 Ana Carolina Pinheiro Campos1†
Ana Carolina Pinheiro Campos1† Geiza Fernanda Antunes1†
Geiza Fernanda Antunes1† Marcio Matsumoto2Rosana Lima Pagano1
Marcio Matsumoto2Rosana Lima Pagano1 Raquel Chacon Ruiz Martinez1,3*
Raquel Chacon Ruiz Martinez1,3*- 1Division of Neuroscience, Hospital Sirio-Libanes, São Paulo, Brazil
- 2Anesthesiology Medical Center, Hospital Sirio-Libanes, São Paulo, Brazil
- 3LIM 23, Institute of Psychiatry, University of São Paulo School of Medicine, São Paulo, Brazil
Chronic pain is a serious public health problem with a strong affective-motivational component that makes it difficult to treat. Most patients with chronic pain suffer from severe depression; hence, both conditions coexist and exacerbate one another. Brain inflammatory mediators are critical for maintaining depression-pain syndrome and could be substrates for it. The goal of our paper was to review clinical and preclinical findings to identify the neuroinflammatory profile associated with the cooccurrence of pain and depression. In addition, we aimed to explore the regulatory effect of neuronal reorganization on the inflammatory response in pain and depression. We conducted a quantitative review supplemented by manual screening. Our results revealed inflammatory signatures in different preclinical models and clinical articles regarding depression-pain syndrome. We also identified that improvements in depressive symptoms and amelioration of pain can be modulated through direct targeting of inflammatory mediators, such as cytokines and molecular inhibitors of the inflammatory cascade. Additionally, therapeutic targets that improve and regulate the synaptic environment and its neurotransmitters may act as anti-inflammatory compounds, reducing local damage-associated molecular patterns and inhibiting the activation of immune and glial cells. Taken together, our data will help to better elucidate the neuroinflammatory profile in pain and depression and may help to identify pharmacological targets for effective management of depression-pain syndrome.
Introduction
Chronic pain is a complex disorder that significantly impacts society and is the leading cause of disability and financial burden worldwide (Global Burden of Disease Study 2016). It is considered a public health problem and affects approximately 20% of the general population (Breivik et al., 2006). This type of pain is defined as that is persistent or intermittent pain that lasts for more than 3 months despite a normal tissue healing time (Merskey and Bogduk, 1994). Nociceptive pain may occur as a consequence of non-neural damage (Cohen and Mao, 2014), while neuropathic pain is induced by lesions or diseases involving the central nervous system (CNS) (Li et al., 2016). The prevalence of chronic pain ranges from 7 to 10% of the general population (van Hecke et al., 2014), and this disorder manifests as spontaneous pain, hyperalgesia and mechanical allodynia, which decrease quality of life (Baron, 2000). Because of its association with poor quality of life, chronic pain often induces depression (von Knorring et al., 1983; Lee et al., 2009; Agüera-Ortiz et al., 2011). Depression can be characterized by psychological and physical symptoms, including low mood or sadness, lack of energy, lack of motivation, insomnia, low sex drive and an inability to enjoy life (Cui, 2015). Depression is also considered a public health problem and is one the major contributors to global disease burden (Collins et al., 2011; Whiteford et al., 2013). Chronic pain may induce depression, and people suffering from depression may also present abnormal pain perception and modulation, which increases the risk of developing chronic pain (Currie and Wang, 2005). It has been estimated that 85% of people affected with chronic pain suffer from severe depression (Bair et al., 2003; Williams et al., 2003), supporting the concept that both conditions coexist and exacerbate one another (Gallagher and Verma, 1999; Blier and Abbott, 2001; Leo, 2005). This association has been labeled depression-pain syndrome or the depression-pain dyad (Lindsay and Wyckoff, 1981; Beckman, 2004; Cox et al., 2017).
A possible reason why these disorders are complex and difficult to treat is that they both involve neuroinflammation. Neuroinflammation is an innate immune response of the nervous system to injury, infection or neurodegenerative disease characterized by the activation of resident glial cells, including microglia and astrocytes; release of cytokines and chemokines; and activation and infiltration of leukocytes (Takeuchi and Akira, 2010; Domercq et al., 2013; Ji et al., 2014, 2016, 2018; Turner et al., 2014; Stephenson et al., 2018; Yang and Zhou, 2019). It has also been proposed that these conditions could be responsible for alterations in the permeability of the blood-brain barrier, immune cell infiltration, and activated microglia (Benatti et al., 2016). Persistent neuroinflammation plays an important role in the induction and maintenance of depression-pain syndrome (Ji et al., 2014; Burke et al., 2015). In this sense, classically activated resident glial cells lose the ability to control the synaptic environment (Liddelow et al., 2017). Specifically, improvements in the synaptic environment could promote symptomatic progress and modify disease progression (Pecina and Zubieta, 2018). However, the relationship between inflammation and the synaptic environment in depression-pain syndrome has been poorly investigated (Liddelow et al., 2017).
In the literature, much effort has been devoted to fully understanding the pathophysiology of chronic pain and depression as well as understanding why comorbidity of these two disorders is so common to guide the development of better treatments. It is not surprising that pain relief medications are still the second-most prescribed drugs (after cardiac-renal drugs) in the United States (Turk, 2002; Van Zee, 2009; Dowell et al., 2016; Volkow and McLellan, 2016). Pharmacological treatment options for chronic pain include opioids, non-steroidal anti-inflammatory drugs, gabapentinoids, tricyclic antidepressants, serotonin-norepinephrine reuptake inhibitors, NMDA antagonists, and topical preparations (Turk, 2002; Cooper et al., 2017; Eccleston et al., 2017; Manchikanti et al., 2017; Riediger et al., 2017; Shanthanna et al., 2017; Szok et al., 2019). The use of opioids and non-steroidal anti-inflammatory drugs is limited by their adverse effects, tolerance, and potential for addiction (Barber and Gibson, 2009; Han et al., 2015; Vowles et al., 2015; Busse et al., 2018). Additionally, considering that chronic pain treatment frequently involves polypharmacy therapy, it is associated with an increased risk of drug interactions and additive adverse effects (Barber and Gibson, 2009; Taylor et al., 2013; Denk et al., 2014; Riediger et al., 2017; Busse et al., 2018). Furthermore, despite the range of drugs available, pharmacological therapies for chronic pain are often inefficient for the management of depression-pain syndrome, and as this condition can affect nearly every aspect of daily life; thus, the development of novel treatment strategies is critical (Bentley et al., 2014; Cohen and Mao, 2014; Crofford, 2015; Brant et al., 2017; Shamji et al., 2017). Treatment focusing on neuroinflammatory substances could help to inhibit glial cells in their activated state, improve the neuronal network in specific regions and induce alleviation of chronic pain and depression (Walker et al., 2013a; Ji et al., 2014, 2018). On the other hand, it has been hypothesized that treatments that modulate the neuronal network may decrease the formation of glial cell-recognized patterns and decrease inflammation, consequently modulating the symptoms of depression-pain syndrome. In this sense, the goal of our paper was to review clinical and preclinical findings to identify the inflammatory signature associated with depression-pain syndrome and the different types of treatments that modulate neuroinflammation. We hope to discuss the anti-inflammatory mechanisms observed in the studied articles and improvements in the neurocircuitry, which may be responsible for the optimal modulation of these symptoms.
Methods
Search Strategy and Inclusion and Exclusion Criteria
A quantitative review search of PubMed, ScienceDirect and MEDLINE for original research articles supplemented by manual screening was conducted between November and December 2019. As this study aimed to review many published articles on the topics of neuroinflammation and pain-depression syndrome, there were no restrictions placed on the publication date for the search. Thus, we opted to not conduct a formal systematic review or meta-analysis. The studies were required to contain the relevant terms “neuroinflammation AND chronic pain AND depression OR glial cells AND chronic pain AND depression OR chronic pain AND depression OR negative effects AND patients AND animal model OR chronic pain AND depression AND animal models AND patients.” The selection criteria included studies with (1) clinical and preclinical data; (2) human and rodent data; and (3) cooccurrence of pain, depression and neuroinflammation. Studies of all sample sizes were included in the analysis. Studies were excluded if they (1) were reviews or meta-analyses; (2) presented repeated data from previously included studies or (3) were not published in English.
As shown in Figure 1, the search yielded 2,548 records. We eliminated articles that did not investigate the cooccurrence of painful and depressive behaviors or did not investigate any inflammatory biomarkers. Ultimately, 35 studies were included in this review.
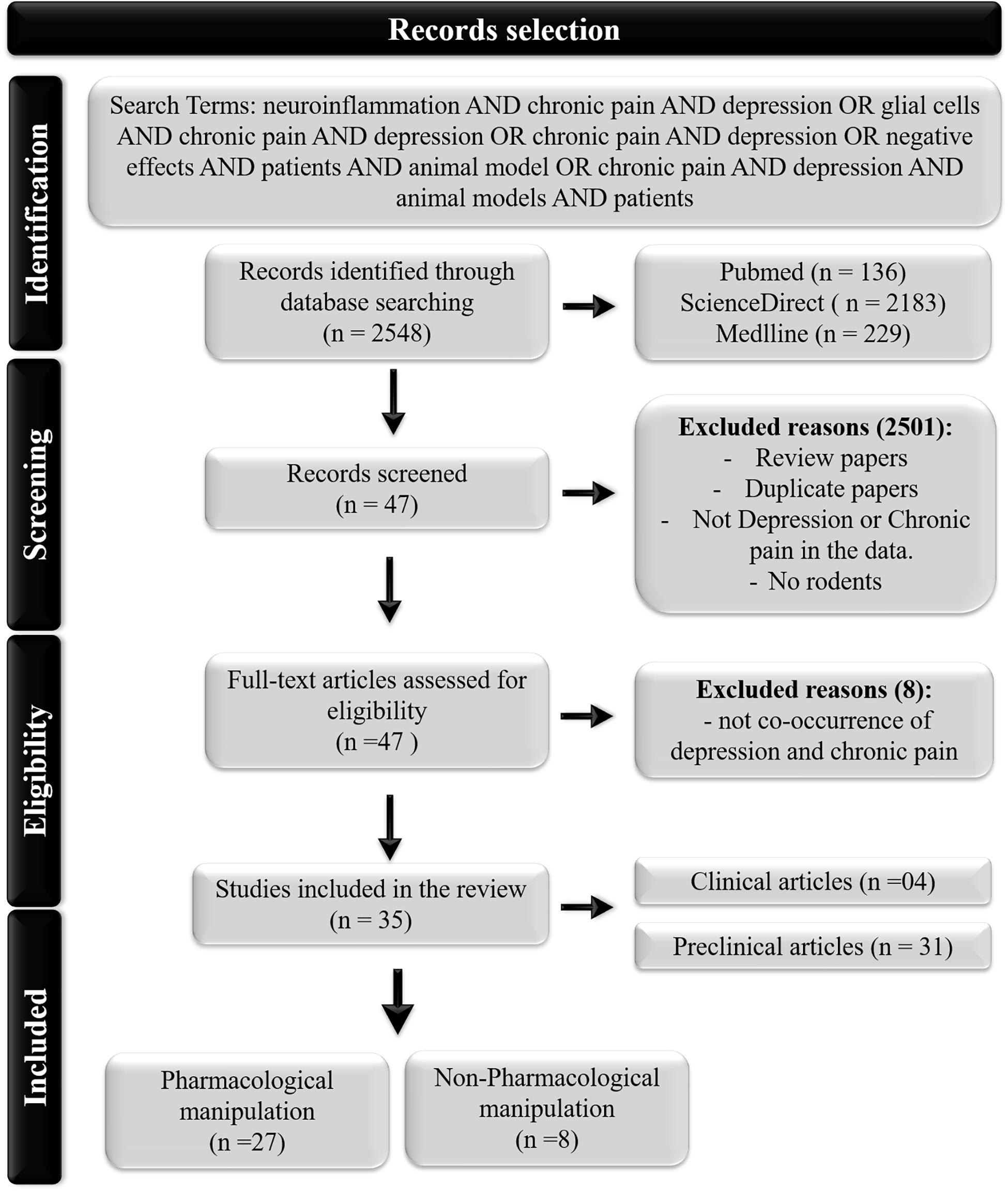
Figure 1. Flowchart of the study.
Results
Target Structures Involved in Pain and Depression
From the 35 selected papers, we found 31 and 4 reports of preclinical models and clinical investigations, respectively. Eight of the selected articles evaluated the inflammatory signature in the presence of painful and depressive behavior, while 27 articles evaluated the inflammatory response related to different types of treatments. The structures that play an important role in pain and depression states listed in the selected papers included the cerebral cortex and its subdivisions, the prefrontal cortex (PFC), the anterior cingulate cortex (ACC), the amygdala, the hippocampus, and the raphe nuclei, as alterations in plasma were observed in human subjects.
The main general markers of the cooccurrence of chronic pain and depression were related to central inflammation, particularly, tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, as well as peripheral discrepancies in cortisol levels. Regarding treatment for depression-pain syndrome, we found that therapies with different mechanisms may attenuate central and peripheral inflammation, raising a discussion about the relationship between the neuronal network and inflammation modulation.
Discussion
Target Structures Involved in Pain and Depression
During pain processing, structures such as the PFC, ACC, nucleus accumbens (NAc), thalamus, hippocampus and amygdala send projections to the periaqueductal gray (PAG) to activate the descending analgesic pathway, including the raphe nucleus and locus ceruleus (Fields and Basbaum, 1999; Apkarian et al., 2005; Seminowicz et al., 2019). These areas are connected and often modulate one another, as first described by Papez (1937). The original Papez circuit consists of the hippocampus, thalamus, cingulate gyrus and cerebral cortex; however, further structures have been added over the years, such as the insular cortex, amygdala, and other subcortical nuclei (Yakovlev, 1948; Nakano, 1998). In this sense, the limbic system, a network of brain structures involved in emotion, memory, learning and homeostatic processes that strongly affect behavior (Arora et al., 2011; Catani et al., 2013), suggesting a major correlation between pain and depression.
Therefore, many authors have searched for inflammatory signatures in limbic system structures, especially the PFC and hippocampus. It has been proposed that the expression of genes encoding proinflammatory cytokines are increased in these limbic structures responsible for pain processing and depression (Arora et al., 2011; Kim et al., 2012). A structure mentioned in 24% of the articles was the PFC. The medial PFC (mPFC), which is composed of the granular and agranular areas, including the ACC (Ong et al., 2019), was also investigated in the articles we found. Additionally, another 12% of the articles described the signature in the cerebral cortex without specifying which regions were involved. Some authors probably preferred to address the structures more generally due to the methodological difficulty of ensuring the extraction if the specific area of the brain from fresh tissue. The mPFC sends projections to the PAG (Bragin et al., 1984; An et al., 1998) that project to the nuclei raphe and are involved in the analgesic descending pathway (Fields and Basbaum, 1999; Apkarian et al., 2005; Seminowicz et al., 2019). The mPFC is important for pain processing and mediates antinociceptive effects due to its connections with other cortical areas and the PAG (Ong et al., 2019). However, increased and persistent activation of the thalamus, which projects to the somatosensory cortex and the PFC, mediates the chronification of pain, possibly via corticostriatal projections, dopaminergic system dysfunction and ventral tegmental area (VTA)-NAc reward pathways (Ong et al., 2019). Additionally, although the PFC is often thought of as the center of thinking and decision making, it is also associated with depression (Treadway et al., 2015). Lesions of the mPFC, which is responsible for affection and negative emotion (Miller and Cohen, 2001), may attenuate depression by decreasing the direct activation of the amygdala (Ellenbogen et al., 2005; Sachdev and Sachdev, 2005; Connolly et al., 2017).
It has been proposed that environmental stress can contribute to the development of depression through inflammatory and epigenetic mechanisms (Slavich and Irwin, 2014; Park et al., 2019). Specifically, the PFC and amygdala have opposite responses to stress since synaptic plasticity is enhanced in the amygdala after depression and is decreased in the PFC (Marsden, 2013). Inhibition of the amygdala by local GABAergic agonists changes pain sensitivity and depressive-like behavior (Seno et al., 2018). Additionally, the volume of the amygdala has been suggested to be correlated with the severity of depression (Li et al., 2014), demonstrating the importance of its activation in the pathophysiology of depression.
