the plant kingdom, and provides plants with a means to adapt rapidly to a new environmental niche and to speciate (Otto, 2007; Ainouche and Jenczewski, 2010; Wang et al., 2013a). It has been estimated that at least 70% of angiosperm species have undergone at least one polyploidization event during their evolution (Masterson, 1994; Soltis and Soltis, 1999). While autopolyploids represent the doubling of a given species' chromosome complement, allopolyploids derive via the chromosome doubling of a hybrid between sexually compatible, but non-identical species. Once such an event has occurred, a number of both genetic and epigenetic modifications are typically induced (Comai, 2000; Osborn et al., 2003; Chen, 2007; Wang et al., 2014). These occur rather frequently in de novo allopolyploids, while in autopolyploids, only a limited amount of genomic reorganization appears to be induced (Rieseberg and Willis, 2007).
The class of small (20–24 nt) non-coding RNA molecules referred to as microRNAs (miRNAs) are generated from a stem-loop structure precursor. Their biological significance lies in their ability to regulate gene expression by targeting a specific transcript for degradation (Bartel, 2004; He and Hannon, 2004). A number of miRNAs are known to play an important role in plant growth and development (Rubio-Somoza et al., 2009; Cao et al., 2016) and in the response to biotic and abiotic stress (Ruiz-Ferrer and Voinnet, 2009; Gao et al., 2016). Furthermore, miRNAs can affect gene expression by reducing stochastic noise, buffer cross-species variation and constrain evolutionary gene expression variation (Cui et al., 2007). In allopolyploids involving Arabidopsis thaliana, certain miRNAs have been shown to act as a buffer against the genomic shock experienced by de novo interspecific allopolyploids (Ha et al., 2009). So far, their effect in autopolyploids has received little attention.
The diploid species Chrysanthemum nankingense is a native of China (Wang et al., 2013b; Sun et al., 2014). Just as polyploids are frequently phenotypically superior to their lower ploidy progenitor (Otto, 2007), a number of the organs formed by autotetraploid C. nankingense are significantly larger than those of the diploid form (Liu et al., 2011). Given the important roles played by miRNAs in regulating plant growth and development in polyploid plants, a program was initiated to contrasting the miRNA content of the diploid and the autotetraploid, with a view both to revealing how polyploidization influenced the content and distribution of miRNAs, and to exploring whether miRNA activity could be responsible for the improved vigor of the autotetraploid.
Materials and Methods
Plant Materials and Growing Conditions
Diploid (2n = 2x = 18) and autotetraploid C. nankingense (2n = 4x = 36) plants were raised in a greenhouse under a ~16 h photoperiod, a constant temperature of ~25°C and a relative humidity of 70–75%. The materials were sourced from the Nanjing Agricultural University Chrysanthemum Germplasm Resource Preserving Centre. At the vegetative state (10–15 leaf old), measurements were made of plant height, leaf length, leaf width, leaf area, and stem length using caliper (Mitutoyo, Japan). Leaf area was estimated from digital images using ImageJ software (Schneider et al., 2012).
RNA Extraction and Small RNA (sRNA) Sequencing
Plants were grown up to the 10–15 leaf stage, at which point leaf and stem was harvested from 3–5 plants. Total RNA was isolated from 0.5 g plant tissue using the RNAiso Plus reagent (TaKaRa, Dalian, China) then treated with RNase-free DNase I (TaKaRa) to remove contaminating DNA. A 5 μg aliquot of pooled RNA was obtained by combining, on an equimolar basis, the RNA extracted from three biological replicates. The quality and production of the RNA were detected using, respectively, an Aglilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and the ABI Step One Plus-Time PCR System (Applied Biosystems, Foster City, CA, USA). The sequencing of the sRNA fraction was carried out on an Illumina 2000 device housed at the Beijing Genomics Institute (http://www.genomics.cn). The datasets are available in the NCBI repository, (http://trace.ncbi.nlm.nih.gov/Traces/sra_sub/sub.cgi?) under accession number SRP080104. Before analysis, adapter sequences, 10% of the nitrogen sequences and low quality (Q < 5) reads were removed, leaving two separate sRNA libraries of “clean” reads (one from diploid C. nankingense and the other from the autotetraploid form). Then the length distribution of clean reads, total, and unique sequences was summarized.
miRNA Annotation and Target Gene Prediction
The set of sRNAs (17–30 nt) represented in the two libraries were filtered to remove ribosomal RNAs, transfer RNAs, small nucleolar RNAs and small nuclear RNAs, and the remaining sequences were then compared with equivalent plant non-coding sRNA sequences deposited in either the GenBank (http://www.ncbi.nlm.nih.gov/genbank/) or the Rfam (http://www.sanger.ac.uk/science/tools/rfam) database. The set of filtered sRNA sequences were aligned via BlastN with those represented in the miRNA database (http://www.mirbase.org). Because the miRNA data of chrysanthemum is absent in miRbase, the general criteria is that small RNAs align to the precursor or mature sequences of corresponding species and obtain miRNA count in miRbase. The novel miRNAs were predicted using software Mireap (http://sourceforge.net/projects/mireap/). The copy number of individual miRNAs was expressed in the form of reads per million reads (RPM), and the criterion set for differential abundance between the two libraries was that the log2 of the ratio between the two RPMs was either >+1 or < −1, along with P-value of < 0.001. To enable this, miRNAs with an RPM of zero were assigned a nominal RPM of 0.01. Targets for each selected C. nankingense miRNA were defined by using its sequence as a query term against the C. indicum transcriptome described by Wang et al. (2013b) and lodged in GenBank as accession number PRJNA245057. Target prediction was based on the Allen et al. (2005) criteria using psRNATarget server (Dai and Zhao, 2011). The predicted targets were functionally classified following the Gene Ontology (http://www.geneontology.org) method, and assigned a pathway using the KEGG database (Kanehisa et al., 2016; http://www.genome.jp/kegg/kegg1.html).
