Abstract
Toxicity, defined as the potential harm a substance can cause to living organisms, requires the implementation of stringent regulatory standards to ensure public safety. These standards involve comprehensive testing frameworks, including hazard identification, dose-response evaluation, exposure assessment, and risk characterization. In drug discovery and development, these processes are often complex, time-consuming, and also resource-intensive. Toxicity-related failures in the later stages of drug development can lead to substantial financial losses, underscoring the need for reliable toxicity prediction during the early discovery phases. The advent of computational approaches has accelerated a shift toward in silico modeling, virtual screening, and, notably, artificial intelligence (AI) to identify potential toxicities earlier in the pipeline. Ongoing advances in databases, algorithms, and computational power have further expanded AI’s role in pharmaceutical research. Today, AI models are capable of predicting wide range of toxicity endpoints, such as hepatotoxicity, cardiotoxicity, nephrotoxicity, neurotoxicity, and genotoxicity, based on diverse molecular representations ranging from traditional descriptors to graph-based methods. This review provides an in-depth examination of AI-driven toxicity prediction, emphasizing its transformative impact on drug discovery and its growing importance in improving safety assessments.
1 Introduction
Toxicity refers to the extent to which a substance can cause harm to living organisms, including animals, plants, bacteria, and humans (Duffus, 1993; McNaught and Wilkinson, 2025). While many chemicals enhance our quality of life, they can also pose significant toxic risks. To ensure public safety, various regulatory frameworks have been established to mitigate these hazards. Given the potential health risks associated with chemical exposure, thorough evaluation of such substances in the environment is essential. Regulatory standards typically mandate toxicity testing, encompassing hazard identification, dose-response assessment, exposure evaluation, and risk characterization (Krewski et al., 2010). As part of hazard identification, it is necessary to determine the specific toxicity endpoints associated with each chemical. In parallel, in vitro and in vivo studies aim to elucidate the conditions under which these toxic effects may occur in humans, often drawing on epidemiological insights. Dose-response assessments examine the relationship between chemical exposure and adverse effects, using benchmarks such as the no-observed-adverse-effect level (NOAEL), lowest-observed-adverse-effect level (LOAEL), and potential carcinogenicity (NRC, 1994). While this approach focuses on the magnitude of exposure required to produce harmful effects, the adverse outcome pathway (AOP) framework provides a complementary mechanistic perspective (Ankley et al., 2010). AOPs begin with a molecular initiating event, such as a chemical binding to a receptor, and proceed through a series of causally connected key events (KEs) until an adverse outcome (AO) is reached at the organism level (Villeneuve et al., 2014). By linking mechanistic insights with experimental data, AOPs exemplify how diverse information sources can be integrated to better understand chemical toxicity (Villeneuve et al., 2014). This growing emphasis on data integration has also driven the development of AI-based models with both experimental and computational inputs to support early-stage toxicity prediction.
The advent of computational approaches, combined with the growing availability of experimental data, has paved the way for more cost-effective, time-efficient strategies in early-stage drug discovery (Mak and Pichika, 2019; Vamathevan et al., 2019). By incorporating AI-based toxicity prediction models into virtual screening pipelines, compounds likely to exhibit toxicity can be filtered out before in vitro assays. This strategy increases the success rate of candidates advancing through toxicity evaluations, thereby enhancing the overall efficiency of drug development (Figure 1A). AI models can be trained on large-scale public databases such as ChEMBL (Gaulton et al., 2017), DrugBank (Wishart et al., 2018), and BindingDB (Liu et al., 2007), which contain in vitro and in vivo experimental results. In addition to open-source datasets, proprietary data generated from in vitro assays, in vivo studies, clinical trials, and post-marketing surveillance can further enrich these models (Pognan et al., 2023). Integrating AI-based toxicity prediction into virtual screening and then feeding back the experimental outcomes from downstream studies (in vitro, in vivo, and clinical), creates a virtuous cycle. This feedback process includes prospective and external validations, which evaluate model performance using newly generated or independent datasets and are essential for demonstrating generalizability and robustness in regulatory submissions. This continuous feedback loop improves model performance over time and supports more informed decision-making in early toxicity assessment (Pognan et al., 2023).
FIGURE 1

Overview of the AI-based toxicity prediction pipeline. (A) Integration of AI-based toxicity prediction into the drug development process. (B) Workflow of model development, including data collection, preprocessing, algorithm selection, and performance evaluation.
To develop such models, a systematic workflow is essential, typically consisting of four key stages: data collection, data preprocessing, model development, and evaluation (Figure 1B). The first step involves gathering drug toxicity data from a variety of sources. These data sources, including both public databases and proprietary collections, provide extensive information on chemical structures, bioactivity, and associated toxicity profiles, forming a rich foundation for supervised learning (Pognan et al., 2023). Once the data is collected, preprocessing is carried out to transform raw experimental results into formats suitable for machine learning. This includes handling missing values, standardizing molecular representations (e.g., SMILES strings or molecular graphs), and performing feature engineering such as calculating molecular descriptors (e.g., molecular weight, clogP, number of rotatable bonds) (Wigh et al., 2022). Toxicity labels are also encoded appropriately. These steps ensure data consistency and help extract informative features for training predictive models. The next stage involves selecting and training appropriate modeling techniques. Depending on the data structure and task complexity, a variety of algorithms can be applied, including Random Forest, XGBoost, Support Vector Machines (SVMs), neural networks, as well as more recent approaches such as Graph Neural Networks (GNNs). GNNs align well with the graph-based nature of molecular structures, which contributes to their strong predictive performance in various molecular property prediction tasks (Jiang et al., 2021; Reiser et al., 2022). In addition, they facilitate the identification of substructures or motifs associated with specific biological effects, thereby enhancing both the accuracy and interpretability of toxicity prediction models (Jiang et al., 2021; Reiser et al., 2022; Wu Z. X. et al., 2023). Transformer-based models, originally developed for natural language processing, have also shown strong potential in cheminformatics (Schwaller et al., 2019; Tibo et al., 2024).
In the evaluation phase, performance metrics are selected based on the type of prediction task. For classification models, metrics such as accuracy, precision, recall, F1-score, and area under ROC curve (AUROC) are used to evaluate the model’s ability to correctly distinguish toxic from non-toxic compounds. For regression models that predict continuous values like LD50 or IC50, commonly used metrics include MSE, RMSE, MAE, and R2. In addition to these quantitative measures, interpretability techniques such as SHAP or attention-based visualizations can provide insights into the features driving model predictions, supporting both model validation and decision-making in drug development (Rodríguez-Pérez and Bajorath, 2020; Wang Y. M. et al., 2023).
Driven by the growing need for early toxicity screening, advances in AI model architectures, and the emergence of robust development frameworks, a number of AI-based toxicity prediction models have recently been proposed. These models vary in scope and specificity, often categorized based on the target organ or the type of assay data used for training. This review summarizes representative toxicity prediction models that cover a broad range of toxicological endpoints. In particular, it focuses on models developed for ADMET profiling, hepatotoxicity, cardiotoxicity, neurotoxicity, and mutagenicity/genotoxicity prediction. Each category reflects distinct biological concerns and methodological approaches. Model development within these domains has evolved in response to challenges such as data scarcity, protocol heterogeneity, and class imbalance (Cavasotto and Scardino, 2022; Liu et al., 2023). To address these issues, various strategies have been employed, including multi-task learning, multimodal integration, and active learning. These strategies are discussed in more detail in later sections. In addition, scaffold-based data splitting is also commonly used to evaluate model generalizability across novel chemical structures while minimizing data leakage. In summarizing these models, this review also highlights differences in data sources, input representations, model architectures, and evaluation strategies and interpretability techniques used across toxicity endpoints. These aspects reflect how AI models are tailored to meet the distinct challenges of each toxicological domain.
2 Benchmark datasets
A wide range of publicly available datasets have been developed to support toxicity prediction using machine learning and deep learning approaches (Table 1). Among the most widely used is Tox21, which comprises qualitative toxicity measurements of 8,249 compounds across 12 biological targets, primarily focused on nuclear receptor and stress response pathways (Richard et al., 2021). A related resource, ToxCast provides high-throughput screening data for approximately 4,746 chemicals tested across hundreds of biological endpoints, offering broad mechanistic coverage for in vitro toxicity profiling (Richard et al., 2016). These datasets are frequently employed as benchmarks for evaluating classification models in predictive toxicology.
TABLE 1
| Dataset Name | Task Type | Description | Ref. |
|---|---|---|---|
| Tox21SL | Binary Classification | Predicts toxicity across 12 biological targets using qualitative measurements (based on the EPA CompTox Dashboard) | Richard et al. (2021) |
| ToxCast | Binary Classification | Provides high-throughput screening (HTS) data across hundreds of assays to evaluate the potential toxicity of chemicals (based on the EPA CompTox Dashboard) | Richard et al. (2016) |
| ClinTox | Binary Classification | Predicts clinical trial toxicity outcomes, distinguishing between approved and failed drugs | Gayvert et al. (2016) |
| hERG (Karim et al., 2021) | Binary Classification | Integrated dataset predicting hERG channel blockade (<10 µM) or not (≥10 µM) | Karim et al. (2021) |
| hERG blockers | Binary Classification | Predicts if a compound blocks the hERG channel, which is crucial for heart rhythm | Wang et al. (2016) |
| hERG Central | Binary & Regression | Provides multiple assays: hERG_at_1uM, hERG_at_10uM (regression), and hERG_inhib (classification) | Du et al. (2011) |
| DILIrank | Binary Classification | Predicts drug-induced liver injury, a common cause of drug withdrawal | Chen et al. (2016) |
| SIDER | Multi-label | Predicts clinical adverse drug reactions associated with marketed drugs | Kuhn et al. (2016) |
| Skin Reaction | Binary Classification | Predicts if a compound can cause skin sensitization reactions | Alves et al. (2015) |
| AMES (Xu et al., 2012) | Binary Classification | Predicts mutagenicity based on the Ames test, indicating potential genetic alterations | Xu et al. (2012) |
| Carcinogens (Lagunin et al., 2009) | Binary Classification | Predicts if a compound is carcinogenic | Lagunin et al. (2009) |
| LD50 (Zhu et al., 2009) | Regression | Predicts acute toxicity (LD50) values, indicating lethal dose levels | Zhu et al. (2009) |
Summary of publicly available benchmark datasets for toxicity prediction.