Persistent pain may cause dystrophy of hippocampal areas by reducing volume, increasing abnormal cytokine expression, and inducing deficits in long-term potentiation as well as impairing neurogenesis (Duric and McCarson, 2005; Kodama et al., 2007; Terada et al., 2008; Al-Amin et al., 2011; del Rey et al., 2011; Erickson et al., 2011; Mutso et al., 2012). These alterations can induce anxiety and stress since the hippocampus and PFC play critical roles in regulating the hypothalamus-pituitary-adrenal (HPA) axis (Jacobson and Sapolsky, 1991). After HPA activation, glucocorticoid hormones (corticosterone in rodents and cortisol in humans) are released from the adrenal gland in response to stress and easily penetrate into the human brain (Karssen et al., 2001). The hippocampus, PFC and brainstem monoaminergic nuclei strongly respond to the increased levels of these glucocorticoids (Reul and de Kloet, 1985). In this sense, dopamine release is increased in acute stress (Castro and Zigmond, 2001), while it is downregulated after chronic exposure, causing modulation of the VTA-NAc reward pathway (Thompson et al., 2004). Additionally, chronic exposure to corticosterone/cortisol induces hippocampal dendritic atrophy (Landfield et al., 1981; Meaney et al., 1988), suggesting that depression and anxiety can induce changes in neural plasticity in areas also involved in controlling the nociceptive system and may “predispose” the brain to persistent pain sensitivity. The majority of the articles we found investigated the hippocampus, demonstrating the importance of this region in pain, depression, and depression-pain-syndrome. We also found that some authors investigated the dorsal root of the ganglia (DRG) and the spinal cord, structures strongly related to pain control (Todd, 2010; Krames, 2014), especially in models of pain induced by nerve damage.
Regarding the assessment of systemic samples, some articles evaluated corticosterone levels and some inflammatory biomarkers in the serum or plasma. Plasma and serum are more related to the neuroendocrine response and peripheral inflammation since the blood-brain barrier (BBB) can prevent the ability of substances in the blood circulation to access the CNS (Daneman and Prat, 2015); however, some studies have noted that neuroinflammatory settings may compromise the integrity of the blood-nerve barrier and alter the BBB (Skaper, 2016; Schaefer et al., 2017). A schematic review of target structures and inflammatory signatures is shown in Figure 2.
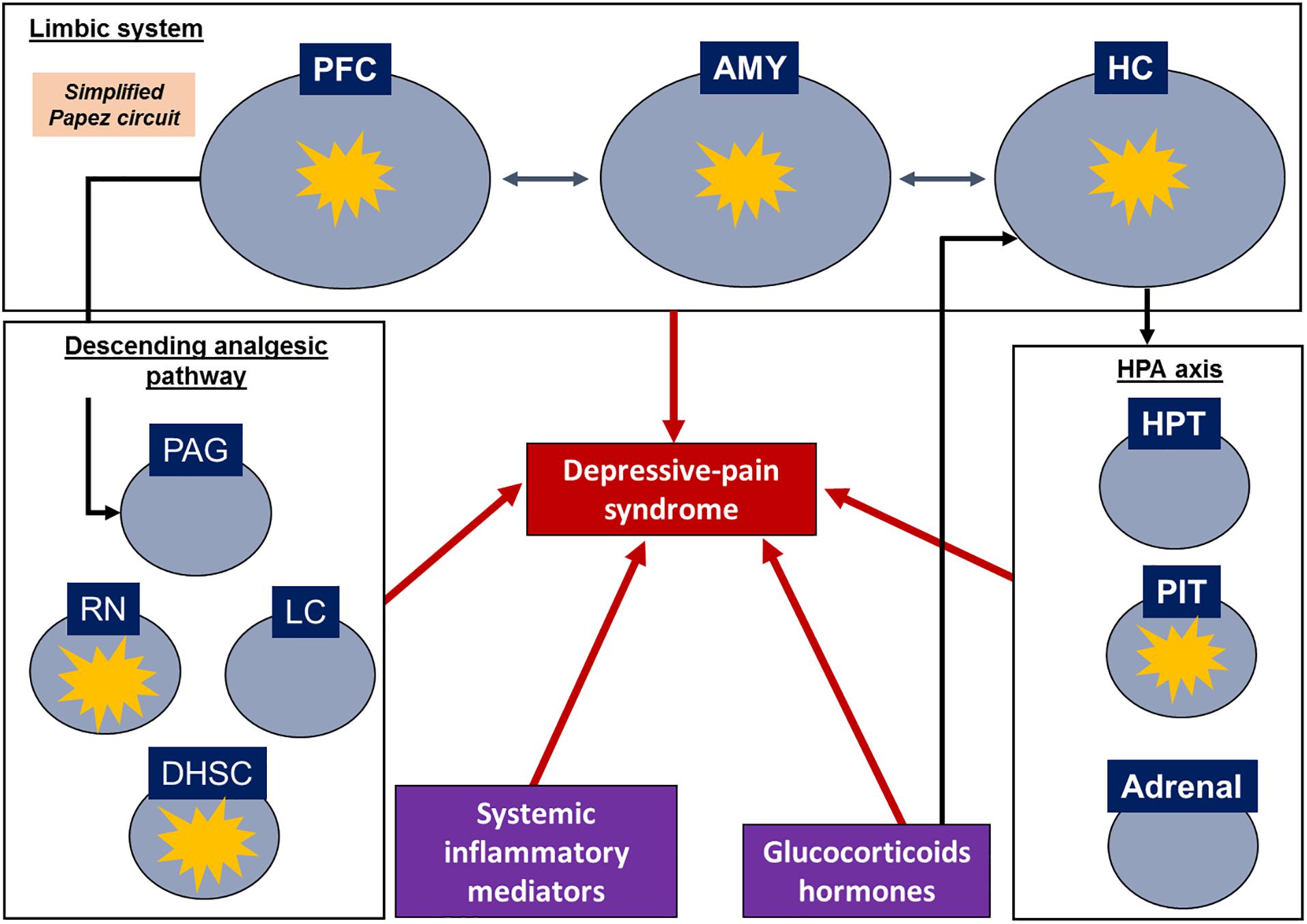
Figure 2. Schematic representation of depressive-pain syndrome and the relationship between target structures and mediators. Target structures with inflammatory signatures in the identified articles. AMY, amygdala; DHSC, dorsal horn of the spinal cord; HC, hippocampus; HPT, hypothalamus; LC, locus coeruleus; PAG, periaqueductal gray matter; PFC, prefrontal cortex; PIT, pituitary; RN, raphe nuclei.
Neuroinflammatory Signature of Preclinical Models (Table 1)
In the peripheral nervous system, classic inflammatory modulators are secreted by neutrophils in a more acute stage of neuroinflammation and then by macrophages and T cells after the persistence of inflammation sensitizes either the nociceptor sensory receptors (Basbaum et al., 2009) or peripheral glial cells, such as Schwann cells and satellite glia cells (Ellis and Bennett, 2013), to potentially noxious stimuli in primary afferent neurons, modulating pain sensitivity (Ji R. R. et al., 2013). Infiltrating macrophages and Schwann cells can secrete proinflammatory cytokines, such as IL-1β and TNF-α (Nathan, 1987; Palmer and Weaver, 2010), which is consistent with the increase in plasma proinflammatory cytokines in people suffering from chronic pain (Vendrusculo-Fangel et al., 2019) and preclinical pain models (Table 1). Both peripheral and central inflammation are associated with symptoms of pain and depression (Walker et al., 2013b), as characterized by dysregulation of the immune system (Burke et al., 2014), neurotransmitters such as noradrenaline and serotonin (Stahl and Briley, 2004; Goldenberg et al., 2010), neuropeptides such as brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) (Kramer et al., 1998; Werner and Coveñas, 2010), oxidative stress (Arora et al., 2011) and cytokines (Wallace, 2006; Dowlati et al., 2010).
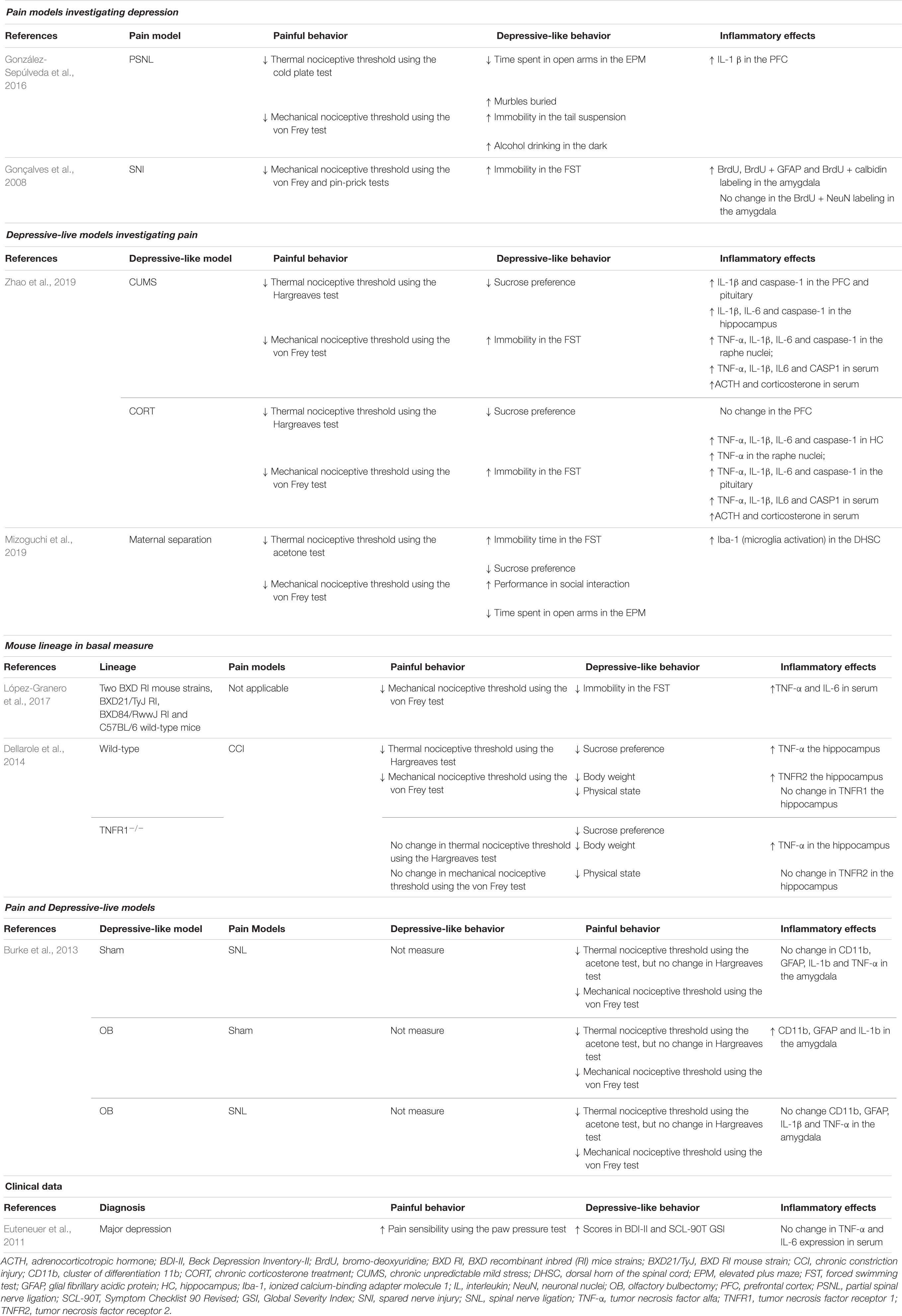
Table 1. Inflammatory signature upon presence of painful and depressive behavior in preclinical and clinical research.
Where and how neuroinflammation starts in the absence of a pathogen or classic injury may be a complex question. In this sense, direct injury of peripheral nerves, as investigated in 57% of the articles regarding neuroinflammation in preclinical models without treatment (Table 1), begins with peripheral inflammation due to Wallerian degeneration that occurs in the distal axonal segment following nerve injury and is characterized by a process of demyelination and disintegration, release of proinflammatory cytokines and macrophage recruitment to the nerve (Toews et al., 1998; Shamash et al., 2002; Zuo et al., 2003; Perrin et al., 2005). In peripheral nerve injury, myelinating Schwann cells start proliferating and produce proinflammatory cytokines, such as TNF-α, IL-1β and monocyte chemoattractant protein 1 (MCP-1), which together with substance P (SP) and calcitonin gene-related peptide (CGRP) act as damage-associated molecular patterns (DAMPs) that activate innate immune resident cells (Zhang and An, 2007). These patterns activate different receptors present within the membrane of resident cells to induce the release of more proinflammatory cytokines and chemokines (Takeuchi and Akira, 2010).
Increased serum levels of TNF-α and IL-6 have been shown in two different BXD mouse lineages that are commonly used as genetic resources to study neuropharmacological phenomenon (Philip et al., 2010). In BXD21/TyJ RI mice, nuclear factor erythroid 2 (Nrf2) expression is upregulated, while BXD84/RwwJ RI mice exhibit low Nrf2 expression (López-Granero et al., 2017). Considering the role of Nrf2 in activating antioxidative mechanisms (Zhang et al., 2013), these mice show greater depressive-like behavior than wild-type mice (López-Granero et al., 2017). Interestingly, increases in TNF-α, IL-1β, IL-6 and caspase-1 are found in the serum of animals submitted to chronic stress paradigms that induce depressive behavior (Zhao et al., 2019). Stress models have been used to study the etiological and developmental components of depression and to screen antidepressant drugs (Nollet et al., 2013).
Increased systemic proinflammatory cytokines can also be found in people affected with major depression before any treatment (Zou et al., 2018), but the relevance of cytokine expression in the blood of people with depressive disorders is not clear (Himmerich et al., 2019). Additionally, even though the dorsal horn of the spinal cord (DHSC) is poorly related to depression, in a model of maternal separation, there is increased microglial activation in this structure. Interestingly, the same article also showed that animals submitted to maternal separation followed by spinal nerve ligation (SNL) exhibited more relevant activation of spinal microglia (Mizoguchi et al., 2019), corroborating the hypothesis that depression may “predispose” the CNS to the pathologic properties of persistent pain. Astrocytes and microglial cells are critical for the induction and maintenance of chronic pain in a preclinical pain model (Watkins et al., 2001). These cells express Toll-like receptors (TLRs), purinergic receptors (with ATP as ligands) and glutamatergic receptors (Domercq et al., 2013; Siracusa et al., 2019). For instance, a low ATP concentration increases the release of chemokines, including IL-2, MCP-1 and CX3CL1, which act by recruiting immune cells from other brain regions or from the bloodstream to the injured tissue (Domercq et al., 2013; Turner et al., 2014). When ATP or glutamate is increased in the synaptic environment, these cells secrete cytokines, such as TNF-α, IL-1β, IL-6, and interferon (IFN)-γ (Suzuki et al., 2004; Taylor et al., 2005; Di Virgilio et al., 2009), which are pivotal inflammatory mediators that further aggravate the inflammatory reaction and may also potentially serve as biomarkers of diseases and therapeutic efficacy (Goldstein et al., 2009; Miller et al., 2009; Chen et al., 2017).
Peripheral nerve injury models present peripheral inflammation and increased proinflammatory cytokines in spinal and supraspinal structures, such as the PFC (González-Sepúlveda et al., 2016) and amygdala (Gonçalves et al., 2008) as well as with decreased thermal and mechanical nociceptive thresholds and depressive-like behavior. TNF receptor (TNFR) knockout mice are protected against CCI-induced pain; however, inflammation in the hippocampus and depressive-like behavior remain (Fischer et al., 2019). Inflammation of the limbic system also occurs in chronic stress models with increased proinflammatory cytokines in the PFC, hippocampus, raphe nuclei and pituitary (Zhao et al., 2019). Additionally, the olfactory bulbectomy-induced depression model exhibited increased CD11b (macrophage/microglia marker), GFAP (Glial fibrillary acidic protein, astrocyte marker) and IL-1β levels in the amygdala, but the spinal nerve ligation-induced neuropathic pain alone or combined with olfactory bulbectomy does not change the expression of these cytokines (Burke et al., 2013), demonstrating that although pain and depression have several similarities, they may also involve different pathways. Inflammation of the structures of the limbic system often correlates with increased serum ACTH and corticosterone levels (Zhao et al., 2019). Increased expression of these hormones is often used as a biomarker for the activation of the HPA axis, which is strongly related to stress (Abrahams et al., 2012; Stephens and Wand, 2012). Focusing on neuroinflammation, Arora and Chopra (2013) observed increases in TNF-α and IL-1β in the hippocampus and PFC after chronic administration of reserpine. In another study, chronic unpredictable mild stress and chronic corticosterone treatment induced an increase in TNF-α, IL-1β, and IL-6 in different brain structures, as well as in the hippocampus and PFC (Zhao et al., 2019). In combined models of pain and depression, increases in the cytokines IL-1β and IL-6 in the amygdala have also been noted (Burke et al., 2013). In this sense, the main general markers of the cooccurrence of chronic pain and depression are related to central inflammation, particularly TNF-α, IL-1β, and IL-6.