Poly (A) Tailing and Reverse Transcription
Poly (A) tailing and reverse transcription were performed using a PrimeScript® miRNA qPCR Starter Kit v2.0 (TaKaRa), following the manufacturer's protocol. Briefly, each 20 μL reaction contained 10 μL 2 × miRNA Reaction Buffer Mix, 2 μL 0.1% w/v BSA, 2 μL miRNA PrimeScript® RT Enzyme Mix, 1 μL total RNA, and 5 μL RNase-free distilled water. Total RNA were isolated from plant tissue of independent plant materials at the 10–15 leaf stage. The Poly (A) tailing reactions and reverse transcription were incubated at 37°C for 2 h, then inactivated by holding at 85°C for 5 s.
Real Time Quantitative-PCR (qRT-PCR)
Each 20 μL qRT-PCR contained 10 μL SYBR® Premix Ex Taq™ II (Takara), 5 μL cDNA (80 ng/μL), 1.0 μL 10 μM forward primer (designed using primer 5.0 software), and 1.0 μL Uni-miR qPCR Primer. The reactions were held at 95°C for 2 min, then subjected to 40 cycles of 95°C/15 s, 55°C/15 s, and 72°C/20 s. The chrysanthemum gene EF1α (Genbank accession number KF305681) was used as the reference sequence, following the suggestion of Wang et al. (2015). In addition, three biological replicates and three technical replicates were performed for each tissue sample. Amplicon abundances were calculated using the 2−ΔΔCT method (Livak and Schmittgen, 2001).
Results
Phenotypic Effect of Autopolyploidization
As also noted by Liu et al. (2011), the tetraploid form of C. nankingense was phenotypically distinct from the diploid form (Figure 1). In particular, plant height, leaf length, and leaf area were all greater in the autotetraploid than in the diploid at vegetative stage (Table 1). These results imply that autotetraploid plants were more vigorous than the diploid, and higher overall biomass was generated during the growth of autotetraploid.

Figure 1. The improved vigor displayed by the autotetraploid form of C. nankingense. (A) Diploid plant, (B) autotetraploid plant. In both cases the plants had developed 10–15 leaves.
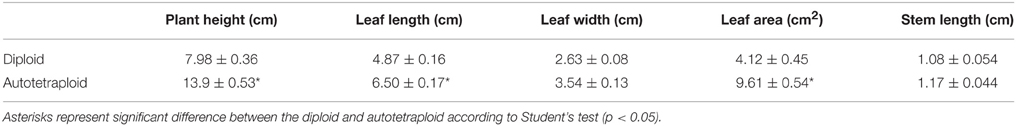
Table 1. Differences in the morphology of the diploid and autotetraploid forms of C. nankingense.
The miRNA Content of Diploid and Autotetraploid C. nankingense
In all 18,217,460 raw reads were acquired from 2x C. nankingense and 20,864,890 from the 4x form (Table 2). The filtering step reduced these numbers to, respectively, 17,984,240 and 20,561,513; of these, 98.77% were classified as clean reads in the 2x library and 99.11% in the 4x one. The combined set of 38,141,274 clean reads included 10,457,666 unique sequences (Figure 2, Table S1). Then total sRNAs and unique sRNAs aligned the NCBI GenBank (Table 3). Except miRNAs, many types of RNA sequences including other non-coding RNAs (tRNAs, rRNAs, snoRNAs, and snRNAs etc.), sRNAs of unknown origin and degraded fragments were removed (Table S2). In addition, the length range of the small RNAs were summarized in Figure 3, the reads of 21 and 24 nt long was abundant in the 2x and 4x library.
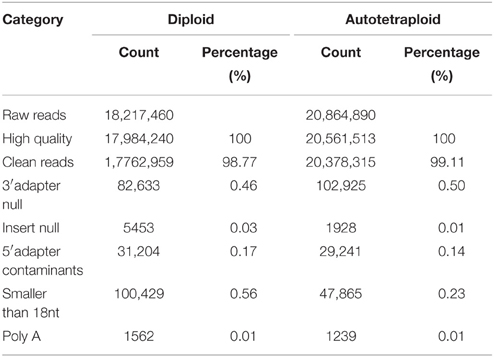
Table 2. The number of sequencing reads obtained from the diploid and autotetraploid forms of C. nankingense.
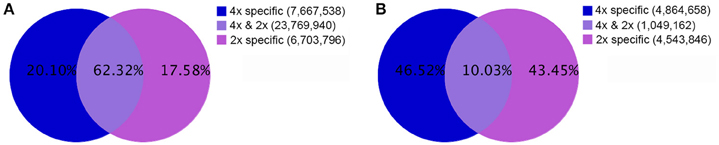
Figure 2. The population of sRNAs in diploid and autotetraploid C. nankingense. (A) The set of total sequences, and (B) the set of unique sequences in diploid and autotetraploid library.
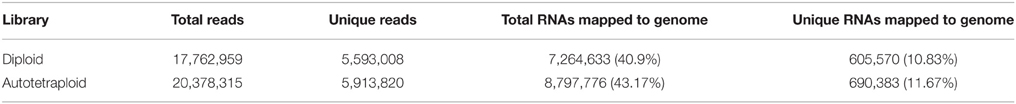
Table 3. Analysis of sequencing reads mapping of sRNA in diploid and autopolyploid library of C. nankingense.