To assess clinical toxicity risks, the ClinTox dataset offers labeled data differentiating compounds that were approved by regulatory agencies from those that failed in clinical trials due to toxicity (Gayvert et al., 2016). Several datasets have been curated for evaluating cardiotoxicity associated with the human Ether-à-go-go–related gene (hERG) channel blockade. The hERG dataset (Wang et al., 2016; Karim et al., 2021) includes over 13,000 compounds annotated with binary labels based on a 10 µM inhibition threshold, while the hERG blockers dataset provides a smaller set of 648 compounds (Wang et al., 2016; Karim et al., 2021). A more extensive resource, hERG Central, encompasses over 300,000 experimental records and supports both classification and regression tasks based on various hERG inhibition assays (Du et al., 2011). Liver toxicity is addressed in the DILIrank (Drug-Induced Liver Injury) dataset, which contains 475 compounds annotated for their hepatotoxic potential, an important factor in post-market drug withdrawals (Xu et al., 2015). The SIDER dataset presents multi-label side effect annotations for more than 1,400 marketed drugs, enabling the prediction of clinically observed adverse drug reactions (Kuhn et al., 2016). For dermatological toxicity, the Skin Reaction dataset includes 404 compounds evaluated for their potential to cause skin sensitization (Alves et al., 2015). Genotoxicity is commonly assessed using the AMES dataset, which comprises 7,255 compounds labeled based on the Ames test—a standard assay for detecting mutagenic potential (Xu et al., 2012). The Carcinogens dataset contains 278 compounds classified as carcinogenic or non-carcinogenic, serving as a benchmark for cancer risk prediction (Lagunin et al., 2009). Finally, acute systemic toxicity is represented by the LD50_Zhu dataset, which includes LD50 values for 7,385 compounds and supports regression modeling of lethal dose responses (Zhu et al., 2009). Collectively, these datasets span a broad range of toxicological endpoints and data modalities and have become foundational resources for the development, validation, and comparison of AI-driven toxicity prediction models.
At the same time, their widespread adoption has revealed several practical challenges that impact real-world applications. For instance, data scarcity in certain toxicity endpoints can hinder the performance of machine learning models that depend on sufficient training data. In some cases, limited data may fail to represent diverse chemical scaffolds, reducing model generalizability. When class imbalance is also present, such as a higher proportion of non-toxic compounds, the effects of data scarcity can be further amplified (Cavasotto and Scardino, 2022). Since toxicity labels are typically derived from experimental measurements, inconsistencies across assay protocols often lead to a lack of data uniformity. This protocol heterogeneity can make it difficult to merge datasets from different sources. Furthermore, annotation noise resulting from experimental variability or ambiguous labeling can introduce additional challenges during model training (Liu et al., 2023).
To overcome these issues, expanding datasets through newly generated experimental data and literature-based curation can help improve coverage and diversity. In parallel, standardizing toxicity testing protocols and documentation practices may enhance data consistency and interoperability. These efforts are expected to contribute meaningfully to the development of more robust and reliable AI-based toxicity prediction models in drug discovery.
3 Computational models for ADMET and toxicity prediction
Several publicly accessible ADMET prediction tools, including ADMETLab 3.0, Deep-PK, ProTox 3.0, Helix-ADMET, FP-ADMET, and admetSAR 2.0 (Yang et al., 2019; Venkatraman, 2021; Zhang et al., 2022; Banerjee et al., 2024; Fu et al., 2024; Myung et al., 2024), provide a wide array of toxicity prediction models, each differing in scope, algorithmic strategy, and coverage. ADMETLab 3.0 offers predictive models for 119 endpoints, including toxicity-related properties such as hERG inhibition, carcinogenicity, and respiratory toxicity. These models are built using directed message-passing neural networks (DMPNNs) and incorporate uncertainty estimation features. The toxicity models, such as the one for hERG inhibition, have demonstrated strong performance with AUROC values approaching 0.94. In terms of interpretability, ADMETLab 3.0 provides uncertainty scores alongside predictions, uses colored indicators to represent empirical decision states, and highlights structural alerts contributing to toxicity (Fu et al., 2024). Deep-PK is a deep learning–based framework that predicts 73 endpoints, including 35 toxicity-related endpoints, 29 other ADMET properties, and 9 general molecular descriptors. While its primary focus lies in pharmacokinetic regression tasks and ADMET optimization, it offers comprehensive support for toxicity assessment through GNN-based pipelines that accept SMILES, SDF, and molecular descriptor inputs. The model also provides interpretability by identifying key molecular subgraphs that contribute to prediction outcomes (Myung et al., 2024). ProTox 3.0 is particularly comprehensive in its treatment of toxicity, providing 61 predictive models covering a broad spectrum of endpoints. These include organ-specific toxicities such as hepatotoxicity, neurotoxicity, cardiotoxicity, and nephrotoxicity, along with models for clinical, immunological, and nutritional toxicities. The platform integrates mechanistic insights through AOPs, molecular initiating events, and target-specific toxicities, and supports ontology-driven, systems-level interpretation (Banerjee et al., 2024). Helix-ADMET is a flexible ADMET prediction platform that combines self-supervised and multi-task learning to enhance generalizability across diverse chemical scaffolds. It supports fine-tuning on user-defined endpoints and classifies toxicity into macro- and micro-level categories (Zhang et al., 2022). FP-ADMET is an open-source tool that focuses on over 50 ADMET-related endpoints, including drug-induced liver injury, hERG inhibition, hemolytic toxicity, mitochondrial toxicity, and cell-specific cytotoxicity. The models are constructed using random forest classifiers trained on 20 different types of chemical fingerprints, enabling broad chemical space coverage and compound exploration (Venkatraman, 2021). admetSAR 2.0 provides 47 curated endpoints, including Ames mutagenicity, carcinogenicity, immunotoxicity, and hERG inhibition. It employs traditional machine learning algorithms such as random forest, SVM, and k-nearest neighbors (KNNs) applied to molecular descriptors and fingerprints (Yang et al., 2019).
The comprehensiveness of these tools not only facilitates broad ADMET screening but also enables prioritization of drug candidates with favorable safety profiles. The development of such general-purpose prediction tools has been largely driven by advances in molecular representations that effectively capture compound features, along with the availability of benchmark datasets annotated with a wide range of ADMET endpoints. On the other hand, tools that focus on specific toxicity types such as hepatotoxicity, cardiotoxicity, nephrotoxicity, neurotoxicity, and genotoxicity/carcinogenicity often require task-specific datasets and tailored feature engineering strategies to enhance predictive performance. The following sections introduce these organ- and mechanism-specific toxicity models and discuss how specialized data and domain-informed approaches contribute to their effectiveness.
4 Endpoint-specific toxicity prediction
Each endpoint is characterized by differences in data properties, sources including databases, and overall data volume. Furthermore, depending on the specific toxicity pathways involved, areas of interest such as the level of interpretability required can also vary. As a result, models for each endpoint have been designed to reflect these unique characteristics, leading to differences in the features used and the methodological approaches adopted (Figure 2 and Table 2). While many of these models share a common foundation in molecular data, it is important to note that the choice of features and modeling techniques is often tailored to the distinct goals and nature of each endpoint.
FIGURE 2
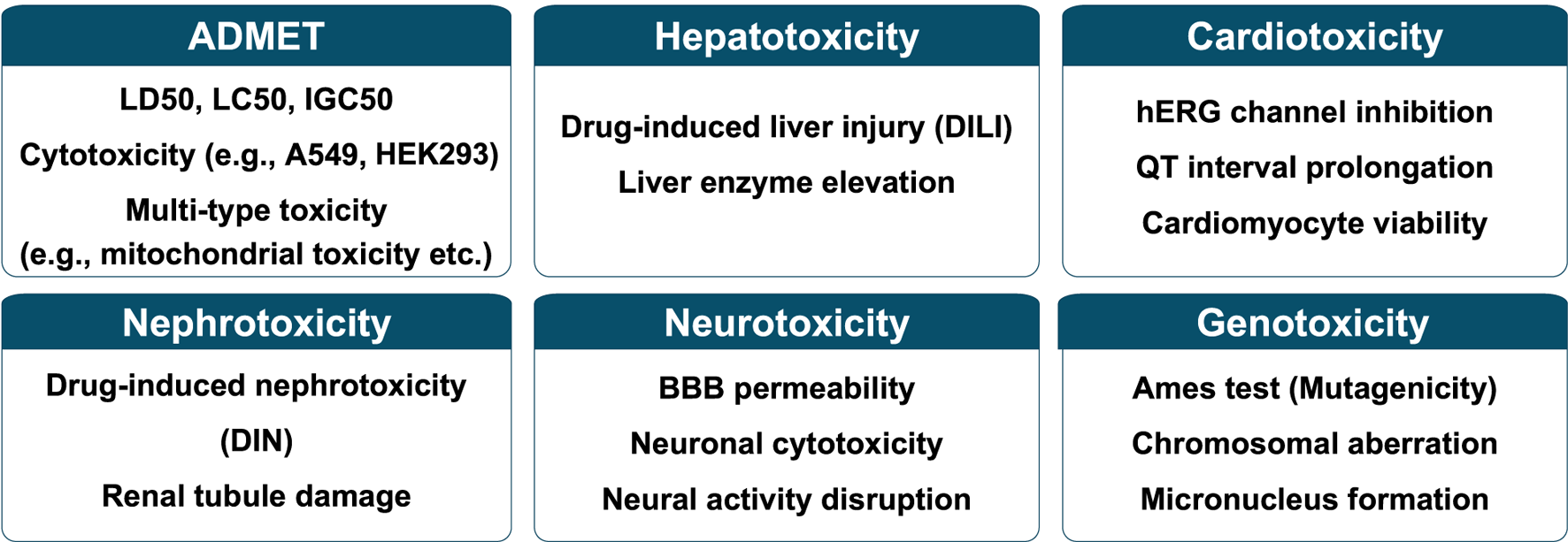
Representative toxicity endpoints categorized into six major classes.