Understanding Treatments for Persistent Pain and Depression
Since chronic pain and depression-pain-syndrome are complex disorders, an extensive class of medications has been used to improve the quality of life of individuals suffering from depressive-pain syndrome. Classic non-steroidal anti-inflammatory drugs (NSAIDs) and analgesics are not efficient in attenuating persistent pain sensitivity (Johannes et al., 2010); hence, the use of benzodiazepines, tricyclic antidepressants, monoamine reuptake inhibitors, glutamatergic antidepressant drugs, and adjuvant psychotherapy has been investigated to attenuate pain sensitivity. In pharmacological treatment, there is also an overlap between pain and depression: much of the medication that can be used to treat persistent pain is also used to treat major depression (Bair et al., 2003). These treatments are often related to the control of monoamines (such as serotonin, noradrenaline and dopamine), as well as glutamate and GABA, which are pivotal in the modulation of pain and depression (Benson et al., 2015). However, as described above, inflammation plays an important role in both comorbidities and, in this sense, may be a successful target for treating individuals with these symptoms.
Different types of non-pharmacological treatments can be used considering the importance of social support and the environment demonstrated in depressed subjects with chronic pain (Trief et al., 1995). Additionally, there is a strong correlation between pain, depression, and poor quality of sleep (Wilson et al., 2002; Smith and Haythornthwaite, 2004; Ohayon, 2005; Staner, 2010; Koffel et al., 2016) that is directly correlated with the level of stress. It has been shown that a stressful environment can induce hyperalgesia in rats (Rivat et al., 2007), while an enriched environment can alleviate chronic pain symptoms (Vachon et al., 2013). In addition to environmental enrichment, another non-pharmacological treatment used to investigate the inflammatory signature is pulsed radiofrequency. Non-pharmacological treatments such as acupuncture, massage and transcutaneous electrical nerve stimulation (TENS) have been administered to people affected with spinal cord injury, and they were shown to improve pain sensitivity when combined with pharmacological treatments (Norrbrink and Lundeberg, 2004). Pulsed radiofrequency uses radiofrequency to generate electrical fields that can affect neuronal membranes, altering the transmission of pain pathways (Byrd and Mackey, 2008) and is a safe and effective treatment for radicular pain, trigeminal neuralgia, occipital neuralgia, shoulder and knee pain (Vanneste et al., 2017).
NSAIDs are the most consumed drugs worldwide (Bozimowski, 2015), and their efficacy, especially in acute and postsurgical pain has been established (KuKanich et al., 2012). NSAIDs inhibit cyclooxygenase enzyme isotypes (COX1-COX4) and prostaglandins, decreasing pain, fever and inflammation by disrupting the arachidonic acid pathway (Osafo et al., 2017). This inhibition may mediate peripheral nociceptor sensitization (Vane, 1971) and the central nociceptive pathway (Hunskaar, 1987; Björkman, 1995; Wang et al., 1995). Thus, in a model of acute pain (induced by formalin) and a stressful depressive (SD) model, the effect of aspirin was compared to that of benzodiazepines and a cholecystokinin (CCK) antagonist (Rivat et al., 2010). Cholecystokinin reduces the antinociceptive effects of opioids, and its antagonist may be an effective tool to prevent opioidergic tolerance (Wiesenfeld-Hallin et al., 2002). In this article, only benzodiazepine was unable to improve the mechanical and thermal nociceptive threshold; however, aspirin was not effective in improving anxious-depressive behavior (Rivat et al., 2010), demonstrating that NSAIDs may not be helpful in the comorbidity of pain and depression comorbidity. Another class of medications used for neuropathic pain is steroidal drugs, such as corticosteroids and dexamethasone (Watanabe and Bruera, 1994). Atkinson and coworkers showed that dexamethasone is not able to control cortisol levels in individuals with chronic pain and major depression (Atkinson et al., 1986), which is related to painful and depressive symptoms (France and Krishnan, 1985). In addition, different fatty acids have been studied in an attempt to attenuate pain and depression. Omega-3 improves anxiety and depression in people affected with chronic pain (Cortes et al., 2013), while omega-3, palmatine and β-caryophyllene attenuate pain and depression in a preclinical diabetic model (Redivo et al., 2016; Shen et al., 2018; Aguilar-Ávila et al., 2019). Berberine, a quaternary ammonium salt derived from a protoberberine group of alkaloids, is the principal bioactive compound found in Coptis chinensis, Berberis vulgaris and other medicinal plants that are effective in treating pain and depression (Tang et al., 2009; Ye et al., 2009). In addition to significantly reducing hypercholesterolemia in rats (Kim and Chung, 2008; Hu and Davies, 2010; Wang et al., 2014), berberine also exerts a depressant action in the central system and has beneficial antinociceptive effects on pain threshold (Shanbhag et al., 1970). Berberine reduces pain- and depressive-like behavior in a reserpine-induced persistent pain model, and this response is related to the inhibition of SP and reactive species in the cerebral cortex and hippocampus (Arora and Chopra, 2013). Another natural compound that has been studied is triptolide, which is extracted from the medicinal plant Tripterygium wilfordii and can be used to suppress different proinflammatory factors from macrophages and T cells (Qiu et al., 1999; Qiu and Kao, 2003). Triptolide shows the same effectiveness in providing pain relief and attenuating depression as fluoxetine, a classical antidepressant (Hu et al., 2017).
As previously described, one of the similarities between the pathophysiology of pain and depression is the importance of monoamines. Hence, treatment with monoamine reuptake inhibitors is very common and was found in 22% of the articles that investigated treatments for pain and depression involving inflammation. Since major depression is associated with decreased serotonin levels (Haase and Brown, 2015), as in the persistent pain state (Bardin, 2011), fluoxetine improves these symptoms by increasing the amount of serotonin in the synaptic environment by selectively inhibiting the reuptake of this monoamine (Fuller et al., 1991). Although fluoxetine is the most studied selective serotonin reuptake inhibitor (SSRI) used to treat chronic pain, its use in clinical trials has been contradictory (Walker et al., 1998); however, it is effective in a preclinical lumbar disk herniation model (Cai et al., 2019). Additionally, fluoxetine plus pioglitazone or metformin, both of which are antidiabetic drugs, attenuates pain and depression with better effects than each treatment individually (Murad and Ayuob, 2015). Pioglitazone is a peroxisome proliferator-activated receptor gamma (PPARy) agonist that inhibits hyperalgesia in a spared nerve injury model (Griggs et al., 2015), while metformin inhibits mammalian target of rapamycin (mTOR) and decreases neuropathic pain and postsurgical pain (Kiałka et al., 2017). Rosiglitazone, another antidiabetic drug that also acts on PPARy, inhibits neuropathic pain induced by sciatic nerve transaction in rats by regulating macrophage polarization (Churi et al., 2008; Hasegawa-Moriyama et al., 2012). Like morphine, rosiglitazone improves mechanical allodynia, but unlike morphine, it improves depressive-like behavior by increasing BDNF and decreasing proinflammatory modulators in the hippocampus (Zong et al., 2018). Interestingly, an AMP-activated protein kinase (AMPK) inhibitor and an agonist of 3-MA (an autophagy inhibitor) block the antinociceptive, antidepressive and anti-inflammatory properties of rosiglitazone (Zong et al., 2018), suggesting that these compounds are involved in the control of pain and depression, probably due to their antidiabetic medication properties. Amitriptyline, similar to fluoxetine, is a monoamine regulator that is used regularly as an antidepressant, acts as a selective serotonin-noradrenalin reuptake inhibitor (SSNRI), and has antinociceptive effects dependent on α2-adrenergic receptors (Hiroki et al., 2017). However, in a spinal nerve ligation and olfactory bulbectomized model of pain and depression, amitriptyline treatment is not able to attenuate thermal hyperalgesia or allodynia but improves depressive-like behavior (Burke et al., 2015). According to the Cochrane Database Reviews (2014), there is not enough information to consider imipramine, which is in the same class of medicines as amitriptyline, a drug for neuropathic pain treatment. α-(Phenylselanyl)acetophenone (PSAP), a new selenium drug, has been tested for its ability to treat neuropathic pain because of its anti-inflammatory and antioxidant properties and its effect on serotonin receptors (Gerzson et al., 2012). According to Sousa et al. (2018), imipramine and PSAP improve pain sensitivity and depressive-like behavior after acute stress restriction (Sousa et al., 2018).
Ketamine is an anesthetic drug with N-methyl-D-aspartate (NMDA) receptor antagonism properties. Since these receptors are related to the amplification of pain signals and opioid tolerance, ketamine acts by reducing these dysfunctions observed in chronic pain conditions (Trujillo and Akil, 1991; Laulin et al., 2002; Lilius et al., 2015). Additionally, ketamine may also regulate monoaminergic receptors (Peltoniemi et al., 2016) and increase the inhibitory serotonergic pathway (Mion and Villevieille, 2013). However, ketamine has been shown to improve depressive-like behavior without attenuating pain in a spared nerve injury model (Pan et al., 2018). Another possible antinociceptive mechanism of ketamine is the inhibition of astrocyte and microglia activation (Sleigh et al., 2014), as minocycline, an antibiotic with high lipid solubility, crosses the BBB and inhibits microglia activation (Yrjänheikki et al., 1998). Minocycline improves pain sensitivity and depressive-like behavior in a model of spinal nerve ligation combined with olfactory bulbectomy (Burke et al., 2014); however, it was shown to be ineffective in patients with lumbar radicular neuropathic pain in a double-blind clinical trial using amitriptyline as a comparator (Vanelderen et al., 2015). A summary of the pharmacological mechanisms of the treatments described in this review is shown in Figure 3 and Table 2.
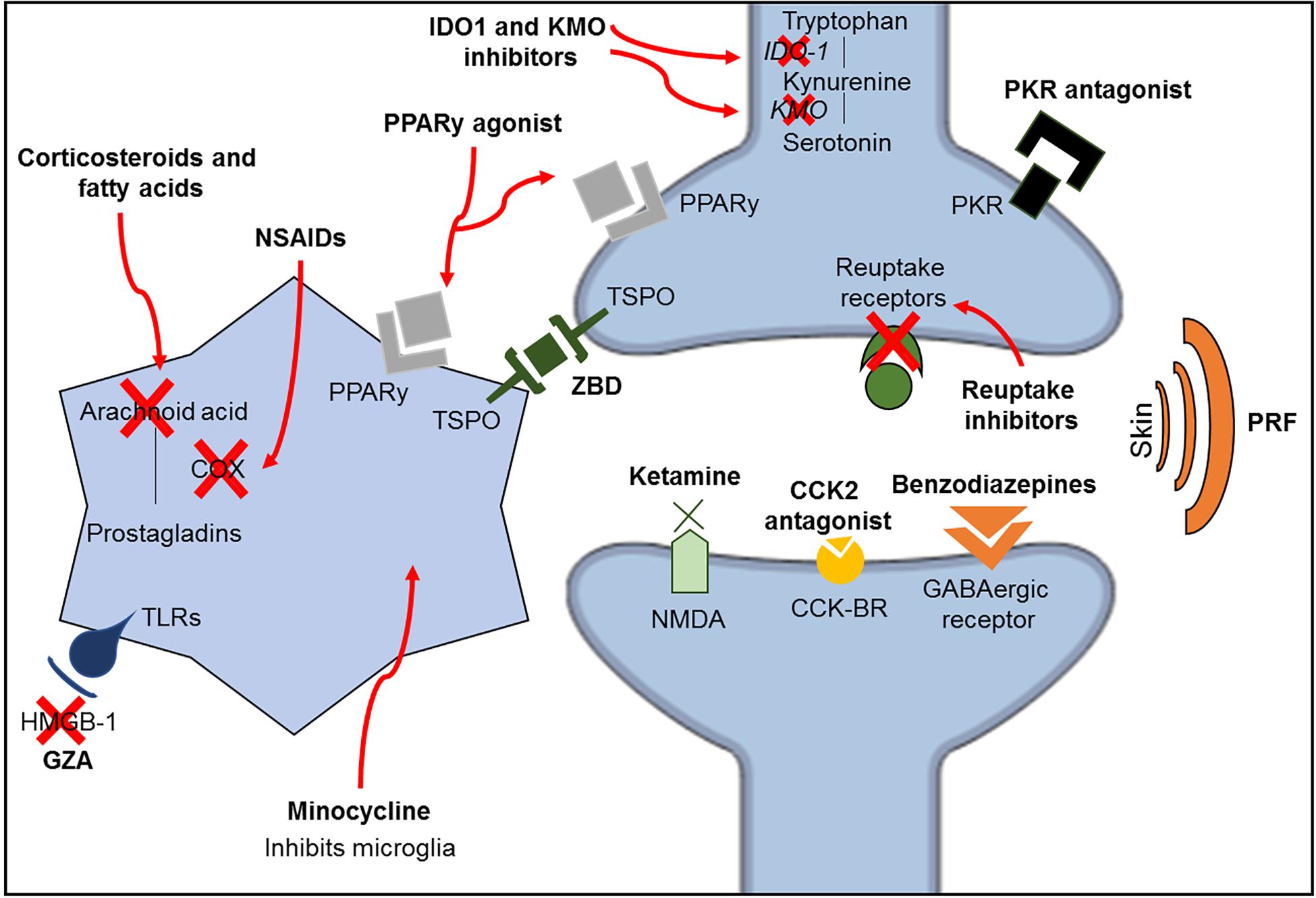
Figure 3. Schematic representation of the classic mechanisms of the different classes of medications investigated in this review. CCK2, cholecystokinin 2; CCK-R, cholecystokinin receptors; GZA, glycyrrhizic acid; HMGB-1, high mobility group box-1; IDO1, indolamine 1,3 deoxygenase; KMO, kynurenine 3-monooxygenase; NMDA, N-methyl-D-aspartate; NSAIDs, non-steroidal anti-inflammatory drugs; PKR, prokineticin family; PPARy, peroxisome proliferator-activated receptor gamma; PRF, pulsed radiofrequency; TLRs, toll-like receptors; TSPO, translocator protein; ZBD, N-benzyl-Nethyl-2-(7,8-dihydro-7-benzyl-8-oxo-2-phenyl-9H-purin-9-yl) acetamide.
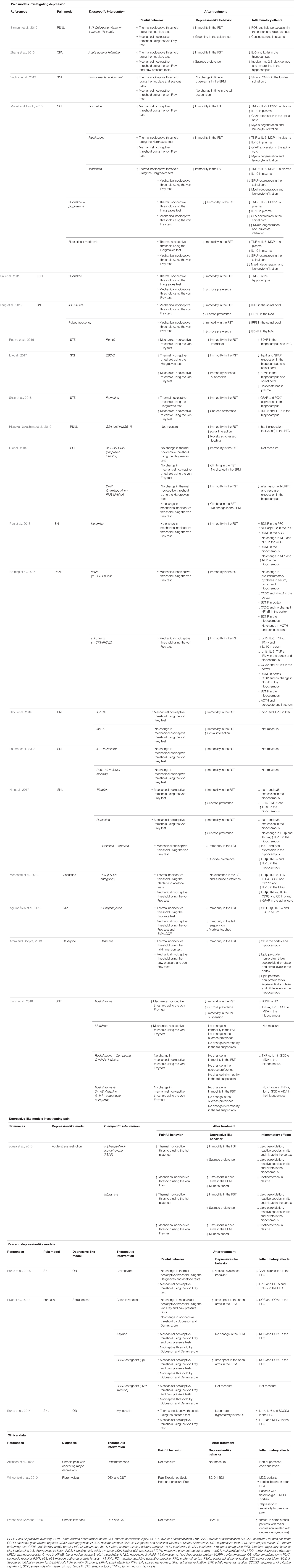
Table 2. Inflammatory response regarding different types of treatments.
Because of the complexity and multifactorial aspects of persistent pain and depression, an extensive number of medications are already used by individuals suffering from depression-pain syndrome but are often not effective. Hence, it is important to investigate new mechanisms of classical drugs to improve their use and to find and develop new drugs and targets to treat these syndromes.