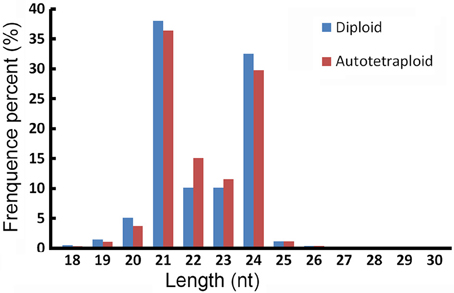
Figure 3. The length distribution of sRNAs in 2x and 4x C. nankingense.
A comparison with the sequences represented in miRBase revealed that the 2x and 4x libraries included, respectively, 162 and 161 known miRNA sequences (Table S3). Based on the criteria applied for differential abundance, 72 miRNAs were classed as significantly more abundant in the 4x form and 71 significantly less abundant, their relative abundances are illustrated in Figures 4A,B. Meanwhile 49 miRNAs were present in the 4x form but were down-regulated in the 2x form, and 52 vice versa (Table S4). 61 and 65 novel miRNAs was identified in 2x and 4x library (Table S5). Of these, 28 were down-regulated in the 4x form but were present in the 2x form and 25 vice versa (Table S6). The differentially abundant showed in Figure 4C, with 14 miRNAs suppressed and 10 miRNAs enhanced in the 4x form.
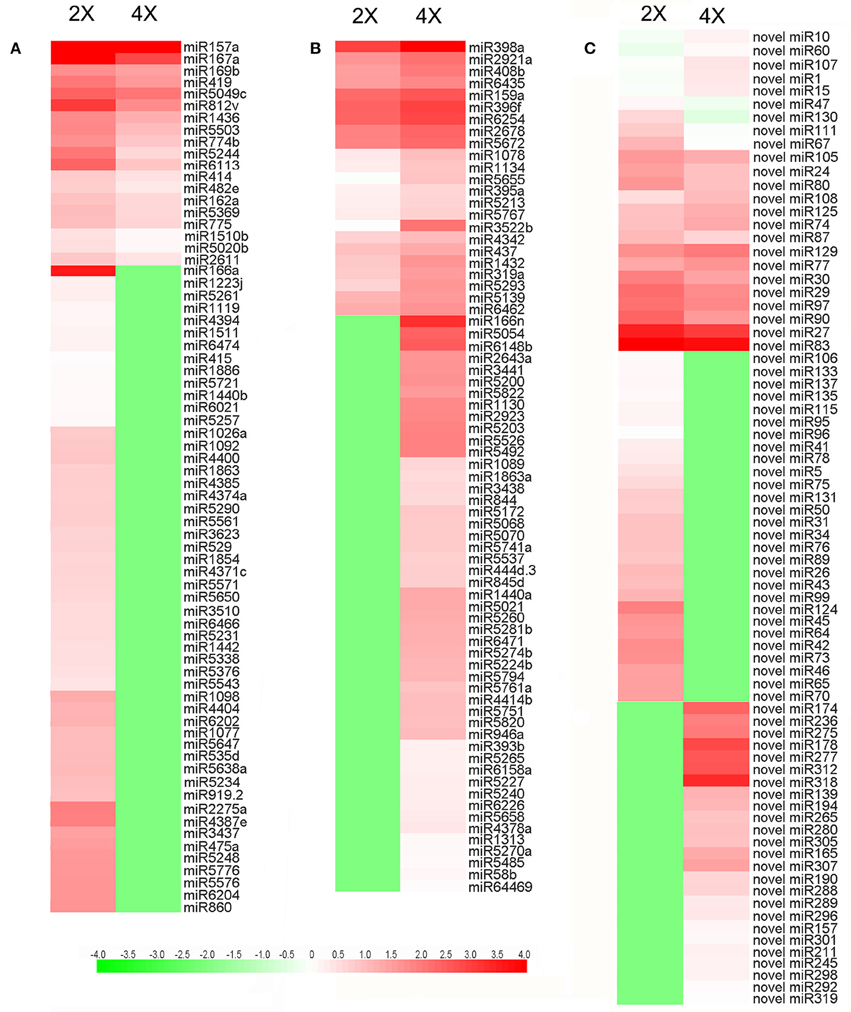
Figure 4. Differential abundance of miRNAs in 2x and 4x C. nankingense. (A,B) Known miRNAs, (C) novel miRNAs. The color intensity reflects the magnitude of the logarithm of the abundance.
Predicted miRNA Targets
The potential targets of the miRNAs were predicted from the C. indicum transcriptome, along with the potential cleavage sites (Sheet 1). For the known miRNAs scored as differentially abundant in the 4x form, 531 potential targets were associated with 78 miRNAs, and were classified into 26 functional Gene Ontology groups (Sheet 2). The predominant biological process was “cellular process,” that of the cellular component was “cell and cell part” and that of function was “binding and catalytic activity” (Figure 5A, Sheet 2). Reference to the KEGG database allowed 302 of the targets to be assigned to 115 pathways (Sheet 3). With respect to 70 novel miRNAs which showed different abundance in the 4x form, 622 potential targets were identified and classified into 27 functional groups (Sheet 2). The predominant biological process was “metabolic process,” that of the cellular component was “cell, cell part, and organelle” and that of function was “binding and catalytic activity” (Figure 5B, Sheet 2). Reference to the KEGG database allowed 187 of the targets to be assigned to 93 pathways (Sheet 3).