TABLE 2
| Endpoint | Data | Featurea | Methodsa |
|---|---|---|---|
| Hepatotoxicity | DILIrank (Chen et al., 2016) DILIst (Thakkar et al., 2020) LiverTox (Hoofnagle et al., 2013), Hepatox (Quinton et al., 1993) Proxy-DILI (Mulliner et al., 2016) SIDER (Kuhn et al., 2016) LTKB (Chen et al., 2011) Greene et al. (2010) Xu et al. (2008) Liew et al. (2011) Yan et al. (2022) |
Molecular structure - Molecular graph - Molecule image - Molecular fingerprints (e.g., ECFP, MACCS keys) Descriptors - Molecular descriptors (e.g., PaDEL) - Physicochemical descriptors Task-specific or specialized features - Predicted PK parameters - Predicted Proxy-DILI labels |
Deep learning models - GeoGNN + ResNet - ResNet - Fully connected neural network (FCNN) Tree-based Models - Random forest - Light gradient boosting machine (LGBM) - Decision trees Bayesian models - Bayesian network |
| Cardiotoxicity | ChEMBL (Gaulton et al., 2017), BindingDB (Liu et al., 2007) PubChem (Kim et al., 2016) Didziapetris and Lanevskij (2016) Wang et al. (2016) Liu et al. (2020) Li et al. (2008) Munawar et al. (2019) Negami et al. (2019) Lanevskij et al. (2022) Ryu et al. (2020) Karim et al. (2021) Zhang Y. M. et al. (2019) Kim et al. (2022) Konda and Kristam (2019) Cai et al. (2019) Doddareddy et al. (2010) |
Molecular structure - Molecular fingerprints - AtomPairs2DFingerprintCount (APC2D) - SMILES embedding vector Descriptors - Molecular descriptors (Mordred, AlvaDesc) - 2D + 3D descriptors (Mordred) |
Deep learning models - GNN (Attentive FP) - Graph attention networks (GAT) + Gated recurrent units (GRU) - GAT - FCNN - GCN - Meta-ensemble model combining GCN, FCNN, and 1D - Convolutional neural network (CNN) Tree-based Models - XGBoost |
| Renal/nephrotoxicity | SIDER (Kuhn et al., 2016) DrugBank (Wishart et al., 2018) ChEMBL (Gaulton et al., 2017) PubChem (Kim et al., 2016) TCM (Chen, 2011) ChemIDplus |
Molecular structure - Molecular fingerprints (Morgan, EstateFP, CDK FP, CDK extended FP, CDK graph -only FP, Klekota–Roth FP, MACCS keys, PubChem FP, Substructure FP) - APC2D - Fragmentor - SMILES embedding vector Descriptors - Molecular descriptors (Chemaxon, Mordred, RDKit, QNPR, alvaDesc, PyDescriptor, GSFrag) |
Deep learning models - CNN - FCNN Tree-based models - Random forest - LGBM - XGBoost SVM Ensemble/hybrid models - Combination of genetic algorithm and Naïve Bayes classifier - Consensus model of random forest, XGBoost, and CNN |
| Neurotoxicity | PubChem (Kim et al., 2016) ChEMBL database (Gaulton et al., 2017) U.S. EPA dataset (Albert, 1994) SIDER (Kuhn et al., 2016) Liu et al. (2021) Mazumdar et al. (2023) Tang et al. (2022) Storchi et al. (2023) ChemIDplus |
Molecular structure - Molecular graph - SMILES embedding - Molecular fingerprints (e.g., ECFP, MACCS keys, PubChem FP, substructure FP, Klekota–Roth FP, Estate FP, CDK FP, CDK ECFP) Descriptors - Molecular descriptors (e.g., PaDEL, CDK, Dragon descriptors) Task-specific or specialized features - MIE predictions |
Deep learning models - DMPNN - MFBERT - MLP -NNET Tree-based Models - Random forest - Extra -trees regressor - C4.5 decision tree SVM kNN Naïve Bayes |
| Genotoxicity/Carcinogenicity | TOXRIC (Wu W. X. et al., 2023) Li’s dataset (Li T. et al., 2021) Xu et al. (2012) Hansen et al. (2009) Benigni et al. (2013) Dimitrov et al. (2016) CPDB (Gold et al., 2005) CCRIS database, ISSCAN (Benigni et al., 2008) |
Molecular structure - Molecular fingerprints (e.g., ECFP2, ECFP4, ECFP6, MACCS keys, PubChem FP, CDK FP, CDK extended FP, Klekota–Roth FP, AP2D, AP2DC, Estate FP, FP4, FP4C) - Mol2vec Descriptors - Molecular descriptors (e.g., RDKit2D, computed molecular descriptors) Task-specific or specialized features - Structural alerts |
Deep Learning Models - 2D-CNN with active learning - Capsule network with self -attention routing - Multitask DNN Tree-based Models - Extremely randomized trees SVM Ensemble/hybrid models - Consensus model (averaging) - Recursive molecular similarity + extremely randomized trees |
Summary comparison table across different toxicity endpoints.
Features and methods refer to those used in the reviewed models; additional options may be applicable.
In hepatotoxicity prediction, physicochemical properties of molecules are known to be influential and are often incorporated into models (Chen et al., 2013a; Kotsampasakou and Ecker, 2017; Lee and Yoo, 2024). Both deep learning and tree-based methods have been used with comparable frequency. For cardiotoxicity, particularly related to hERG channel blockade, the availability of larger datasets has encouraged the use of more data-intensive deep learning approaches. GNNs are frequently applied due to their structural compatibility with molecules and their ability to offer interpretability through substructure-level attention (Jiang et al., 2021; Yang et al., 2024; Lee and Yoo, 2025). In renal or nephrotoxicity prediction, traditional machine learning models are more commonly used, as they tend to perform better than deep learning when data are limited (Xu et al., 2023). In neurotoxicity studies, a single study may develop multiple models to address distinct tasks such as BBB permeability, neuronal cytotoxicity, neural activity interference, and general neurotoxicity, enabling broader predictive coverage (Pang et al., 2025). For genotoxicity and carcinogenicity, multi-task learning has been applied to predict outcomes across several Ames test strains within a single model. This approach outperformed single-task models by leveraging shared parameters across tasks (Martínez et al., 2022). These variations, driven by endpoint-specific requirements, are elaborated in the subsequent sections.
4.1 Hepatotoxicity
The physiological functions of the liver, a fundamental organ in maintaining systemic homeostasis, include detoxification, plasma protein synthesis, regulation of lipid and glucose metabolism, bile production, and immune modulation (Gu and Manautou, 2012). While the liver’s metabolic processes can render many chemicals less toxic, it has the potential to enhance their toxicity as well, thereby exerting a detrimental effect on the liver (Gu and Manautou, 2012). Pathologies of the liver, such as hepatic steatosis and fibrosis, can adversely impact the metabolism of nutrients, endocrine substances and pharmaceuticals resulting in pronounced systemic implications for overall physiological homeostasis (Foulds et al., 2017; Heeren and Scheja, 2021). Due to its multifaceted physiological roles and vulnerability to chemical-induced damage, the liver frequently experiences drug toxicity. Consequently, it becomes imperative to accurately assess the hepatotoxicity of drugs, commonly referred to as drug-induced liver injury (DILI), an area of active research (Regev, 2014). Both in vitro and in vivo methods are employed to evaluate the hepatotoxicity of drugs, although these approaches can be laborious and costly (Ai et al., 2018; Walker et al., 2020). Moreover, the level of agreement between liver toxicity in animals and humans averages approximately 55% (Babai et al., 2021). Consequently, there exists a demand for predictive models that can foresee liver toxicity and help mitigate development risk and late-stage failure.
Various machine learning approaches have been proposed to address the limitations of traditional DILI assessment, particularly in terms of scalability and interpretability (Table 3). InterDILI focused on enhancing interpretability by employing permutation feature importance and attention mechanisms to identify both general and compound-specific substructures and physicochemical properties contributing to DILI, using five publicly available datasets and multiple machine learning algorithms (Lee and Yoo, 2024). DILIPredictor employed a two-stage modeling approach by integrating proxy-DILI labels with chemical structure features. By identifying the most contributing MACCS substructures to DILI toxicity, it further provided insights into species-specific hepatotoxicity and mechanistic causes through substructure interpretation. The model also provides a web interface for easy access to DILI predictions and their interpretation without the need for local installation (Seal et al., 2024). GeoDILI introduced an interpretable graph neural network that leverages 3D molecular geometry and gradient-based attribution to identify atom-level toxicophores, addressing the lack of geometric and mechanistic considerations in previous models (Wu W. X. et al., 2023). It encodes molecular structures using a fine-tuned geometry-based GNN (GeoGNN), with the resulting vector passed through a ResNet for binary DILI classification. Notably, it applies to a rare attention-free interpretation method for GNNs, offering an alternative to attention-based approaches. OvA-QSTR utilized a one-vs-all classification strategy based on PaDEL-derived molecular descriptors and feature selection via correlation heatmaps, aiming to isolate DILI-related features with statistical clarity (Celik and Karaduman, 2023). The model proposed by Rao et al. predicted DILI severity by integrating physicochemical descriptors with off-target profiles, highlighting the importance of drug-target interactions and promiscuity in distinguishing between different levels of hepatotoxicity (Rao et al., 2023). Lastly, ResNet18DNN converted SMILES codes into molecular images and applied deep neural networks to learn abstract chemical features from visual input, offering a novel image-based perspective in DILI prediction (Chen et al., 2022).