Understanding the Anti-inflammatory Signature of Depressive-Pain Treatments
Beyond understanding the neuroinflammatory signature of the pathophysiology of pain and depression, we found a total of 23 articles (39%) that investigated the role of different types of treatments in controlling pain, depressive-like behavior and neuroinflammation. We will address the treatments used in the articles found in our research, the identified neuroinflammatory signatures in response to treatment and the possible neuronal networks involved in the amelioration of depressive-pain symptoms.
In preclinical trials, it is common to investigate molecular inhibitors or antagonists in an attempt to understand the specific role of different neuromodulators. We found that in 39% of the preclinical models, treatments involving different inhibitors or antagonists were investigated. Acute or subchronic treatment with these inhibitors may shed light on the possible mechanisms of pain and depression or may indicate a new target for treating these syndromes.
Neuroimaging and postmortem studies have shown an increase in mitochondrial translocator protein (tryptophan-rich sensory proteins -TSPO) in subjects with major depressive disorder (Enache et al., 2019). It has been shown that during local or systemic inflammation, TSPO is overexpressed in inflammatory cells and can be considered a marker of proinflammatory microglia (Selvaraj and Tu, 2016; Owen et al., 2017; Beckers et al., 2018). Li et al. (2017) showed that N-benzyl-N-ethyl-2-(7,8-dihydro-7-benzyl-8-oxo-2-phenyl-9H-purin-9-yl) acetamide (ZBD-2), a TSPO ligand, decreases microglia and astrocyte activation and increases BDNF expression in the hippocampus and spinal cord in neuropathic rats subjected to spinal cord injury, improving pain sensitivity and depressive-like behavior by decreasing corticosterone levels in the plasma. This finding corroborates the idea that mitochondrial dysfunction plays a pivotal role in inflammation and DAMP formation (Shimada et al., 2012).
Different types of inhibitors of the inflammatory cascade have been used to attenuate painful and depressive-like behavior. The prokineticin family receptor (PKR) is present in the DRG and spinal cord and plays a role in sensitizing nociceptors via transient receptor potential vanilloid receptor 1 (TRPV1), inducing the release of proinflammatory cytokines (Negri et al., 2006; Li et al., 2016). PC-1, a PKR antagonist, was used in a model of chemotherapy-induced neuropathic pain with vincristine and was shown to decrease the levels of proinflammatory cytokines and infiltrating leucocytes without changing the expression of GFAP in the spinal cord (Moschetti et al., 2019), demonstrating that the antinociceptive properties of PKR inhibition may be related to peripheral mechanisms. Reinforcing these results, 2-AP, another PKR inhibitor, was also shown to decrease inflammasome and caspase-1 expression in the hippocampus of rats subjected to chronic constriction injury (Li et al., 2019). The tryptophan-metabolizing enzyme indolamine 1,3 deoxygenase (IDO1) is a direct modulator of inflammatory cytokine release (O’Connor et al., 2009) and is pivotal for the metabolism of tryptophan into kynurenine (KIN), which is further converted to kynurenine 3-monooxygenase (KMO) (Schwarcz and Stone, 2017). In this sense, the effect of inhibition of IDO and KMO, which attenuates depressive-like behavior but weakly ameliorates painful behavior, was compared to that of an inhibitor of IL-1 receptor antagonist (IL-1RA) in two different articles (Zhou et al., 2015; Laumet et al., 2017).
As previously described, glial cells are very important for the maintenance of the inflammatory response within the nervous system. Interferon regulatory factor 8 (IRF8) deficiency prevents the activation of microglia (Liu et al., 2018), and the mechanisms by which IRF8 deficiency improves pain sensitivity and depressive-like behavior are very similar to those of pulsed radiofrequency, which decreases IRF8 in the spinal cord and BDNF in the NAc, demonstrating that non-pharmacological treatments also play an important role in microglial activation and the VTA-NAc reward pathway (Fang et al., 2019). High-mobility group box-1 (HMBG1) is secreted and activates immune cells via Toll-like receptor 4 (TLR4), inducing the production of cytokines and chemokines (Andersson and Tracey, 2011; Agalave and Svensson, 2015). Since HMBG1 is upregulated in the spinal cord and sciatic nerve in neuropathic pain models (Ren et al., 2012; Nakamura et al., 2013), the effect of glycyrrhizic acid (GZA), an inhibitor of HMGB-1, in a partial sciatic nerve ligation model was investigated. GZA attenuates depressive-like behaviors and decreases microglial activation in the PFC (Hisaoka-Nakashima et al., 2019). In relation to microglia and depressive-pain syndrome, minocycline, which also inhibits microglial activation, normalizes thermal and mechanical nociceptive thresholds and improves anxiety-depressive behavior (Burke et al., 2014; Xu et al., 2017; Dai et al., 2019). Additionally, similar to macrophages, microglia is also polarized toward the classical proinflammatory M1 phenotype or the alternative anti-inflammatory M2 phenotype (Jha et al., 2016). Although minocycline is able to increase the expression of IL-10 and mannose receptor (MRC2), both of which are markers of the M2 phenotype, it does not inhibit the increases in IL-1β, IL-6 or suppressor of cytokine signaling 3 (SOCS3) in the PFC in a spinal nerve ligation and olfactory bulbectomy model (Burke et al., 2014).
The compound m-trifluoromethyl-diphenyl diselenide (m-CF3-PhSe)2 permeates the BBB and modulates opioid receptors without inducing opioid tolerance (Brüning et al., 2015; Rosa et al., 2018). Acute and subchronic treatment with this compound improves mechanical nociceptive thresholds and depressive-like behavior, decreases COX-2 and increases BDNF in the cerebral cortex and hippocampus in a partial sciatic nerve ligation model; however, only subchronic administration decreases proinflammatory cytokines and increases IL-10 in the serum, cerebral cortex and hippocampus and decreases corticosterone in the serum (Brüning et al., 2015). Although aspirin, a classic NSAID, is not able to improve depressive-like behavior, like benzodiazepines and a cholecystokinin 2 (CCK2) antagonist, it decreases Inducible nitric oxide synthase (iNOS) and COX2 in the PFC in the formalin test and a social defeat model (Rivat et al., 2010). Fatty acids with anti-inflammatory properties, such as omega-3, which increases BDNF in the hippocampus and PFC (Redivo et al., 2016); palmatine, which decreases GFAP and purinergic receptor P2X7 expression in the hippocampus (Shen et al., 2018); and b-caryophyllene, which suppresses the levels of SP and proinflammatory cytokines in the serum of diabetic animals (Aguilar-Ávila et al., 2019), may be used to attenuate pain and depression. Additionally, like fluoxetine, triptolide decreases microglial activation in the hippocampus, but only triptolide or triptolide combined with fluoxetine decreases proinflammatory cytokines and increases IL-10 in the hippocampus after improving spinal nerve ligation-induced pain and depressive behavior (Hu et al., 2017).
Indeed, manipulation of monoamines may also involve anti-inflammatory mechanisms. In a lumbar disk herniation model, fluoxetine decreases TNF-α in the hippocampus (Cai et al., 2019), while in a chronic constriction injury model, it decreases proinflammatory cytokines in the plasma, increases IL-10, decreases myelin degeneration and leukocyte infiltration in peripheral nerves and decreases astrocyte activation in the spinal cord; additionally, it has better effects when combined with antidiabetic drugs (Murad and Ayuob, 2015). Despite having similar mechanisms, amitriptyline does not improve thermal nociceptive thresholds but decreases astrocyte activation (Burke et al., 2015). Interestingly, this decrease is accompanied by decreased IL-10 and C-C chemokine ligand 5 (CCL5/RANTES) but increased TNF-α in the PFC in a co-model of pain and depression induced by spinal nerve ligation and olfactory bulbectomy model (Burke et al., 2015). A possible explanation for this finding is that CCL5 is released upon TNF-α activation to regulate microglia-astrocyte crosstalk and glutamate reuptake in the PFC (Pittaluga, 2017). The antioxidant α-(phenylselanyl) acetophenone (PSAP) was compared to imipramine, another drug that modulates monoamines. PSAP also seems to interact with serotonin type 1A (5HT-1A) receptor but not with noradrenaline, dopamine or adenosine receptors (Gerzson et al., 2012). In addition to improving behavior, both drugs decrease lipid peroxidation, reactive oxygen species (ROS), nitrite and nitrate levels in the cerebral cortex and hippocampus and inhibit corticosterone levels in the plasma after acute stress restriction (Sousa et al., 2018). Another new compound with monoaminergic properties is 3-(4-chlorphenylselany)-1-methyl-1H-indole, which improves pain sensitivity and depressive-like behavior and, like PSAP and imipramine, also decreases ROS and lipid peroxidation in the cerebral cortex and hippocampus and corticosterone in the plasma (Birmann et al., 2019). In this sense, the regulation of monoamines is clearly pivotal to the control of neuroinflammation. Possibly, improvements in the synaptic environment by the normalization of neurotransmitter levels decreases DAMPs, such as ROS and reactive nitrogen species (NOS), and inhibits the activation of glial cells, decreasing the release of proinflammatory cytokines. Persistent activation of NMDA receptors by glutamate cytotoxicity also plays an important role in DAMP formation and, consequently, in the pathophysiology of pain and depression (Petrenko et al., 2003; Duman and Li, 2012). Acute ketamine decreases proinflammatory cytokines, IDO1 and kynurenine in the hippocampus after improving painful and depressive behavior in a model of complete Freund’s adjuvant-induced acute pain (Zhang et al., 2016). However, in a persistent pain model, such as a sciatic nerve injury model, ketamine decreases depressive-like behavior and increases BDNF in the PFC and ACC but does not change the mechanical nociceptive threshold (Pan et al., 2018). These findings corroborate the strong evidence that ketamine decreases postoperative pain scores in acute nociceptive pain (Bell and Kalso, 2018) but that it seems to be less effective for chronic pain treatment. However, the central mechanism by which ketamine exerts neuroprotection in the PFC and ACC suggests that it is useful for treating major depression disorder (Daly et al., 2018).
Role of Inflammation in Neuronal Networks
Microglia is the most abundant resident immune cell in the CNS, able to recognize DAMPs and pathogen-associated molecular patterns (PAMPs) and respond by producing many inflammatory mediators (Dheen et al., 2007). In addition to being important for the immune response, microglia also play a pivotal role in the pruning of synaptic spines induced by sensory inputs or neuronal injury (Wake et al., 2009; Tremblay et al., 2010). In this sense, microglia-deficient brain slices show an increase in excitatory synapses, while the presence of microglia suppresses the AMPA receptor within the neuronal membrane and synaptic adhesion molecules (Ji K. et al., 2013). In addition, one astrocyte may interact with over 100,000 synapses to form a tripartite synapse (Halassa et al., 2007; Araque, 2008). Astrocytes can coordinate and control synaptic transmission through neurotransmitter receptors, transporters, and cell adhesion molecules (Halassa et al., 2007). The cytotoxicity induced by excessive glutamate in the synaptic environment is prevented by the astrocyte reuptake process (Hertz and Zielke, 2004), suggesting the pivotal role of these cells in the control of synapses.
In pathological conditions after an injury or disease, microglial cells may show the classical activated pattern (M1) characterized by upregulation of nuclear factor-κB (NF-κB), signal transducer and activator of transcription (STAT), TNF-α, IL-1β and different proinflammatory mediators (Jha et al., 2016). M1 microglia have the capacity to induce changes in the phenotype of astrocytes to the classical activation phenotype (A1), which is consistent with the increase in inflammation and tissue damage (Liddelow et al., 2017) seen in depressive pain syndrome. A1 astrocytes decrease the formation of excitatory synapses in an in vitro coculture system and decrease the frequency and amplitude of excitatory postsynaptic currents in the remaining synapses (Liddelow et al., 2017). Indeed, pain and depression conditions induce the downregulation of glutamate type 1 transporter (GLT1) in response to inflammation (Medina et al., 2013; Guo et al., 2019), suggesting that the astrocyte-reuptake process involved in pruning synapses is impaired in depression-pain syndrome. As described above, the cooccurrence of chronic pain and depression is intimately related to increases in TNF-α, IL-1β, and IL-6 in target structures such as the PFC, ACC, amygdala and hippocampus, emphasizing the involvement of glial activation in limbic regions involved in pain and depression processing.
Even though IL-1β is related to resident glial cells, it has been shown that this cytokine plays a role in synaptic plasticity (Kelly et al., 2003; Mandolesi et al., 2013). Once recognized by neurons, IL-1 increases glutamate release through prostaglandin (PGE)-2 production (Sang et al., 2005; Mishra et al., 2012), activating NMDA receptors, increasing glutamatergic sensitivity and exacerbating excitotoxicity (Viviani et al., 2006; Fogal and Hewett, 2008). In the rat hippocampus, IL-1β decreases synaptic connections by increasing both pre- and postsynaptic glutamate release (Mishra et al., 2012). In this sense, inflammation inhibits the ability of astrocytes to reuptake glutamate, and IL1-β increases glutamate levels within the synaptic cleft. TNF-α also plays a role in increasing the exocytosis of AMPA receptors on hippocampal pyramidal cells (Ogoshi et al., 2005) while promoting the endocytosis of GABA A receptors (Stellwagen et al., 2005), triggering an imbalance between excitatory and inhibitory controls. The relationship between inflammation and the neuronal environment investigated by two articles is described in Table 1. González-Sepúlveda et al. (2016) showed that the increase in IL-1β in the PFC relates to an increase in the glutamate/glycine ratio, while Dellarole et al. (2014) demonstrated that TNFR-deficient mice are protected against increases in neurogenesis, neuroplasticity and myelin impairments induced by neuropathic pain. TNF-α also inhibits noradrenaline release in the hippocampus (Spengler et al., 2007), which is postulated as a common target in pain and depression (Fasick et al., 2015). Hence, inhibiting inflammation may be an important tool for restarting the optimal modulation and control of synapses within the CNS, improving depressive pain symptoms. However, ketamine, which acts on NMDA receptors, not only decreases inflammatory markers, as previously described but also inhibits the KYN/tryptophan ratio while increasing the 5-HT/tryptophan ratio (Zhang et al., 2016). Accordingly, fluoxetine increases 5-HT in the hippocampus while decreasing inflammation (Cai et al., 2019), suggesting that improving the synaptic network also affects inflammation status (Figure 4). The same rationale may apply to other treatments, such as benzodiazepines, reuptake inhibitors, and CCK2 and PKR inhibitors, which classically involve pre- and postsynaptic neurons, as described above. In addition to acting through a neuronal mechanism, these drugs are able to inhibit inflammation in depressive-pain models.
