 María Isabel Murillo
María Isabel Murillo Christian Gaiddon
Christian Gaiddon Ronan Le Lagadec
Ronan Le Lagadec- 1Instituto de Química, Universidad Nacional Autónoma de México, Ciudad Universitaria, Ciudad de México, Mexico
- 2Strasbourg Université, Inserm UMR_S U1113, IRFAC, Strasbourg, France
The development of cancers is often linked to the alteration of essential redox processes, and therefore, oxidoreductases involved in such mechanisms can be considered as attractive molecular targets for the development of new therapeutic strategies. On the other hand, for more than two decades, transition metals derivatives have been leading the research on drugs as alternatives to platinum-based treatments. The success of such compounds is particularly due to their attractive redox kinetics properties, favorable oxidation states, as well as routes of action different to interactions with DNA, in which redox interactions are crucial. For instance, the activity of oxidoreductases such as PHD2 (prolyl hydroxylase domain-containing protein) which can regulate angiogenesis in tumors, LDH (lactate dehydrogenase) related to glycolysis, and enzymes, such as catalases, SOD (superoxide dismutase), TRX (thioredoxin) or GSH (glutathione) involved in controlling oxidative stress, can be altered by metal effectors. In this review, we wish to discuss recent results on how transition metal complexes have been rationally designed to impact on redox processes, in search for effective and more specific cancer treatments.
Introduction
Reduction-oxidation (redox) processes are at the center of many functions in chemistry and biology and have become one of the leading research topics in biochemistry and biophysics. (Monteiro and Stern, 1996; Nakamura et al., 1997; Berglund et al., 2002; Guiseppi-Elie et al., 2002; Finkel, 2003; Della et al., 2011). Redox proteins and enzymes can also conduct reactions of industrial and pharmaceutical importance. (Truppo, 2017; Prier and Kosjek, 2019). The fundamental structure of such proteins consists of catalytic sites connected by redox chains, which can be described as multielectron redox centers or clusters of single electron redox centers that interact with substrates and function as sources or sinks of electrons. Most transition metals can display multiple oxidation states and can be found as active sites of many proteins and, as such, playing essential roles in oxidoreduction functions (Sheldon and Woodley, 2018; Turner and Kumar, 2018). Oxidoreductases are considered catalysts for important biological processes that require electron transfers, including photosynthesis, respiration, metabolism, and signaling processes governing gene regulation and expression (Allen et al., 1995). Between 30 and 50 percent of all proteins are considered oxidation/reduction enzymes or metalloproteins. As such, studies aimed at elucidating the molecular and electrochemical properties linked with the chemical and biological electron transport systems displayed by redox proteins have been extensively developed (Prabhulkar et al., 2012).
Redox homeostasis is maintained by the net physiologic balance between reducing and oxidizing equivalents within subcellular compartments, in particular through components like reactive oxygen species (ROS) and antioxidant enzymes (Berglund et al., 2002). Studying and understanding such processes is fundamental for cancer treatment (Narayanan et al., 2020). Traditionally, the free-radical theory of cancer considered that oxidative stress due to reactive oxygen/nitrogen species (ROS/RNS) could generate DNA damage and promote genetic instability (Hussain et al., 2003). However, ROS/RNS are now thought to be involved in not only in direct DNA damage but also in modulations of redox-regulated signaling pathways, which may be both beneficial or detrimental in cancers.
Unlike normal differentiated cells, which rely primarily on mitochondrial oxidative phosphorylation to generate the necessary energy in the form of ATP for cellular processes, most cancer cells rely on aerobic glycolysis. After tumor growth, there are fewer blood vessels, which leads to less oxygen (hypoxia), and cancer cells develop a hypoxic response through the hypoxia-responsive transcription factor HIF1A. This transcription factor plays a key role by inducing the expression of VEGF, a growth factor that stimulates vascularization and the expression of glucose transporters such as GLUT1 and redox enzymes (for instance LDH). This allows the reprogramming of metabolism towards glycolytic metabolism, which does not require as much oxygen. This process is called the Warburg effect (Vaupel and Multhoff, 2021). Cancer cells also exhibit increased ATP production and important levels of ROS, which permits to maintain high cell proliferation through the metabolic resetting. Antioxidant therapy can protect normal cells by activating cell survival signaling cascades, such as the nuclear factor erythroid 2-related factor Nrf2 pathway (Irwin et al., 2013). Nrf2 is a crucial regulator that protects cells from oxidative stress. Adaptations resulting from Nrf2 activation may have beneficial effects under stress conditions through modulation of antioxidant pathways but may also participate in the development of resistance to cancer therapy (Zhang, 2010). Due to their implications in cancer pathogenesis, redox homeostasis and the metabolic switch from glycolysis to oxidative phosphorylation appear as promising targets for cancer therapy (Gaiddon et al., 2021). These pathways include HIF1/2 and NRF2 mechanisms that contribute to the modification of the expression of transporters (e.g. glucose transporters), redox enzymes (e.g. LDH, PDK2), chaperone proteins and antioxidant enzymes (e.g. GSH) (Figure 1).
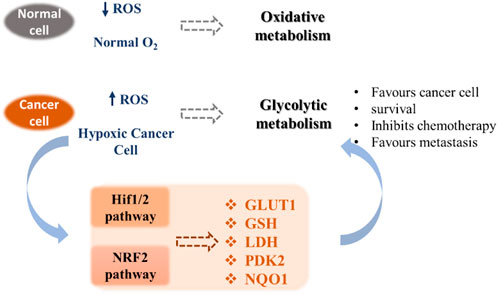
FIGURE 1. Metabolic pathways involved in tumor adaptation to its stressful environment (Gaiddon et al., 2021).
Transition metal-based derivatives have been intensively studied for their attractive anticancer properties (Raymond et al., 1998; Garbutcheon et al., 2011; Ndagi et al., 2017; Parveen, 2020). Platinum-based drugs, mainly cisplatin and its analogs carboplatin and oxaliplatin (Figure 2), have been used worldwide in cancer treatment (Dilruba and Kalayda, 2016). Other platinum-based molecules, such as miriplatin, nedaplatin, lobaplatin, and heptaplatin have also been approved regionally. The mode of action of these compounds is mostly through direct interactions with DNA, inducing DNA damage, which activates series of molecular mechanisms, including induction of the p53 tumor suppressor gene. Consequently, alterations in the p53 pathway, such as mutations in p53, lower the response toward platinum-based drugs (Blanchet et al., 2021). Additionally, the low selectivity of platinum drugs for cancer cells generates serious side effects on various tissues, including the nervous system and the muscles (Benosman et al., 2007; Benosman et al., 2011; Oun et al., 2018; Voisinet et al., 2021).
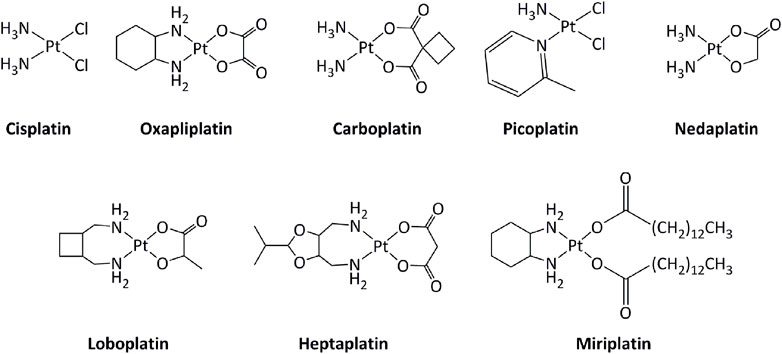
FIGURE 2. Platinum complexes used as anticancer drugs.
In order to limit such severe side-effects caused by platinum compounds, the use of other metals have been extensively explored. Both the redox properties of the metal and of the ligands in transition metal complexes can generate new routes of action that can bypass resistance mechanisms toward platinum or other DNA damaging drugs. Ruthenium derivatives have often been shown to exhibit a lower toxicity, linked with a higher selectivity towards cancer cells, than platinum-based drugs (Oun et al., 2018). In particular, four ruthenium derivatives have been evaluated clinical trials. NAMI-A was successfully studied in phase I, but poor efficacy was obtained in phase II, while the low solubility of related compound KP1019 limited further development (Figure 3). (Alessio and Messori, 2019; Bergamo and Sava, 20072007; Baier et al., 2022; a; Monro et al., 2019)
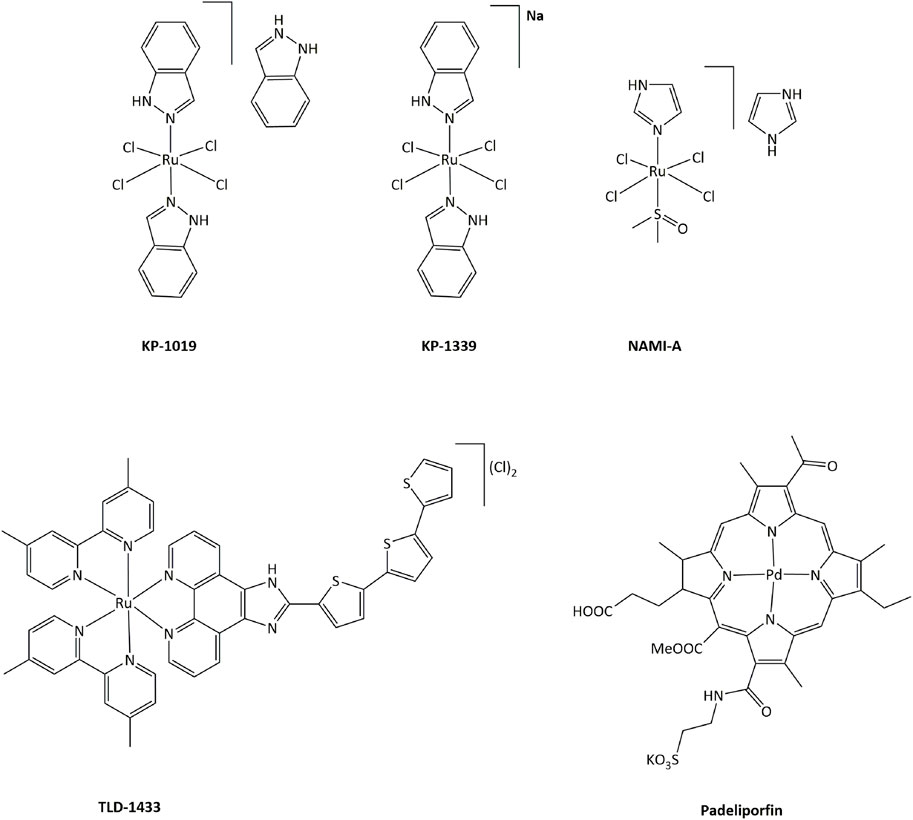
FIGURE 3. Ruthenium complexes studied in clinical trials as candidates for anticancer treatments and palladium compound approved for clinical use.
Ruthenium(III) KP-1339 is currently undergoing clinical trials, delivering promising Ib phase data for anticancer activity. Ruthenium(II) complex TLD-1433 acts as a photosensitizer, and is currently being evaluated in phase II for photodynamic therapy (PDT) against human non-muscle invasive bladder cancer. 1) Furthermore, many other transition metal complexes have been studied for their cytotoxic properties and potential use as anticancer drugs (Meier-Menches et al., 2018; Zamora et al., 2018; McFarland et al., 2019; Anthony et al., 2020). Notably, palladium-based compound Padeliporfin (commercially known as Tookad, Figure 3) has recently been approved by the European Medicines Agency in the European Union for PDT in patients with low-risk prostate cancer (Coleman et al., 2021).
The mechanisms of action for metal complexes can be varied, and in particular can be driven by redox reactions. In 2011, Heffeter et al. reviewed how metal complexes could carry out their cytotoxic activity in cancer cells through interactions with the cellular redox homeostasis (Jungwirth et al., 2011). A review by Sadler et al. in 2013 discusses multiple targets of metal complexes that are able to interfere with the cellular redox state (Romero-Canelón and Sadler, 2013). Redox-based mechanisms have also been successfully exploited, particularly with Ru(III) and Pt(IV) derivatives, in processes where the complexes act as prodrugs that are activated by the reducing environment of cancer cells, as highlighted in a 2012 review by Lippard et al. and in another review by Sadler et al. in 2017 (Graf and Lippard, 2012; Zhang and Sadler, 2017). Catalytic action has also been discussed for ruthenium and iridium compounds (Dougan et al., 2008; Liu and Sadler, 2014). More recently, one review focused on how metal-based drugs could induce anticancer immune responses, and another on transition metal complexes for photodynamic therapy (PDT) and photoactivated chemotherapy (PACT). (Englinger et al., 2019; Imberti et al., 2020).
Within this context and because of the high number of studies published every year on the anticancer activity of transition metal complexes, in this review, we wish to present updated information that highlight the importance of redox processes in cancer metabolic pathways, and how tumor development may be hindered by redox interactions with metal complexes. In addition to platinum and ruthenium compounds, we will discuss representative and recent examples of iron, osmium, iridium, rhodium, copper, silver and gold complexes that show redox-mediated anticancer activity.
The redox landscape in cancer
The redox balance is efficiently regulated in living organisms. For instance, ROS and RNS are generated during normal physiological metabolism and in response to stress, including exposure to xenobiotics, cytokines, growth factors, hormones, and invasion of bacteria (Roy et al., 2017). Although the generation of ROS and RNS is involved in crucial cell signaling functions, excessive amounts can generate malfunctions to proteins, lipids, carbohydrates, and nucleic acids, and disorders such as aging, hypertension, atherosclerosis, ischemia/reperfusion, renal diseases, diabetic neuropathies, Alzheimer’s disease and cancer (Sharifi-Rad et al., 2020; Boy et al., 2021). It is why pharmaceutical exploration aimed at modulating the oxidative response in therapies is a very active field of research (Purohit et al., 2019). Cells in tumors are particularly sensitive to oxidative stress as they commonly present higher levels of ROS due to the dysregulation of the redox balance, and excess of ROS potentially contributes to oncogenesis by oxidative DNA damage (Montero and Jassem, 2011).
The majority of ROS/RNS are hydrogen peroxide (H2O2), hydroxyl radicals (OH•), superoxide radicals (O2˙−), nitric oxide (NO˙), and peroxynitrite (ONOO−). ROS or RNS are able to activate or inactivate proteins by reacting with sulfhydryl (sulfenylation), glutathione (GSH, glutanylation), and cysteine (oxidation) groups (Jones et al., 2004; Defelipe et al., 2015). Antioxidant proteins are important tools for the control of ROS/RNS levels and conduct target-specific transduction of redox signals. The major enzymatic antioxidants include superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione S transferase (GST), and glutaredoxin (Grx), and operate in cooperation with thiol-redox couples to regulate ROS/RNS levels. It is worth noting that all these enzymes are ubiquitous. Six major redox couples are usually present in a cell: NADH/NAD, NADPH/NADP, cysteine (Cys)/cystine (CySS), GSH/glutathione disulfide (GSSG), peroxiredoxin (Prx)-sulfiredoxin (Srx), and thioredoxin (Trx)/thioredoxin disulfide (TrxSS). For instance, thiol systems can adjust the production of H2O2 by limiting its diffusibility and stability in each subcellular compartment, while the pKa of specific residues on proteins determines how sensitive these residues are to the available H2O2 (Thamsen and Jakob, 2011; Jacob et al., 2012). Additionally, thiol groups can in turn be modified (e.g., nitrosylation, sulfhydration, metal ion binding) allowing to act as signaling molecules to control cell function (Marino and Gladyshev, 2011).