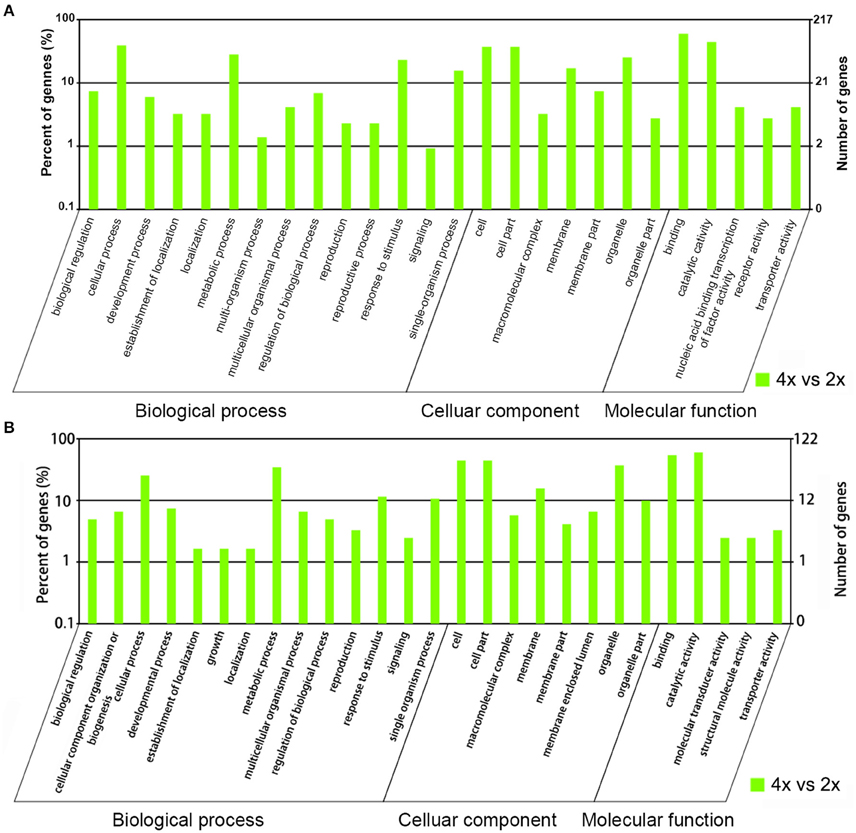
Figure 5. Gene ontology of differentially abundant miRNAs. The targets of (A) known miRNAs, and (B) novel miRNAs which were more abundant in the 4x than the 2x form of C. nankingense.
Targets Involved in Growth- and Development-Related miRNAs
With respect to genes involved in plant growth and development, there were 45 targets among the ones associated with the set of miRNAs scored as more abundant in the 4x than in the 2x form of C. nankingense (Table 4). Three of the novel miRNAs targeted genes of this type: One was a homolog of a gene encoding FLO/LFY-like protein targeted by novel-miR97, the second encoded an EIN4 homolog (ethylene receptor) targeted by novel-miR130, and the third was a MYB transcription factor (MYB46) targeted by novel-miR211 (Table 4).

Table 4. Potential target genes involved in growth and development of miRNAs in the 2x and 4x form of C. nankingense.
qRT-PCR Validation of the Differential Abundance of Selected miRNAs and Their Targets
When a series of qRT-PCRs was performed to validate the differential abundance of ten of the known miRNAs and their targets, full consistency was achieved with the sequencing outcome (Figure 6). The expression of miRNAs and their potential targets involved in growth and development were investigated, 16 known miRNAs were identified in 4x vs. 2x (Figure 7). In addition, 10 potential targets included SPL (Unigene1274, Unigene12160), ARF (CL6948.Contig1, CL6255.Contig2), NAC (CL17795.Contig2, Unigene3214), AP2 (CL17146.Contig2), TIR1 (CL6931.Contig2, Unigene25326), and bHLH (CL2783.Contig2) were identified in diploid and autotetraploid (Figure 8, Table S7).
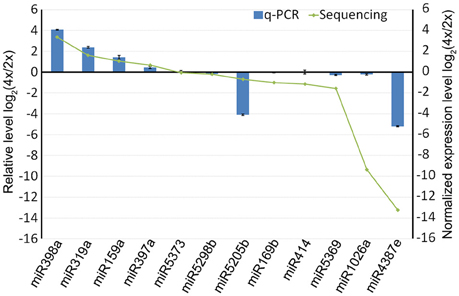
Figure 6. The abundance of selected miRNAs derived from a qRT-PCR experiment compared to the predicted outcome from the sequencing data. Each bar shows the mean ± SE of a triplicated assay.