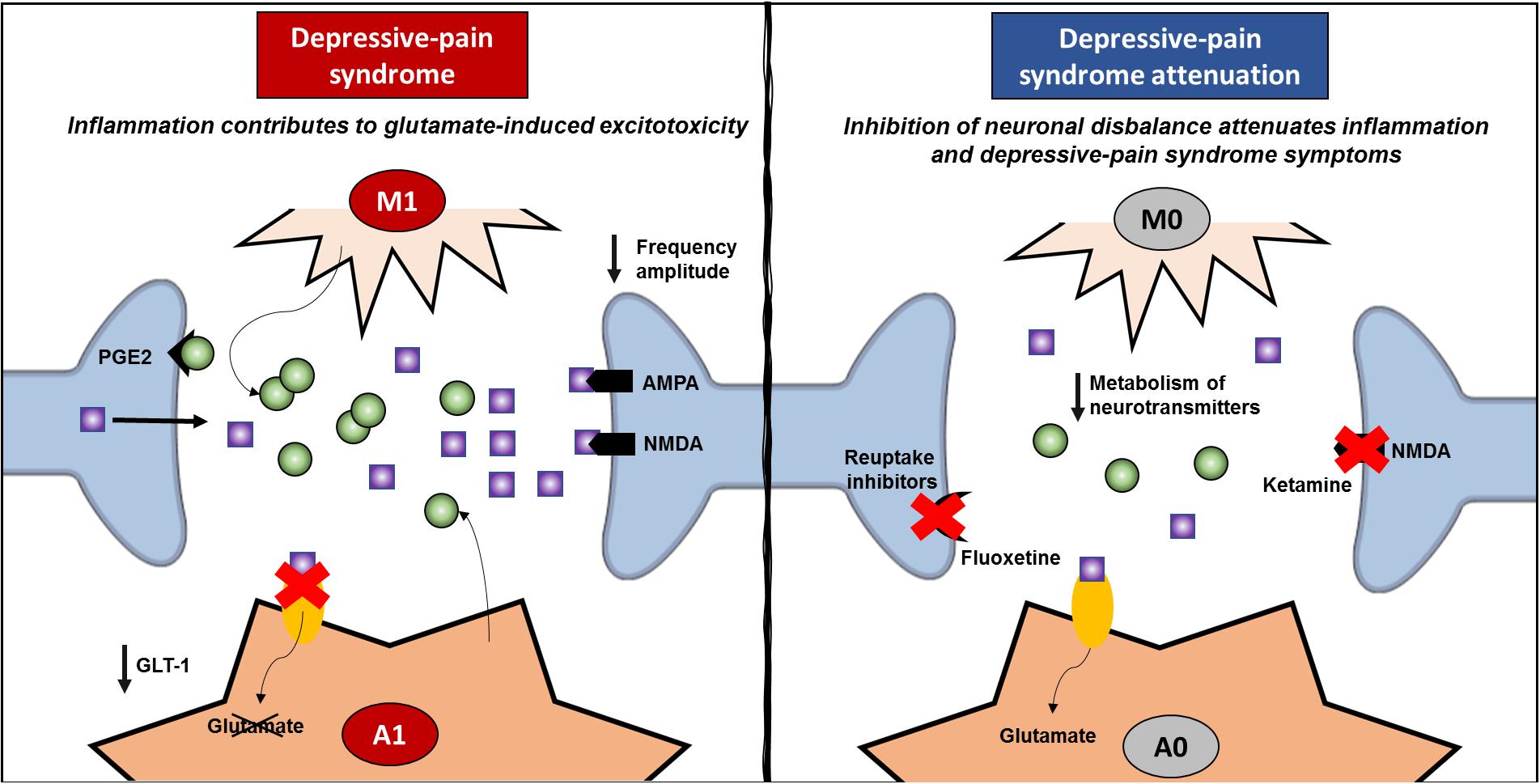
Figure 4. Review of inflammatory signature and neural network modulation. Depressive pain syndrome is related to activation of glial cells, such as microglia and astrocytes. Once classically activated (M1/A1), these cells release proinflammatory cytokines, especially TNF-α and IL-1β, and upon specific receptor activation, increase glutamate release in the synaptic cleft. Classically activated astrocytes induce a decrease in GLT-1, which enables the reuptake of glutamate and decreases glutamate-induced cytotoxicity. Hence, both AMPA and NMDA postsynaptic receptors are strongly activated, decreasing the frequency amplitude and contributing to neural network imbalance. On the other hand, anti-inflammatory treatments inhibit the classic activation of glial cells and the exacerbated release of proinflammatory mediators, decreasing neural network imbalance. In addition, the success of pharmacological treatments that act through mechanisms related to neuronal dysfunction, such as ketamine and fluoxetine, which improve neuronal network dysfunction and attenuating depressive pain syndrome, also corroborate the negative feedback of inflammation. GLT, glutamate transporter; PGE, prostaglandin.
Taken together, our findings suggest that neuronal deficits and the inflammatory response may trigger each other through a feedback mechanism, contributing to the complexity of the control of depression-pain syndrome. In this sense, it is important for treatments to address the role of neurotransmitters and receptors as well as to improve the entire synaptic cleft, including inhibiting classically activated resident cells and the inflammatory response. In other words, these data strongly support the idea that controlling neuroinflammation is closely connected with improving pain and depression states. Direct inhibition of the inflammatory cascade or indirect inhibition by decreasing cytotoxicity and DAMP formation in local synapses concomitant with controlling glial cell activation are important strategies for improving the quality of life of people suffering from depression-pain syndrome.
Conclusion and Future Directions
Pain and depression are often comorbid and decrease the quality of life of many individuals worldwide. Indeed, much effort has been expended to investigate different classes of medications to ameliorate these symptoms. In this sense, preclinical models are a resource for the development of drugs or for better elucidating their peripheral and central mechanisms. It is important to take into consideration that preclinical models are tools to provide a better understanding of pathogenesis and for testing the potential of novel therapeutic approaches. However, there are limitations intrinsic from the experimental models referring to translate research into practice. The choice of the appropriate preclinical model to address pharmacological treatments should be consider as the most reliable and closer to reproduce a persistent pain as observed in clinical practice. Even though the models should be able to reproduce the nociceptive behavior, it is pivotal that they also reproduce depressive-anxiety behavior, mimicking what is observed in patients. Additionally, the experimental design should be carefully addressed taking into consideration the presence of peripheral and central sensibilization before the initiation of the pharmacological treatment as it happens with individuals suffering from persistent pain. In addition to directly targeting inflammation mediators such as cytokines and molecular inhibitors of the inflammatory cascade to improve pain sensitivity and depression, drugs that improve and regulate the synaptic environment and its neurotransmitters may act as anti-inflammatory compounds, reducing local DAMPs and inhibiting the activation of immune and glial cells. In this review, we highlight the inflammatory signature of different preclinical models and clinical articles related to depressive-pain syndrome. Additionally, we discuss the role of therapeutic interventions targeting depression, pain and neuroinflammation. We hope that shedding light on the inflammatory profile will aid in finding new targets and in improving classic treatments to bring benefits to individuals who suffer from these disorders.
Author Contributions
AC and GA contributed to the conception of the study, acquisition of the data, and writing of the manuscript. MM and RP wrote and revised the manuscript. RM conceived the presented review, supervised the project, and wrote the manuscript. All the authors contributed to the article and approved the submitted version.
Funding
The authors are the recipients of grants from the Government of Brazil – São Paulo Research Foundation (FAPESP, ACPC 2018/18695-9) and Coordination for the Improvement of Higher Education Personnel (CAPES, GFA 88882.366209/2019-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank all the research assistants and staff of Hospital Sírio-Libanês.
References
Abrahams, L., Semjonous, N. M., Guest, P., Zielinska, A., Hughes, B., Lavery, G. G., et al. (2012). Biomarkers of hypothalamic-pituitary-adrenal axis activity in mice lacking 11β-HSD1 and H6PDH. J. Endocrinol. 214, 367–372. doi: 10.1530/JOE-12-0178
Agalave, N. M., and Svensson, C. I. (2015). Extracellular high-mobility group box 1 protein (HMGB1) as a mediator of persistent pain. Mol. Med. 20, 569–578. doi: 10.2119/molmed.2014.00176
Agüera-Ortiz, L., Failde, I., Mico, J. A., Cervilla, J., and López-Ibor, J. J. (2011). Pain as a symptom of depression: prevalence and clinical correlates in patients attending psychiatric clinics. J. Affect. Disord. 130, 106–112.
Aguilar-Ávila, D. S., Flores-Soto, M. E., Tapia-Vázquez, C., Pastor-Zarandona, O. A., López-Roa, R. I., and Viveros-Paredes, J. M. (2019). β-Caryophyllene, a Natural Sesquiterpene, Attenuates neuropathic pain and depressive-like behavior in experimental diabetic mice. J. Med. Food 22, 460–468. doi: 10.1089/jmf.2018.0157
Al-Amin, H., Sarkis, R., Atweh, S., Jabbur, S., and Saadé, N. (2011). Chronic dizocilpine or apomorphine and development of neuropathy in two animal models II: effects on brain cytokines and neurotrophins. Exp. Neurol. 228, 30–40. doi: 10.1016/j.expneurol.2010.11.005
An, X., Bandler, R., Ongür, D., and Price, J. L. (1998). Prefrontal cortical projections to longitudinal columns in the midbrain periaqueductal gray in macaque monkeys. J. Comp. Neurol. 401, 455–479.
Andersson, U., and Tracey, K. J. (2011). HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29, 139–162. doi: 10.1146/annurev-immunol-030409-101323
Apkarian, A. V., Bushnell, M. C., Treede, R. D., and Zubieta, J. K. (2005). Human brain mechanisms of pain perception and regulation in health and disease. Eur. J. Pain 9, 463–484.
Araque, A. (2008). Astrocytes process synaptic information. Neuron Glia Biol. 4, 3–10. doi: 10.1017/S1740925X09000064
Arora, V., and Chopra, K. (2013). Possible involvement of oxido-nitrosative stress induced neuro-inflammatory cascade and monoaminergic pathway: underpinning the correlation between nociceptive and depressive behaviour in a rodent model. J. Affect. Disord. 151, 1041–1052. doi: 10.1016/j.jad.2013.08.032
Arora, V., Kuhad, A., Tiwari, V., and Chopra, K. (2011). Curcumin ameliorates reserpine-induced pain-depression dyad: behavioural, biochemical, neurochemical and molecular evidences. Psychoneuroendocrinology 36, 1570–1581. doi: 10.1016/j.psyneuen.2011.04.012
Atkinson, J. H. Jr., Kremer, E. F., Risch, S. C., Dana, R., and Janowsky, D. S. (1986). Neuroendocrine responses in psychiatric and pain patients with major depression. Biol. Psychiatry 21, 612–620.
Bair, M. J., Robinson, R. L., Katon, W., and Kroenke, K. (2003). Depression and pain comorbidity: a literature review. Arch. Intern. Med. 163, 2433–2445.
Barber, J. B., and Gibson, S. J. (2009). Treatment of chronic non-malignant pain in the elderly: safety considerations. Drug Saf. 32, 457–474. doi: 10.2165/00002018-200932060-00003
Bardin, L. (2011). The complex role of serotonin and 5-HT receptors in chronic pain. Behav. Pharmacol. 22, 390–404. doi: 10.1097/FBP.0b013e328349aae4
Baron, R. (2000). Peripheral neuropathic pain: from mechanisms to symptoms. Clin. J. Pain 16, S12–S20.
Basbaum, A. I., Bautista, D. M., Scherrer, G., and Julius, D. (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. doi: 10.1016/j.cell.2009.09.028
Beckers, L., Ory, D., Geric, I., Declercq, L., Koole, M., Kassiou, M., et al. (2018). Increased expression of translocator protein (TSPO) marks pro-inflammatory microglia but does not predict neurodegeneration. Mol. Imaging Biol. 20, 94–102. doi: 10.1007/s11307-017-1099-1
Beckman, T. J. (2004). Discussing the depression-pain dyad with patients. Arch. Intern. Med. 164, 804–805.
Bell, R. F., and Kalso, E. A. (2018). Ketamine for pain management. Pain Rep. 3:e674. doi: 10.1097/PR9.0000000000000674
Benatti, C., Blom, J. M., Rigillo, G., Alboni, S., Zizzi, F., Torta, R., et al. (2016). Disease-induced neuroinflammation and depression. CNS Neurol. Disord. Drug Targets 15, 414–433.
Benson, C., Mifflin, K., Kerr, B., Jesudasan, S. J., Dursun, S., and Baker, G. (2015). Biogenic amines and the amino acids GABA and glutamate: relationships with pain and depression. Mod. Trends Pharmacopsychiatry 30, 67–79. doi: 10.1159/000435933
Bentley, J. N., Viswanathan, A., Rosenberg, W. S., and Patil, P. G. (2014). Treatment of medically refractory cancer pain with a combination of intrathecal neuromodulation and neurosurgical ablation: case series and literature review. Pain Med. 15, 1488–1495. doi: 10.1111/pme.12481
Birmann, P. T., Sousa, F. S. S., Domingues, M., Brüning, C. A., Vieira, B. M., Lenardão, E. J., et al. (2019). 3-(4-Chlorophenylselanyl)-1-methyl-1H-indole promotes recovery of neuropathic pain and depressive-like behavior induced by partial constriction of the sciatic nerve in mice. J. Trace Elem. Med. Biol. 54, 126–133. doi: 10.1016/j.jtemb.2019.04.014
Björkman, R. (1995). Central antinociceptive effects of non-steroidal anti-inflammatory drugs and paracetamol. Experimental studies in the rat. Acta Anaesthesiol. Scand. Suppl. 103, 1–44.
Blier, P., and Abbott, F. V. (2001). Putative mechanisms of action of antidepressant drugs in affective and anxiety disorders and pain. J. Psychiatry Neurosci. 26, 37–43.
Bragin, E. O., Yeliseeva, Z. V., Vasilenko, G. F., Meizerov, E. E., Chuvin, B. T., and Durinyan, R. A. (1984). Cortical projections to the periaqueductal grey in the cat: a retrograde horseradish peroxidase study. Neurosci. Lett. 51, 271–275.
Brant, J. M., Keller, L., McLeod, K., Yeh, C., and Eaton, L. H. (2017). Chronic and refractory pain: a systematic review of pharmacologic management in oncology. Clin. J. Oncol. Nurs. 21, 31–53. doi: 10.1188/17.CJON.S3.31-53
Breivik, H., Collett, B., Ventafridda, V., Cohen, R., and Gallacher, D. (2006). Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur. J. Pain 10, 287–333.
Brüning, C. A., Martini, F., Soares, S. M., Sampaio, T. B., Gai, B. M., Duarte, M. M., et al. (2015). m-Trifluoromethyl-diphenyl diselenide, a multi-target selenium compound, prevented mechanical allodynia and depressive-like behavior in a mouse comorbid pain and depression model. Prog. Neuropsychopharmacol. Biol. Psychiatry 63, 35–46. doi: 10.1016/j.pnpbp.2015.05.011
Burke, N. N., Finn, D. P., and Roche, M. (2015). Neuroinflammatory mechanisms linking pain and depression. Mod. Trends Pharmacopsychiatry 30, 36–50. doi: 10.1159/000435931
Burke, N. N., Geoghegan, E., Kerr, D. M., Moriarty, O., Finn, D. P., and Roche, M. (2013). Altered neuropathic pain behaviour in a rat model of depression is associated with changes in inflammatory gene expression in the amygdala. Genes Brain Behav. 12, 705–713. doi: 10.1111/gbb.12080
Burke, N. N., Kerr, D. M., Moriarty, O., Finn, D. P., and Roche, M. (2014). Minocycline modulates neuropathic pain behaviour and cortical M1-M2 microglial gene expression in a rat model of depression. Brain Behav. Immun. 42, 147–156. doi: 10.1016/j.bbi.2014.06.015
Busse, J. W., Wang, L., Kamaleldin, M., Craigie, S., Riva, J. J., Montoya, L., et al. (2018). Opioids for chronic noncancer pain: a systematic review and meta-analysis. JAMA 320, 2448–2460. doi: 10.1001/jama.2018.18472
Byrd, D., and Mackey, S. (2008). Pulsed radiofrequency for chronic pain. Curr. Pain Headache Rep. 12, 37–41.
Cai, L., He, Q., Lu, Y., Hu, Y., Chen, W., Wei, L., et al. (2019). Comorbidity of pain and depression in a lumbar disc herniation model: biochemical alterations and the effects of fluoxetine. Front. Neurol. 24:1022. doi: 10.3389/fneur.2019.01022
Castro, S. L., and Zigmond, M. J. (2001). Stress-induced increase in extracellular dopamine in striatum: role of glutamatergic action via N-methyl-D-aspartate receptors in substantia nigra. Brain Res. 901, 47–54.
Catani, M., Dell’acqua, F., and Thiebaut de Schotten, M. (2013). A revised limbic system model for memory, emotion and behaviour. Neurosci. Biobehav. Rev. 37, 1724–1737. doi: 10.1016/j.neubiorev.2013.07.001
Chen, L., Deng, H., Cui, H., Fang, J., Zuo, Z., Deng, J., et al. (2017). Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9, 7204–7218. doi: 10.18632/oncotarget.23208
Churi, S. B., Abdel-Aleem, O. S., Tumber, K. K., Scuderi-Porter, H., and Taylor, B. K. (2008). Intrathecal rosiglitazone acts at peroxisome proliferator-activated receptor-gamma to rapidly inhibit neuropathic pain in rats. J. Pain 9, 639–649. doi: 10.1016/j.jpain.2008.02.002
Cochrane Database Reviews (2014). Neuropathic Pain. Available online at: https://www.cochranelibrary.com/cdsr/table-of-contents?volume=2014&issue=5$¿$ (assessed May 20, 2020).