Metabolic abnormalities and ROS generation in cancer cells
A critical point in the metabolic–redox mechanisms in cancer is the “hypermetabolism” required for growth and proliferation of tumor cells which results in intracellular ROS production in the mitochondria, NADPH oxidases (NOXs), peroxisomes, and endoplasmic reticulum (ER) (Batinic-Haberle et al., 2011; Barrera, 2012). These mechanisms are ubiquitous, such as NOX originally described in leukocytes, but found throughout the body. NOX has seven different isoforms, NOX1-5, DUOX1 and DUOX2, each isoform characterized by the specific catalytic subunit, the interacting proteins and the localization in the different cells of the body (Rastogi et al., 2017). Mitochondrial ROS are byproducts of metabolic processes during which electrons escape from the mitochondrial electron transport chain (Mito-ETC) and react with molecular oxygen to generate superoxide anions (O2−) (Batinic-Haberle et al., 2012). In addition, metabolic enzymes, such as 2-oxoglutarate dehydrogenase (OGDH), pyruvate dehydrogenase (PDH), glycerol-3-phosphate dehydrogenase (GPDH), and flavoprotein-ubiquinone oxidoreductase (FQR), also contribute to O2− production (Antunes and Cadenas, 2000; Baud and Karin, 2009; Becuwe et al., 2014). Along with Mito-ETC, oncogenic activation triggers the production of ROS through NOX-mediated NADPH oxidation (Figure 4) (Yoboue et al., 2018)
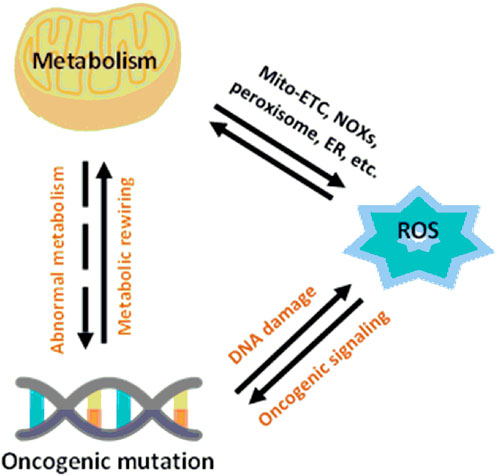
FIGURE 4. Relationship between metabolism and redox signaling in cancer cells (Wang et al., 2019).
Another fundamental process for the intracellular production of ROS is the cooperation between mitochondria, the endoplasmic reticulum (ER), and peroxisomes (Wang et al., 2019).
NADPH oxidases
NADPH oxidases (NOXs) play a fundamental role in a wide range of physiological processes, such as gene expression regulation, cell signaling and differentiation, but are also involved in many pathological processes, including cancer. Several studies have demonstrated that cancer cells often display mutations which can increase ROS generation from NOX enzymes, which in turn can lead to tumorigenesis (Jaramillo et al., 2012; Jaramillo et al., 2015). A particular type of mutation involves the GTPase KRAS, a member of the RAS oncogene family. KRAS mutations induce NOX1-mediated ROS formation and metastasis.63-
Catalases
The CAT enzymes are present in most of cells exposed to oxygen and are involved in lowering high concentrations of H2O2 (Nakabeppu et al., 2006; Munro and Treberg, 2017). CAT can also react with peroxynitrite, a strong oxidizing agent produced by the reaction between nitric oxide (NO˙) and O2−, associated with pathological events (ONOO−/ONOOH). In cancer cells, CAT can be found in high concentrations in the plasma membrane and occasionally released in the extracellular matrix, and can act as a tumor suppressor and as a survival agent during tumor progression (Naranjo-Suarez et al., 2013; Narayanan et al., 2020). However, higher catalase levels have been associated with more aggressive cancers when compared to lower CAT concentrations (Glorieux and Calderon, 2018; Galasso et al., 2021).
Glutathione
When compared with normal cells, cancer cells contain higher GSH levels, as GSH metabolism appears to be involved in protecting cancer cells from apoptosis (Gamcsik et al., 2012). Furthermore, increased levels of GSH within tumor cells are associated with resistance to platinum-containing anticancer compounds, due to the formation of GSH-platinum conjugates mediated by glutathione S-transferase P1 (GSP1) (Peklak-Scott et al., 2008). The GSH related metabolism genes are regulated by Nrf2 genes, which have been used as redox state index for platinum resistant cancers (Galluzzi et al., 2012). The overall cellular redox state is regulated by three systems, two of which are glutathione-dependent: the reduced glutathione (GSH)/oxidized glutathione (GSSG) system (Figure 5), the glutaredoxin (Grx) system and the thioredoxin (Trx)/Trx reductase system (Giles, 2006; Townsend, 2007; Tew and Townsend, 2011). GSH acts directly as an electron donor, whereas Grx uses GSH or GR as an electron donor and depends on the intracellular concentration of GSH. On the other hand, Trx uses nicotinamide adenine dinucleotide phosphate (NADPH) as an electron donor, independently of GSH (Tew and Townsend, 2011).
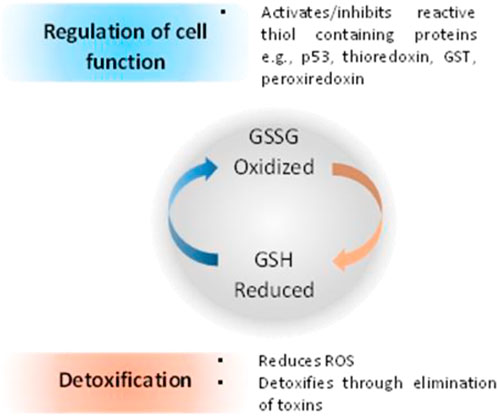
FIGURE 5. Relationship between redox state of glutathione and regulation of cell function and cell detoxification (Montero and Jassem, 2011).
NADPH dehydrogenases (quinone)
Quinone reductase 1 (NQO1) can be considered the redox barrier between the organism and its environment (Oberley et al., 19801980). NQO1 detoxifies ROS-generating quinones to hydroquinones through a back and forth route, using NAD(P)H to reduce FAD and then catalyzing a two-electron reduction to generate FAD and hydroquinone (Oshikawa et al., 2010).
Redox enzymes as a target of drugs for cancer treatment
In addition to their activity on cell division, many cytotoxic drugs are able to induce oxidative stress by modulating the concentration of ROS (Giles, 2006; Townsend, 2007). Furthermore, the susceptibility of some cancer cells towards redox enzymes has been considered as a therapeutic target for the rational design of new anticancer agents (Xiong et al., 2021). As many drugs currently applied in chemotherapies have an impact on redox pathways, probably contributing to their antitumor activity, evaluating the possibility of precisely affecting the cellular redox balance has become a leading trend in anticancer research (Table 1) (Chen and Chang, 2019)
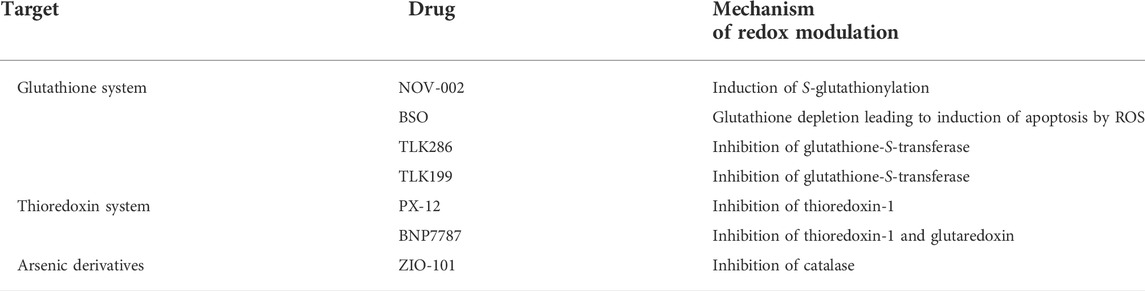
TABLE 1. Redox modulation by cytotoxic anticancer drugs currently used clinically (Chen and Chang, 2019).
Metal complexes can also affect the cellular redox chemistry, directly through metal- or ligand-based redox processes or indirectly by interacting with biomolecules implicated in cellular redox pathways (Bandeira et al., 2017; Ortega et al., 2021).
Iron complexes
Iron(II) complexes bearing triapine-type heterocyclic thiosemicarbazone ligands (triapine = 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, a molecule studied in the treatment of cancers) have been reported to inhibit ribonucleotide reductase (RNR), an enzyme which catalyzes the reduction of ribose to deoxyribose in nucleotides for DNA synthesis (Plamthottam et al., 2019). Inhibition of RNR by triapine results in depletion of DNA precursors, selectively depriving replicating cancer cells of nucleotides for survival. The redox-active form of triapine responsible for RNR inhibition is the Fe(II) (triapine)2 fragment. Iron complexes with triapine analogs (1 and 2, Figure 6) have shown in vitro that redox events are crucial for RNR inhibition, and were able to inhibit cell proliferation at similar or lower concentrations (250 nM - 0.7 μM) than triapine alone (Plamthottam et al., 2019). The reductive activation of Fe(III)-triapine by thioredoxin reductase-1 (TrxR1) and glutathione reductase (GR), leading to the generation of reactive species has been demonstrated. In particular, TrxR1 displayed high activity with Fe(III)-thiosemicarbazone derivatives, and a specificity between the Fe(III) complexes and the redox centers of TrxR has been observed. (b; Myers et al., 2013; Lovejoy et al., 2011; Richardson et al., 2009). Iron(III) complex 3 (Figure 6) with thiosemicarbazone-derived ligands is reduced by ascorbate to iron(II), increasing lipid peroxidation. The formation of ascorbyl radical anion (Asc˙−) has been detected after adding ascorbate to the iron(III) complex, resulting in the production of ROS (Selyutina et al., 2022). The use of ascorbate to promote the redox activity of these potential anticancer agents was demonstrated in vitro. Complex 3 showed antiproliferative action on the human melanoma cell line SK-MEL-28 at concentrations of 3.125–25 μM, in the presence of 1,000 μM of ascorbate (Kontoghiorghes et al., 2020). Upon introduction of a methoxy group, compound 4c displayed elevated cytotoxicity towards CaSki cancer cells. The IC50 values for complex 4c were 0.75, 6.73, 7.32 and 23.71 µM for Caski, SiHa, HeLa and L02 cells, respectively. Studies on the cell death mechanisms induced by complex 4c showed that cancer cell growth was suppressed by apoptosis, and the TrxR activity of Caski, SiHa, and HeLa cells decreased to 48.92, 84.51 and 86.01% respectively (Xie et al., 2017). Evaluation of the relationship between the inhibition of TrxR and the cytotoxic activity suggests that compound 4c carries out its activity through TrxR inhibition, affecting cellular redox balance and leading to cell death.
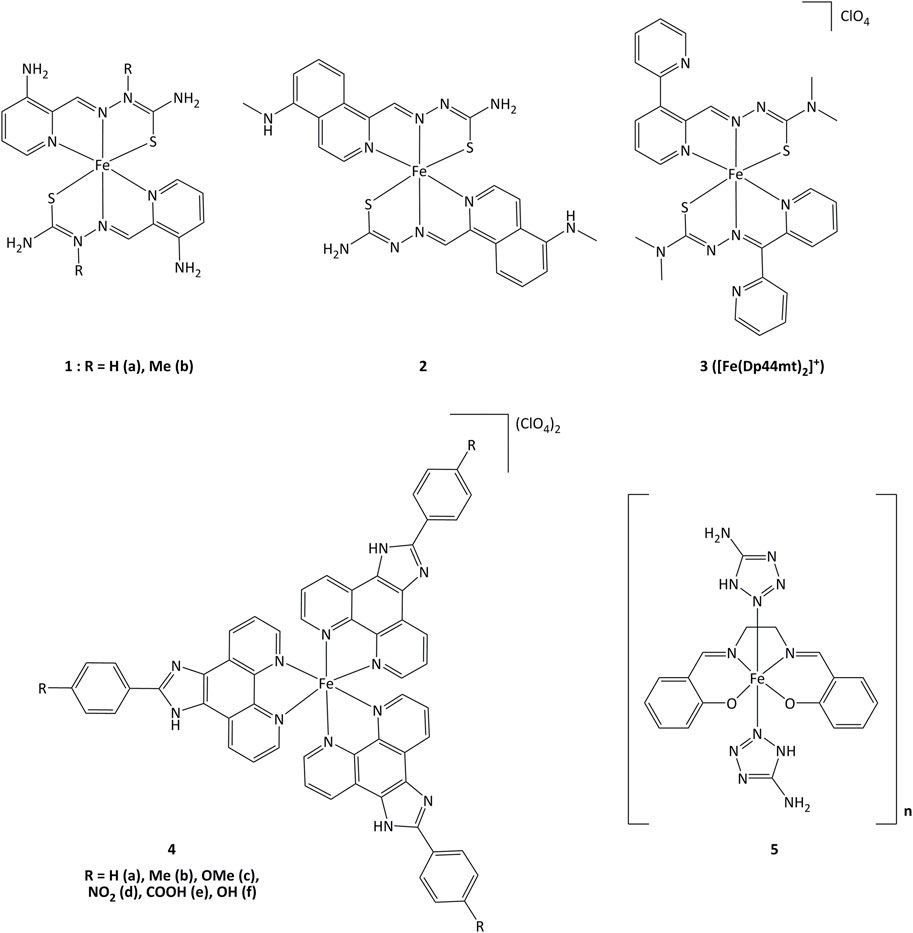
FIGURE 6. Iron complexes able to alter redox enzymatic activity.
Iron(III) complexes bearing salen-type ligands (salen = N,N′-ethylenebis(salicylaldimate) dianion) have been studied for their anticancer activity. The cell death induced by complexes like 5 was related to DNA cleavage and superoxide dismutase (SOD) mimicking activity, probably generating local imbalance in superoxide/hydrogen peroxide levels, leading to cell apoptosis. Complex 5 was highly active against K562 and MCF-7 (IC50 = 6.4 and 13.1 µM, respectively) with IC50 value of 1.89 µM for the inhibition of SOD (Herchel et al., 2009).