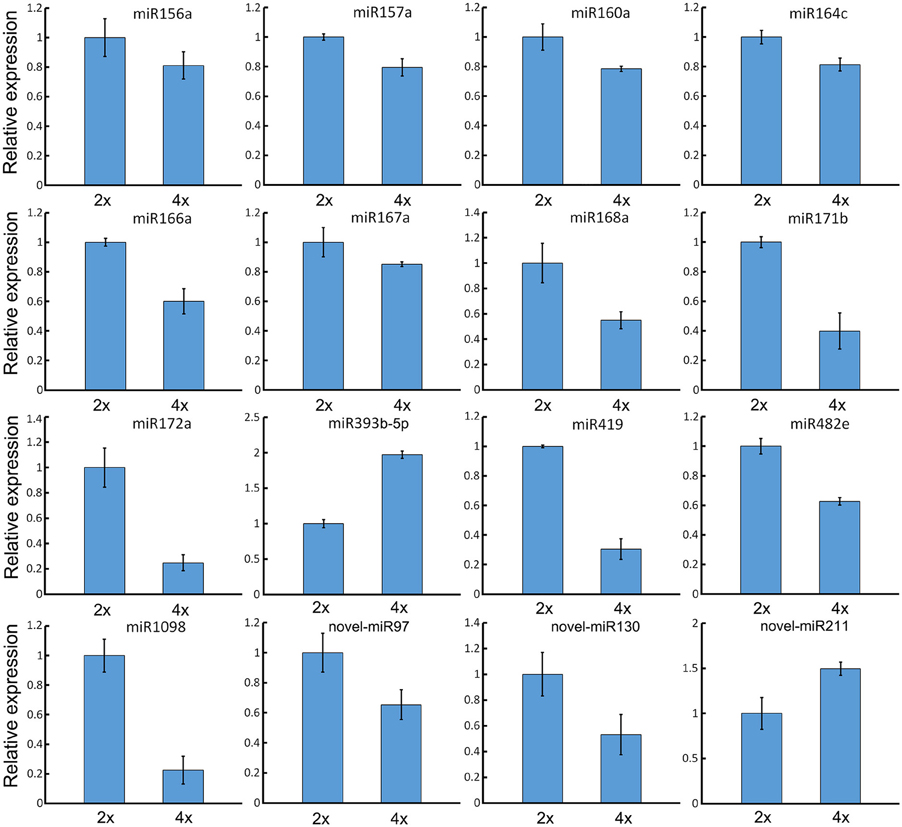
Figure 7. Validation of expression patterns of miRNAs involved in growth and development by qRT-PCR. Each bar shows the mean ± SE of a triplicated assay.
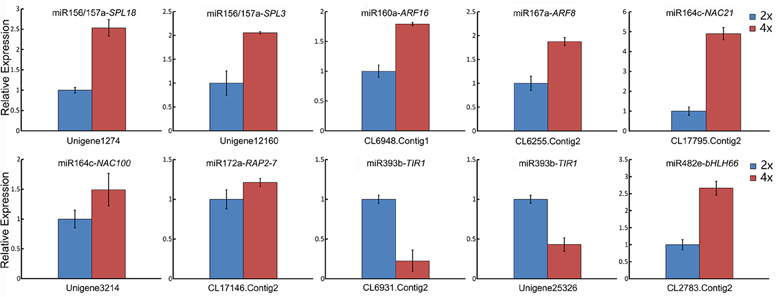
Figure 8. qRT-PCR-estimated abundance of transcript of selected gene targets involved in growth and development and associated with miRNAs in the 2x and 4x form of C. nankingense. Each bar shows the mean ± SE of a triplicated assay.
Discussion
Since the majority of angiosperm species are recognizably polyploid or, like both maize and A. thaliana, have decayed from a polyploid progenitor (“cryptic” polyploids), it has been concluded that polyploidization has been a major driver of plant evolution (Adams and Wendel, 2005). Evidence is accumulating that the activity of miRNAs is important in shaping newly polyploidized genomes (Vazquez et al., 2008; Haa et al., 2009). Thus, the intention here was to explore how they influence the growth and development of the autotetraploid form of C. nankingense.
Autopolyploidization Has Immediate Effect on the Range of miRNAs Produced
The founder pool of miRNAs, at least in monocotyledonous species, is very ancient, and the pace of birth and death of conserved miRNAs is slow (Abrouk et al., 2012). In both the 2x and 4x forms of C. nankingense, the known miRNA distribution was dominated by molecules in the length range 20–25 nt (Figure S1, Table S8), 22 nt (56.17%), and 25 nt (26.0%) known miRNAs displayed the predominant population in the 2x C. nankingense, but 21 nt (50.93%) and 24 nt (24.84%) known miRNAs dominated in the 4x C. nankingense. In the A. thaliana diploid a variety of miRNAs (21–25 nt in length) is biologically significant (Vazquez et al., 2008), it is mainly the 21 nt group which is responsible for differential gene expression in Arabidopsis spp. allopolyploids (Haa et al., 2009). However, in tomato hybrids, Li J. et al. (2014) have shown that it is the 24 nt group which most strongly influences gene expression. At least four Dicer-like (DCL)-encoded enzymes (DCL1–DCL4) catalyze the formation of miRNAs in Arabidopsis (Kasschau et al., 2007), DCL1 have an important role in miRNA accumulation (Kurihara and Watanabe, 2004). Those of length 21 nt and 23–25 nt are generated by DCL1 and DCL3 (Kurihara and Watanabe, 2004; Vazquez et al., 2008). The accumulation of 23–25 nt miRNAs has been correlated with the organ-specific expression of DCL (Vazquez et al., 2008). When the transcription of the various Dicer homolog DCLs was quantified in both diploid and autotetraploid C. nankingense, there was no evidence of any up- or down-regulation (Figure S2). Thus, autopolyploidization is thought likely to have a significant immediate effect on the distribution of miRNAs.
The Effect of Autopolyploidization on miRNA Abundances
Despite there being little evidence for changes induced by autopolyploidization in the range of miRNAs present, there was a distinct effect on the abundance of some of them. Thus, the copy number of 72 known and 35 novel miRNAs was higher in the 4x than in the 2x form of C. nankingense. Of these, 49 known and 25 novel miRNAs were not detected in the diploid, but were highly abundant in the autotetraploid (Tables S4, S6). At the same time, the abundance of 71 known and 42 novel miRNAs was reduced by autopolyploidization; of these 52 known and 28 novel ones were undetectable in the autotetraploid (Tables S4, S6). In some Brassica spp. allopolyploids, it has been claimed that genome redundancy may reflect the increased frequency of changes in miRNA abundance (Shen et al., 2014). In wheat, the proportion of sRNAs classifiable as miRNAs increases with ploidy level (Kenan-Eichler et al., 2011), while according to Li A. et al. (2014), in wheat hexaploids, the abundance of many miRNAs does not reflect additivity. Autopolyploidization is also relatively common in plants, its immediate result is genetic redundancy and process genome flexibility (Parisod et al., 2010). It is suggested that autopolyploidization as well as allopolyploidization has a considerable effect on the population of miRNAs, and thus on gene expression. In addition, miRNAs are associated with non-additive expression of target genes, the expression variation of miRNAs can lead to changes in gene expression, growth vigor, and adaptation (Chen, 2007; Ha et al., 2009). The accumulations of miRNAs contribute to their targets diversity resulting in different growth vigor in autotetraploid relative to diploid. This process could confer a benefit in terms of greater fitness and/or adaptation to an autopolyploid such as 4x C. nankingense.