Cohen, S. P., and Mao, J. (2014). Neuropathic pain: mechanisms and their clinical implications. BMJ 348:f7656. doi: 10.1136/bmj.f7656
Collins, P. Y., Patel, V., Joestl, S. S., March, D., Insel, T. R., Daar, A. S., et al. (2011). Grand challenges in global mental health. Nature 475, 27–30. doi: 10.1038/475027a
Connolly, C. G., Ho, T. C., Blom, E. H., LeWinn, K. Z., Sacchet, M. D., Tymofiyeva, O., et al. (2017). Resting-state functional connectivity of the amygdala and longitudinal changes in depression severity in adolescent depression. J. Affect. Disord. 207, 86–94. doi: 10.1016/j.jad.2016.09.026
Cooper, T. E., Heathcote, L. C., Clinch, J., Gold, J. I., Howard, R., Lord, S. M., et al. (2017). Antidepressants for chronic non-cancer pain in children and adolescents. Cochrane Database. Syst. Rev. 8:CD012535. doi: 10.1002/14651858.CD012535.pub2
Cortes, M. L., Castro, M. M. C., de Jesus, R. P., Neto, J. A. B., and Kraychete, D. C. (2013). Therapy with omega-3 fatty acids for patients with chronic pain and anxious and depressive symptoms. Rev. Dor. 14, 48–51.
Cox, D. R., Ashby, S., Mace, J. C., DelGaudio, J. M., Smith, T. L., Orlandi, R. R., et al. (2017). The pain-depression dyad and the association with sleep dysfunction in chronic rhinosinusitis. Int. Forum Allergy Rhinol. 7, 56–63. doi: 10.1002/alr.21843
Crofford, L. J. (2015). Chronic pain: Where the body meets the brain. Trans. Am. Clin. Climatol. Assoc. 126, 167–183.
Currie, S. R., and Wang, J. (2005). More data on major depression as an antecedent risk factor for first onset of chronic back pain. Psychol. Med. 35, 1275–1282.
Dai, J., Ding, Z., Zhang, J., Xu, W., Guo, Q., Zou, W., et al. (2019). Minocycline relieves depressive-like behaviors in rats with bone cancer pain by inhibiting microglia activation in hippocampus. Anesth. Analg. 129, 1733–1741. doi: 10.1213/ANE.0000000000004063
Daly, E. J., Singh, J. B., Fedgchin, M., Cooper, K., Lim, P., Shelton, R. C., et al. (2018). Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry 75, 139–148. doi: 10.1001/jamapsychiatry.2017.3739
Daneman, R., and Prat, A. (2015). The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 7:a020412. doi: 10.1101/cshperspect.a020412
del Rey, A., Yau, H. J., Randolf, A., Centeno, M. V., Wildmann, J., Martina, M., et al. (2011). Chronic neuropathic pain-like behavior correlates with IL-1β expression and disrupts cytokine interactions in the hippocampus. Pain 152, 2827–2835. doi: 10.1016/j.pain.2011.09.013
Dellarole, A., Morton, P., Brambilla, R., Walters, W., Summers, S., Bernardes, D., et al. (2014). Neuropathic pain-induced depressive-like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. Brain Behav. Immun. 41, 65–81. doi: 10.1016/j.bbi.2014.04.003
Denk, F., McMahon, S. B., and Tracey, I. (2014). Pain vulnerability: a neurobiological perspective. Nat. Neurosci. 17, 192–200. doi: 10.1038/nn.3628
Dheen, S. T., Kaur, C., and Ling, E. A. (2007). Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 14, 1189–1197.
Di Virgilio, F., Ceruti, S., Bramanti, P., and Abbracchio, M. P. (2009). Purinergic signalling in inflammation of the central nervous system. Trends Neurosci. 32, 79–87. doi: 10.1016/j.tins.2008.11.003
Domercq, M., Vázquez-Villoldo, N., and Matute, C. (2013). Neurotransmitter signaling in the pathophysiology of microglia. Front. Cell. Neurosci. 19:49. doi: 10.3389/fncel.2013.00049
Dowell, D., Haegerich, T. M., and Chou, R. (2016). CDC Guideline for prescribing opioids for chronic pain–united states, 2016. JAMA 315, 1624–1645. doi: 10.1001/jama.2016.1464
Dowlati, Y., Herrmann, N., Swardfager, W., Liu, H., Sham, L., Reim, E. K., et al. (2010). A meta-analysis of cytokines in major depression. Biol. Psychiatry 67, 446–457. doi: 10.1016/j.biopsych.2009.09.033
Duman, R. S., and Li, N. (2012). A neurotrophic hypothesis of depression: role of synaptogenesis in the actions of NMDA receptor antagonists. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2475–2484. doi: 10.1098/rstb.2011.0357
Duric, V., and McCarson, K. E. (2005). Hippocampal neurokinin-1 receptor and brain-derived neurotrophic factor gene expression is decreased in rat models of pain and stress. Neuroscience 133, 999–1006.
Eccleston, C., Cooper, T. E., Fisher, E., Anderson, B., and Wilkinson, N. M. (2017). Non-steroidal anti-inflammatory drugs (NSAIDs) for chronic non-cancer pain in children and adolescents. Cochrane Database. Syst. Rev. 8:CD012537.
Ellenbogen, J. M., Hurford, M. O., Liebeskind, D. S., Neimark, G. B., and Weiss, D. (2005). Ventromedial frontal lobe trauma. Neurology 64:757.
Ellis, A., and Bennett, D. L. (2013). Neuroinflammation and the generation of neuropathic pain. Br. J. Anaesth. 111, 26–37. doi: 10.1093/bja/aet128
Enache, D., Pariante, C. M., and Mondelli, V. (2019). Markers of central inflammation in major depressive disorder: a systematic review and meta-analysis of studies examining cerebrospinal fluid, positron emission tomography and post-mortem brain tissue. Brain Behav. Immun. 81, 24–40. doi: 10.1016/j.bbi.2019.06.015
Erickson, K. I., Voss, M. W., Prakash, R. S., Basak, C., Szabo, A., Chaddock, L., et al. (2011). Exercise training increases size of hippocampus and improves memory. Proc. Natl. Acad. Sci. U.S.A. 108, 3017–3022. doi: 10.1073/pnas.1015950108
Euteneuer, F., Schwarz, M. J., Hennings, A., Riemer, S., Stapf, T., Selberdinger, V., et al. (2011). Depression, cytokines and experimental pain: evidence for sex-related association patterns. J. Affect. Disord. 131, 143–149. doi: 10.1016/j.jad.2010.11.017
Fang, X., Xu, X., Lin, X., and Liu, R. (2019). Downregulated spinal IRF8 and BDNF in NAC are involved in neuropathic pain-induced depression relief via pulsed radiofrequency on dorsal root ganglion in rat SNI model. Brain Res. Bull. 146, 192–200. doi: 10.1016/j.brainresbull.2019.01.008
Fasick, V., Spengler, R. N., Samankan, S., Nader, N. D., and Ignatowski, T. A. (2015). The hippocampus and TNF: common links between chronic pain and depression. Neurosci. Biobehav. Rev. 53, 139–159. doi: 10.1016/j.neubiorev.2015.03.014
Fields, H. L., and Basbaum, A. I. (1999). “Central nervous system mechanisms of pain modulation,” in Textbook of Pain, eds P. D. Wall R. Melzack (Edinburgh: Churchill Livingstone), 309–329.
Fischer, R., Sendetski, M., Del Rivero, T., Martinez, G. F., Bracchi-Ricard, V., Swanson, K. A., et al. (2019). TNFR2 promotes Treg-mediated recovery from neuropathic pain across sexes. Proc. Natl. Acad. Sci. U.S.A. 116, 17045–17050. doi: 10.1073/pnas.1902091116
Fogal, B., and Hewett, S. J. (2008). Interleukin-1β: a bridge between inflammation and excitotoxicity? J. Neurochem. 106, 1–23.
France, R. D., and Krishnan, K. R. (1985). The dexamethasone suppression test as a biologic marker of depression in chronic pain. Pain 21, 49–55. doi: 10.1016/0304-3959(85)90076-4
Fuller, R. W., Wong, D. T., and Robertson, D. W. (1991). Fluoxetine, a selective inhibitor of serotonin uptake. Med. Res. Rev. 11, 17–34.
Gallagher, R. M., and Verma, S. (1999). Managing pain and comorbid depression: a public health challenge. Semin. Clin. Neuropsychiatry 4, 203–220.
Gerzson, M. F., Victoria, F. N., Radatz, C. S., de Gomes, M. G., Boeira, S. P., Jacob, R. G., et al. (2012). In vitro antioxidant activity and in vivo antidepressant-like effect of α-(phenylselanyl) acetophenone in mice. Pharmacol. Biochem. Behav. 102, 21–29. doi: 10.1016/j.pbb.2012.03.016
Goldenberg, D. L., Clauw, D. J., Palmer, R. H., Mease, P., Chen, W., and Gendreau, R. M. (2010). Durability of therapeutic response to milnacipran treatment for fibromyalgia. Results of a randomized, double-blind, monotherapy 6-month extension study. Pain Med. 11, 180–194. doi: 10.1111/j.1526-4637.2009.00755.x
Goldstein, B. I., Kemp, D. E., Soczynska, J. K., and McIntyre, R. S. (2009). Inflammation and the phenomenology, pathophysiology, comorbidity, and treatment of bipolar disorder: a systematic review of the literature. J. Clin. Psychiatry 70, 1078–1090. doi: 10.4088/JCP.08r04505
Gonçalves, L., Silva, R., Pinto-Ribeiro, F., Pêgo, J. M., Bessa, J. M., Pertovaara, A., et al. (2008). Neuropathic pain is associated with depressive behaviour and induces neuroplasticity in the amygdala of the rat. Exp. Neurol. 213, 48–56. doi: 10.1016/j.expneurol.2008.04.043
González-Sepúlveda, M., Pozo, O. J., Marcos, J., and Valverde, O. (2016). Chronic pain causes a persistent anxiety state leading to increased ethanol intake in CD1 mice. J. Psychopharmacol. 30, 188–203. doi: 10.1177/0269881115622238
Griggs, R. B., Donahue, R. R., Morgenweck, J., Grace, P. M., Sutton, A., Watkins, L. R., et al. (2015). Pioglitazone rapidly reduces neuropathic pain through astrocyte and nongenomic PPARγ mechanisms. Pain 156, 469–482. doi: 10.1097/01.j.pain.0000460333.79127.be
Guo, W., Imai, S., Zou, S., Yang, J., Watanabe, M., Wang, J., et al. (2019). Altered glial glutamate transporter expression in descending circuitry and the emergence of pain chronicity. Mol. Pain 15:1744806918825044. doi: 10.1177/1744806918825044
Haase, J., and Brown, E. (2015). Integrating the monoamine, neurotrophin and cytokine hypotheses of depression–a central role for the serotonin transporter? Pharmacol Ther. 147, 1–11. doi: 10.1016/j.pharmthera.2014.10.002
Halassa, M. M., Fellin, T., and Haydon, P. G. (2007). The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol. Med. 13, 54–63.
Han, B., Compton, W. M., Jones, C. M., and Cai, R. (2015). Nonmedical prescription opioid use and use disorders among adults aged 18 through 64 years in the United States, 2003-2013. JAMA 314, 1468–1478. doi: 10.1001/jama.2015.11859
Hasegawa-Moriyama, M., Ohnou, T., Godai, K., Kurimoto, T., Nakama, M., and Kanmura, Y. (2012). Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates postincisional pain by regulating macrophage polarization. Biochem. Biophys. Res. Commun. 426, 76–82. doi: 10.1016/j.bbrc.2012.08.039
Hertz, L., and Zielke, H. R. (2004). Astrocytic control of glutamatergic activity: astrocytes as stars of the show. Trends Neurosci. 27, 735–743.
Himmerich, H., Patsalos, O., Lichtblau, N., Ibrahim, M. A. A., and Dalton, B. (2019). Cytokine research in depression: principles, challenges, and open questions. Front. Psychiatry 10:30. doi: 10.3389/fpsyt.2019.00030
Hiroki, T., Suto, T., Saito, S., and Obata, H. (2017). Repeated administration of amitriptyline in neuropathic pain: modulation of the noradrenergic descending inhibitory system. Anesth. Analg. 125, 1281–1288. doi: 10.1213/ANE.0000000000002352
Hisaoka-Nakashima, K., Tomimura, Y., Yoshii, T., Ohata, K., Takada, N., Zhang, F. F., et al. (2019). High-mobility group box 1-mediated microglial activation induces anxiodepressive-like behaviors in mice with neuropathic pain. Prog. Neuropsychopharmacol. Biol. Psychiatry 92, 347–362. doi: 10.1016/j.pnpbp.2019.02.005
Hu, X., Dong, Y., Jin, X., Zhang, C., Zhang, T., Zhao, J., et al. (2017). The novel and potent anti-depressive action of triptolide and its influences on hippocampal neuroinflammation in a rat model of depression comorbidity of chronic pain. Brain Behav. Immun. 64, 180–194. doi: 10.1016/j.bbi.2017.03.005
Hu, Y., and Davies, G. E. (2010). Berberine inhibits adipogenesis in high-fat diet-induced obesity mice. Fitoterapia 81, 358–366. doi: 10.1016/j.fitote.2009.10.010
Hunskaar, S. (1987). Similar effects of acetylsalicylic acid and morphine on immediate responses to acute noxious stimulation. Pharmacol. Toxicol. 60, 167–170.
Jacobson, L., and Sapolsky, R. (1991). The role of the hippocampus in feedback regulation of the hypothalamic-pituitary-adrenocortical axis. Endocr. Rev. 12, 118–134.
Jha, M. K., Lee, W. H., and Suk, K. (2016). Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem. Pharmacol. 103, 1–16. doi: 10.1016/j.bcp.2015.11.003
Ji, K., Akgul, G., Wollmuth, L. P., and Tsirka, S. E. (2013). Microglia actively regulate the number of functional synapses. PLoS One 8:e56293. doi: 10.1371/journal.pone.0056293
Ji, R. R., Berta, T., and Nedergaard, M. (2013). Glia and pain: is chronic pain a gliopathy? Pain. 154, S10–S28. doi: 10.1016/j.pain.2013.06.022
Ji, R. R., Chamessian, A., and Zhang, Y. Q. (2016). Pain regulation by non-neuronal cells and inflammation. Science 354, 572–577.
Ji, R. R., Nackley, A., Huh, Y., Terrando, N., and Maixner, W. (2018). Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 129, 343–366. doi: 10.1097/ALN.0000000000002130
Ji, R. R., Xu, Z. Z., and Gao, Y. J. (2014). Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 13, 533–548. doi: 10.1038/nrd4334
Johannes, C. B., Le, T. K., Zhou, X., Johnston, J. A., and Dworkin, R. H. (2010). The prevalence of chronic pain in United States adults: results of an Internet-based survey. J. Pain 11, 1230–1239. doi: 10.1016/j.jpain.2010.07.002
Karssen, A. M., Meijer, O. C., van der Sandt, I. C., Lucassen, P. J., de Lange, E. C., de Boer, A. G., et al. (2001). Multidrug resistance P-glycoprotein hampers the access of cortisol but not of corticosterone to mouse and human brain. Endocrinology 142, 2686–2694.
Kelly, A., Vereker, E., Nolan, Y., Brady, M., Barry, C., Loscher, C. E., et al. (2003). Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1β on long term potentiation in rat dentate gyrus. J. Biol. Chem. 278, 19453–19462.
Kiałka, M., Doroszewska, K., Janeczko, M., and Milewicz, T. (2017). [Metformin-new potential medicine in pain treatment?]. Przegl Lek. 74, 81–83.
Kim, H., Chen, L., Lim, G., Sung, B., Wang, S., McCabe, M. F., et al. (2012). Brain indoleamine 2, 3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Invest. 122, 2940–2954. doi: 10.1172/JCI61884
Kim, S., and Chung, J. H. (2008). Berberine prevents UV-induced MMP-1 and reduction of type I procollagen expression in human dermal fibroblasts. Phytomedicine 15, 749–753. doi: 10.1016/j.phymed.2007.11.004
Kodama, D., Ono, H., and Tanabe, M. (2007). Altered hippocampal long-term potentiation after peripheral nerve injury in mice. Eur. J. Pharmacol. 574, 127–132.
Koffel, E., Kroenke, K., Bair, M. J., Leverty, D., Polusny, M. A., and Krebs, E. E. (2016). The bidirectional relationship between sleep complaints and pain: analysis of data rom a randomized trial. Health Psychol. 35, 41–49. doi: 10.1037/hea0000245
Kramer, M. S., Cutler, N., Feighner, J., Shrivastava, R., Carman, J., Sramek, J. J., et al. (1998). Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science 281, 1640–1645.