Ruthenium complexes
Ruthenium derivatives are among the most studied and promising compounds for potential anticancer treatments. The success of ruthenium is notably due to specific redox kinetics properties and the relevant oxidation states (II) and (III). In studies aimed at the development of biosensors, our group has shown that cyclometalated ruthenium complexes can alter the activity of purified oxidoreductases, such as glucose oxidase, horseradish peroxidase, lactate dehydrogenase or PHD2 (Ryabov et al., 2001; Saavedra-Diaz et al., 2013; Bautista et al., 2016; Vidimar et al., 2019). Such compounds were used as mediators (electron shuttles) in the electron transfer to or from oxidized or reduced active sites of redox enzymes. The ruthenium complexes 7–11 shown in Figure 7 mediate the electron transfer and display high reactivity with respect to horseradish peroxidase (HRP) and glucose oxidase (GO) (Ryabova et al., 1999). Organometallic ruthenium(II) derivatives bearing cyclometalated 2-phenylpyridine (phpyH), (11 and 12, Figure 7), function as noncompetitive inhibitors of glucose oxidase in the oxidation of β-D-glucose by O2. The analogous coordination compound 13 behaves, in contrast, as a competitive inhibitor. Oxidation of Ru(II) to Ru(III) compounds 14 and 15 does not make the complexes competitive inhibitors (Saavedra-Diaz et al., 2013). Interestingly, if ruthenium complexes are able to inhibit redox enzyme activity, the reverse can also occur. For instance, ruthenium(III) compound 14 (oxidized form of 11) promotes the enzymatic activity of glucose oxidase (Saavedra-Diaz et al., 2013). Bis-cyclometalated complex 15 is able to transport electrons from the reduced active site of PQQ-dependent alcohol dehydrogenase (PQQ-ADH) to an electrode with 1,2-propanediol as substrate (Le Lagadec et al., 2006). Our group also studied how modifications in the ligand structure could affect the ability of the metallacycles to interact with their direct biological targets. The activity of two purified oxidoreductases, glucose oxidase and horseradish peroxidase, was evaluated in the presence of the cyclometalated derivatives (Licona et al., 2020). The calculation of the k3 rate constant for the electron transfer between the active site of the enzyme and the complexes showed that the ability to alter the activity of both enzymes is related to their oxidoreduction potentials. The coordination of a second phenanthroline ligand in 7 (RDC34) lowered the redox potential by c.a. 100 mV and increased the lipophilicity when compared with 6 (RDC11). Such results showed that the modification of the spatial structure of the complexes may also be responsible for their capacity to alter the redox enzyme function (Anand et al., 2009).
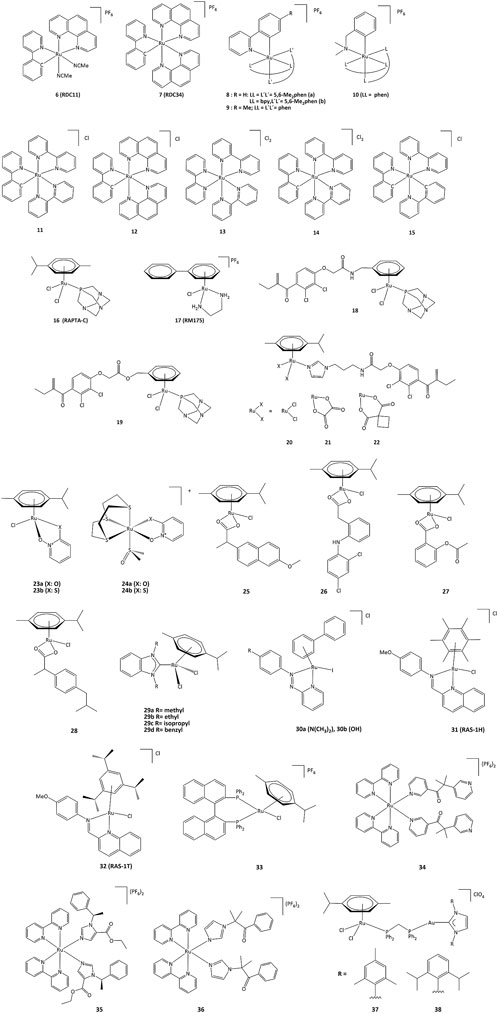
FIGURE 7. Ruthenium compounds capable to interact with redox enzymes.
To understand the role of the RDC11 complex in cancer metabolism, studies were performed on the HIF1A (hypoxia-inducible factor) pathway (Vidimar et al., 2019). At the molecular level, RDC11 can affect redox enzyme activities and intracellular redox state by increasing the NAD+/NADH ratio and ROS levels, and at the metabolic level, the HIF1A pathway is affected by inducing the activity of the iron redox enzyme PHD2, an enzyme that controls HIF1A protein levels (Vidimar et al., 2019). Notably, unlike cisplatin, the activity of RDC11 was not affected by the presence of mutations in p53 (Gaiddon et al., 2005). As such, PHD2 could be considered a direct target of RDC11, which could activate the PHD2 activity through a mechanism possibly involving the redox activity of the ruthenium complex. Inhibition of HIF1A led to decreased angiogenesis in patient-derived xenografts using fragments of primary human colon tumors (Alpeeva et al., 2003).
An important feature of cancer cells is their elevated lactate production due to high glucose consumption and the switch to glycolytic metabolism. Lactate dehydrogenase (LDH), which catalyzes the production of lactate in the final step of the glycolytic pathway, is a fundamental enzyme in such process (Netanya and Robert, 2019). To get a better understanding on how cyclometalated compounds could impact on the activity of LDH in vitro and in cancer cells, a comparative study was performed using polypyridine ruthenium(II) complex 13 and its structurally related cyclometalated-phenylpyridine counterparts 11 and 12 (Bautista et al., 2016). The cytotoxicity in gastric and colon cancer cells induced by 11 and 12 is significantly higher when compared to 13. The inhibition mechanisms on purified LDH were evaluated and kinetic studies allowed the calculation of the corresponding inhibition constants. Though complexes 11 and 13 are structurally similar, their inhibition modes are different. Cyclometalated complex 11 behaves as a non-competitive inhibitor of LDH, suggesting no interaction with LDH in the vicinities of lactate/pyruvate or NAD+/NADH binding sites (Bautista et al., 2016).
Such results suggested that ruthenium complexes might affect the redox state of cancer cells by altering the activity of redox enzymes (Meng et al., 2009). This could induce the oxidation of proteins causing misfolding and activation of the unfolded protein response (UPR), also called the endoplasmic reticulum stress (ER stress) pathway (King and Wilson, 2020). The UPR pathway helps cancer cells to survive under drastic conditions and contributes to resistance in chemotherapy and radiotherapy (Limia et al., 2019). However, despite the role of UPR in promoting cancer progression and resistance to chemotherapy, artificial induction of ER stress has been suggested as a potential anticancer strategy. This approach has been successfully demonstrated with RDC11 and RDC34 which were able to strongly induce CHOP, a transcription factor that mediates apoptosis in response to ER stress (Meng et al., 2009). Interestingly, RDC34 displayed a higher expression of CHOP than RDC11, which can be explained by a greater retention in the endoplasmic reticulum due to its higher lipophilicity (Klajner et al., 2014). Similarly, structure-activity studies of RDC complexes revealed that complexes with a relatively significant lipophilicity and redox potentials in a specific 0.4–0.6 V (vs. SCE) region were the most active. Such dependence on the redox potential probably indicates that electron transfer to/from Ru(II) should play a role in their UPR-inducing activity (Klajner et al., 2014; Gaiddon et al., 2021).
Derivatives 18 and 19 in which the arene ligand is substituted by ethacrynic acid through an amide or an ester moiety were able to inhibit GST P1-1, with IC50 values in the 5.9–13.7 μM range (Townsend et al., 2005; Ang and Dyson, 2006; Suss-Fink, 2010). Other ruthenium–arene complexes bearing EA-modified imidazoles (20–22) are also efficient inhibitors of GST P1-1 and can inhibit cells growth of cisplatin resistant human ovarian cancer cells with IC50 from 9 to 15 μM (Ang et al., 2007).
Complex 17 (RM175), which specifically binds to guanine bases of DNA, can also react with the thiol group of GSH to form [Ru(η (Nakamura et al., 1997)-biphenyl) (en) (GS]+ (en = ethylenediamine, GS = glutathione). Further addition of oxygen to the thiolate ligand produces the sulfenate complex. Finally, the sulfinate adduct can be generated by oxidation (Novakova et al., 2003; Wang et al., 2005). Such combination of GSH and oxidation reactions contribute to the binding to guanine in DNA. Substitution of the sulfenate ligand by guanine N7 generates a redox-mediated pathway to DNA binding (Wu et al., 2013).
The development of hormone-dependent forms of cancers of lung, larynx, and bladder cancers have been associated with isozymes from the aldo–keto reductase 1C subfamily (AKR1C) (Penning and Byrns, 2009; Lanišnik and Penning, 2014). Furthermore, AKR1C isozymes are related to the resistance to many anticancer drugs, including platinum-based (Chen et al., 2008; Chen et al., 2013). Ruthenium complexes bearing as ligands the zinc ionophores pyrithione and its oxygen-containing analog (23–24) have been studied against AKR1C isozymes. If compounds 23a and 23b were able to efficiently inhibit AKR1C1, the inhibitory activity was much lower for 24a and 24b. In addition, 23b also displayed high cytotoxicity (EC50 = 3.8 μM) on the hormone-dependent breast cancer cell line MCF-7, when complex 24b bearing a sulfur macrocycle was almost inactive (EC50 = 200 μM). On the other hand, 23a and 24a did not show cytotoxic effects against the same cancer cell line (Kljun et al., 2016).
Nonsteroidal anti-inflammatory drugs (NSAIDs) have been able to display chemopreventive properties in cancer cells due to their ability to block cyclooxygenase (COX-1 and COX-2) and lipoxygenase (LOX) enzymes which are often upregulated in malignant tumors (Feng et al., 2014; Banti and Hadjikakou, 2016; Boodram et al., 2016). The coordination of NSAID to a [ruthenium(arene)] moiety in complexes 25–28 allowed the inhibition of COX and LOX activity, and antiproliferative activity against series of cancer cell lines.120.
N-heterocyclic carbene (NHC) metal complexes have also been studied as potential metal-based drugs. For instance, ruthenium complexes 29a-29d can react with biologically relevant thiols and selenols. TrxR enzymes activity could be inhibited by such complexes, with IC50 values ranging from 0.30 to 3.74 μM. The compounds are also cytotoxic against several cancer lines, with IC50 values of 2.06 and 51.67 μΜ for MCF-7 breast cancer cells and >100 and 2.40 μΜ for HT-29 colon cancer cells for 29c and 29d respectively (Oehninger et al., 2013).
The N=N azo bonds in complexes 30a and 30b bearing azpy-type ligands (azpy = 2-phenylazopyridine) generate redox potentials that are biologically accessible, and oxidation of GSH to GSSG is observed under physiological conditions and important levels of ROS in A549 lung cancer cells have been detected (Chow et al., 2014). Unlike RM175 which forms a key intermediate with GSH for subsequent DNA binding, ruthenium-arene complexes 30a and 30b can catalyze the oxidation of GSH to GSSG, with the first step being the reduction of the azo bond (-N=N-) by GSH, followed by the elimination of GSSG and the catalytic cycle is completed by the reduction of O2 to H2O2 and the subsequent oxidation of the ligand to regenerate the azo bond. Such ligand-based redox reactions provide new concepts for catalytic drug design (Dougan et al., 2008).
The two ruthenium Schiff base complexes, RAS-1H and RAS-1T (31, 32), induced non-apoptotic programmed cell death through the ER stress mechanism (Chow et al., 2016). Interestingly, RAS-1T shows a ROS-mediated ER stress pathway, while RAS-1H is independent of ROS. However, both complexes are more active against apoptosis-resistant cell lines than clinical drugs.
Complex 33 displayed a cytotoxic activity 15 and 7.5 times higher than cisplatin against A549 and HeLa cells, respectively (Li et al., 2018). This ruthenium complex reacted with the NAD+/NADH couple through transfer hydrogenation reactions and also induced ROS in cells (Li et al., 2018). As overexpression of P450 enzymes in tumors is often associated with resistance to various drugs, the use of P450 inhibitors as ligands allowed the preparation of ruthenium prodrugs 34–36, that can be triggered to controllably release the inhibitors. Activation of the compounds by light provides the free ligands that can inhibit the P450 enzymes, while the remaining ruthenium center can damage DNA (Zamora et al., 2017).
Heterobimetallic ruthenium-gold complexes 37 and 38 were highly active against series of cancer cells, displaying a better selectivity than their mononuclear counterparts. The TrxR activity of HCT116 cells was inhibited by compound 37 (IC50 = 5.22 µM), while cisplatin was inactive. Complex 38 presented cytotoxicity with IC50 values of 5.2, 73.2 and 8.1 µM towards Caki-1, HEK-293T and HTC116 cancer cells, respectively (Férnandez-Gallardo et al., 2016).
Osmium complexes
Cyclometalated osmium complexes synthesized by our research group have shown high cytotoxic activity, with IC50 below 1 μM on various series of cancer cell lines, driven by the level of lipophilicity and low reduction potential (Boff et al., 2013). For instance, the ODC2 (39, Figure 8) and ODC3 complexes (40) cause cell death by inducing the transcription factor CHOP and the ER stress pathway (Suntharalingam et al., 2013; Oakes, 2017; Gaiddon et al., 2021).
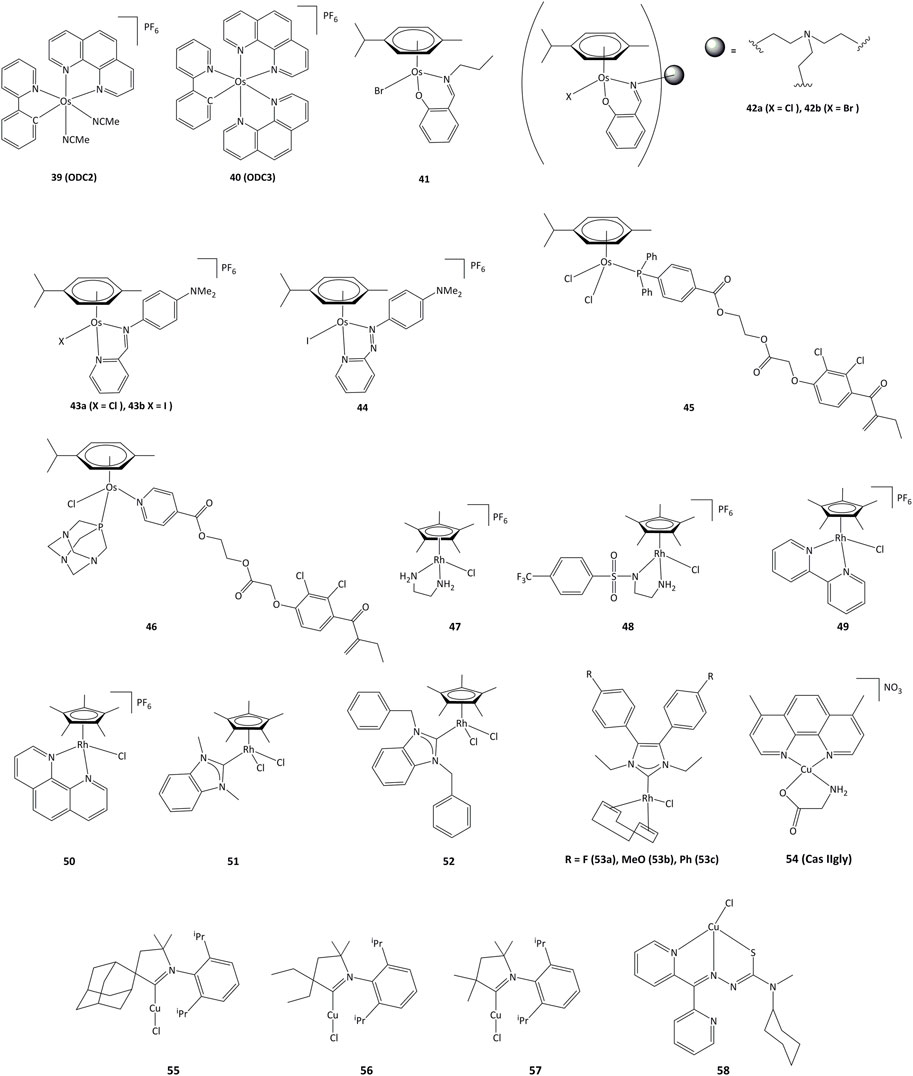
FIGURE 8. Osmium, rhodium and copper complexes studied as anticancer agents.