Differentially Abundant miRNAs Targeting Genes Involved in Growth and Development
MiRNAs are ubiquitous in both plant and animal genomes (Ha et al., 2008). In plants, miRNAs can influence a number of aspects of phenotype, growth, and development and the stress response (Dugas and Bartel, 2004; Zhang et al., 2006). Several of the potential miRNA target genes have been related to the regulation of growth and development (Table 4, Figure 8). miR156a/157a, which was less abundant in the autotetraploid than in the diploid, targets five members of SQUAMOSA PROMOTER BINDING PROTEIN-LIKE (SPL) family, genes which play fundamental roles in growth, development and metabolism (Lei et al., 2016) and may associate with the vigor of the autotetraploid. miR160a and miR167a interact with AUXIN RESPONSE FACTOR (ARF) genes, the products of which showed more abundant in autotetraploid relative to diploid, regulate a variety of physiological processes, especially plant growth and development (Guilfoyle and Hagen, 2007). MiR393b negatively mediate the gene TRANSPORT INHIBITOR RESPONSE 1 (TIR1), the product of which limits the plant's sensitivity to auxin, with a knock-on effect on development (Chen et al., 2011). However, the gene of TIR1 down-regulated after autopolyploidization, suggesting that auxin may contribute to the vigor of autotetraploid C. nankingense. miR172a targets three members of APETALA2 (AP2) genes and one, the former is a class of transcription factors which includes a key regulator of floral development (Riechmann and Meyerowitz, 1998; Spanudakis and Jackson, 2014), and the latter involved in stress responses, secondary metabolism, growth and developmental programs (Licausi et al., 2013). The transcription factor is important for environmental stimuli to enhance the adaption of autotetraploid C. nankingense. The potential target of miR419 and miR1098 is the UPL family of genes encoding E3 ubiquitin-protein ligases, which are an important component of ubiquitination, a post-translational modification delivering plasticity and environmental adaptation (Stone and Callis, 2007). Finally, miR482e interacts with a member of the bHLH transcription factor family, many members of which act as key regulators of gene expression (Toledo-Ortiz et al., 2003). Taken together, miRNAs play important roles in plant growth and development in C. nankingense by negatively mediating targets. The involvement of miRNAs in such genes may explain why the autotetraploid form of C. nankingense is more vigorous than its diploid progenitor.
Conclusion
Here, a comparison was drawn between the miRNA content of diploid and autotetraploid C. nankingense with the intention of exploring the basis of the latter's more vigorous phenotype. Thus autopolyploidization seems likely to have had an immediate effect the distribution of miRNAs, and it have a marked effect on the abundance of many of the miRNAs, including a number which target genes of known importance to growth and development in other plants. The suggestion is that miRNAs may well-contribute to the greater vigor displayed by the autotetraploid form of C. nankingense.
Author Contributions
BD and JJ contributed to bioinformatics analysis and writing of the manuscript. HW, ZG, and FC conceived of the study, participated in its design, and contributed to revisions of the manuscript. WF and TL participated in experiment materials preparation. YC and SC helped with the RNA extraction and real-time PCR, all authors read and approved the final manuscript.
Funding
Fundamental Research Funds for the National Nature Science Foundation of China (31272203), the Central Universities (KYTZ201401), “Programs of Innovation and Entrepreneurship Talents” of Jiangsu Province Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01412
Figure S1. The length distribution of miRNAs in 2x and 4x C. nankingense.
Figure S2. The expression of DCLs in the 2x and 4x form of C. nankingense. Each bar shows the mean ± SE of a triplicated assay. The results showed insignificant differences between the 2x and 4x form of C. nankingense. Significant differences were determined by t-test (P < 0.05).
Table S1. The full set and the set of unique sRNAs present in the diploid and autotetraploid forms of C. nankingense.
Table S2. Variation in the distribution of sRNAs present in the diploid and autotetraploid forms of C. nankingense.
Table S3. The count and sequences of known miRNAs in the diploid and autotetraploid forms of C. nankingense.
Table S4. The differential abundance of known miRNAs in the diploid and autotetraploid forms of C. nankingense.
Table S5. The count and sequences of novel miRNAs in the diploid and autotetraploid forms of C. nankingense.
Table S6. The differential abundance of novel miRNAs in the diploid and autotetraploid forms of C. nankingense.
Table S7. Primer sequences used to amplify target genes by qRT-PCR.
Table S8. The distribution of miRNA in 2x and 4x form C. nankingense.
Sheet 1. The predicted targets of miRNAs which differed in abundance between the diploid and autotetraploid forms of C. nankingense.
Sheet 2. Gene ontology classification of the targets of miRNAs which differed in abundance between the diploid and autotetraploid forms of C. nankingense.
Sheet 3. KEGG pathway annotation of the targets of miRNAs which differed in abundance between the diploid and autotetraploid forms of C. nankingense.