Krames, E. S. (2014). The role of the dorsal root ganglion in the development of neuropathic pain. Pain Med. 15, 1669–1685. doi: 10.1111/pme.12413
KuKanich, B., Bidgood, T., and Knesl, O. (2012). Clinical pharmacology of nonsteroidal anti-inflammatory drugs in dogs. Vet. Anaesth. Analg. 39, 69–90. doi: 10.1111/j.1467-2995.2011.00675.x
Landfield, P. W., Baskin, R. K., and Pitler, T. A. (1981). Brain aging correlates: retardation by hormonal-pharmacological treatments. Science 214, 581–584.
Laulin, J. P., Maurette, P., Corcuff, J.-B., Rivat, C., Chauvin, M., and Simonnet, G. (2002). The role of ketamine in preventing fentanyl-induced hyperalgesia and subsequent acute morphine tolerance. Anesth. Analg. 94, 1263–1269. doi: 10.1097/00000539-200205000-00040
Laumet, G., Edralin, J. D., Chiang, A. C.-A., Dantzer, R., Heijnen, C. J., and Kavelaars, A. (2018). Resolution of inflammation-induced depression requires T lymphocytes and endogenous brain interleukin-10 signaling. Neuropsychopharmacology 43, 2597–2605. doi: 10.1038/s41386-018-0154-1
Laumet, G., Zhou, W., Dantzer, R., Edralin, J. D., Huo, X., Budac, D. P., et al. (2017). Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain. Brain Behav. Immun. 66, 94–102. doi: 10.1016/j.bbi.2017.07.008
Lee, P., Zhang, M., Hong, J. P., Chua, H. C., Chen, K. P., Tang, S. W., et al. (2009). Frequency of painful physical symptoms with major depressive disorder in Asia: relationship with disease severity and quality of life. J. Clin. Psychiatry 70, 83–91.
Li, Q., Liu, S., Zhu, X., Mi, W., Maoying, Q., Wang, J., et al. (2019). Hippocampal PKR/NLRP1 inflammasome pathway is required for the depression-like behaviors in rats with neuropathic pain. Neuroscience 412, 16–28. doi: 10.1016/j.neuroscience.2019.05.025
Li, X. M., Meng, J., Li, L. T., Guo, T., Yang, L. K., Shi, Q. X., et al. (2017). Effect of ZBD-2 on chronic pain, depressive-like behaviors, and recovery of motor function following spinal cord injury in mice. Behav. Brain Res. 322, 92–99. doi: 10.1016/j.bbr.2017.01.025
Li, X. Y., Wan, Y., Tang, S. J., Guan, Y., Wei, F., and Ma, D. (2016). Maladaptive plasticity and neuropathic pain. Neural Plast. 2016:4842159. doi: 10.1155/2016/4842159
Li, Y., Yan, J., Wang, D., Sun, M., Zhu, Y., Zhu, X., et al. (2014). Magnetic resonance study of the structure and function of the hippocampus and amygdala in patients with depression. Chin. Med. J. 127, 3610–3615.
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lilius, T. O., Jokinen, V., Neuvonen, M. S., Niemi, M., Kalso, E. A., and Rauhala, P. V. (2015). Ketamine coadministration attenuates morphine tolerance and leads to increased brain concentrations of both drugs in the rat. Br. J. Pharmacol. 172, 2799–2813. doi: 10.1111/bph.12974
Lindsay, P. G., and Wyckoff, M. (1981). The depression-pain syndrome and its response to antidepressants. Psychosomatics 22, 571–577. doi: 10.1016/S0033-3182(81)73478-9
Liu, R., Xu, X., Xu, Y., Fang, X., and Lin, X. (2018). Pulsed radiofrequency on dorsal root ganglion relieved neuropathic pain associated with downregulation of the spinal interferon regulatory factor 8, microglia, p38MAPK expression in a CCI rat model. Pain Physician 21, E307–E322.
López-Granero, C., Antunes Dos Santos, A., Ferrer, B., Culbreth, M., Chakraborty, S., Barrasa, A., et al. (2017). BXD recombinant inbred strains participate in social preference, anxiety and depression behaviors along sex-differences in cytokines and tactile allodynia. Psychoneuroendocrinology 80, 92–98. doi: 10.1016/j.psyneuen.2017.03.006
Manchikanti, L., Kaye, A. M., Knezevic, N. N., McAnally, H., Slavin, K., Trescot, A. M., et al. (2017). Responsible, safe, and effective prescription of opioids for chronic non-cancer pain: American Society of Interventional Pain Physicians (ASIPP) Guidelines. Pain Physician 20, S3–S92.
Mandolesi, G., Musella, A., Gentile, A., Grasselli, G., Haji, N., Sepman, H., et al. (2013). Interleukin-1β alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosci. 33, 12105–12121.
Marsden, W. N. (2013). Synaptic plasticity in depression: molecular, cellular and functional correlates. Prog. Neuropsychopharmacol. Biol. Psychiatry 43, 168–184. doi: 10.1016/j.pnpbp.2012.12.012
Meaney, M. J., Viau, V., Aitken, D. H., and Bhatnagar, S. (1988). Stress-induced occupancy and translocation of hippocampal glucocorticoid receptors. Brain Res. 445, 198–203.
Medina, A., Burke, S., Thompson, R. C., Bunney, W. Jr., Myers, R. M., Schatzberg, A., et al. (2013). Glutamate transporters: a key piece in the glutamate puzzle of major depressive disorder. J. Psychiatr. Res. 47, 1150–1156. doi: 10.1016/j.jpsychires.2013.04.007
Merskey, H., and Bogduk, N. (eds) (1994). IASP Task Force on Taxonomy, Part III: Pain Terms, A Current List with Definitions and Notes on Usage. Seattle, WA: IASP Press, 209–214.
Miller, A. H., Maletic, V., and Raison, C. L. (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65, 732–741. doi: 10.1016/j.biopsych.2008.11.029
Miller, E. K., and Cohen, J. D. (2001). An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 24, 167–202.
Mion, G., and Villevieille, T. (2013). Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci. Ther. 19, 370–380. doi: 10.1111/cns.12099
Mishra, A., Kim, H. J., Shin, A. H., and Thayer, S. A. (2012). Synapse loss induced by interleukin-1β requires pre-and post-synaptic mechanisms. J. Neuroimmune Pharmacol. 7, 571–578.
Mizoguchi, H., Fukumoto, K., Sakamoto, G., Jin, S., Toyama, A., Wang, T., et al. (2019). Maternal separation as a risk factor for aggravation of neuropathic pain in later life in mice. Behav. Brain Res. 359, 942–949. doi: 10.1016/j.bbr.2018.06.015
Moschetti, G., Amodeo, G., Paladini, M. S., Molteni, R., Balboni, G., Panerai, A., et al. (2019). Prokineticin 2 promotes and sustains neuroinflammation in vincristine treated mice: focus on pain and emotional like behavior. Brain Behav. Immun. 82, 422–431. doi: 10.1016/j.bbi.2019.09.012
Murad, H., and Ayuob, N. (2015). Co-administration of pioglitazone improves fluoxetine’s antinociceptive, neuroprotective, and antidepressant effects in chronic constriction injury in rats. Pain Physician 18, 609–620.
Mutso, A. A., Radzicki, D., Baliki, M. N., Huang, L., Banisadr, G., Centeno, M. V., et al. (2012). Abnormalities in hippocampal functioning with persistent pain. J Neurosci. 32, 5747–5756. doi: 10.1523/JNEUROSCI.0587-12.2012
Nakamura, Y., Morioka, N., Abe, H., Zhang, F. F., Hisaoka-Nakashima, K., Liu, K., et al. (2013). Neuropathic pain in rats with a partial sciatic nerve ligation is alleviated by intravenous injection of monoclonal antibody to high mobility group box-1. PLoS One 8:e73640. doi: 10.1371/journal.pone.0073640
Nakano, I. (1998). The limbic system: an outline and brief history of the concept. Neuropathology 18, 211–214.
Negri, L., Lattanzi, R., Giannini, E., Colucci, M., Margheriti, F., Melchiorri, P., et al. (2006). Impaired nociception and inflammatory pain sensation in mice lacking the prokineticin receptor PKR1: focus on interaction between PKR1 and the capsaicin receptor TRPV1 in pain behavior. J. Neurosci. 26, 6716–6727.
Nollet, M., Le Guisquet, A. M., and Belzung, C. (2013). Models of depression: unpredictable chronic mild stress in mice. Curr. Protoc. Pharmacol. Chapter 5, 5.65.1–5.65.17. doi: 10.1002/0471141755.ph0565s61
Norrbrink, B. C., and Lundeberg, T. (2004). Non-pharmacological pain-relieving therapies in individuals with spinal cord injury: a patient perspective. Complement. Ther. Med. 12, 189–197.
O’Connor, J. C., Lawson, M. A., André, C., Moreau, M., Lestage, J., Castanon, N., et al. (2009). Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 14, 511–522. doi: 10.1038/sj.mp.4002148
Ogoshi, F., Yin, H. Z., Kuppumbatti, Y., Song, B., Amindari, S., and Weiss, J. H. (2005). Tumor necrosis-factor-alpha (TNF-alpha) induces rapid insertion of Ca2+-permeable alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA)/kainate (Ca-A/K) channels in a subset of hippocampal pyramidal neurons. Exp. Neurol. 193, 384–393.
Ohayon, M. M. (2005). Relationship between chronic painful physical condition and insomnia. J. Psychiatr. Res. 39, 151–159.
Ong, W. Y., Stohler, C. S., and Herr, D. R. (2019). Role of the prefrontal cortex in pain processing. Mol. Neurobiol. 56, 1137–1166. doi: 10.1007/s12035-018-1130-9
Osafo, N., Agyare, C., Obiri, D. D., and Antwi, A. O. (2017). “Mechanism of action of nonsteroidal anti-inflammatory drugs,” in Nonsteroidal Anti-Inflammatory Drugs, ed. A. G. A. Al-kaf (London: Intech Open). Available online at: https://www.intechopen.com/books/nonsteroidal-anti-inflammatory-drugs/mechanism-of-action-of-nonsteroidal-anti-inflammatory-drugs
Owen, D. R., Narayan, N., Wells, L., Healy, L., Smyth, E., Rabiner, E. A., et al. (2017). Pro-inflammatory activation of primary microglia and macrophages increases 18 kDa translocator protein expression in rodents but not humans. J. Cereb. Blood Flow Metab. 37, 2679–2690. doi: 10.1177/0271678X17710182
Palmer, M. T., and Weaver, C. T. (2010). Autoimmunity: increasing suspects in the CD4+ T cell lineup. Nat. Immunol. 11, 36–40. doi: 10.1038/ni.1802
Pan, W., Zhang, G. F., Li, H. H., Ji, M. H., Zhou, Z. Q., Li, K. Y., et al. (2018). Ketamine differentially restores diverse alterations of neuroligins in brain regions in a rat model of neuropathic pain-induced depression. Neuroreport 29, 863–869. doi: 10.1097/WNR.0000000000001045
Papez, J. W. (1937). A proposed mechanism of emotion. J. Neuropsychiatry Clin. Neurosci. 7, 103–112.
Park, C., Rosenblat, J. D., Brietzke, E., Pan, Z., Lee, Y., Cao, B., et al. (2019). Stress, epigenetics and depression: a systematic review. Neurosci. Biobehav. Rev. 102, 139–152. doi: 10.1016/j.neubiorev.2019.04.010
Pecina, M., and Zubieta, J. K. (2018). Expectancy modulation of opioid neurotransmission. Int. Rev. Neurobiol. 138, 17–37. doi: 10.1016/bs.irn.2018.02.003
Peltoniemi, M. A., Hagelberg, N. M., Olkkola, K. T., and Saari, T. I. (2016). Ketamine: a review of clinical pharmacokinetics and pharmacodynamics in anesthesia and pain therapy. Clin. Pharmacokinet. 55, 1059–1077. doi: 10.1007/s40262-016-0383-6
Perrin, F. E., Lacroix, S., Avilés-Trigueros, M., and David, S. (2005). Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain 128, 854–866.
Petrenko, A. B., Yamakura, T., Baba, H., and Shimoji, K. (2003). The role of N-methyl-D-aspartate (NMDA) receptors in pain: a review. Anesth. Analg. 97, 1108–1116.
Philip, V. M., Gomero, D. B., Ansah, T. A., Blaha, C. D., Cook, M. N., Hamre, K. M., et al. (2010). High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes Brain Behav. 2, 129–159.
Pittaluga, A. (2017). CCL5-glutamate cross-talk in astrocyte-neuron communication in multiple sclerosis. Front. Immunol. 8:1079. doi: 10.3389/fimmu.2017.01079
Qiu, D., and Kao, P. N. (2003). Immunosuppressive and anti-inflammatory mechanisms of triptolide, the principal active diterpenoid from the Chinese medicinal herb Tripterygium wilfordii Hook. f. Drugs R D. 4, 1–18.
Qiu, D., Zhao, G., Aoki, Y., Shi, L., Uyei, A., Nazarian, S., et al. (1999). Immunosuppressant PG490 (triptolide) inhibits T-cell interleukin-2 expression at the level of purine-box/nuclear factor of activated T-cells and NF-kappa B transcriptional activation. J. Biol. Chem. 274, 13443–13450.
Redivo, D. D., Schreiber, A. K., Adami, E. R., Ribeiro, D. E., Joca, S. R., Zanoveli, J. M., et al. (2016). Effect of omega-3 polyunsaturated fatty acid treatment over mechanical allodynia and depressive-like behavior associated with experimental diabetes. Behav. Brain Res. 298, 57–64. doi: 10.1016/j.bbr.2015.10.058
Ren, P. C., Zhang, Y., Zhang, X. D., An, L. J., Lv, H. G., He, J., et al. (2012). High-mobility group box 1 contributes to mechanical allodynia and spinal astrocytic activation in a mouse model of type 2 diabetes. Brain Res. Bull. 88, 332–337. doi: 10.1016/j.brainresbull.2012.03.002
Reul, J. M., and de Kloet, E. R. (1985). Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology 117, 2505–2511.
Riediger, C., Schuster, T., Barlinn, K., Maier, S., Weitz, J., and Siepmann, T. (2017). Adverse effects of antidepressants for chronic pain: a systematic review and meta-analysis. Front. Neurol. 8:307. doi: 10.3389/fneur.2017.00307
Rivat, C., Becker, C., Blugeot, A., Zeau, B., Mauborgne, A., Pohl, M., et al. (2010). Chronic stress induces transient spinal neuroinflammation, triggering sensory hypersensitivity and long-lasting anxiety-induced hyperalgesia. Pain 150, 358–368. doi: 10.1016/j.pain.2010.05.031
Rivat, C., Laboureyras, E., Laulin, J. P., Le Roy, C., Richebé, P., and Simonnet, G. (2007). Non-nociceptive environmental stress induces hyperalgesia, not analgesia, in pain and opioid-experienced rats. Neuropsychopharmacology 32, 2217–2228.
Rosa, S. G., Pesarico, A. P., and Nogueira, C. W. (2018). m-Trifluoromethyl-diphenyl diselenide promotes resilience to social avoidance induced by social defeat stress in mice: contribution of opioid receptors and MAPKs. Prog. Neuropsychopharmacol. Biol. Psychiatry 82, 123–135. doi: 10.1016/j.pnpbp.2017.11.021
Sachdev, P. S., and Sachdev, J. (2005). Long-term outcome of neurosurgery for the treatment of resistant depression. J. Neuropsychiatry Clin. Neurosci. 17, 478–485.
Sang, N., Zhang, J., Marcheselli, V., Bazan, N. G., and Chen, C. (2005). Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J. Neurosci. 25, 9858–9870.
Schaefer, C. P., Tome, M. E., and Davis, T. P. (2017). The opioid epidemic: a central role for the blood brain barrier in opioid analgesia and abuse. Fluids Barriers CNS 14:32. doi: 10.1186/s12987-017-0080-3
Schwarcz, R., and Stone, T. W. (2017). The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 112, 237–247. doi: 10.1016/j.neuropharm.2016.08.003
Selvaraj, V., and Tu, L. N. (2016). Current status and future perspectives: TSPO in steroid neuroendocrinology. J. Endocrinol. 231, R1–R30.
Seminowicz, D. A., Remeniuk, B., Krimmel, S. R., Smith, M. T., Barrett, F. S., Wulff, A. B., et al. (2019). Pain-related nucleus accumbens function: modulation by reward and sleep disruption. Pain 160, 1196–1207. doi: 10.1097/j.pain.0000000000001498
Seno, M. D. J., Assis, D. V., Gouveia, F., Antunes, F. F., Kuroki, M., Oliveira, C. C., et al. (2018). The critical role of amygdala subnuclei in nociceptive and depressive-like behaviors in peripheral neuropathy. Sci Rep. 8:13608. doi: 10.1038/s41598-018-31962-w
Shamash, S., Reichert, F., and Rotshenker, S. (2002). The cytokine network of Wallerian degeneration: tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. J. Neurosci. 22, 3052–3060.