Mononuclear and trinuclear arene Os(II) complexes bearing pyridylimine or phenoxyimine derived ligands (41–42) were active against cisplatin-resistant cancer lines, and it has been shown that they were able to inhibit the topoisomerase I (Pommier, 2006; Banothile et al., 2014). The activity of related osmium complexes bearing iminopyridine ligands (43a and 43b) has also been evaluated (Fu et al., 2012). Complexes 43a and 43b were active against ovarian and lung cancer cell lines, and their activity associated with the production of ROS and oxidation of NADH. Contrary to their ruthenium azopyridine analogues (30a and 30b), the complexes cannot oxidize GSH, but can oxidize NADH to NAD+ through a hydride transfer to the osmium(II) center (Romero-Canelón et al., 2013). Related azo derivative 44 acts through a ROS-dependent pathway, and its cytotoxicity is inversely related to the intracellular concentration of GSH (Needham et al., 2017). Complexes 45 and 46 bearing the EA fragment were able to inhibit between 20 and 30% of GST enzyme activity, even in cisplatin-resistant cancer cell lines (Agonigi et al., 2016; Allocati et al., 2018).
Rhodium complexes
In recent years, interest in potential rhodium(III) drugs has flourished due to their enzymes inhibition capacity (Sohrabi et al., 2021). Rhodium(III) complexes 47–50 (Figure 8) can reduce NAD+ to NADH using formate as the hydride source. The competition reactions between NAD+ and pyruvate for formate-catalyzed reduction showed a preference for NAD+ reduction (Soldevila-Barreda et al., 2015).
Rhodium(III) complexes bearing NHC ligands have been studied as inhibitors of TrxR. The study of IC50 values for various human cancer cell lines showed that the presence of a benzyl substituent on the nitrogen atoms of the NHC affected the activity, as 51 presented a lower cytotoxicity than 52 towards cancer cells. However, both complexes exhibited strong inhibition of TrxR (IC50 values of ∼1 μM for 51 and 52) (Truong et al., 2020). Rhodium(I) complexes 53a-c also showed cytotoxic activity towards MCF-7, HT-29 y HepG2 cancer cell lines, where the lowest IC50 values were obtained for HepG2 cells (1.33, 5.84 y 4.96 μM for 53a - 53c, respectively). Complex 53a was able to inhibit TrxR both in vitro and in vivo and showed an IC50 value of 2.5 μM for TrxR in HepG2. It is proposed that TrxR could be a possible biological target for the 53a complex (Fan et al., 2019).
Copper complexes
Copper plays an important role in the development of cancer, through the generation of angiogenesis and metastasis, and effective cellular uptake of copper by malignant cells has been observed (Gaur et al., 2018; Quiles et al., 2020). Copper shows high redox activity as it can easily switch between I and II oxidation states in intracellular medium, allowing potential interaction with redox enzymes. Copper(II) chelates (cassiopeins) have been evaluated as cytotoxic agents towards human lung cancer cells H157 and A549. Complex 54 (Cas IIgly, Figure 8) was shown to inhibit glutathione through redox cycling, generating ROS and inducing apoptosis. In both cell lines, Cas IIgly induced a dramatic decrease in intracellular GSH levels, most of which was oxidized to GSSG (IC50 values were in the 2.5–5 μM range) presumably through the reduction of Cu(II) to Cu(I) (Kachadourian et al., 2010). Cyclic(alkyl) (amino)carbene (CAAC) copper complexes 55–57 have been evaluated against series of cancer cells, displaying IC50 values around 0.14–17.4 µM on all cancer lines and TrxR inhibition of up to 52.3% at 10 μM (Bertrand et al., 2017).
Copper complexes bearing thiosemicarbazone ligands have also been studied as cytotoxic agents (Richardson et al., 2006; c; Kalinowski et al., 2009). Compound 58 allowed for a notable GSH depletion and lysosomal damage causing apoptosis, and was able to modify the GSH/GSSG ratio from 0 to 7% of the control, corroborating an important redox activity. Complex 58 showed an IC50 of 5 μM towards SK-N-MC cells (Lovejoy et al., 2011; Park et al., 2016).
Platinum complexes
The anticancer activity of platinum(II) complexes has generally been associated to cross-linking with the nitrogen bases of DNA, forming adducts that inhibit replication and generate strand breaks and miscoding, causing apoptosis and inhibition of RNA and protein synthesis (Ortega et al., 2021). However, DNA interactions are not the only mechanisms and targeting cytosolic proteins is also important for inducing apoptosis (Zhou et al., 2002; Ortega et al., 2021). For instance, TrxR can interact with platinum compounds and cisplatin-derivatized TrxR can provoke apoptosis in cancer cells. 4) In order to reduce the side effects and drug resistance caused by Pt(II), the use of Pt(IV) complexes has been evaluated (Hall et al., 2007). Such platinum(IV) derivatives are pro-drugs that can be reduced intracellularly to the corresponding active Pt(II) compound (Johnstone et al., 2016; Olszewski et al., 2011; e). Thus, the design of new Pt(IV) compounds displaying high cellular uptake and sensitivity to reduction by enzymes overexpressed in cancer has been highlighted (Czarnomysy et al., 2021; Zhong et al., 2020; 1; Wexelblatt and Gibson, 2012). Four octahedral Pt(IV) compounds have entered clinical trials (tetraplatin, iproplatin, satraplatin, and LA-12, Figure 9). Unfortunately, LA-12 failed in phase I trials, while tetraplatin showed high neurotoxicity and was not investigated after phase I. Iproplatin showed limited benefits in phase II trials, and studies on the orally available satraplatin were dropped in phase III (Nagyal et al., 2020; Chunyan et al., 2021).
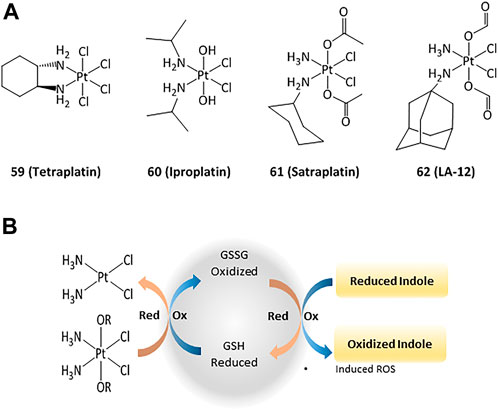
FIGURE 9. Pt(IV) anticancer drugs that have entered clinical trials (A) and redox interaction of Pt(IV) complexes and glutathione (B).
Studies on the possible routes of action of such platinum derivatives showed that the coordination of carboxylic acid ligands as redox modulators in the axial positions of the Pt(IV) center enhanced the antiproliferative effects through simultaneous DNA interactions and generation of ROS (Figure 9) (Wangpaichitr et al., 2021; Tolan et al., 2016; f)
Platinum(II) derivatives can also exhibit redox activity in biological systems. For example, cisplatin and transplatin monochlorido analogs with heterocyclic acylhydrazones (63, Figure 10) inhibited bovine GPx-1 and murine TrxR-1 and exhibited higher cytotoxicity than cisplatin and transplatin (Lemmerhirt et al., 2018). The IC50 towards various cancer cell lines were in the 0.7–22.8 μM range. Complexes 63a - 63f exhibited higher activity than cisplatin and transplatin, with inhibition higher than 50% towards TrxR observed at 25 μM. In addition to their DNA-intercalating capacity, terpyridine-platinum(II) complexes 64 also targeted TrxR (Lo et al., 2009).
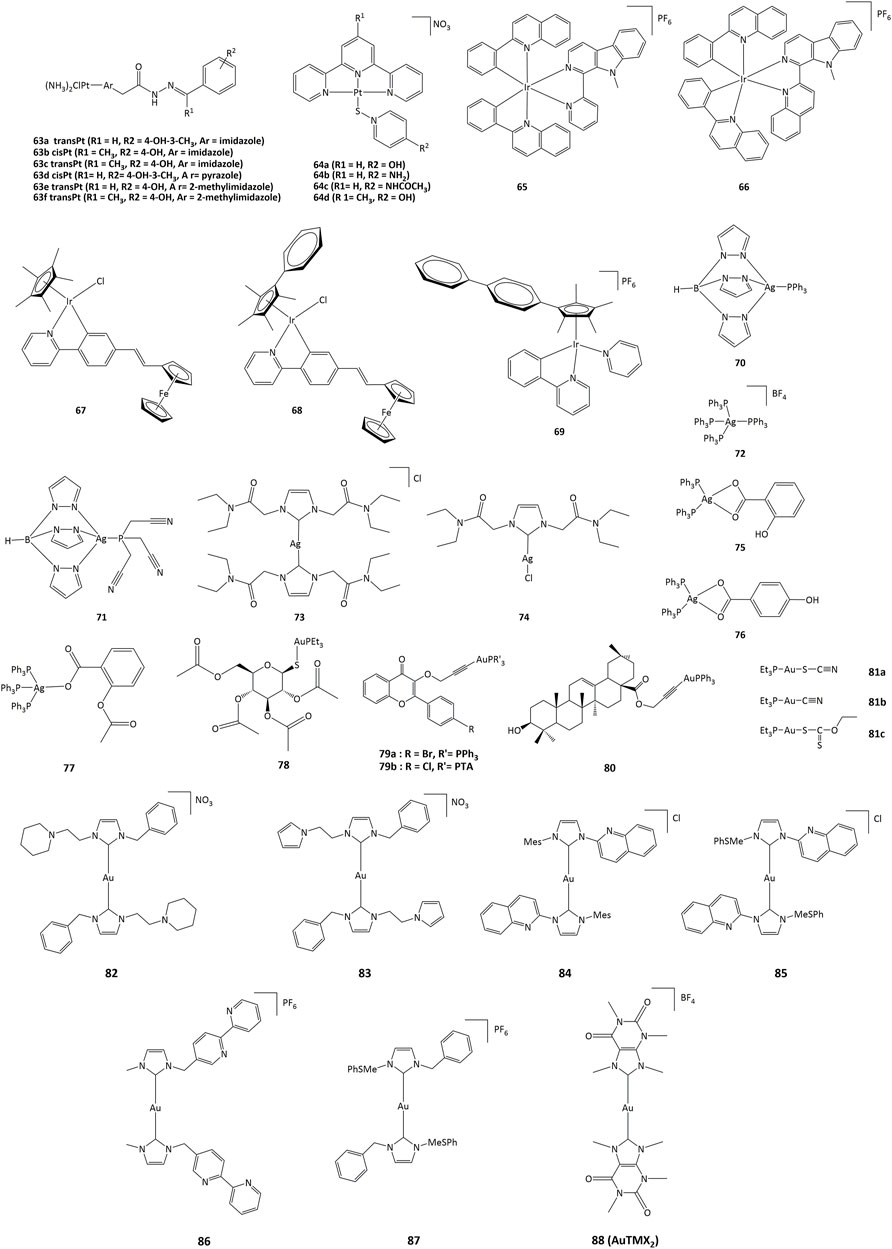
FIGURE 10. Platinum, iridium, silver and gold compounds studied as potential enzymes inhibitors.
Iridium complexes
Iridium(III) derivatives can participate in cellular redox reactions and inhibit proteins, and the use of cyclometalated ligands or the substitution of small counter-anions by larger ones allowed for the synthesis of highly cytotoxic compounds (Wang et al., 2021; g; Mou et al., 2017; Zhang et al., 2018a; Pettinari et al., 2015; Venkatesh et al., 2017) (Cao et al., 2013; Leung et al., 2013; g) (Mou et al., 2017; Prieto-Castañeda et al., 2022). The anticancer activities of such iridium complexes are generated by different mechanisms, such as catalytic interference with cellular redox balance, (Li et al., 2017), interactions with protein kinases, (Du et al., 2018), and regulation of non-apoptotic pathways (Novakova et al., 2003; Wilbuer et al., 2010). Additionally, cyclometalated iridium(III) complexes are efficient photosensitizers (PSs) capable to generate singlet oxygen (1O2), allowing for their potential application in PDT. (Lv et al., 2016; Nam et al., 2016). Other cytotoxic ROS such as superoxide anion (O2˙−) and hydroxyl radicals (˙OH) have also been produced (Novohradsky et al., 2019). The activity of cyclometalated complexes 65 and 66 (Figure 10) against lung cancer cells (A549) increased remarkably after irradiation at 425 nm, with a phototoxicity index between 93 and 120 (Qin et al., 2020). Cell-based assays showed that 66 produced a rise in intracellular ROS concentrations, reduction in ATP production, mitochondrial DNA damage, increase in lipid peroxidation, and inhibition of proteasomal activity (Qin et al., 2020).
Ferrocenyl-substituted half-sandwich iridium(III) cyclometalated-phenylpyridine complexes showed a higher cytotoxic activity than cisplatin. Notably, these bimetallic iridium–iron (67–69) derivatives were more active against A549, Hela, and HepG2 cells than their respective monometallic iridium and ferrocene compounds (Ge et al., 2019). Such activity has been explained by the easy conversion of NADH to NAD+ through hydride transfer by the Ir(III)Cp* group to form iridium-hydride species. The hydride can further be transferred to oxygen to form H2O2 (Lu and Holmgren, 2012; Liu et al., 2014).
Silver complexes
The activity of silver complexes against bacteria and cancer cells can be associated to their solubility and stability in water, lipophilicity, redox properties and rate of release of silver ions (Gandin and Fernandes, 2015; Medici et al., 2016; Chenga and Qi, 2017). Homoleptic and heteroleptic phosphine silver(I) complexes 70–72 (Figure 10) selectively inhibit the selenoenzyme thioredoxin reductase both as an purified enzyme and in human ovarian cancer cells, with inhibition concentration values in the nanomolar range, causing disruption of cellular thiol-redox homeostasis and apoptosis (Dammak et al., 2020). Silver complexes 73 and 74 bearing NHC ligands have also been studied against series of cancer cell lines, with IC50 values in the range of 16–24 μM in cisplatin-resistant cells. Such silver(I) complexes also displayed TrxR inhibition with concentrations in the nanomolar range (Pellei et al., 2012).
Another series of triphenylphosphine complexes (75–77) inhibited the lipoxygenase enzyme (LOX) with IC50 values of 2.3, 7.6 and 7.2 µM for 75–77 complexes, respectively. Complex 75 presented IC50 values of 1.6 μM against leiomyosarcoma cells (LMS) and 2.5 μM for human breast adenocarcinoma (MCF-7). Compound 76 presented IC50 values of 1.6 and 2.0 μM for LMS and MCF-7, respectively, while IC50 for compound 77 were 1.5 and 1.6 μM for LMS and MCF-7 (Poyraz et al., 2011; Banti et al., 2012).
Gold complexes
Among gold complexes, auranofin (78, Figure 10), is of special importance. Auranofin was approved by the FDA for the treatment of rheumatoid arthritis in 1985 and is currently evaluated for applications in neurodegenerative diseases, acquired immunodeficiency syndrome, parasitic and bacterial infection, as well as anticancer agent. The routes of action of gold compounds often involve enzyme inhibition, and the anticancer activity of auronafin has mainly been attributed to the inhibition of TrxR enzyme (IC50 = 82.6 nM) (Marzano et al., 2007).