TABLE 3
| Approach | Year | Dataset | Features | Algorithm | Performance | Ref. | Availability |
|---|---|---|---|---|---|---|---|
| InterDILI | 2024 | FDA NCTR (Chen et al., 2013b) Greene et al. (2010) Xu et al. (2008) Liew et al. (2011) DILIrank (Chen et al., 2016) |
Morgan fingerprints, Physicochemical descriptors (RDKit) |
Random forest, LGBM, Logistic regression (LR), FCNN |
DILI prediction [Hold-out] - AUROC: 0.97 - AUPRC: 0.95 - ACC: 0.90 [10-fold CV] - AUROC: 0.87 - ACC: 0.78 - AUPRC: 0.87 |
Lee and Yoo (2024) | https://github.com/bmil-jnu/InterDILI |
| DILI Predictor | 2024 | DILIst (Thakkar et al., 2020) DILIrank (Chen et al., 2016) Proxy-DILI (Mulliner et al., 2016) |
Morgan fingerprints, MACCS keys, Physicochemical descriptors (RDKit), Predicted PK parameters, Predicted proxy-DILI labels |
Random forest | DILI prediction - AUROC: 0.63 - LR+: 1.40 |
Seal et al. (2024) |
https://dili.serve.scilifelab.se/
https://github.com/srijitseal/DILI_Predictor?tab=readme-ov-file |
| GeoDILI | 2023 | DILIst (Thakkar et al., 2020) DILIrank (Chen et al., 2016) Yan et al. (2022) |
Molecular graph | GeoGNN + ResNet | DILI prediction - AUROC: 0.908 - ACC: 0.975 - F1-score: 0.905 - MCC: 0.732 |
Wu W. X. et al. (2023) | https://github.com/CSU-QJY/GeoDILI |
| OvA-QSTR | 2023 | LiverTox (Hoofnagle et al., 2013), PubChem (Kim et al., 2016) | Molecular descriptors (PaDEL) | Bayesian network, Decision trees, Random forest |
DILI prediction BayesNet - AUPRC: 0.718 to 0.869 |
Celik and Karaduman (2023) | |
| Rao et al. | 2023 | DILIrank (Chen et al., 2016) | Physicochemical descriptors (RDKit + QikProp) | Random forest, SVM, FCNN, LR | DILI prediction - AUROC: 0.88 - Sensitivity: 0.73 - Specificity: 0.9 |
Rao et al. (2023) | |
| ResNet18DNN | 2022 | DILIrank (Chen et al., 2016), LiverTox (Hoofnagle et al., 2013), Hepatox (Quinton et al., 1993) SIDER (Kuhn et al., 2016), LTKB (Chen et al., 2011), Literature (Chen et al., 2013b; Xu et al., 2015) |
Smiles converted in images by RDKit | ResNet | DILI prediction - AUROC: 0.958 |
Chen et al. (2022) |
Summary of recently published prediction tools of DILI.
4.2 Cardiotoxicity prediction
Cardiotoxicity is a major concern in drug development, often leading to late-stage failures or market withdrawals. Compounds posing cardiovascular risks have been withdrawn, while others face increasing regulatory scrutiny, underscoring the need for early risk assessment strategies. An illustrative case involves Janus kinase (JAK) inhibitors, namely, tofacitinib, baricitinib, and upadacitinib, used to treat rheumatoid arthritis. In 2021, the U.S. FDA issued a boxed warning for these agents due to elevated risks of cardiovascular events, malignancies, thrombosis, and mortality (Kragstrup et al., 2022). Such examples highlight the importance of identifying cardiotoxic compounds early in the drug discovery process. A common mechanism of cardiotoxicity involves QT interval prolongation and ventricular arrhythmias, often resulting from inhibition of the hERG potassium channel, which is critical for cardiac repolarization (Yang et al., 2020). To mitigate these risks, evaluation of hERG liability is required at the preclinical stage per ICH S7B guidelines (FDA, 2005), and is increasingly recommended during earlier stages, including lead optimization. Early identification enables structural refinement to avoid cardiotoxicity before costly development steps.
Several recently developed computational tools for cardiotoxicity prediction are summarized in Table 4, with a particular focus on assessing hERG channel blockade—a critical concern in early drug development. hERGBoost presents a quantitative modeling approach using gradient boosting to predict IC50 values of potential hERG inhibitors, allowing a more nuanced evaluation of cardiotoxic risk beyond binary classification. Although the model is easily accessible through a web interface, it does not provide interpretability for its predictions (Yu et al., 2025). The following models, though not web-accessible, are designed to provide interpretability. hERGAT employs a hybrid architecture combining GAT and GRU to capture both atomic-level and molecule-level interactions, enhancing interpretability through attention-based substructure identification (Lee and Yoo, 2025). AttenhERG incorporates uncertainty estimation within a graph neural network framework, aiming to improve the reliability of predictions and assist compound optimization. It provides interpretability through atom-level attention weight visualizations, highlighting which molecular substructures contribute to hERG inhibition (Yang et al., 2024). DMFGAM integrates both fingerprint-derived and graph-based features using a SMILES graph attention network and fully connected neural layers, reflecting the advantage of multimodal input representations (Wang T. Y. et al., 2023). CardioTox Net utilizes a meta-ensemble strategy that merges outputs from multiple deep learning architectures (GCN, FCNN, 1D-CNN), each trained on diverse molecular encodings, to enhance prediction robustness across varying datasets and evaluation criteria (Karim et al., 2021). Lastly, DeepHIT focuses on minimizing false negatives by training multiple deep neural networks on a large gold-standard dataset, and includes a chemical transformation module for generating safer analogs based on known cardiotoxic compounds (Ryu et al., 2020).
TABLE 4
| Approach | Year | Dataset | Features | Algorithm | Performance | Ref. | Availability |
|---|---|---|---|---|---|---|---|
| hERGBoost | 2025 | ChEMBL (Gaulton et al., 2017) BindingDB (Liu et al., 2007) Didziapetris and Lanevskij (2016) Wang et al. (2016) Liu et al. (2020) Li et al. (2008) Munawar et al. (2019) Negami et al. (2019) Lanevskij et al. (2022) Ryu et al. (2020) Karim et al. (2021) |
Descriptors (AlvaDesc), Molecular fingerprints |
XGBoost | hERG channel inhibition [Regression] - R2: 0.622 - RMSE: 0.595 - MAE: 0.383 [Classification] - ACC: 0.814 - MCC: 0.614 |
Yu et al. (2025) | http://ssbio.cau.ac.kr/software/hergboost/ |
| hERGAT | 2025 | ChEMBL (Gaulton et al., 2017) PubChem (Kim et al., 2016) Li et al. (2008) Wang et al. (2016) Zhang H. et al. (2019) Kim et al. (2022) |
Descriptors, Molecular fingerprints, Molecular graph |
GAT + GRU | hERG channel inhibition - AUROC: 0.907 - AUPRC: 0.904 |
Lee and Yoo (2025) | https://github.com/bmil-jnu/hERGAT |
| AttenhERG | 2024 | ChEMBL (Gaulton et al., 2017), PubChem (Kim et al., 2016), BindingDB (Liu et al., 2007) Kim et al. (2022) |
Molecular graph | GNN (Attentive FP) | hERG channel inhibition - AUROC: 0.835 - BAC: 0.767 - MCC: 0.543 |
Yang et al. (2024) | https://github.com/Tianbiao-Yang/AttenhERG |
| DMFGAM | 2023 | CHEMBL (Gaulton et al., 2017) Liu et al. (2020) Konda and Kristam (2019) Munawar et al. (2019) Negami et al. (2019) |
Morgan fingerprints (ECFP2) AtomPairs2DFingerprintCount (APC2D) |
SMILES graph attention network (SGAT), FCNN |
hERG channel inhibition - AUROC: 0.894 - ACC: 0.817 - MCC: 0.630 - Sensitivity: 0.847 |
Wang Y. M. et al. (2023) | https://github.com/zhaoqi106/DMFGAM |
| CardioTox net | 2021 | BindingDB (Liu et al., 2007) ChEMBL (Gaulton et al., 2017) Cai et al. (2019) Didziapetris and Lanevskij (2016) Doddareddy et al. (2010) |
Molecular graph, Morgan fingerprints (ECFP2), 2D+3D Descriptors (Mordred), SMILES embedding vector, Fingerprint embedding vector |
meta-ensemble combining, GCN, FCNN and 1D-CNN |
hERG channel inhibition [Test set-I] - ACC: 0.810 - BAC: 0.810 - MCC: 0.599 Sensitivity: 0.833 [Test set-II] - ACC: 0.755 - BAC: 0.754 - MCC: 0.452 - Sensitivity: 0.909 [Test set-III] - ACC: 0.746 - BAC: 0.746 - MCC: 0.220 - Sensitivity: 0.794 |
Karim et al. (2021) | https://github.com/Abdulk084/CardioTox |
| DeepHIT | 2020 | BindingDB (Liu et al., 2007) ChEMBL (Gaulton et al., 2017) Cai et al. (2019) Didziapetris and Lanevskij (2016) Doddareddy et al. (2010) In-house dataset |
Molecular Fingerprints (PyBioMed), Descriptors (Mordred), Molecular Graph |
GCN, FCNN |
hERG channel inhibition - ACC: 0.773 - Sensitivity: 0.833 - BAC: 0.738 - MCC: 0.476 |
Ryu et al. (2020) | https://academic.oup.com/bioinformatics/article/36/10/3049/5727757 |
Recently published prediction tools of cardiotoxicity.