References
Abrouk, M., Zhang, R., Murat, F., Li, A., Pont, C., Mao, L., et al. (2012). Grass microRNA gene paleohistory unveils new insights into gene dosage balance in subgenome partitioning after whole-genome duplication. Plant Cell 24, 1776–1792. doi: 10.1105/tpc.112.095752
Adams, K. L., and Wendel, J. F. (2005). Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 8, 135–141. doi: 10.1016/j.pbi.2005.01.001
Ainouche, M. L., and Jenczewski, E. (2010). Focus on polyploidy. New Phytol. 186, 1–4. doi: 10.1111/j.1469-8137.2010.03215.x
Allen, E., Xie, Z., Gustafson, A. M., and Carrington, J. C. (2005). microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 121, 207–221. doi: 10.1016/j.cell.2005.04.004
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. doi: 10.1016/S0092-8674(04)00045-5
Cao, D., Wang, J., Ju, Z., Liu, Q., Li, S., Tian, H., et al. (2016). Regulations on growth and development in tomato cotyledon, flower and fruit via destruction of miR396 with short tandem target mimic. Plant Sci. 247, 1–12. doi: 10.1016/j.plantsci.2016.02.012
Chen, Z.-H., Bao, M.-L., Sun, Y.-Z., Yang, Y.-J., Xu, X.-H., Wang, J.-H., et al. (2011). Regulation of auxin response by miR393-targeted transport inhibitor response protein 1 is involved in normal development in Arabidopsis. Plant Mol. Biol. 77, 619–629. doi: 10.1007/s11103-011-9838-1
Chen, Z. J. (2007). Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Annu. Rev. Plant Biol. 58, 377. doi: 10.1146/annurev.arplant.58.032806.103835
Comai, L. (2000). Genetic and epigenetic interactions in allopolyploid plants. Plant Mol. Biol. 43, 387–399. doi: 10.1023/A:1006480722854
Cui, Q., Yu, Z., Purisima, E. O., and Wang, E. (2007). MicroRNA regulation and interspecific variation of gene expression. Trends Genet. 23, 372–375. doi: 10.1016/j.tig.2007.04.003
Dai, X., and Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Dugas, D. V., and Bartel, B. (2004). MicroRNA regulation of gene expression in plants. Curr. Opin. Plant Biol. 7, 512–520. doi: 10.1016/j.pbi.2004.07.011
Gao, S., Yang, L., Zeng, H. Q., Zhou, Z. S., Yang, Z. M., Li, H., et al. (2016). A cotton miRNA is involved in regulation of plant response to salt stress. Sci. Rep. 6:19736. doi: 10.1038/srep19736
Guilfoyle, T. J., and Hagen, G. (2007). Auxin response factors. Curr. Opin. Plant Biol. 10, 453–460. doi: 10.1016/j.pbi.2007.08.014
Haa, M. B. C., Lua, J. B., Tiana, L., Ramachandrand, V., Kasschaue, K. D., Chapmane, E. J., et al. (2009). Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Natl. Acad. Sci. U.S.A. 106, 17835–17840. doi: 10.1073/pnas.0907003106
Ha, M., Lu, J., Tian, L., Ramachandran, V., Kasschau, K. D., Chapman, E. J., et al. (2009). Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc. Nat.l Acad. Sci.U.S.A. 106, 17835–17840. doi: 10.1073/pnas.0907003106
Ha, M., Pang, M., Agarwal, V., and Chen, Z. J. (2008). Interspecies regulation of microRNAs and their targets. Biochim. Biophys. Acta 1779, 735–742. doi: 10.1016/j.bbagrm.2008.03.004
He, L., and Hannon, G. J. (2004). MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531. doi: 10.1038/nrg1379
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kasschau, K. D., Fahlgren, N., Chapman, E. J., Sullivan, C. M., Cumbie, J. S., Givan, S. A., et al. (2007). Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 5:e57. doi: 10.1371/journal.pbio.0050057
Kenan-Eichler, M., Leshkowitz, D., Tal, L., Noor, E., Melamed-Bessudo, C., Feldman, M., et al. (2011). Wheat hybridization and polyploidization results in deregulation of small RNAs. Genetics 188, 263–272. doi: 10.1534/genetics.111.128348
Kurihara, Y., and Watanabe, Y. (2004). Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. U.S.A. 101, 12753–12758. doi: 10.1073/pnas.0403115101
Lei, K.-J., Lin, Y.-M., Ren, J., Bai, L., Miao, Y.-C., An, G.-Y., et al. (2016). Modulation of the Phosphate-Deficient Responses by MicroRNA156 and its Targeted SQUAMOSA PROMOTER BINDING PROTEIN-LIKE 3 in Arabidopsis. Plant Cell Physiol. 57, 192–203. doi: 10.1093/pcp/pcv197
Li, A., Liu, D., Wu, J., Zhao, X., Hao, M., Geng, S., et al. (2014). mRNA and small RNA transcriptomes reveal insights into dynamic homoeolog regulation of allopolyploid heterosis in nascent hexaploid wheat. Plant Cell 26, 1878–1900. doi: 10.1105/tpc.114.124388
Li, J., Sun, Q., Yu, N., Zhu, J., Zou, X., Qi, Z., et al. (2014). The role of small RNAs on phenotypes in reciprocal hybrids between Solanum lycopersicum and S. pimpinellifolium. BMC Plant Biol. 14:296. doi: 10.1186/s12870-014-0296-1
Licausi, F., Ohme-Takagi, M., and Perata, P. (2013). APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: mediators of stress responses and developmental programs. New Phytol. 199, 639–649. doi: 10.1111/nph.12291
Liu, S., Chen, S., Chen, Y., Guan, Z., Yin, D., and Chen, F. (2011). In vitro induced tetraploid of Dendranthema nankingense (Nakai) Tzvel. shows an improved level of abiotic stress tolerance. Sci. Hortic. 127, 411–419. doi: 10.1016/j.scienta.2010.10.012
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2 −ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Masterson, J. (1994). Stomatal size in fossil plants: evidence for polyploidy in majority of angiosperms. Science 264, 421–423. doi: 10.1126/science.264.5157.421
Osborn, T. C., Pires, J. C., Birchler, J. A., Auger, D. L., Chen, Z. J., Lee, H.-S., et al. (2003). Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19, 141–147. doi: 10.1016/S0168-9525(03)00015-5
Otto, S. P. (2007). The evolutionary consequences of polyploidy. Cell 131, 452–462. doi: 10.1016/j.cell.2007.10.022
Parisod, C., Holderegger, R., and Brochmann, C. (2010). Evolutionary consequences of autopolyploidy. New Phytol. 186, 5–17. doi: 10.1111/j.1469-8137.2009.03142.x
Riechmann, J. L., and Meyerowitz, E. M. (1998). The AP2/EREBP family of plant transcription factors. Biol. Chem. 379, 633–646.