Gold(I) complexes 79 bearing flavone-derived ligands displayed anticancer activity towards undifferentiated Caco-2 and MCF-7 cells with IC50 values lower than cisplatin and similar to auranofin. The IC50 values for compounds 78 and 79 towards undifferentiated Caco-2 cancer cell line were 1.52 and 2.33 μM, respectively. The cytotoxicity of complexes 79 can be associated with the inhibition of cyclooxygenase 1/2 enzyme and alteration of the activity of thioredoxin reductase and glutathione reductase (Mármol et al., 2019). Gold(I) complex 80 with oleanolic acid-derived ligand can provoke apoptosis in ovarian cancer A2780 cells through different mechanisms, such as induction of ER stress and inhibition of TrxR. Gold(I) compounds bearing pentacyclic triterpene ligand are able to inhibit the TrxR enzyme with an IC50 value of 2.61 μM, while free pentacyclic triterpene showed an IC50 > 50 μM. Complex 80 was active against A2780 cells, with IC50 of 10.24 μM (Mianli et al., 2020).
Linear gold(I) complexes bearing triethylphosphine and cyanate (81a), thiocyanate (81b) or ethylxanthate (81c) ligands were able to inhibit TrxR1 and TrxR2. The IC50 values towards TrxR1 were 1.1, 1.8 and 0.7 nM, and 7.8, 5.0 and 3.6 nM for TrxR2, for 81a—81c, respectively. Complexes 81a—81c presented IC50 values of approximately 80 and 2-fold lower than those of cisplatin and auranofin, respectively, towards different cancer cell lines such as HCT-15 (IC50 = 0.32, 0.08 and 0.61 μM for 81a-c) and HeLa (IC50 = 0.18, 0.09 and 0.13 μM) (Gandin et al., 2010). On the other hand, gold(I) NHC complexes 82–87 were active against A2780cis, A2780, HepG2, HepAD38 and MDCK cancer cell lines with IC50 values in the 0.11–5 μM range (Schuh et al., 2012; Hickey et al., 2008; Karaca et al., 2017; h). The cytotoxic activity of these compounds has been associated to the ability to generate cell cycle arrest through different pathways, particularly via the inhibition of TrxR. The cellular activity of TrxR was reduced by about 55–60% in A549 lung cancer cells upon treatment with 2.5 μM of the gold derivatives (82–87) (Zhang et al., 2018b).
Recently, Gerner et al. showed that the cationic bis-NHC gold(I) complex [Au(9-methylcaffeine-8-ylidene)2]+ 88) can display multimodal activity in ovarian cancer cells. It was demonstrated that 88 affects nuclear and telomeric proteins. It also affects actin, leading to the induction of Nrf2 genes, in parallel with the production of GSH. Treatment of cancer cells with 88 also led to a 2-fold reduction in the ratio of reduced to oxidized glutathione (Meier-Menches et al., 2020).
Conclusions
In this review, we highlighted recent developments on the use of transition metal complexes as anticancer agents acting through changes in the intracellular redox balance and interaction with redox enzymes. The tuning of the redox properties of the complexes through the rational design of the ligands and judicious choice of metal and oxidation state is crucial for their ability to interact with redox active enzymes, resulting in increased biological and anticancer activities. Although the exact mechanisms of action for the cytotoxicity exerted by such metal derivatives are not always unquestionably determined, evaluating the roles played by redox interactions provides essential information that would allow to prepare more effective and selective antineoplastic drugs. To reach this goal, an extensive effort has to be taken using unbiased approaches (proteomic, transcriptomic, metabolomic) to compare the activity of a wide range of metal complexes and identify the direct interactants, regulated pathways and metabolites that are impacted by those compounds. Such methodology will allow to decipher without bias the physico-chemical determinants that drive the cytotoxicity and redox impact of metal complexes on cells. In parallel, biologists and oncologists need to further elucidate how cancer cells adapt to the metabolic challenges raised by tumor growth, aiming at identifying novel druggable targets for metal-based molecules.
Author contributions
MM, CG, and RL contributed in the literature research, design, writing and revision of the manuscript.
Acknowledgments
We are grateful to all colleagues and students who have been involved in our works on the anticancer activity of cyclometalated complexes. Their names are listed in the references. We also wish to thank DGAPA–UNAM (projects IN-207419 and IN-211522), and CONACyT (A1-S-15068), ITI Innovec, Ligue contre le Cancer, Itmo Cancer, ARC, INCa for funding our research projects on elucidating the mechanisms of action of transition metals complexes.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agonigi, G., Riedel, T., Gay, M. P., Biancalana, L., Oñate, E., Dyson, P. J., et al. (2016). Arene osmium complexes with ethacrynic acid-modified ligands: Synthesis, characterization, and evaluation of intracellular glutathione S-transferase inhibition and antiproliferative activity. Organometallics 35, 1046–1056. doi:10.1021/acs.organomet.6b00197
Alessio, E., and Messori, L. (2019). NAMI-A and KP1019/1339, two iconic ruthenium anticancer drug candidates face-to-face: A case story in medicinal inorganic chemistry. Molecules 24, 1995. doi:10.3390/molecules24101995
Allen, J. F., Alexciev, K., and Håkansson, G. (1995). Photosynthesis: Regulation by redox signalling. Curr. Biol. 5, 869–872. doi:10.1016/S0960-9822(95)00176-X
Allocati, N., Masulli, M., Ilio, C. D., and Federici, L. (2018). Glutathione transferases: Substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis 7, 8. doi:10.1038/s41389-017-0025-3
Alpeeva, I. S., Soukharev, V. S., Alexandrova, L., Shilova, N. V., Bovin, N. V., Csoregi, E., et al. (2003). Cyclometalated ruthenium(II) complexes as efficient redox mediators in peroxidase catalysis. J. Biol. Inorg. Chem. 8, 683–688. doi:10.1007/s00775-003-0467-2
Anand, R., Maksimoska, J., Pagano, N., Wong, E. Y., Gimotty, P. A., Diamond, S. L., et al. (2009). Toward the development of a potent and selective organoruthenium mammalian sterile 20 kinase inhibitor. J. Med. Chem. 52, 1602–1611. doi:10.1021/jm8005806
Anesta, K., and Arnér, E. S. J. (2003). Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine. J. Biol. Chem. 278, 15966–15972. doi:10.1074/jbc.M210733200
Ang, W. H., De Luca, A., Chapuis-Bernasconi, C., Juillerat-Jeanneret, L., Bello, M., Dyson, P. J., et al. (2007). Organometallic ruthenium inhibitors of glutathione-S-transferase P1-1 as anticancer drugs. ChemMedChem 2, 1799–1806. doi:10.1002/cmdc.200700209
Ang, W. H., and Dyson, P. J. (2006). Classical and non-classical ruthenium-based anticancer drugs: Towards targeted chemotherapy. Eur. J. Inorg. Chem. 2006, 4003–4018. doi:10.1002/ejic.200600723
Anthony, E. J., Bolitho, E. M., Bridgewater, H. E., Carter, O. W. L., Donnelly, J. M., Imberti, C., et al. (2020). Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 48, 12888–12917. doi:10.1039/d0sc04082g
Antunes, F., and Cadenas, E. (2000). Estimation of H2O2 gradients across biomembranes. FEBS Lett. 475, 121–126. doi:10.1016/s0014-5793(00)01638-0
Baier, D., Schoenhacker-Alte, B., Rusz, M., Pirker, C., Mohr, T., Mendrina, T., et al. (2022). The anticancer ruthenium compound bold-100 targets glycolysis and generates a metabolic vulnerability towards glucose deprivation. Pharmaceutics 14, 238. doi:10.3390/pharmaceutics14020238
Bandeira, S., Gonzalez-Garcia, J., Pensa, E., Albrecht, T., and Vilar, R. (2017). A redox-activated g-quadruplex DNA binder based on a platinum(IV)–salphen complex. Angew. Chem. Int. Ed. 57, 310–313. doi:10.1002/anie.201709968
Banothile, C. E., Makhubela, M. M., and Smith, G. S. (2014). Evaluation of trimetallic Ru(II)- and Os(II)-Arene complexes as potential anticancer agents. J. Organomet. Chem. 772-773, 229–241. doi:10.1016/j.jorganchem.2014.08.034
Banti, C. N., Giannoulis, A. D., Kourkoumelis, N., Owczarzak, A. M., Poyraz, M., Kubicki, M., et al. (2012). Mixed ligand–silver(I) complexes with anti-inflammatory agents which can bind to lipoxygenase and calf-thymus DNA, modulating their function and inducing apoptosis. Metallomics 4, 545. doi:10.1039/c2mt20039b
Banti, C. N., and Hadjikakou, S. K. (2016). Non-steroidal anti-inflammatory drugs (NSAIDs) in metal complexes and their effect at the cellular level. Eur. J. Inorg. Chem. 19, 3048–3071. doi:10.1002/ejic.201501480
Barrera, G. (2012). Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 1–21. doi:10.5402/2012/137289
Batinic-Haberle, I., Rajic, Z., Tovmasyan, A., Reboucas, J. S., Ye, X., Leong, K. W., et al. (2011). Diverse functions of cationic Mn(III) Nsubstituted pyridylporphyrins, recognized as SOD mimics. Free Radic. Biol. Med. 51, 1035–1053. doi:10.1016/j.freeradbiomed.2011.04.046
Batinic-Haberle, I., Spasojevic, I., Tse, H. M., Tovmasyan, A., Rajic, Z., Clair, D. K., et al. (2012). Design of Mn porphyrins for treating oxidative stress injuries and their redox-based regulation of cellular transcriptional activities. Amino Acids 42, 95–113. doi:10.1007/s00726-010-0603-6
Baud, V., and Karin, M. (2009). Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8, 33–40. doi:10.1038/nrd2781
Bautista, H. R., Saavedra-Diaz, R. O., Shen, L. Q., Orvain, C., Gaiddon, C., Le Lagadec, R., et al. (2016). Impact of cyclometalated ruthenium(II) complexes on lactate dehydrogenase activity and cytotoxicity in gastric and colon cancer cells. J. Inorg. Biochem. 163, 28–38. doi:10.1016/j.jinorgbio.2016.07.014
Becuwe, P., Ennen, M., Klotz, R., Barbieux, C., and Grandemange, S. (2014). Manganese superoxide dismutase in breast cancer: From molecular mechanisms of gene regulation to biological and clinical significance. Free Radic. Biol. Med. 77, 139–151. doi:10.1016/j.freeradbiomed.2014.08.026
Benosman, S., Gross, I., Clarke, N., Jochemsen, N. J., Okamoto, K., Loeffler, J.-P., et al. (2007). Multiple neurotoxic stresses converge on MDMX proteolysis to cause neuronal apoptosis. Cell. Death Differ. 14, 2047–2057. doi:10.1038/sj.cdd.4402216
Benosman, S., Meng, X., Von Grabowiecki, Y., Palamiuc, L., Hritcu, L., Gross, I., et al. (2011). Complex regulation of p73 isoforms after alteration of amyloid precursor polypeptide (APP) function and DNA damage in neurons. J. Biol. Chem. 286, 43013–43025. doi:10.1074/jbc.M111.261271
Bergamo, A., and Sava, G. (20072007). Ruthenium complexes can target determinants of tumour malignancy. Dalton Trans., 1267–1272. doi:10.1039/b617769g
Berglund, G. I., Carlsson, G. H., Smith, A. T., Szoke, H., Henriksen, A., Hajdu, J., et al. (2002). The catalytic pathway of horseradish peroxidase at high resolution. Nature 417, 463–468. doi:10.1038/417463a
Bertrand, B., Romanov, A. S., Davis, J., Bochmann, M., Brooks, M., O'Connell, M., et al. (2017). Synthesis, structure and cytotoxicity of cyclic (alkyl)(amino) carbene and acyclic carbene complexes of group 11 metals. Dalton Trans. 46, 15875–15887. doi:10.1039/c7dt03189k
Blanchet, A., Bourgmayer, A., Kurtz, J.-E., Mellitzer, G., and Gaiddon, C. (2021). Isoforms of the p53 Family and Gastric Cancer: A Ménage à Trois for an Unfinished Affair. Cancers 13, 916. doi:10.3390/cancers13040916
Boff, B., Gaiddon, C., and Pfeffer, M. (2013). Cancer cell cytotoxicity of cyclometalated compounds obtained with osmium(II) complexes. Inorg. Chem. 52, 2705–2715. doi:10.1021/ic302779q
Boodram, J. N., Mcgregor, I. J., Bruno, P. M., Cressey, P. B., Hemann, M. T., Suntharalingam, K., et al. (2016). Breast cancer stem cell potent copper(II)-Non-Steroidal anti-inflammatory drug complexes. Angew. Chem. Int. Ed. 55, 2845–2850. doi:10.1002/anie.201510443
Boy, N., Kaixin, L., Yixian, Z., Ting, W., Guilan, Q., Xin, P., et al. (2021). Application of glutathione depletion in cancer therapy: Enhanced ROS-based therapy, ferroptosis, and chemotherapy. Biomaterials 277, 121110. doi:10.1016/j.biomaterials.2021.121110
Cao, R., Jia, J. L., Ma, X. C., Zhou, M., and Fei, H. (2013). Membrane localized iridium(III) complex induces endoplasmic reticulum stress and mitochondria-mediated apoptosis in human cancer cells. J. Med. Chem. 56, 3636–3644. doi:10.1021/jm4001665
Chen, C. C., Chu, C.-B., Liu, K.-J., Huang, C.-Y. F., Chang, J.-Y., Pan, W. Y., et al. (2013). Gene expression profiling for analysis acquired oxaliplatin resistant factors in human gastric carcinoma TSGH-S3 cells: The role of IL-6 signaling and Nrf2/AKR1C axis identification. Biochem. Pharmacol. 86, 872–887. doi:10.1016/j.bcp.2013.07.025
Chen, J., Adikari, M., Pallai, R., Parekh, H. K., and Simpkins, H. (2008). Dihydrodiol dehydrogenases regulate the generation of reactive oxygen species and the development of cisplatin resistance in human ovarian carcinoma cells. Cancer Chemother. Pharmacol. 61, 979–987. doi:10.1007/s00280-007-0554-0
Chen, S. H., and Chang, J. Y. (2019). New insights into mechanisms of cisplatin resistance: From tumor cell to microenvironment. Int. J. Mol. Sci. 20, 4136. doi:10.3390/ijms20174136