4.3 Renal/nephrotoxicity prediction
The kidneys are vital excretory organs that maintain homeostasis by producing urine, eliminating waste, and regulating water, electrolytes, and acid–base balance. During renal clearance, pharmaceutical compounds undergo filtration, reabsorption, and secretion, contributing to their metabolism and excretion (Gong et al., 2022). However, this process also increases the kidneys’ exposure to potentially harmful substances, giving rise to drug-induced nephrotoxicity (DIN). The prevalence of DIN in the adult population has been reported to range from 14% to 26% (Shi et al., 2022). Drug-induced renal failure accounts for approximately 25% of acute kidney injury (AKI) cases in hospitalized patients, with aminoglycoside antibiotics, NSAIDs, contrast agents, and angiotensin-converting enzyme inhibitors (ACEi) among the most common causative drugs (Ghane Shahrbaf and Assadi, 2015; Gong et al., 2022). The underlying mechanisms of DIN are multifactorial, involving damage to tubular epithelial cells, ureteral obstruction, interstitial nephritis, and disruption of intra-glomerular hemodynamics (Shi et al., 2022).
Assessing DIN risk remains challenging due to the vast diversity of pharmaceutical agents with nephrotoxic potential. Many compounds, beyond the commonly recognized nephrotoxic drugs, can elicit kidney injury through distinct mechanisms and at varying sites within the renal architecture (Shi et al., 2022). These include selective damage to proximal or distal tubules, glomeruli, or the renal interstitium, depending on the drug’s chemical properties, metabolites, and mechanisms of accumulation or transport within renal tissues. Given these complexities, traditional toxicological methods remain indispensable; however, they are often impractical for efficiently screening large number of compounds in the early stages of drug development. As a result, computational approaches that integrate diverse molecular features are increasingly recognized as valuable tools for the early identification of nephrotoxic risk (Table 5).
TABLE 5
| Approach | Year | Dataset | Features | Algorithm | Performance | Ref. | Availability |
|---|---|---|---|---|---|---|---|
| Gong et al. | 2022 | SIDER (Kuhn et al., 2016), DrugBank (Wishart et al., 2018), ChEMBL (Gaulton et al., 2017) TCM (Chen, 2011) |
Atom Pair 2D, fingerprint, Estate FP, CDK extended FP, CDK FP, CDK graph only FP, Klekota–Roth FP, MACCS keys, PubChem FP, substructure FP |
FCNN, LGBM, SVM |
DIN prediction [Herbal Medicines (Test set-I)] - ANN_PubChemFP - AUROC: 0.911 - ACC: 0.867 - MCC: 0.740 - SVM_GraphFP - AUROC:0.902 - ACC: 0.867 - MCC: 0.761 [Chemical Medicines (Test set-II)] - LGBM _KRFP - AUROC:0.896 - ACC: 0.861 - MCC: 0.721 - SVM_GraphFP - AUROC:0.894 - ACC: 0.814 - MCC: 0.629 [Mixed Medicines (Test set-III)] - SVM_GraphFP - AUROC:0.915 - ACC: 0.857 - MCC: 0.723 - ANN_PubChemFP - AUROC:0.903 - ACC: 0.857 - MCC: 0.718 |
Gong et al. (2022) | |
| Shi et al. | 2022 | SIDER (Kuhn et al., 2016), Pubchem (Kim et al., 2016) | Chemaxon descriptors, Fragmentor, GSFrag descriptors, Mordred descriptors, PyDescriptor, QNPR descriptors, RDKit descriptors, alvaDesc descriptors |
Consensus model of random forest XGBoost CNN |
DIN prediction - AUROC: 0.93 - MCC: 0.72 - Accuracy (Q): 0.86 - Sensitivity: 0.85 - Specificity: 0.87 - Enrichment Factor (EF): 1.72 |
Shi et al. (2022) | http://www.sapredictor.cn/ |
| Zhang et al. | 2019 | ChemIDplus | Morgan fingerprints (ECFP6), Molecular descriptors |
Naïve Bayes classifier, Genetic algorithm |
Chemical-induced urinary tract toxicity - ACC: 0.84 |
Zhang Y. M et al. (2019) |
Recent examples of nephrotoxicity prediction tools.
The predictive model proposed by Gong et al. (Gong et al., 2022) was developed by utilizing the technique of fingerprinting chemical drugs and Chinese herbal medicines. This model aimed to provide a comprehensive prediction of nephrotoxicity. On the other hand, Shi et al. (Shi et al., 2022) developed a nephrotoxicity prediction model based on physicochemical property analysis. Among the approaches tested, the model utilizing QNPR descriptors with a random forest algorithm achieved the highest accuracy of 87.16%. Notably, the consensus model outperformed individual models, attaining a superior AUROC of 0.93. The model is accessible via a web interface and provides interpretability by identifying structural alerts associated with nephrotoxicity, using f-score and positive rate analysis of each fragment derived from KRFP fingerprints. Lastly, Zhang et al. (Zhang H. et al., 2019) categorized molecular features based on factors such as the number of nitrogen atoms, AlogP, molecular weight, hydrogen bond acceptors and donors, and fractional polar surface area. Among the evaluated algorithms, the Naïve Bayes classifier demonstrated superior performance and was ultimately selected for nephrotoxicity prediction.
4.4 Neurotoxicity prediction
Neurotoxicity refers to the toxicity that affects both central and peripheral nervous systems leading to their impaired function and structure (Legradi et al., 2018). The mechanisms of neurotoxicity are broadly categorized into neuronopathy, axonopathy, myelinopathy, and neurotransmission-associated toxicity (Valentine, 2020; Kocot-Kepska et al., 2021). Even therapeutic drugs can exhibit neurotoxic effects; for instance, vincristine, a plant-derived chemotherapeutic alkaloid, is known to cause peripheral neuropathy, which manifests as numbness, tingling, and motor weakness. Given these risks, it is essential to screen for neurotoxicity during drug development to ensure the safety of new chemical entities. To this end, the OECD Test Guidelines 418, 419, and 424 are internationally recognized as standard protocols for assessing neurotoxic effects. However, these in vivo testing methods are time-consuming, costly, and reliant on animal use. Consequently, there is a growing demand for faster and more efficient in silico approaches to complement traditional testing methods in predicting neurotoxicity (Jiang et al., 2020).
In response to this need, several computational models have recently been developed to improve the prediction of neurotoxicity (Table 6). NeuTox 2.0 employs a hybrid deep learning framework that integrates molecular fingerprints, descriptors, and GNNs through multimodal feature fusion. It was trained on four neurotoxicity-related datasets and demonstrated strong generalizability and robustness, enabling its use in large-scale chemical screening. This design allows the model to predict various facets of neurotoxicity, offering a broader perspective on neurotoxic effects. However, since all input features are derived from the same molecular structure, the model’s multimodal nature is limited in scope (Pang et al., 2025). DINeuroTpredictor is a web-based model built on clinical neurotoxicity data using multiple machine learning algorithms and molecular fingerprints. It also provides insights into key physicochemical features and structural alerts associated with neurotoxic potential (Zhao et al., 2022). Gadaleta et al. proposed a QSAR-based approach linked to AOPs, modeling molecular initiating events to support mechanistic neurotoxicity prediction (Gadaleta et al., 2022). Lastly, Jiang et al. developed regression models using PyBioMed descriptors and ensemble learning methods, focusing on chemical diversity and model applicability domains to enhance prediction reliability (Jiang et al., 2020).
TABLE 6
| Approach | Year | Dataset | Features | Algorithm | Performance | Ref. | Availability |
|---|---|---|---|---|---|---|---|
| NeuTox 2.0 | 2025 | PubChem Bioassay database (Kim et al., 2016) ChEMBL database (Gaulton et al., 2017) U.S. EPA dataset (Albert, 1994) SIDER (Kuhn et al., 2016) Liu et al. (2021), Mazumdar et al. (2023) Tang et al. (2022) Storchi et al. (2023) |
Molecular graph, Molecular fingerprint (ECFP), Molecular descriptor (Padel) |
DMPNN, MFBERT |
Blood–Brain Barrier Penetration - AUROC: 0.9708 - ACC: 0.9120 - MCC: 0.8157 - F1 Score: 0.9274 Neuronal Cytotoxicity - AUROC: 0.9637 - ACC: 0.9093 - MCC: 0.8171 - F1 Score: 0.8969 Neural Activity Interference - AUROC: 0.8509 - ACC: 0.8007 - MCC: 0.5292 - F1 Score: 0.6651 Neurotoxicity - AUROC: 0.8297 - ACC: 0.7945 - MCC: 0.5140 - F1 Score: 0.8539 |
Pang et al. (2025) | https://github.com/xuejunhe/NeuTox-2.0 |
| DINeuro Tpredictor | 2022 | SIDER (Kuhn et al., 2016), PubChem (Kim et al., 2016) | Estate FP, CDK FP, CDK ECFP, Klekota–Roth FP, MACCS keys, PubChem FP, substructure FP |
Random Forest, SVM, C4.5 decision, tree, kNN, Naïve Bayes |
Neurotoxicity 5-fold CV - AUROC: 0.83 - BAC: 0.7651 - MCC: 0.52 |
Zhao et al. (2022) | http://dineurot.sapredictor.cn/ |
| Gadaleta et al. (2022) | 2022 | ChEMBL (Kim et al., 2016), Literature (Kosnik et al., 2020) | MIE predictions, Dragon descriptors, ECFP |
Random forest, kNN, MLP-NNET |
Neurotoxicity - AUROC: 0.91 - MCC: 0.66 |
Gadaleta et al. (2022) | |
| Jiang et al. (2020) | 2020 | ChemIDplus | MATSp2, bcutv10, MRVSA5, GATSe2, Rpc, EstateVSA1, Geto, Smax15, MTPSA, bcute2, J, Chiv10, Chiv9, mChi1, Smin8, Hy, Smin32, MATSv3, MATSe3, MACCS keys | Extra-trees regressor | Autonomic Neurotoxicity (pLD50) - q2: 0.784 - RMSE: 0.201 - MAE: 0.159 |
Jiang et al. (2020) |
Recent examples of neurotoxicity prediction tools.