Rieseberg, L. H., and Willis, J. H. (2007). Plant speciation. Science 317, 910–914. doi: 10.1126/science.1137729
Rubio-Somoza, I., Cuperus, J. T., Weigel, D., and Carrington, J. C. (2009). Regulation and functional specialization of small RNA–target nodes during plant development. Curr. Opin. Plant Biol. 12, 622–627. doi: 10.1016/j.pbi.2009.07.003
Ruiz-Ferrer, V., and Voinnet, O. (2009). Roles of plant small RNAs in biotic stress responses. Annu. Rev. Plant Biol. 60, 485–510. doi: 10.1146/annurev.arplant.043008.092111
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Shen, Y., Zhao, Q., Zou, J., Wang, W., Gao, Y., Meng, J., et al. (2014). Characterization and expression patterns of small RNAs in synthesized Brassica hexaploids. Plant Mol. Biol. 85, 287–299. doi: 10.1007/s11103-014-0185-x
Soltis, D. E., and Soltis, P. S. (1999). Polyploidy: recurrent formation and genome evolution. Trends Ecol. Evol. 14, 348–352. doi: 10.1016/S0169-5347(99)01638-9
Spanudakis, E., and Jackson, S. (2014). The role of microRNAs in the control of flowering time. J. Exp. Bot. 65, 365–380. doi: 10.1093/jxb/ert453
Stone, S. L., and Callis, J. (2007). Ubiquitin ligases mediate growth and development by promoting protein death. Curr. Opin. Plant Biol. 10, 624–632. doi: 10.1016/j.pbi.2007.07.010
Sun, J., Ren, L., Cheng, Y., Gao, J., Dong, B., Chen, S., et al. (2014). Identification of differentially expressed genes in Chrysanthemum nankingense (Asteraceae) under heat stress by RNA Seq. Gene 552, 59–66. doi: 10.1016/j.gene.2014.09.013
Toledo-Ortiz, G., Huq, E., and Quail, P. H. (2003). The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 15, 1749–1770. doi: 10.1105/tpc.013839
Vazquez, F., Blevins, T., Ailhas, J., Boller, T., and Meins, F. (2008). Evolution of Arabidopsis MIR genes generates novel microRNA classes. Nucleic Acids Res. 36, 6429–6438. doi: 10.1093/nar/gkn670
Wang, H., Chen, S., Jiang, J., Zhang, F., and Chen, F. (2015). Reference gene selection for cross-species and cross-ploidy level comparisons in Chrysanthemum spp. Sci. Rep. 5:8094. doi: 10.1038/srep08094
Wang, H., Jiang, J., Chen, S., Fang, W., Guan, Z., Liao, Y., et al. (2013a). Rapid genomic and transcriptomic alterations induced by wide hybridization: Chrysanthemum nankingense × Tanacetum vulgare and C. crassum × Crossostephium chinense (Asteraceae). BMC Genomics 14:902. doi: 10.1186/1471-2164-14-902
Wang, H., Jiang, J., Chen, S., Qi, X., Peng, H., Li, P., et al. (2013b). Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PLoS ONE 8:e62293. doi: 10.1371/journal.pone.0062293
Wang, H., Qi, X., Gao, R., Wang, J., Dong, B., Jiang, J., et al. (2014). Microsatellite polymorphism among Chrysanthemum sp. polyploids: the influence of whole genome duplication. Sci. Rep. 4:6730. doi: 10.1038/srep06730
Keywords: microRNA, autopolyploidization, vigor, growth and development, adaptation
Citation: Dong B, Wang H, Song A, Liu T, Chen Y, Fang W, Chen S, Chen F, Guan Z and Jiang J (2016) miRNAs Are Involved in Determining the Improved Vigor of Autotetrapoid Chrysanthemum nankingense. Front. Plant Sci. 7:1412. doi: 10.3389/fpls.2016.01412
Received: 20 April 2016; Accepted: 05 September 2016;
Published: 28 September 2016.
Edited by:
Jianjun Chen, University of Florida, USAReviewed by:
Jose Luis Reyes, National Autonomous University of Mexico, MexicoLing Yuan, University of Kentucky, USA
Copyright © 2016 Dong, Wang, Song, Liu, Chen, Fang, Chen, Chen, Guan and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiafu Jiang, jiangjiafu@njau.edu.cn
Zhiyong Guan, guanzy@njau.edu.cn