Shamji, M. F., De Vos, C., and Sharan, A. (2017). The advancing role of neuromodulation for the management of chronic treatment-refractory pain. Neurosurgery 80, S108–S113. doi: 10.1093/neuros/nyw047
Shanbhag, S. M., Kulkarni, H. J., and Gaitonde, B. B. (1970). Pharmacological actions of berberine on the central nervous system. Jpn. J. Pharmacol. 20, 482–487.
Shanthanna, H., Gilron, I., Rajarathinam, M., AlAmri, R., Kamath, S., Thabane, L., et al. (2017). Benefits and safety of gabapentinoids in chronic low back pain: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 14:e1002369. doi: 10.1371/journal.pmed.1002369
Shen, Y., Guan, S., Ge, H., Xiong, W., He, L., Liu, L., et al. (2018). Effects of palmatine on rats with comorbidity of diabetic neuropathic pain and depression. Brain Res. Bull. 139, 56–66. doi: 10.1016/j.brainresbull.2018.02.005
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Siracusa, R., Fusco, R., and Cuzzocrea, S. (2019). Astrocytes: role and functions in brain pathologies. Front. Pharmacol. 10:1114. doi: 10.3389/fphar.2019.01114
Skaper, S. D. (2016). Mast cell - Glia dialogue in chronic pain and neuropathic pain: blood-brain barrier implications. CNS Neurol. Disord. Drug Targets 15, 1072–1078.
Slavich, G. M., and Irwin, M. R. (2014). From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol. Bull. 140, 774–815. doi: 10.1037/a0035302
Sleigh, J., Harvey, M., Voss, L., and Denny, B. (2014). Ketamine-more mechanisms of action than just NMDA blockade. Trends Anaesth. Crit. Care 4, 76–81.
Smith, M. T., and Haythornthwaite, J. A. (2004). How do sleep disturbance and chronic pain inter-relate? Insights from the longitudinal and cognitive-behavioral clinical trials literature. Sleep Med. Rev. 8, 119–132.
Sousa, F. S. S., Birmann, P. T., Baldinotti, R., Fronza, M. G., Balaguez, R., Alves, D., et al. (2018). α- (phenylselanyl) acetophenone mitigates reserpine-induced pain-depression dyad: behavioral, biochemical and molecular docking evidences. Brain Res. Bull. 142, 129–137. doi: 10.1016/j.brainresbull.2018.07.007
Spengler, R. N., Sud, R., Knight, P. R., and Ignatowski, T. A. (2007). Antinociception mediated by alpha(2)-adrenergic activation involves increasing tumor necrosis factor alpha (TNFalpha) expression and restoring TNFalpha and alpha(2)-adrenergic inhibition of norepinephrine release. Neuropharmacology 52, 576–589. doi: 10.1016/j.neuropharm.2006.08.027
Stahl, S., and Briley, M. (2004). Understanding pain in depression. Hum. Psychopharmacol. 1, S9–S13.
Staner, L. (2010). Comorbidity of insomnia and depression. Sleep Med. Rev. 14, 35–46. doi: 10.1016/j.smrv.2009.09.003
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228.
Stephens, M. A., and Wand, G. (2012). Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res. 34, 468–483.
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S. (2018). Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219. doi: 10.1111/imm.12922
Suzuki, T., Hide, I., Ido, K., Kohsaka, S., Inoue, K., and Nakata, Y. (2004). Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J. Neurosci. 24, 1–7.
Szok, D., Tajti, J., Nyári, A., and Vécsei, L. (2019). Therapeutic approaches for peripheral and central neuropathic pain. Behav. Neurol. 2019:8685954. doi: 10.1155/2019/8685954
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tang, J. S., Qu, C. L., and Huo, F. Q. (2009). The thalamic nucleus submedius and ventrolateral orbital cortex are involved in nociceptive modulation: a novel pain modulation pathway. Prog. Neurobiol. 89, 383–389. doi: 10.1016/j.pneurobio.2009.10.002
Taylor, D. L., Jones, F., Kubota, E. S., and Pocock, J. M. (2005). Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor alpha-induced neurotoxicity in concert with microglial-derived Fas ligand. J. Neurosci. 25, 2952–2964.
Taylor, R. Jr., V Pergolizzi, J. Jr., Puenpatom, R. A., and Summers, K. H. (2013). Economic implications of potential drug-drug interactions in chronic pain patients. Expert Rev. Pharmacoecon. Outcomes Res. 13, 725–734. doi: 10.1586/14737167.2013.851006
Terada, M., Kuzumaki, N., Hareyama, N., Imai, S., Niikura, K., Narita, M., et al. (2008). Suppression of enriched environment-induced neurogenesis in a rodent model of neuropathic pain. Neurosci. Lett. 440, 314–318. doi: 10.1016/j.neulet.2008.05.078
Thompson, J. L., Pogue-Geile, M. F., and Grace, A. A. (2004). Developmental pathology, dopamine, and stress: a model for the age of onset of schizophrenia symptoms. Schizophr. Bull. 30, 875–900.
Todd, A. J. (2010). Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 11, 823–836. doi: 10.1038/nrn2947
Toews, A. D., Barrett, C., and Morell, P. (1998). Monocyte chemoattractant protein 1 is responsible for macrophage recruitment following injury to sciatic nerve. J. Neurosci. Res. 53, 260–267.
Treadway, M. T., Waskom, M. L., Dillon, D. G., Holmes, A. J., Park, M. T. M., Chakravarty, M. M., et al. (2015). Illness progression, recent stress, and morphometry of hippocampal subfields and medial prefrontal cortex in major depression. Biol. Psychiatry 77, 285–294. doi: 10.1016/j.biopsych.2014.06.018
Tremblay, M. È., Lowery, R. L., and Majewska, A. K. (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 8:e1000527. doi: 10.1371/journal.pbio.1000527
Trief, P. M., Carnrike, C. L. Jr., and Drudge, O. (1995). Chronic pain and depression: is social support relevant? Psychol. Rep. 76, 227–236.
Trujillo, K. A., and Akil, H. (1991). Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 251, 85–87.
Turk, D. C. (2002). Clinical effectiveness and cost-effectiveness of treatments for patients with chronic pain. Clin. J. Pain 18, 355–365.
Turner, M. D., Nedjai, B., Hurst, T., and Pennington, D. J. (2014). Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 1843, 2563–2582. doi: 10.1016/j.bbamcr.2014.05.014
Vachon, P., Millecamps, M., Low, L., Thompsosn, S. J., Pailleux, F., Beaudry, F., et al. (2013). Alleviation of chronic neuropathic pain by environmental enrichment in mice well after the establishment of chronic pain. Behav. Brain Funct. 9:22. doi: 10.1186/1744-9081-9-22
van Hecke, O., Austin, S. K., Khan, R. A., Smith, B. H., and Torrance, N. (2014). Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 155, 654–662. doi: 10.1016/j.pain.2013.11.013
Van Zee, A. (2009). The promotion and marketing of oxycontin: commercial triumph, public health tragedy. Am. J. Public Health 99, 221–227. doi: 10.2105/AJPH.2007.131714
Vane, J. R. (1971). Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 231, 232–235.
Vanelderen, P., Van Zundert, J., Kozicz, T., Puylaert, M., De Vooght, P., Mestrum, R., et al. (2015). Effect of minocycline on lumbar radicular neuropathic pain: a randomized, placebo-controlled, double-blind clinical trial with amitriptyline as a comparator. Anesthesiology 122, 399–406. doi: 10.1097/ALN.0000000000000508
Vanneste, T., Van Lantschoot, A., Van Boxem, K., and Van Zundert, J. (2017). Pulsed radiofrequency in chronic pain. Curr. Opin. Anaesthesiol. 30, 577–582. doi: 10.1097/ACO.0000000000000502
Vendrusculo-Fangel, L. M., Fangel, R., and Marqueti, R. D. C. (2019). Systemic alterations in plasma biomarkers levels in patients with chronic pain. Br. J. Pain 2, 274–278. doi: 10.5935/2595-0118.20190048
Viviani, B., Gardoni, F., Bartesaghi, S., Corsini, E., Facchi, A., Galli, C. L., et al. (2006). Interleukin-1β released by gp120 drives neural death through tyrosine phosphorylation and trafficking of NMDA receptors. J. Biol. Chem. 281, 30212–30222.
Volkow, N. D., and McLellan, A. T. (2016). Opioid abuse in chronic pain–misconceptions and mitigation strategies. N. Engl. J. Med. 374, 1253–1263. doi: 10.1056/NEJMra1507771
von Knorring, L., Perris, C., Eisemann, M., Eriksson, U., and Perris, H. (1983). Pain as a symptom in depressive disorders. II. Relationship to personality traits as assessed by means of KSP. Pain 17, 377–384.
Vowles, K. E., McEntee, M. L., Julnes, P. S., Frohe, T., Ney, J. P., and van der Goes, D. N. (2015). Rates of opioid misuse, abuse, and addiction in chronic pain: a systematic review and data synthesis. Pain 156, 569–576. doi: 10.1097/01.j.pain.0000460357.01998.f1
Wake, H., Moorhouse, A. J., Jinno, S., Kohsaka, S., and Nabekura, J. (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009
Walker, A. K., Budac, D. P., Bisulco, S., Lee, A. W., Smith, R. A., Beenders, B., et al. (2013a). NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 38, 1609–1616. doi: 10.1038/npp.2013.71
Walker, A. K., Kavelaars, A., Heijnen, C. J., and Dantzer, R. (2013b). Neuroinflammation and comorbidity of pain and depression. Pharmacol. Rev. 66, 80–101. doi: 10.1124/pr.113.008144
Walker, Z., Walker, R. W., Robertson, M. M., and Stansfeld, S. (1998). Antidepressant treatment of chronic tension-type headache: a comparison between fluoxetine and desipramine. Headache 38, 523–528. doi: 10.1046/j.1526-4610.1998.3807523.x
Wallace, D. J. (2006). Is there a role for cytokine based therapies in fibromyalgia. Curr. Pharm. Des. 12, 17–22.
Wang, B. C., Li, D., Hiller, J. M., Hillman, D. E., Pasternack, B. S., and Turndorf, H. (1995). The antinociceptive effect of S-(+)-ibuprofen in rabbits: epidural versus intravenous administration. Anesth. Analg. 80, 92–96.
Wang, Z., Chen, Z., Yang, S., Wang, Y., Huang, Z., Gao, J., et al. (2014). Berberine ameliorates collagen-induced arthritis in rats associated with anti-inflammatory and anti-angiogenic effects. Inflammation 37, 1789–1798. doi: 10.1007/s10753-014-9909-y
Watanabe, S., and Bruera, E. (1994). Corticosteroids as adjuvant analgesics. J. Pain Symptom Manage. 9, 442–445.
Watkins, L. R., Milligan, E. D., and Maier, S. F. (2001). Glial activation: a driving force for pathological pain. Trends Neurosci. 24, 450–455.
Werner, F. M., and Coveñas, R. (2010). Classical neurotransmitters and neuropeptides involved in major depression: a review. Int. J. Neurosci. 120, 455–470. doi: 10.3109/00207454.2010.483651
Whiteford, H. A., Degenhardt, L., Rehm, J., Baxter, A. J., Ferrari, A. J., Erskine, H. E., et al. (2013). Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet 382, 1575–1586. doi: 10.1016/S0140-6736(13)61611-6
Wiesenfeld-Hallin, Z., Xu, X. J., and Hökfelt, T. (2002). The role of spinal cholecystokinin in chronic pain states. Pharmacol. Toxicol. 91, 398–403.
Williams, L. S., Jones, W. J., Shen, J., Robinson, R. L., Weinberger, M., and Kroenke, K. (2003). Prevalence and impact of depression and pain in neurology outpatients. J. Neurol. Neurosurg. Psychiatry 74, 1587–1589.
Wilson, K. G., Eriksson, M. Y., D’Eon, J. L., Mikail, S. F., and Emery, P. C. (2002). Major depression and insomnia in chronic pain. Clin. J. Pain 18, 77–83.
Wingenfeld, K., Nutzinger, D., Kauth, J., Hellhammer, D. H., and Lautenbache, S. (2010). Salivary cortisol release and hypothalamic pituitary adrenal axis feedback sensitivity in fibromyalgia is associated with depression but not with pain. J. Pain 11, 1195–1202. doi: 10.1016/j.jpain.2010.02.011
Xu, N., Tang, X. H., Pan, W., Xie, Z. M., Zhang, G. F., Ji, M. H., et al. (2017). Spared nerve injury increases the expression of microglia M1 markers in the prefrontal cortex of rats and provokes depression-like behaviors. Front. Neurosci. 11:209. doi: 10.3389/fnins.2017.00209
Yakovlev, P. I. (1948). Motility behavior and the brain: stereodynamical organization and the neural coordinates of behavior. J. Nerv. Ment. Dis. 107, 313–335.
Yang, Q. Q., and Zhou, J. W. (2019). Neuroinflammation in the central nervous system: symphony of glial cells. Glia 67, 1017–1035. doi: 10.1002/glia.23571
Ye, M., Fu, S., Pi, R., and He, F. (2009). Neuropharmacological and pharmacokinetic properties of berberine: a review of recent research. J. Pharm. Pharmacol. 61, 831–837. doi: 10.1211/jpp/61.07.0001
Yrjänheikki, J., Keinänen, R., Pellikka, M., Hökfelt, T., and Koistinaho, J. (1998). Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc. Natl. Acad. Sci. U.S.A. 95, 15769–15774.
Zhang, G. F., Wang, J., Han, J. F., Guo, J., Xie, Z. M., Pan, W., et al. (2016). Acute single dose of ketamine relieves mechanical allodynia and consequent depression-like behaviors in a rat model. Neurosci. Lett. 631, 7–12. doi: 10.1016/j.neulet.2016.08.006
Zhang, J. M., and An, J. (2007). Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 45, 27–37.
Zhang, M., An, C., Gao, Y., Leak, R. K., Chen, J., and Zhang, F. (2013). Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 100, 30–47. doi: 10.1016/j.pneurobio.2012.09.003
Zhao, J., Gao, X., Wang, A., Wang, Y., Du, Y., Li, L., et al. (2019). Depression comorbid with hyperalgesia: different roles of neuroinflammation induced by chronic stress and hypercortisolism. J. Affect. Disord. 256, 117–124. doi: 10.1016/j.jad.2019.05.065
Zhou, W., Dantzer, R., Budac, D. P., Walker, A. K., Mao-Ying, Q. L., Lee, A. W., et al. (2015). Peripheral indoleamine 2,3-dioxygenase 1 is required for comorbid depression-like behavior but does not contribute to neuropathic pain in mice. Brain Behav. Immun. 46, 147–153. doi: 10.1016/j.bbi.2015.01.013
Zong, J., Liao, X., Ren, B., and Wang, Z. (2018). The antidepressant effects of rosiglitazone on rats with depression induced by neuropathic pain. Life Sci. 203, 315–322. doi: 10.1016/j.lfs.2018.04.057
Zou, W., Feng, R., and Yang, Y. (2018). Changes in the serum levels of inflammatory cytokines in antidepressant drug-naïve patients with major depression. PLoS One 13:e0197267. doi: 10.1371/journal.pone.0197267
Keywords: neuroinflammation, pain, depression, depression-pain syndrome, glial cells
Citation: Campos ACP, Antunes GF, Matsumoto M, Pagano RL and Martinez RCR (2020) Neuroinflammation, Pain and Depression: An Overview of the Main Findings. Front. Psychol. 11:1825. doi: 10.3389/fpsyg.2020.01825
Received: 27 February 2020; Accepted: 02 July 2020;
Published: 31 July 2020.
Edited by:
Qing Zhao, Second Affiliated Hospital of Guangzhou Medical University, ChinaReviewed by:
Ming Yi, Peking University, ChinaJixin Liu, Xidian University, China
Patricia Barton Crane, East Carolina University, United States
Copyright © 2020 Campos, Antunes, Matsumoto, Pagano and Martinez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raquel Chacon Ruiz Martinez, cXVlbG1hcnRpbmV6QHlhaG9vLmNvbS5icg==
†These authors have contributed equally to this work