Chenga, Y., and Qi, Y. (2017). Current progresses in metal-based anticancer complexes as mammalian TrxR inhibitors. Anticancer. Agents Med. Chem. 17, 1046–1069. doi:10.2174/1871520617666170213150217
Chow, M. J., Licona, C., Wong, D. Y. Q., Pastorin, G., Gaiddon, C., Ang, W. H., et al. (2014). Discovery and investigation of anticancer ruthenium-arene schiff-base complexes via water-promoted combinatorial three-component assembly. J. Med. Chem. 57, 6043–6059. doi:10.1021/jm500455p
Chow, M. J., Licona, C., Wong, D. Y. Q., Pastorin, G., Gaiddon, C., Ang, W. H., et al. (2016). Structural tuning of organoruthenium compounds allows oxidative switch to control ER stress pathways and bypass multidrug resistance. Chem. Sci. 7, 4117–4124. doi:10.1039/c6sc00268d
Chunyan, J., Glen, B. D., Yingjie, Z., and Chuanzhu, G. (2021). Platinum(IV) antitumor complexes and their nano-drug delivery. Coord. Chem. Rev. 429, 213640. doi:10.1016/j.ccr.2020.213640
Coleman, J., Sjoberg, D. D., Demac, Q., Odea, C., McGill, C., Tracey, C., et al. (2021). Mp46-04 Results of the pcm-204 phase 2b trial of partial-gland ablation for men with intermediate-risk prostate cancer with Padeliporfin (Wst11 or Tookad) vascular-targeted photodynamic therapy. J. Urology 39, 17006. doi:10.1097/JU.0000000000002067.04
Czarnomysy, R., Radomska, D., Szewczyk, O. K., Roszczenko, P., and Bielawski, K. (2021). Platinum and palladium complexes as promising sources for antitumor treatments. Int. J. Mol. Sci. 22, 8271. doi:10.3390/ijms22158271
Dammak, K., Porchia, M., De Franco, M., Zancato, M., Naïli, H., Gandin, V., et al. (2020). Antiproliferative homoleptic and heteroleptic phosphino silver(I) complexes: Effect of ligand combination on their biological mechanism of action. Molecules 25, 5484. doi:10.3390/molecules25225484
Defelipe, L. A., Lanzarotti, E., Gauto, D., Marti, M. A., and Turjanski, A. G. (2015). Protein topology determines cysteine oxidation fate: The case of sulfenyl amide formation among protein families. PLoS Comput. Biol. 11, 1004051. doi:10.1371/journal.pcbi.1004051
Della, . P. E. A., Elliott, M., Jones, D. D., and Macdonald, J. E. (2011). Orientation-dependent electron transport in a single redox protein. ACS Nano 6, 355–361. doi:10.1021/nn2036818
Dilruba, S., and Kalayda, G. V. (2016). Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 77, 1103–1124. doi:10.1007/s00280-016-2976-z
Dougan, S. J., Habtemariam, A., McHale, S. E., Parsons, S., and Sadler, P. J. (2008). Catalytic organometallic anticancer complexes. Proc. Natl. Acad. Sci. U. S. A. 105, 11628–11633. doi:10.1073/pnas.0800076105
Du, Q., Guo, L. H., Tian, M., Ge, X. X., Yang, Y. L., Jian, X. Y., et al. (2018). Potent half-sandwich iridium(III) and ruthenium(II) anticancer complexes containing a P^O-chelated ligand. Organometallics 37, 2880–2889. doi:10.1021/acs.organomet.8b00402
Englinger, B., Pirker, C., Heffeter, P., Terenzi, A., Kowol, C. R., Keppler, B. K., et al. (2019). Metal drugs and the anticancer immune response. Chem. Rev. 119, 1519–1624. doi:10.1021/acs.chemrev.8b00396
Fan, R., Bian, M., Hu, L., and Liu, W. (2019). A new rhodium(I) NHC complex inhibits TrxR: In vitro cytotoxicity and in vivo hepatocellular carcinoma suppression. Eur. J. Med. Chem. 183, 111721. doi:10.1016/j.ejmech.2019.111721
Feng, J., Du, X., Liu, H., Sui, X., Zhang, C., Tang, Y., et al. (2014). Manganese-mefenamic acid complexes exhibit high lipoxygenase inhibitory activity. Dalton Trans. 43, 10930–10939. doi:10.1039/C4DT01111B
Férnandez-Gallardo, J., Elie, B. T., Sanaú, M. S., and Contel, M. (2016). Versatile synthesis of cationic N-heterocyclic carbene–gold(I) complexes containing a second ancillary ligand. Design of heterobimetallic ruthenium–gold anticancer agents. Chem. Commun. 52, 3155–3158. doi:10.1039/C5CC09718E
Finkel, T. (2003). Oxidant signals and oxidative stress. Curr. Opin. Cell. Biol. 15, 247–254. doi:10.1016/s0955-0674(03)00002-4
Fu, Y., Romero, M. J., Habtemariam, A., Snowden, M. E., Song, L., Clarkson, G. J., et al. (2012). The contrasting chemical reactivity of potent isoelectronic iminopyridine and azopyridine osmium(II) arene anticancer complexes. Chem. Sci. 3, 2485. doi:10.1039/C2SC20220D
Gaiddon, C., Gross, I., Meng, X., Sidhoum, M., Mellitzer, G., Romain, B., et al. (2021). Bypassing the resistance mechanisms of the tumor ecosystem by targeting the endoplasmic reticulum stress pathway using ruthenium- and osmium-based organometallic compounds: An exciting long-term collaboration with dr. Michel pfeffer. Molecules 26, 5386. doi:10.3390/molecules26175386
Gaiddon, C., Jeannequin, P., Bischoff, P., Pfeffer, M., Sirlin, C., Loeffler, J. P., et al. (2005). Ruthenium(II)-derived organometallic compounds induce cytostatic and cytotoxic effects on mammalian cancer cell lines through p53-dependent and p53-independent mechanisms. J. Pharmacol. Exp. Ther. 315, 1403–1411. doi:10.1124/jpet.105.089342
Galasso, M., Gambino, S., Romanelli, M. G., Donadelli, M., and Scupoli, M. T. (2021). Browsing the oldest antioxidant enzyme: Catalase and its multiple regulation in cancer. Free Radic. Biol. Med. 172, 264–272. doi:10.1016/j.freeradbiomed.2021.06.010
Galluzzi, L., Senovilla, L., Vitale, I., Michels, J., Martins, I., Keep, O., et al. (2012). Molecular mechanisms of cisplatin resistance. Oncogene 31, 1869–1883. doi:10.1038/onc.2011.384
Gamcsik, M. P., Kasibhatla, M. S., Teeter, S. D., and Colvin, O. M. (2012). Glutathione levels in human tumors. Biomarkers 17, 671–691. doi:10.3109/1354750X.2012.715672
Gandin, V., and Fernandes, A. P. (2015). Metal- and semimetal-containing inhibitors of thioredoxin reductase as anticancer agents. Molecules 20, 12732–12756. doi:10.3390/molecules200712732
Gandin, V., Fernandes, A. P., Rigobello, M. P., Dani, B., Sorrentino, F., Tisato, F., et al. (2010). Cancer cell death induced by phosphine gold(I) compounds targeting thioredoxin reductase. Biochem. Pharmacol. 90–101. doi:10.1016/j.bcp.2009.07.023
Garbutcheon, B. S. K., Grant, M. P., Harper, B. W., Krause, A. M., Manohar, M., Orkey, N., et al. (2011). Transition metal based anticancer drugs. Curr. Top. Med. Chem. 11, 521–542. doi:10.2174/156802611794785226
Gaur, K., Vazquez-Salgado, A. M., Duran-Camacho, G., Dominguez-Martinez, I., Benjamin-Rivera, J. A., Fernandez, V. L., et al. (2018). Iron and copper intracellular chelation as an anticancer drug strategy. Inorganics 6, 126. doi:10.3390/inorganics6040126
Ge, X., Chen, S., Liu, X., Wang, Q., Gao, L., Zhao, L., et al. (2019). Ferrocene-appended iridium(III) complexes: Configuration regulation, anticancer application, and mechanism research. Inorg. Chem. 58, 14175–14184. doi:10.1021/acs.inorgchem.9b02227
Giles, G. I. (2006). The redox regulation of thiol dependent signaling pathways in cancer. Curr. Pharm. Des. 12, 4427–4443. doi:10.2174/138161206779010549
Glorieux, C., and Calderon, P. B. (2018). Catalase down-regulation in cancer cells exposed to arsenic trioxide is involved in their increased sensitivity to a pro-oxidant treatment. Cancer Cell. Int. 18, 24. doi:10.1186/s12935-018-0524-0
Graf, N., and Lippard, S. J. (2012). Redox activation of metal-based prodrugs as a strategy for drug delivery. Adv. Drug Deliv. Rev. 64, 993–1004. doi:10.1016/j.addr.2012.01.007
Guiseppi-Elie, A., Lei, C. H., and Baughman, R. H. (2002). Direct electron transfer of glucose oxidase on carbon nanotubes. Nanotechnology 13, 559–564. doi:10.1088/0957-4484/13/5/303
Hall, M. D., Mellor, H. R., Callaghan, R., and Hambley, T. W. (2007). Basis for design and development of platinum(IV) anticancer complexes. J. Med. Chem. 50, 3403–3411. doi:10.1021/jm070280u
Herchel, R., Sindelar, Z., Travnicek, Z., Zboril, R., and Vanco, J. (2009). Novel 1D chain Fe(III)-salen-like complexes involving anionic heterocyclic N-donor ligands. Synthesis, X-ray structure, magnetic, 57Fe Mössbauer, and biological activity studies. Dalton Trans. 49, 9870–9880. doi:10.1039/b912676g
Hickey, J. L., Ruhayel, R. A., Barnard, P. J., Baker, M. V., Berners-Price, S. J., Filipovska, A., et al. (2008). Mitochondria-targeted chemotherapeutics: The rational design of gold(I) N-heterocyclic carbene complexes that are selectively toxic to cancer cells and target protein selenols in preference to thiols. J. Am. Chem. Soc. 130, 12570–12571. doi:10.1021/ja804027j
Hussain, S. P., Hofseth, L. J., and Harris, C. C. (2003). Radical causes of cancer. Nat. Rev. Cancer 3, 276–285. doi:10.1038/nrc1046
Imberti, C., Zhang, P., Huang, H., and Sadler, P. J. (2020). New designs for phototherapeutic transition metal complexes. Angew. Chem. Int. Ed. 59, 61–73. doi:10.1002/anie.201905171
Irwin, M. E., Valle, N. R. D., and Chandra, J. (2013). Redox control of leukemia: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 18, 1349–1383. doi:10.1089/ars.2011.4258
Jacob, C., Battaglia, E., Burkholz, T., Peng, D., Bagrel, D., Montenarh, M., et al. (2012). Control of oxidative posttranslational cysteine modifications: From intricate chemistry to widespread biological and medical applications. Chem. Res. Toxicol. 25, 588–604. doi:10.1021/tx200342b
Jaramillo, M. C., Briehl, M. M., Batinic-Haberle, I., and Tome, M. E. (2015). Manganese(III) meso-tetrakis N-ethylpyridinium-2-yl porphyrin acts as a pro-oxidant to inhibit electron transport chain proteins, modulate bioenergetics, and enhance the response to chemotherapy in lymphoma cells. Free Radic. Biol. Med. 83, 89–100. doi:10.1016/j.freeradbiomed.2015.01.031
Jaramillo, M. C., Briehl, M. M., Crapo, J. D., Batinic-Haberle, I., and Tome, M. E. (2012). Manganese porphyrin, MnTE-2-PyP5+ acts as a pro-oxidant to potentiate glucocorticoid-induced apoptosis in lymphoma cells. Free Radic. Biol. Med. 52, 1272–1284. doi:10.1016/j.freeradbiomed.2012.02.001
Jaramillo, M. C., and Zhang, D. D. (2013). The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes. Dev. 27, 2179–2191. doi:10.1101/gad.225680.113
Jin, M. S., Oldham, M. L., Zhang, Q., and Chen, J. (2012). Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569. doi:10.1038/nature11448
Johnstone, T. C., Suntharalingam, K., and Lippard, S. J. (2016). The next generation of platinum drugs: Targeted Pt(II) agents, nanoparticle delivery, and Pt(IV) prodrugs. Chem. Rev. 116, 3436–3486. doi:10.1021/acs.chemrev.5b00597
Jones, D. P., Go, Y. M., Anderson, C. L., Ziegler, T. R., Kinkade, J. M., Kirlin, W. G., et al. (2004). Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J. 18, 1246–1248. doi:10.1096/fj.03-0971fje
Jungwirth, U., Kowol, C. R., Keppler, B. K., Hartinger, C. G., Berger, W., Heffeter, P., et al. (2011). Anticancer activity of metal complexes: Involvement of redox processes. Antioxid. Redox Signal. 15, 1085–1127. doi:10.1089/ars.2010.3663
Kachadourian, R., Brechbuhl, H. M., Ruiz-Azuara, L., Gracia-Mora, I., and Day, B. J. (2010). Casiopeína IIgly-induced oxidative stress and mitochondrial dysfunction in human lung cancer A549 and H157 cells. Toxicology 268, 176–183. doi:10.1016/j.tox.2009.12.010
Kalinowski, D. S., Quach, P., and Richardson, D. R. (2009). Thiosemicarbazones: The new wave in cancer treatment. Future Med. Chem. 1, 1143–1151. doi:10.4155/fmc.09.80
Karaca, Ö., Scalcon, V., Meier-Menches, S. M., Bonsignore, R., Brouwer, J., Tonolo, F., et al. (2017). Characterization of hydrophilic gold(I) N-heterocyclic carbene (NHC) complexes as potent TrxR inhibitors using biochemical and mass spectrometric approaches. Inorg. Chem. 56, 14237–14250. doi:10.1021/acs.inorgchem.7b02345
King, A. P., and Wilson, J. J. (2020). Endoplasmic reticulum stress: An arising target for metal-based anticancer agents. Chem. Soc. Rev. 49, 8113–8136. doi:10.1039/D0CS00259C
Klajner, M., Licona, C., Fetzer, L., Hebraud, P., Mellitzer, G., Pfeffer, M., et al. (2014). Subcellular localization and transport kinetics of ruthenium organometallic anticancer compounds in living cells: A dose-dependent role for amino acid and iron transporters. Inorg. Chem. 53, 5150–5158. doi:10.1021/ic500250e
Kljun, J., Anko, M., Traven, K., Sinreih, M., Pavlič, R., Peršič, S., et al. (2016). Pyrithione-based ruthenium complexes as inhibitors of aldo-keto reductase 1C enzymes and anticancer agents. Dalton Trans. 45, 11791–11800. doi:10.1039/c6dt00668j
Kontoghiorghes, G. J., Kolnagou, A., Kontoghiorghe, C. N., Mourouzidis, L., Timoshnikov, V. A., Polyakov, N. E., et al. (2020). Trying to solve the puzzle of the interaction of ascorbic acid and iron: Redox, chelation and therapeutic implications. Medicines 7, 45. doi:10.3390/medicines7080045
Lanišnik, T., and Penning, T. M. (2014). Role of aldo–keto reductase family 1 (AKR1) enzymes in human steroid metabolism. Steroids 79, 49–63. doi:10.1016/j.steroids.2013.10.012