4.5 Genotoxicity/carcinogenicity
Genotoxicity is defined as the capacity of deleterious agents to induce harm to the genetic material within cells (Ren et al., 2017). Mutagenicity pertains to the capacity of a substance to induce alterations in genetic material, which could potentially instigate diverse ailments, such as cancer (Ferguson, 2010; Basu, 2018). Carcinogenicity is the potential of a compound to cause cancer (Schrenk, 2018). These three concepts, i.e., genotoxicity, mutagenicity, and carcinogenicity, exhibit a strong correlation due to the fact that substances that possess genotoxic properties frequently result in mutations, and these mutations can induce the development of cancer (Barnes et al., 2018; Nohmi, 2018). Given the fact that cancer is one of the most prominent reasons contributing to mortality on a global scale, it becomes imperative to thoroughly scrutinize the plausible factors that give rise to this ailment. Unlike other forms of toxicity, carcinogenicity is distinct in that it does not exhibit a threshold in the assessment of dose-response. This phenomenon arises from the fact that a lone anomaly through interactions with DNA, instigated by a specific compound, can yield a protracted consequence and engender the formation of neoplastic growth (Nohmi, 2018). Numerous principles are established in light of this, particularly the guidelines of S1B(R1) (ICH, 2022), S2 (R1) (ICH, 2020), and M7 (R1) (ICH, 2023) outlined by the ICH. The course of action typically takes 2 years and involves around 500 rodents, making it a rigorous, time-consuming, and resource-intensive task (Li T. et al., 2021). Furthermore, the test’s complexity depends on whether it is analyzing the genetic, DNA, or chromosomal level, and whether it is intended for somatic or germline cells (Ren et al., 2017). Due to these obstacles, there is a growing demand for AI-assisted prediction to overcome these challenges.
Currently available AI prediction tools of mutagenicity and genotoxicity are summarized in Table 7 muTOX-AL proposed a deep active learning framework to address the challenge of limited labeled data in mutagenicity prediction. By actively selecting the most informative molecules from a vast chemical space and presenting them for annotation, the model significantly reduces the number of training samples required. It also demonstrates strong discriminative power by identifying structurally similar molecules with opposing mutagenic properties (Xu et al., 2024). Fournier et al. introduced a model capable of predicting genotoxicity across various assays, including Ames test results, chromosomal aberrations, and gene mutations, thereby expanding the scope of genotoxicity evaluation. Despite its broad predictive scope, the model introduced by Fournier et al. does not provide executable tools or source code, limiting its immediate applicability and reproducibility (Fournier et al., 2023). DCAMCP employed a self-attention routing capsule network to improve generalizability while reducing the number of trainable parameters, demonstrating balanced performance across multiple evaluation metrics (Chen et al., 2023). Shinada et al. constructed a model using descriptors derived from density functional theory (DFT). Although its performance was modest, the study highlighted opportunities to improve computational approaches (Shinada et al., 2022). Martínez et al. developed the first predictive model based on Ames test standards (OECD TA98, TA100, TA1535, TA1537, and TA102), setting a precedent for mutagenicity prediction using standardized experimental protocols. The multi-task learning framework with shared parameters enabled information transfer across tasks, improving mutagenicity prediction for each strain (Martínez et al., 2022).
TABLE 7
| Approach | Year | Dataset | Features | Algorithm | Performance | Ref. | Availability |
|---|---|---|---|---|---|---|---|
| muTOX-AL | 2024 | TOXRIC (Wu Z. X. et al., 2023) Li’s dataset (Li S. M. et al., 2021) |
Molecular fingerprints (ECFP2, ECFP4, ECFP6) MACCS keys Molecular descriptors (RDkit2D) |
2D-CNN Active learning |
Mutagenicity (Ames test) Full training (5,988 samples) - AUROC: 0.9093 - ACC: 0.8476 - F1 Score: 0.8383 - Recall: 0.8350 Active learning (1,438 samples) − 95% of the full-model accuracy using 1,438 samples (∼24% of the training data) |
Xu et al. (2024) | https://github.com/Felicityxuhy/muTOX-AL |
| Fournier et al. (2023) | 2023 |
Xu et al. (2012)
Hansen et al. (2009) Benigni et al. (2013) Dimitrov et al. (2016) |
2D descriptors MACCS keys |
Recursive molecular similarity, Extremely randomized trees | Mutagenicity (Ames test) - AUROC: 0.9208 chromosomal abnormalities - AUROC: 0.9191 mammalian cell gene mutation test - AUROC: 0.9722 |
Fournier et al. (2023) | |
| DCAMCP | 2023 | CPDB (Gold et al., 2005), CCRIS database, ISSCAN (Benigni et al., 2008) | Various molecular fingerprints MACCS keys PubChem FP CDK FP CDK extended FP Klekota-Roth FP AP2D Klekota-Roth count AP2DC Substructure (FP4) Substructure count (FP4C) Estate FP |
A capsule network with a self-attention routing algorithm | Carcinogenicity - AUROC: 0.793 - ACC: 0.718 - Sensitivity: 0.721 - Specificity: 0.715 |
Chen et al. (2023) | https://github.com/zhaoqi106/DCAMCP |
| Shinada et al. (2022) | 2022 | Hansen et al. (2009) | MACCS, ECFP, Mol2vec, Computed molecular descriptors, structural alerts relevant to mutagenicity, DFT-based descriptors | SVM | Mutagenicity (Ames test) AUROC: 0.926 |
Shinada et al. (2022) | https://bitbucket.org/sbx-publication/enhanced_representation_mutagenicity/src/master/ |
| Martínez et al. (2022) | 2022 | ISSSTY v1-a database (Benigni et al., 2013) | 0D, 1D, and 2D molecular descriptors | Multitask-FCNN Consensus model (averaging) |
Mutagenicity (Ames test) Balanced ACC: 0.93 MCC: 0.89 H1 Score: 0.92 Specificity: 0.86 Sensitivity: 0.99 |
Martínez et al. (2022) | https://github.com/VirSabando/MTL_DNN_Ames |
Recent examples of Genotoxicity/carcinogenicity prediction tools.
5 Emerging AI innovations in toxicity prediction
As previously discussed, AI model design is significantly affected by both the characteristics and the volume of data available for training. In the context of toxicity prediction for drug discovery, input data typically comprises molecular structures, physicochemical properties, and task-specific features. However, these tasks are often constrained by the limited availability of labeled data. To address this challenge, a variety of data-efficient learning strategies have been developed to maximize predictive performance under label-scarce conditions.
In data-scarce settings, transfer learning strategies use pre-trained parameters to boost toxicity prediction performance. For instance, HelixADMET employs a three-stage training framework that incorporates self-supervised pretraining on large-scale unlabeled molecular data, followed by multi-task and fine-tuning stages to transfer learned chemical knowledge to various ADMET endpoints, significantly improving extrapolation to novel chemical scaffolds (Zhang et al., 2022). Multimodal models ingest diverse data types (e.g., chemical structures, omics profiles, and bioactivity assays) simultaneously to capture complementary information. For example, M2REMAP is a multimodal deep learning framework that predicts drug indications, mono-drug side effects, and drug–drug interaction side effects by integrating molecular chemical structures with clinical semantic embeddings derived from large-scale electronic health records (EHR). By learning joint representations across these heterogeneous modalities, M2REMAP achieves superior predictive accuracy and generalizability over unimodal baselines (Wen et al., 2023). Martínez et al. developed multi-task deep neural networks to simultaneously predict Ames mutagenicity across multiple Salmonella typhimurium strains (Martínez et al., 2022). They demonstrated that shared representations improved performance, especially on the strains with limited training data. Active learning enhances data efficiency by strategically selecting the most informative samples, enabling high model performance even with limited labeled data. For example, the muTOX-AL framework integrates structure-based and activity-based selection strategies to guide experimental toxicology, significantly improving model performance with fewer labeled compounds compared to random sampling (Xu et al., 2024). Federated learning enables multiple institutions to collaboratively train a global toxicity prediction model on decentralized datasets in which each party keeps its raw data locally and only shares model updates, thus preserving data privacy and regulatory compliance while benefiting from a much larger, heterogeneous training pool. The MELLODDY project exemplifies this approach, demonstrating that federated QSAR models trained across ten pharmaceutical companies achieved comparable or superior predictive performance to local models, while maintaining strict data confidentiality (Heyndrickx et al., 2023).
In parallel, interpretability techniques are also advancing to better inform and guide decision-making in drug discovery based on model predictions. SHAP estimates the contribution of each input feature to the output, providing insight into which molecular properties influence model decisions (Lundberg and Lee, 2017). For graph-based models, methods like EdgeSHAPer extend this concept by identifying important substructures within molecular graphs (Mastropietro et al., 2022). Attention-based visualizations, commonly used in transformer and graph neural network models, highlight which parts of the input the model focuses on during prediction (Ying et al., 2019; Zheng et al., 2019). For example, in SMILES-based models, attention heatmaps can reveal which atoms or functional groups are most influential in predicting toxicity. Counterfactual explanations, on the other hand, offer intuitive and sparse insights by showing the smallest alteration to input features that would change a model’s prediction, particularly useful for understanding how minimal molecular changes affect outcomes. In drug design, small structural modifications can often result in counterfactual cases with significant impact on activity or toxicity, leading to growing interest in counterfactual explanation methods to better capture such subtle yet meaningful variations (Wellawatte et al., 2022).