Le Lagadec, R., Alexandrova, L., Estevez, H., Pfeffer, M., Laurinavičius, V., Razumiene, J., et al. (2006). Bis-Ruthena(III)cycles [Ru(C∩N)2(N∩N)]PF6 as low-potential mediators for PQQ alcohol dehydrogenase (C∩N = 2-phenylpyridinato or 4-(2-tolyl)pyridinato, N∩N = bpy or phen). Eur. J. Inorg. Chem. 14, 2735–2738. doi:10.1002/ejic.200600375
Lemmerhirt, H., Behnisch, S., Bodtke, A., Lillig, C. H., Pazderova, L., Kasparkova, J., et al. (2018). Effects of cytotoxic cis- and trans-diammine monochlorido platinum(II) complexes on selenium-dependent redox enzymes and DNA. J. Inorg. Biochem. 178, 94–105. doi:10.1016/j.jinorgbio.2017.10.011
Leung, C. H., Zhong, H. J., Chan, D. S. H., and Ma, D. L. (2013). Bioactive iridium and rhodium complexes as therapeutic agents. Coord. Chem. Rev. 257, 1764–1776. doi:10.1016/j.ccr.2013.01.034
Li, J. J., Guo, L., Tian, Z., Tian, M., Zhang, S., Xu, K., et al. (2017). Novel half-sandwich iridium(III) imino-pyridyl complexes showing remarkable in vitro anticancer activity. Dalton Trans. 46, 15520–15534. doi:10.1039/C7DT03265J
Li, J. J., Tian, M., Tian, Z., Zhang, S., Yan, C., Shao, C., et al. (2018). Half-sandwich iridium(III) and ruthenium(II) complexes containing P^P-chelating ligands: A new class of potent anticancer agents with unusual redox features. Inorg. Chem. 57, 1705–1716. doi:10.1021/acs.inorgchem.7b01959
Licona, C., Delhorme, J. -B., Riegel, G., Vidimar, V., Cerón-Camacho, R., Boff, B., et al. (2020). Anticancer activity of ruthenium and osmium cyclometalated compounds: Identification of ABCB1 and EGFR as resistance mechanisms. Inorg. Chem. Front. 7, 678–688. doi:10.1039/C9QI01148J
Limia, C. M., Sauzay, C., Urra, H., Hetz, C., Chevet, E., Avril, T., et al. (2019). Emerging roles of the endoplasmic reticulum associated unfolded protein response in cancer cell migration and invasion. Cancers 11, 631. doi:10.3390/cancers11050631
Liu, Z., Romero-Canelón, I., Qamar, B. J., Hearn, M., Habtemariam, A., Barry, N. P. E., et al. (2014). The potent oxidant anticancer activity of organoiridium catalysts. Angew. Chem. Int. Ed. 53, 3941–3946. doi:10.1002/anie.201311161
Liu, Z., and Sadler, P. J. (2014). Organoiridium complexes: Anticancer agents and catalysts. Acc. Chem. Res. 47, 1174–1185. doi:10.1021/ar400266c
Lo, Y.-C., Ko, T.-P., Su, W.-C., Su, T.-L., and Wang, A. H.-J. (2009). Terpyridine-platinum(II) complexes are effective inhibitors of mammalian topoisomerases and human thioredoxin reductase 1. J. Inorg. Biochem. 103, 1082–1092. doi:10.1016/j.jinorgbio.2009.05.006
Lovejoy, D. B., Jansson, P. J., Brunk, U. T., Wong, J., Ponka, P., Richardson, D. R., et al. (2011). Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 71, 5871–5880. doi:10.1158/0008-5472.CAN-11-1218
Lu, J., and Holmgren, A. (2012). Thioredoxin system in cell death progression. Antioxid. Redox Signal. 7, 1738–1747. doi:10.1089/ars.2012.4650
Lv, W., Zhang, Z., Zhang, K. Y., Yang, H., Liu, S., Xu, A., et al. (2016). A mitochondria-targeted photosensitizer showing improved photodynamic therapy effects under hypoxia. Angew. Chem. Int. Ed. 55, 9947–9951. doi:10.1002/anie.201604130
Mandal, P., Kundu, B. K., Vyas, K., Sabu, V., Helen, A., Dhankhar, S. S., et al. (2018). Ruthenium(II) arene NSAID complexes: Inhibition of cyclooxygenase and antiproliferative activity against cancer cell lines. Dalton Trans. 47, 517–527. doi:10.1039/C7DT03637J
Marino, S. M., and Gladyshev, V. N. (2011). Redox biology: Computational approaches to the investigation of functional cysteine residues. Antioxid. Redox Signal. 15, 135–146. doi:10.1089/ars.2010.3561
Mármol, I., Castellnou, P., Alvarez, R., Gimeno, M. C., Rodríguez-Yoldi, M. J., Cerrada, E., et al. (2019). Alkynyl Gold(I) complexes derived from 3-hydroxyflavones as multi-targeted drugs against colon cancer. Eur. J. Med. Chem. 183, 111661. doi:10.1016/j.ejmech.2019.111661
Marzano, C., Gandin, V., Folda, A., Scutari, G., Bindoli, A., Rigobello, M. P., et al. (2007). Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic. Biol. Med. 42, 872–881. doi:10.1016/j.freeradbiomed.2006.12.021
McFarland, S. A., Mandel, A., Dumoulin-White, R., and Gasser, G. (2019). Metal-based photosensitizers for photodynamic therapy: The future of multimodal oncology? Curr. Opin. Chem. Biol. 56, 23–27. doi:10.1016/j.cbpa.2019.10.004
Medici, S., Peana, M., Crisponi, G., Nurchi, V. M., Lachowicz, J. I., Remelli, M., et al. (2016). Silver coordination compounds: A new horizon in medicine. Coord. Chem. Rev. 328, 349–359. doi:10.1016/j.ccr.2016.05.015
Meier-Menches, S. M., Gerner, C., Berger, W., Hartinger, C. G., and Keppler, B. K. (2018). Structure-activity relationships for ruthenium and osmium anticancer agents-towards clinical development. Chem. Soc. Rev. 47, 909–928. doi:10.1039/c7cs00332c
Meier-Menches, S. M., Neuditschko, B., Zappe, K., Schaier, M., Gerner, M. C., Schmetterer, K. G., et al. (2020). An organometallic gold(I) bis-N-heterocyclic carbene complex with multimodal activity in ovarian cancer cells. Chem. Eur. J. 26, 15528–15537. doi:10.1002/chem.202003495
Meng, X., Leyva, M. L., Jenny, M., Gross, I., Benosman, S., Fricker, B., et al. (2009). A Ruthenium-containing organometallic compound reduces tumor growth through induction of the endoplasmic reticulum stress gene CHOP. Cancer Res. 69, 5458–5466. doi:10.1158/0008-5472.CAN-08-4408
Mianli, B., Ying, S., Yuanhao, L., Zhongren, X., Rong, F., Ziwen, L., et al. (2020). A gold(I) complex containing an oleanolic acid derivative as a potential anti-ovarian-cancer agent by inhibiting TrxR and activating ROS-mediated ERS. Chem. Eur. J. 26, 7092–7108. doi:10.1002/chem.202000045
Monro, S., Colón, K. L., Yin, H., Roque, H., Konda, P., Gujar, S., et al. (2019). Transition metal complexes and photodynamic therapy from a tumor-centered approach: Challenges, opportunities, and highlights from the development of TLD1433. Dalton Trans. 119, 797–7823. doi:10.1039/c0dt01816c
Monteiro, H. P., and Stern, A. (1996). Redox modulation of tyrosine phosphorylation-dependent signal transduction pathways. Free Radic. Biol. Med. 21, 323–333. doi:10.1016/0891-5849(96)00051-2
Montero, A. J., and Jassem, J. (2011). Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs 71, 1385–1396. doi:10.2165/11592590-000000000-00000
Mou, Z., Deng, N., Zhang, F., Zhang, J., Cen, J., Zhang, X., et al. (2017). Half-sandwich” Schiff-base Ir(III) complexes as anticancer agents. Eur. J. Med. Chem. 138, 72–82. doi:10.1016/j.ejmech.2017.06.027
Munro, D., and Treberg, J. R. (2017). A radical shift in perspective: Mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 220, 1170–1180. doi:10.1242/jeb.132142
Myers, J. M., Cheng, Q. A., William, E., Kalyanaraman, B., Filipovska, A., Arnér, E. S. J., et al. (2013). Redox activation of Fe(III)–thiosemicarbazones and Fe(III)–bleomycin by thioredoxin reductase: Specificity of enzymatic redox centers and analysis of reactive species formation by ESR spin trapping. Free Radic. Biol. Med. 60, 183–194. doi:10.1016/j.freeradbiomed.2013.02.016
Nagyal, L., Kumar, A., Sharma, R., Yadav, R., Chaudhary, P., Singh, R., et al. (2020). Bioinorganic chemistry of platinum(IV) complexes as platforms for anticancer agents. Curr. Bioact. Compd. 16, 726–737. doi:10.2174/1573407215666190409105351
Nakabeppu, Y., Sakumi, K., Sakamoto, K., Tsuchimoto, D., Tsuzuki, T., Nakatsu, Y., et al. (2006). Mutagenesis and carcinogenesis caused by the oxidation of nucleic acids. Biol. Chem. 387, 373–379. doi:10.1515/BC.2006.050
Nakamura, H., Nakamura, K., and Yodoi, J. (1997). Redox regulation of cellular activation. Annu. Rev. Immunol. 15, 351–369. doi:10.1146/annurev.immunol.15.1.351
Nam, J. S., Kang, M. G., Kang, J., Park, S. Y., Lee, S. J. C., Kim, H. T., et al. (2016). Endoplasmic reticulum-localized iridium(III) complexes as efficient photodynamic therapy agents via protein modifications. J. Am. Chem. Soc. 138, 10968–10977. doi:10.1021/jacs.6b05302
Naranjo-Suarez, S., Carlson, B. A., Tobe, R., Yoo, M. H., Tsuji, P. A., Gladyshev, V. N., et al. (2013). Regulation of HIF-1a activity by overexpression of thioredoxin is independent of thioredoxin reductase status. Mol. Cells 36, 151–157. doi:10.1007/s10059-013-0121-y
Narayanan, D., Ma, S., and Özcelik, D. (2020). Targeting the redox landscape in cancer therapy. Cancers 12, 1706. doi:10.3390/cancers12071706
Ndagi, U., Mhlongo, N., and Soliman, M. E. (2017). Metal complexes in cancer therapy – an update from drug design perspective. Drug Des. devel. Ther. 11, 599–616. doi:10.2147/DDDT.S119488
Needham, R. J., Sanchez-Cano, C., Zhang, X., Romero-Canelón, I., Habtemariam, A., Cooper, M. S., et al. (2017). In-cell activation of organo-osmium(II) anticancer complexes. Angew. Chem. Int. Ed. 56, 1017–1020. doi:10.1002/anie.201610290
Netanya, Y. S., and Robert, C. S. (2019). The warburg effect, lactate, and nearly a century of trying to cure cancer. Semin. Nephrol. 39, 380–393. doi:10.1016/j.semnephrol.2019.04.007
Novakova, O., Chen, H., Vrana, O., Rodger, A., Sadler, P. J., Brabec, V., et al. (2003). DNA interactions of monofunctional organometallic ruthenium(II) antitumor complexes in cell-free media. Biochemistry 42, 11544–11554. doi:10.1021/bi034933u
Novohradsky, V., Rovira, A., Hally, C., Galindo, A., Vigueras, G., Gandioso, A., et al. (2019). Towards novel photodynamic anticancer agents generating superoxide anion radicals: A cyclometalated Ir(III) complex conjugated to a far-red emitting coumarin. Angew. Chem. Int. Ed. 58, 6311–6315. doi:10.1002/anie.201901268
Oakes, S. A. (2017). Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiology-Cell Physiology 312, 93–102. doi:10.1152/ajpcell.00266.2016
Oberley, L. W., Oberley, T. D., and Buettner, G. R. (19801980). Cell differentation, aging and cancer: The possible roles of superoxide and superoxide dismutases. Med. Hypotheses 6, 249–268. doi:10.1016/0306-9877(80)90123-1
Oehninger, L., Stefanopoulou, M., Alborzinia, H., Schur, J., Ludewig, S., Namikawa, S., et al. (2013). Evaluation of arene ruthenium(II) N-heterocyclic carbene complexes as organometallics interacting with thiol and selenol containing biomolecules. Dalton Trans. 42, 1657–1666. doi:10.1039/C2DT32319B
Olszewski, U., Ulsperger, E., Geissler, K., and Hamilton, G. (2011). Comparison of the effects of the oral anticancer platinum(IV) complexes oxoplatin and metabolite cis-diammine-tetrachlorido-platinum(IV) on global gene expression of NCI-H526 Cells. J. Exp. Pharmacol. 3, 43–50. doi:10.2147/JEP.S13630
Ortega, E., Vigueras, G., Ballester, F. J., and Ruiz, J. (2021). Targeting translation: A promising strategy for anticancer metallodrugs. Coord. Chem. Rev. 446, 214129. doi:10.1016/j.ccr.2021.214129
Oshikawa, J., Urao, N., Kim, H. W., Kaplan, N., Razvi, M., McKinney, R., et al. (2010). Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS One 5, 10189. doi:10.1371/journal.pone.0010189
Oun, R., Moussa, Y. E., and Wheate, N. J. (2018). The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 47, 6645–6653. doi:10.1039/c8dt00838h
Park, K. C., Fouani, L., Jansson, P. J., Wooi, D., Sahni, S., Lane, D. J. R., et al. (2016). Copper and conquer: Copper complexes of di-2-pyridylketone thiosemicarbazones as novel anti-cancer therapeutics. Metallomics 8, 874–886. doi:10.1039/c6mt00105j
Parveen, S. (2020). Recent advances in anticancer ruthenium Schiff base complexes. Appl. Organomet. Chem. 34, 5687–5711. doi:10.1002/aoc.5687
Peklak-Scott, C., Smitherman, P. K., Townsed, A. J., and Morrow, C. S. (2008). Role of glutathione S-transferase P1-1 in the cellular detoxification of cisplatin. Mol. Cancer Ther. 1, 3247–3255. doi:10.1158/1535-7163.MCT-08-0250
Pellei, M., Gandin, V., Marinelli, M., Marzano, C., Yousufuddin, M., Dias, H. V. R., et al. (2012). Synthesis and biological activity of ester- and amide-functionalized imidazolium salts and related water-soluble coinage metal N-heterocyclic carbene complexes. Inorg. Chem. 51, 9873–9882. doi:10.1021/ic3013188
Penning, T. M., and Byrns, M. C. (2009). Human aldo–keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch. Biochem. Biophys. 1155, 241–250. doi:10.1016/j.abb.2007.04.024
Pettinari, R., Marchetti, F., Pettinari, C., Condello, F., Petrini, A., Scopelliti, R., et al. (2015). Organometallic rhodium(III) and iridium(III) cyclopentadienyl complexes with curcumin and bisdemethoxycurcumin co-ligands. Dalton Trans. 44, 20523–20531. doi:10.1039/C5DT03037D
Plamthottam, S., Sun, D., Van Valkenburgh, J., Valenzuela, J., Ruehle, B., Steele, D., et al. (2019). Activity and electrochemical properties: Iron complexes of the anticancer drug triapine and its analogs. J. Biol. Inorg. Chem. 24, 621–632. doi:10.1007/s00775-019-01675-0
Pommier, Y. (2006). Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 6, 789–802. doi:10.1038/nrc1977
Poyraz, M., Banti, C. N., Kourkoumelis, N., Dokorou, V., Manos, M. J., Simčič, M., et al. (2011). Synthesis, structural characterization and biological studies of novel mixed ligand Ag(I) complexes with triphenylphosphine and aspirin or salicylic acid, Inorganica Chim. Acta 375, 114–121. doi:10.1016/j.ica.2011.04.032