Building on these recent advances, the next phase of toxicity prediction may be driven by foundation models and large-scale language-based systems. Looking ahead, emerging foundation models such as MoleculeGPT (Liu et al., 2024), BioT5 (Pei et al., 2023), and ChemCrow (Bran et al., 2024) could be applied to toxicity prediction. Even before the advent of large language models (LLMs), Papamokos and Silins demonstrated that integrating QSAR modeling with text mining improved the mechanistic understanding of carcinogenicity and helped compensate for limited structure–activity data on non-genotoxic compounds (Papamokos and Silins, 2016). By linking chemical structures with literature-derived modes of action, their hybrid approach offered more biologically meaningful interpretations to support mechanism-based toxicity evaluation. Today, with the advent of powerful LLMs, such strategies can be further scaled and generalized. Fine-tuning these large, pre-trained models enables researchers to integrate broad and transferable chemical and biological knowledge into downstream toxicity prediction tasks, while producing mechanistically interpretable results even in data-scarce domains. These AI-driven methods not only improve predictive accuracy in data-scarce scenarios but are also continuing to advance rapidly, expanding the possibilities for mechanism-informed toxicology.
6 AI in regulatory toxicology
The use of AI-based models in regulatory toxicology is drawing growing interest, especially as agencies seek alternatives to animal testing. Yet, adoption remains limited due to the absence of clear validation standards and acceptance criteria. ICH guidelines, including M7 (R2), S2 (R1), and S1B(R1), provide frameworks for using in silico approaches such as AI-based models and advanced QSAR tools (ICH, 2022; 2023). These can support mutagenic impurity screening, genotoxicity testing, and carcinogenicity assessment, provided they are properly justified and validated. In this context, the FDA NCTR’s AI4TOX program is specifically aimed at applying AI to toxicology to develop new tools that support FDA regulatory science and strengthen the safety review of FDA-regulated products (An FDA Artificial Intelligence, 2024). It focuses on leveraging AI for tasks like developing virtual animal models, evaluating toxicological endpoints, and analyzing complex data from FDA documents and histopathology. For broader adoption, AI models must align with regulatory expectations, demonstrate consistent performance, and offer interpretability.
As the use of AI in regulatory toxicology continues to expand, it becomes increasingly important to consider how existing validation principles can be adapted or extended to ensure these models meet regulatory standards. To enhance the reliability and regulatory acceptance of AI-based toxicity prediction models, it is useful to apply the OECD QSAR validation principles (OECD, 2014). Originally developed for traditional QSAR models, these principles outline key elements such as defined endpoints, transparent algorithms, applicability domains, performance metrics, and mechanistic interpretation when possible. While these criteria remain broadly relevant, the guidance was established before the advent of modern AI techniques. Given the rapid development of AI and its increasing integration into the drug discovery process, there is a growing need for updated validation frameworks that explicitly address the unique challenges and opportunities presented by AI-based modeling approaches.
7 Limitations, challenges, and future directions
The efficacy and safety of chemical compounds are fundamental considerations in drug discovery, with toxicity representing a key determinant of clinical success or failure. AI-based prediction models have emerged as powerful tools for toxicity assessment during the early stages of drug discovery. As databases continue to grow, computational resources become more accessible, and AI architectures evolve, these models have significantly advanced beyond traditional computational methods and enable reliable predictions across various toxicological endpoints, including hepatotoxicity, cardiotoxicity, nephrotoxicity, neurotoxicity, and genotoxicity. This review has systematically examined both general-purpose ADMET prediction tools and endpoint-specific toxicity models, highlighting rapid progress, increasing methodological sophistication, and expanding diversity within the field of computational toxicology. Nevertheless, several critical challenges persist. First of all, despite significant advances, current AI models frequently struggle with accurately predicting complex and rare toxicity events due to intrinsic biological complexities. The scarcity of high-quality labeled data, particularly data that accurately reflects clinical outcomes or rare toxicological events, severely constrains model training and validation. Also, generalizability to novel chemical scaffolds remains uncertain, limiting confidence in AI predictions for structurally diverse or innovative drug candidates. Finally, interpretability also remains a crucial bottleneck; although advanced AI models offer powerful predictive capabilities, their complex inner workings often limit the clarity and transparency required by regulatory bodies and clinical practitioners.
To overcome these limitations, future research can be focused on the integration of diverse data types, including detailed chemical structures, comprehensive biological assay outcomes, multi-omics profiles, and real-world clinical datasets. Such integration will enable AI models to capture the multifaceted nature of toxicological responses in a better way. Harmonizing toxicity annotations across multiple databases will also significantly enhance data interoperability, enabling more extensive and efficient utilization of available data resources. In parallel, fostering deeper cross-disciplinary collaboration among computational scientists, toxicologists, medicinal chemists, clinical pharmacologists, and regulatory experts is essential. Such collaborations can facilitate the development of predictive models that are not only robust and accurate but also practically interpretable, ensuring that model insights can directly inform discovery and regulatory decisions.
As AI technologies continue to evolve, it would be definite that they hold significant potential for enhancing early-stage decision-making, substantially reducing late-stage drug development failures, and accelerating the delivery of safer, more effective therapeutic solutions to patients. To fully employ this potential, it is crucial to foster a deeper understanding of the real-world implications and limitations of predictive outcomes. Practical integration requires not just technological advances but also a comprehensive awareness of pharmaceutical, clinical realities and regulatory standards. Thus, ongoing dialogue and knowledge-sharing between computational developers, experimental toxicologists, clinical researchers, and regulatory stakeholders will be indispensable in shaping the next-generation of AI-driven predictive toxicology tools that meaningfully improve drug discovery outcomes in both academic research and industry practice.
Statements
Author contributions
HL: Investigation, Writing – original draft. JK: Investigation, Visualization, Writing – original draft. J-WK: Conceptualization, Project administration, Writing – review and editing. YL: Conceptualization, Project administration, Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the Korean Ministry of Environment under the Environmental Health R&D Program (No. 2021003310005) and the Chung-Ang University Research Grants in 2023.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
- ACC
Accuracy
- AEs
Adverse Events
- ADME
Absorption, Distribution, Metabolism, and Excretion
- ADMET
Absorption, Distribution, Metabolism, Excretion, and Toxicity
- AKI
Acute Kidney Injury
- AI
Artificial Intelligence
- AlogP
Logarithm of Partition Coefficient
- ANN
Artificial Neural Network
- AO
Adverse Outcome
- AOPs
Adverse Outcome Pathways
- AUROC
Area Under the Receiver Operating Characteristic Curve
- AUPRC
Area Under the Precision–Recall Curve
- BAC
Balanced Accuracy
- BBB
Blood–Brain Barrier
- CDK
Chemistry Development Kit fingerprint
- C4.5 DT
C4.5 Decision Tree
- CYP
Cytochrome P450
- CNN
Convolutional Neural Network
- DILI
Drug-Induced Liver Injury
- DIN
Drug-Induced Nephrotoxicity
- DMPNN
Directed Message-Passing Neural Networks
- DNN
Deep Neural Network
- ECFP
Extended-Connectivity Fingerprint
- EPA
The U.S. Environmental Protection Agency
- EHR
Electronic Health Records
- FCNN
Fully Connected Neural Network
- FDA
The U.S. Food and Drug Administration
- FP
Fingerprint
- GAT
Graph Attention Networks
- GCN
Graph Convolutional Networks
- GCNN
Graph Convolutional Neural Network;
- GeoGNN
Geometry-based Graph Neural Network
- GNN
Graph Neural Network
- GPU
Graphics Processing Unit
- GRU
Gated Recurrent Unit
- hERG
Human Ether-à-go-go Related Gene
- HTS
High-Throughput Screening
- IC50
Half Maximal Inhibitory Concentration
- ICH
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- JAK
Janus Kinase
- KEs
Key Events
- kMedoids
k-Medoids Clustering Algorithm
- kNN
k-Nearest Neighbor
- LD50
Lethal Dose for 50% of the Population
- LGBM
Light Gradient-Boosting Machine
- LOAEL
Lowest-Observed-Adverse-Effect Level
- LR
Linear Regression
- LTKB
Liver Toxicity Knowledge Base
- MACCS
Molecular ACCess System
- MAE
Mean Absolute Error
- MAPE
Mean Absolute Percentage Error
- MCC
Matthews Correlation Coefficient
- MIEs
Molecular initiating events
- MMP
Matrix metalloproteinase
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide Assay
- NCTR
National Center for Toxicological Research
- NOAEL
No-Observed-Adverse-Effect Level
- NSAIDs
Non-Steroidal Anti-Inflammatory Drugs
- OECD
Organization for Economic Co-operation and Development
- OECD TA
OECD Toxicity Assays
- PK
Pharmacokinetic
- PPB
Plasma Protein Binding
- PRC
Precision–Recall Curve
- QNPR
Quantitative Name–Property Relationship
- QSAR
Quantitative Structure–Activity Relationship
- RF
Random Forest
- RMSE
Root Mean Square Error
- SGAT
SMILES Graph Attention Network
- SHAP
SHapley Additive exPlanations
- SIDER
Side Effect Resource
- SMILES
Simplified Molecular Input Line Entry System
- SMOTE
Synthetic Minority Over-sampling Technique
- SVM
Support Vector Machine
- XGBoost
Extreme Gradient Boosting
References
1
Ai H. X. Chen W. Zhang L. Huang L. C. Yin Z. M. Hu H. et al (2018). Predicting drug-induced liver injury using ensemble learning methods and molecular fingerprints. Toxicol. Sci.165 (1), 100–107. 10.1093/toxsci/kfy121
2
Albert R. E. (1994). Carcinogen risk assessment in the us environmental-protection-agency. Crit. Rev. Toxicol.24 (1), 75–85. 10.3109/10408449409017920
3
Alves V. M. Muratov E. Fourches D. Strickland J. Kleinstreuer N. Andrade C. H. et al (2015). Predicting chemically-induced skin reactions. Part I: QSAR models of skin sensitization and their application to identify potentially hazardous compounds. Toxicol. Appl. Pharmacol.284 (2), 262–272. 10.1016/j.taap.2014.12.014
4
An FDA Artificial Intelligence (AI) (2024). Program for toxicology. Available online at: https://www.fda.gov/about-fda/nctr-research-focus-areas/artificial-intelligence (Accessed June 13, 2025).