Prabhulkar, S., Tian, H., Wang, X., Zhu, J. J., and Li, C. Z. (2012). Engineered proteins: Redox properties and their applications. Antioxid. Redox Signal. 17, 1796–1822. doi:10.1089/ars.2011.4001
Prier, C. K., and Kosjek, B. (2019). Recent preparative applications of redox enzymes. Curr. Opin. Chem. Biol. 49, 105–112. doi:10.1016/j.cbpa.2018.11.011
Prieto-Castañeda, A., Lerida-Viso, A., Avellanal-Zaballa, E., Sola-Llano, R., Bañuelos, J., Agarrabeitia, A., et al. (2022). Phosphorogenic dipyrrinato-iridium(III) complexes as photosensitizers for photodynamic therapy. Inorganica Chim. Acta 197, 109886–119943. doi:10.1016/j.ica.2020.119925
Purohit, V., Simeone, D. M., and Lyssiotis, C. A. (2019). Metabolic regulation of redox balance in cancer. Cancers 11, 955. doi:10.3390/cancers11070955
Qin, W. W., Pan, Z. Y., Cai, D. H., Li, Y., and He, L. (2020). Cyclometalated iridium(III) complexes for mitochondria-targeted combined chemophotodynamic therapy. Dalton Trans. 49, 3562–3569. doi:10.1039/D0DT00180E
Quiles, J. L., Sánchez-González, C., Vera-Ramírez, L., Giampieri, F., Navarro-Hortal, M. D., Xiao, J., et al. (2020). Reductive stress, bioactive compounds, redox-active metals, and dormant tumor cell biology to develop redox-based tools for the treatment of cancer. Antioxid. Redox Signal. 20, 860–881. doi:10.1089/ars.2020.8051
Rastogi, R., Geng, X., Li, F., and Ding, Y. (2017). NOX activation by subunit interaction and underlying mechanisms in disease. Front. Cell. Neurosci. 10, 301. doi:10.3389/fncel.2016.00301
Raymond, E., Chaney, S., Taamma, A., and Cvitkovic, E. (1998). Oxaliplatin: A review of preclinical and clinical studies. Ann. Oncol. 9, 1053–1071. doi:10.1023/a:1008213732429
Richardson, D. R., Kalinowski, D. S., Richardson, V., Sharpe, P. C., Lovejoy, D. B., Islam, M., et al. (2009). 2-Acetylpyridine thiosemicarbazones are potent iron chelators and antiproliferative agents: Redox activity, iron complexation and characterization of their antitumor activity. J. Med. Chem. 52, 1459–1470. doi:10.1021/jm801585u
Richardson, D. R., Sharpe, P. C., Lovejoy, D. B., Senaratne, D., Kalinowski, D. S., Islam, M., et al. (2006). Dipyridyl thiosemicarbazone chelators with potent and selective antitumor activity form iron complexes with redox activity. J. Med. Chem. 49, 6510–6521. doi:10.1021/jm0606342
Romero-Canelón, I., and Sadler, P. J. (2013). Next-generation metal anticancer complexes: Multitargeting via redox modulation. Inorg. Chem. 52, 12276–12291. doi:10.1021/ic400835n
Romero-Canelón, I., Salassa, L., and Sadler, P. J. (2013). The contrasting activity of iodido versus chlorido ruthenium and osmium arene azo- and imino-pyridine anticancer complexes: Control of cell selectivity, cross-resistance, p53 dependence, and apoptosis pathway. J. Med. Chem. 56, 1291–1300. doi:10.1021/jm3017442
Roy, J., Galano, J. M., Durand, T., LeGuennec, J. Y., and Lee, J. C. (2017). Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 31, 3729–3745. doi:10.1096/fj.201700170R
Ryabov, A. D., Sukharev, V. S., Alexandrova, L., Le Lagadec, R., and Pfeffer, M. (2001). New synthesis and new bio-application of cyclometalated ruthenium(II) complexes for fast mediated electron transfer with peroxidase and glucose oxidase. Inorg. Chem. 40, 6529–6532. doi:10.1021/ic010423h
Ryabova, E. S., Goral, V. N., Csoregi, E., Mattiasson, B., and Ryabov, A. D. (1999). Coordinative approach to mediated electron transfer: Ruthenium complexed to native glucose oxidase. Angew. Chem. Int. Ed. Engl. 38, 804–807. doi:10.1002/(SICI)1521-3773(19990315)38:6<804::AID-ANIE804>3.0.CO;2-6
Saavedra-Diaz, R. O., Le Lagadec, R., and Ryabov, A. D. (2013). 2-Phenylpyridine ruthenacycles as effectors of glucose oxidase activity: Inhibition by Ru(II) and activation by Ru(III). J. Biol. Inorg. Chem. 18, 547–555. doi:10.1007/s00775-013-0999-z
Schuh, E., Pflüger, C., Citta, A., Folda, A., Rigobello, M. P., Bindoli, A., et al. (2012). Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. J. Med. Chem. 55, 5518–5528. doi:10.1021/jm300428v
Selyutina, O. Y., Kononova, P. A., Koshman, V. E., Shelepova, E. A., Azad, M., Gholam, A. R., et al. (2022). Ascorbate-and iron-driven redox activity of Dp44mT and Emodin facilitates peroxidation of micelles and bicelles. Biochimica Biophysica Acta - General Subj. 1866, 130078. doi:10.1016/j.bbagen.2021.130078
Sharifi-Rad, M., Kumar, N. V., Zucca, P., Varoni, E. M., Dini, L., Panzarini, E., et al. (2020). Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 11, 694. doi:10.3389/fphys.2020.00694
Sheldon, A., and Woodley, J. M. (2018). Role of biocatalysis in sustainable chemistry. Chem. Rev. 118, 801–838. doi:10.1021/acs.chemrev.7b00203
Sohrabi, M., Saeedi, M., Larijani, B., and Mahdavi, M. (2021). Recent advances in biological activities of rhodium complexes: Their applications in drug discovery research. Eur. J. Med. Chem. 216, 113308. doi:10.1016/j.ejmech.2021.113308
Soldevila-Barreda, J. J., Habtemariam, A., Romero-Canelón, I., and Sadler, P. J. (2015). Half-sandwich rhodium(III) transfer hydrogenation catalysts: Reduction of NAD+ and pyruvate, and antiproliferative activity. J. Inorg. Biochem. 153, 322–333. doi:10.1016/j.jinorgbio.2015.10.008
Suntharalingam, K., Johnstone, T. C., Bruno, P. M., Lin, W., Hemann, M. T., Lippard, S. J., et al. (2013). Bidentate ligands on osmium(VI) nitrido complexes control intracellular targeting and cell death pathways. J. Am. Chem. Soc. 135, 14060–14063. doi:10.1021/ja4075375
Suss-Fink, G. (2010). Arene ruthenium complexes as anticancer agents. Dalton Trans. 39, 1673–1688. doi:10.1039/B916860P
Tew, K. D., and Townsend, D. M. (2011). Redox platforms in cancer drug discovery and development. Curr. Opin. Chem. Biol. 15, 156–161. doi:10.1016/j.cbpa.2010.10.016
Thamsen, M., and Jakob, U. (2011). The redoxome: Proteomic analysis of cellular redox networks. Curr. Opin. Chem. Biol. 15, 113–119. doi:10.1016/j.cbpa.2010.11.013
Tolan, D., Gandin, V., Morrison, L., Nahan, A. E., Marzano, C., Montagner, D., et al. (2016). Oxidative stress induced by Pt(IV) pro-drugs based on the cisplatin scaffold and indole carboxylic acids in axial position. Sci. Rep. 6, 29367. doi:10.1038/srep29367
Townsend, D. M., Findlay, V. L., and Tew, K. D. (2005). Glutathione S‐transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 401, 287–307. doi:10.1016/S0076-6879(05)01019-0
Townsend, D. M. (2007). S-Glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Mol. Interv. 7, 313–324. doi:10.1124/mi.7.6.7
Truong, D., Sullivan, M. P., Tong, K. K., Steel, T. R., Prause, A., Lovett, J. H., et al. (2020). Potent inhibition of thioredoxin reductase by the Rh derivatives of anticancer M(areno/Cp*)(NHC)Cl2 complexes. Inorg. Chem. 59, 3281–3289. doi:10.1021/acs.inorgchem.9b03640
Truppo, M. D. (2017). Biocatalysis in the pharmaceutical industry: The need for speed. ACS Med. Chem. Lett. 8, 476–480. doi:10.1021/acsmedchemlett.7b00114
Turner, N. J., and Kumar, R. (2018). Editorial overview: Biocatalysis and biotransformation: The golden age of biocatalysis. Curr. Opin. Chem. Biol. 43, A1–A3. doi:10.1016/j.cbpa.2018.02.012
Vaupel, P., and Multhoff, G. (2021). Revisiting the warburg effect: Historical dogma versus current understanding. J. Physiol. 599, 1745–1757. doi:10.1113/jp278810
Venkatesh, V., Martin, R. V., Wedge, C. J., Canelon, I. R., Cano, C. S., Song, J. I., et al. (2017). Mitochondria-targeted spin-labelled luminescent iridium anticancer complexes. Chem. Sci. 8, 8271–8278. doi:10.1039/c7sc03216a
Vidimar, V., Licona, C., Ceron-Camacho, R., Guerin, E., Coliat, P., Venkatasamy, A., et al. (2019). A redox ruthenium compound directly targets PHD2 and inhibits the HIF1 pathway to reduce tumor angiogenesis independently of p53. Cancer Lett. 440-441, 145–155. doi:10.1016/j.canlet.2018.09.029
Voisinet, M., Venkatasamy, A., Alratrout, H., Delhorme, J.-B., Brigand, C., Rohr, S., et al. (2021). How to prevent sarcopenia occurrence during neoadjuvant chemotherapy for oesogastric adenocarcinoma? Nutr. Cancer 73 (5), 802–808. doi:10.1080/01635581.2020.1770813
Wang, F., Xu, F., Habtemariam, A., Bella, J., and Sadler, P. J. (2005). Competition between glutathione and guanine for a ruthenium(II) arene anticancer complex: Detection of a sulfenato intermediate. J. Am. Chem. Soc. 127, 17734–17743. doi:10.1021/ja053387k
Wang, K., Jiang, J., Lei, Y., Zhou, S., Wei, Y., Huang, C., et al. (2019). Targeting metabolic-redox circuits for cancer therapy. Trends biochem. Sci. 44, 401–414. doi:10.1016/j.tibs.2019.01.001
Wang, L., Guan, R., Xie, L., Liao, X., Xong, K., Rees, T. W., et al. (2021). An ER-targeting iridium(III) complex that induces immunogenic cell death in non-small-cell lung cancer. Angew. Chem. Int. Ed. 133, 4657–4665. doi:10.1002/anie.202013987
Wangpaichitr, M., Theodoropoulos, G., Nguyen, D. J. M., Wu, C., Spector, S. A., Feun, L. G., et al. (2021). Cisplatin resistance and redox-metabolic vulnerability: A second alteration. Int. J. Mol. Sci. 22, 7379. doi:10.3390/ijms22147379
Wexelblatt, E., and Gibson, D. (2012). What do we know about the reduction of Pt(IV) pro-drugs? J. Inorg. Biochem. 117, 220–229. doi:10.1016/j.jinorgbio.2012.06.013
Wilbuer, A., Vlecken, D. H., Schmitz, D. J., Kraling, K., Harms, K., Bagowski, C. P., et al. (2010). Iridium complex with antiangiogenic properties. Angew. Chem. Int. Ed. 49, 3839–3842. doi:10.1002/anie.201000682
Witte, A. B., Anesta, K., Jerremalm, E., Ehrsson, H., and Arnér, E. S. J. (2005). Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic. Biol. Med. 39, 696–703. doi:10.1016/j.freeradbiomed.2005.04.025
Wu, K., Hu, W., Luo, Q., Li, X., Xiong, S., Sadler, P. J., et al. (2013). Competitive binding sites of a ruthenium arene anticancer complex on oligonucleotides studied by mass spectrometry: Ladder-sequencing versus top-down. J. Am. Soc. Mass Spectrom. 24, 410–420. doi:10.1007/s13361-012-0539-z
Xie, L., Luo, Z., Zhao, Z., and Chen, T. (2017). Anticancer and antiangiogenic iron(II) complexes that target thioredoxin reductase to trigger cancer cell apoptosis. J. Med. Chem. 60, 202–214. doi:10.1021/acs.jmedchem.6b00917
Xiong, X., Liu, L.-Y., Mao, Z.-W., and Zou, T. (2021). Approaches towards understanding the mechanism-of-action of metallodrugs. Coord. Chem. Rev. 453, 214311. doi:10.1016/j.ccr.2021.214311
Yoboue, E. D., Sitia, R., and Simmen, T. (2018). Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell. Death Dis. 9, 331. doi:10.1038/s41419-017-0033-4
Zamora, A., Denning, C. A., Heidary, D. K., Wachter, E., Nease, L. A., Ruiz, J., et al. (2017). Ruthenium-containing P450 inhibitors for dual enzyme inhibition and DNA damage. Dalton Trans. 46, 2165–2173. doi:10.1039/C6DT04405K
Zamora, A., Vigueras, G., Rodríguez, V., Santana, M. D., and Ruiz, J. (2018). Cyclometalatediridium(III) luminescent complexes in therapy and phototherapy. Coord. Chem. Rev. 360, 34–76. doi:10.1016/j.ccr.2018.01.010
Zhang, C., Hemmert, C., Gornitzka, H., Cuvillier, O., Zhang, M., Domingo, R. W-Y., et al. (2018). Cationic and neutral N-heterocyclic carbene gold(I) complexes: Cytotoxicity, NCI-60 screening, cellular uptake, inhibition of mammalian thioredoxin reductase, and reactive oxygen species formation. ChemMedChem 13, 1218–1229. doi:10.1002/cmdc.201800181
Zhang, D. D. (2010). The nrf2-keap1-ARE signaling pathway: The regulation and dual function of Nrf2 in cancer. Antioxid. Redox Signal. 13, 1623–1626. doi:10.1089/ars.2010.3301
Zhang, H. R., Guo, L. H., Tian, Z. Z., Tian, M., Zhang, S. M., Xu, Z. S., et al. (2018). Significant effects of counteranions on the anticancer activity of iridium(III) complexes. Chem. Commun. 54, 4421–4424. doi:10.1039/c8cc01326h
Zhang, P., and Sadler, P. J. (2017). Redox-active metal complexes for anticancer therapy. Eur. J. Inorg. Chem. 839, 5–14. doi:10.1002/ejic.201600908
Zhong, Y., Jia, C., Zhang, X., Liao, X., Yang, B., Cong, Y., et al. (2020). Targeting drug delivery system for platinum(Ⅳ)-Based antitumor complexes. Eur. J. Med. Chem. 194, 112229. doi:10.1016/j.ejmech.2020.112229
Keywords: anticancer therapy, oxidoreductases, redox balance, transition metals, tumor metabolism
Citation: Murillo MI, Gaiddon C and Le Lagadec R (2022) Targeting of the intracellular redox balance by metal complexes towards anticancer therapy. Front. Chem. 10:967337. doi: 10.3389/fchem.2022.967337
Received: 12 June 2022; Accepted: 29 June 2022;
Published: 11 August 2022.
Edited by:
Dinorah Gambino, Universidad de la República, UruguayReviewed by:
Alzir Azevedo Batista, Federal University of São Carlos, BrazilSamuel Meier-Menches, University of Vienna, Austria
Copyright © 2022 Murillo, Gaiddon and Le Lagadec. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronan Le Lagadec, cm9uYW5AdW5hbS5teA==