5
Ankley G. T. Bennett R. S. Erickson R. J. Hoff D. J. Hornung M. W. Johnson R. D. et al (2010). Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem.29 (3), 730–741. 10.1002/etc.34
6
Babai S. Auclert L. Le-Louet H. (2021). Safety data and withdrawal of hepatotoxic drugs. Therapie76 (6), 715–723. 10.1016/j.therap.2018.02.004
7
Banerjee P. Kemmler E. Dunkel M. Preissner R. (2024). ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res.52 (W1), W513–W520. 10.1093/nar/gkae303
8
Barnes J. L. Zubair M. John K. Poirier M. C. Martin F. L. (2018). Carcinogens and DNA damage. Biochem. Soc. Trans.46 (5), 1213–1224. 10.1042/Bst20180519
9
Basu A. K. (2018). DNA damage, mutagenesis and cancer. Int. J. Mol. Sci.19 (4), 970. 10.3390/ijms19040970
10
Benigni R. Battistelli C. L. Bossa C. Tcheremenskaia O. Crettaz P. (2013). New perspectives in toxicological information management, and the role of ISSTOX databases in assessing chemical mutagenicity and carcinogenicity. Mutagenesis28 (4), 401–409. 10.1093/mutage/get016
11
Benigni R. Bossa C. Richard A. M. Yang C. H. (2008). A novel approach: chemical relational databases, and the role of the ISSCAN database on assessing chemical carcinogenicity. Ann. Dell Ist. Super. Di Sanita44 (1), 48–56.
12
Bran A. M. Cox S. Schilter O. Baldassari C. White A. D. Schwaller P. (2024). Augmenting large language models with chemistry tools. Nat. Mach. Intell.6 (5), 525–535. 10.1038/s42256-024-00832-8
13
Cai C. P. Guo P. F. Zhou Y. D. Zhou J. W. Wang Q. Zhang F. X. et al (2019). Deep learning-based prediction of drug-induced cardiotoxicity. J. Chem. Inf. Model.59 (3), 1073–1084. 10.1021/acs.jcim.8b00769
14
Cavasotto C. N. Scardino V. (2022). Machine learning toxicity prediction: latest advances by toxicity end point. Acs Omega7 (51), 47536–47546. 10.1021/acsomega.2c05693
15
Celik F. K. Karaduman G. (2023). Machine learning-based prediction of drug-induced hepatotoxicity: an OvA-QSTR approach. J. Chem. Inf. Model.63 (15), 4602–4614. 10.1021/acs.jcim.3c00687
16
Chen C. Y. C. (2011). TCM Database@Taiwan: the world's largest traditional Chinese medicine database for drug screening in silico. Plos One6 (1), e15939. 10.1371/journal.pone.0015939
17
Chen M. J. Borlak J. Tong W. D. (2013a). High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology58 (1), 388–396. 10.1002/hep.26208
18
Chen M. J. Hong H. X. Fang H. Kelly R. Zhou G. X. Borlak J. et al (2013b). Quantitative structure-activity relationship models for predicting drug-induced liver injury based on FDA-approved drug labeling annotation and using a large collection of drugs. Toxicol. Sci.136 (1), 242–249. 10.1093/toxsci/kft189
19
Chen M. J. Suzuki A. Thakkar S. Yu K. Hu C. C. Tong W. D. (2016). DILIrank: the largest reference drug list ranked by the risk for developing drug-induced liver injury in humans. Drug Discov. Today21 (4), 648–653. 10.1016/j.drudis.2016.02.015
20
Chen M. J. Vijay V. Shi Q. Liu Z. C. Fang H. Tong W. D. (2011). FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov. Today16 (15-16), 697–703. 10.1016/j.drudis.2011.05.007
21
Chen Z. Jiang Y. Zhang X. Y. Zheng R. Qiu R. J. Sun Y. et al (2022). ResNet18DNN: prediction approach of drug-induced liver injury by deep neural network with ResNet18. Briefings Bioinforma.23 (1), bbab503. 10.1093/bib/bbab503
22
Chen Z. Zhang L. Sun J. Q. Meng R. Yin S. D. Zhao Q. (2023). DCAMCP: a deep learning model based on capsule network and attention mechanism for molecular carcinogenicity prediction. J. Cell. Mol. Med.27 (20), 3117–3126. 10.1111/jcmm.17889
23
Didziapetris R. Lanevskij K. (2016). Compilation and physicochemical classification analysis of a diverse hERG inhibition database. J. Computer-Aided Mol. Des.30 (12), 1175–1188. 10.1007/s10822-016-9986-0
24
Dimitrov S. D. Diderich R. Sobanski T. Pavlov T. S. Chankov G. V. Chapkanov A. S. et al (2016). QSAR Toolbox - workflow and major functionalities. Sar Qsar Environ. Res.27 (3), 203–219. 10.1080/1062936x.2015.1136680
25
Doddareddy M. R. Klaasse E. C. Ijzerman A. P. Bender A. (2010). Prospective validation of a comprehensive in silico hERG model and its applications to commercial compound and drug databases. Chemmedchem5 (5), 716–729. 10.1002/cmdc.201000024
26
Du F. Yu H. B. Zou B. Y. Babcock J. Long S. Y. Li M. (2011). hERGCentral: a large database to store, retrieve, and analyze compound-human ether-à-go-go related gene channel interactions to facilitate cardiotoxicity assessment in drug development. Assay Drug Dev. Technol.9 (6), 580–588. 10.1089/adt.2011.0425
27
Duffus J. (1993). Glossary for chemists of terms used in toxicology (IUPAC Recommendations 1993). Pure Appl. Chem.65 (9), 2003–2122. 10.1351/pac199365092003
28
FDA (2005). International conference on harmonisation; guidance on S7B nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals; availability. Notice. Fed. Regist.70, 61133–61134. Available online at: https://www.ncbi.nlm.nih.gov/pubmed/16237859.
29
Ferguson L. R. (2010). Chronic inflammation and mutagenesis. Mutat. Research/Fundamental Mol. Mech. Mutagen.690 (1-2), 3–11. 10.1016/j.mrfmmm.2010.03.007
30
Foulds C. Trevino L. York B. Walker C. L. (2017). Endocrine-disrupting chemicals and fatty liver disease. Nat. Rev. Endocrinol.13 (8), 445–457. 10.1038/nrendo.2017.42
31
Fournier M. Vroland C. Megy S. Aguero S. Chemelle J. A. Defoort B. et al (2023). In silico genotoxicity prediction by similarity search and machine learning algorithm: optimization and validation of the method for high energetic materials. Propellants Explos. Pyrotech.48 (4), e202200259. 10.1002/prep.202200259
32
Fu L. Shi S. H. Yi J. C. Wang N. N. He Y. H. Wu Z. X. et al (2024). ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res.52 (W1), W422–W431. 10.1093/nar/gkae236
33
Gadaleta D. Spînu N. Roncaglioni A. Cronin M. T. D. Benfenati E. (2022). Prediction of the neurotoxic potential of chemicals based on modelling of molecular initiating events upstream of the adverse outcome pathways of (developmental) neurotoxicity. Int. J. Mol. Sci.23 (6), 3053. 10.3390/ijms23063053
34
Gaulton A. Hersey A. Nowotka M. Bento A. P. Chambers J. Mendez D. et al (2017). The ChEMBL database in 2017. Nucleic Acids Res.45 (D1), D945–D954. 10.1093/nar/gkw1074
35
Gayvert K. M. Madhukar N. S. Elemento O. (2016). A data-driven approach to predicting successes and failures of clinical trials. Cell Chem. Biol.23 (10), 1294–1301. 10.1016/j.chembiol.2016.07.023
36
Ghane Shahrbaf F. Assadi F. (2015). Drug-induced renal disorders. J. Ren. Inj. Prev.4 (3), 57–60. 10.12861/jrip.2015.12
37
Gold L. S. Manley N. B. Slone T. H. Rohrbach L. Garfinkel G. B. (2005). Supplement to the carcinogenic potency database (CPDB): results of animal bioassays published in the general literature through 1997 and by the national toxicology program in 1997-1998. Toxicol. Sci.85 (2), 747–808. 10.1093/toxsci/kfi161
38
Gong Y. N. Teng D. Wang Y. M. Gu Y. X. Wu Z. R. Li W. H. et al (2022). In silico prediction of potential drug-induced nephrotoxicity with machine learning methods. J. Appl. Toxicol.42 (10), 1639–1650. 10.1002/jat.4331
39
Greene N. Fisk L. Naven R. T. Note R. R. Patel M. L. Pelletier D. J. (2010). Developing structure-activity relationships for the prediction of hepatotoxicity. Chem. Res. Toxicol.23 (7), 1215–1222. 10.1021/tx1000865
40
Gu X. S. Manautou J. E. (2012). Molecular mechanisms underlying chemical liver injury. Expert Rev. Mol. Med.14, e4. 10.1017/S1462399411002110
41
Hansen K. Mika S. Schroeter T. Sutter A. ter Laak A. Steger-Hartmann T. et al (2009). Benchmark data set for in silico prediction of ames mutagenicity. J. Chem. Inf. Model.49 (9), 2077–2081. 10.1021/ci900161g
42
Heeren J. Scheja L. (2021). Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab.50, 101238. 10.1016/j.molmet.2021.101238
43
Heyndrickx W. Mervin L. Morawietz T. Sturm N. Friedrich L. Zalewski A. et al (2023). MELLODDY: cross-pharma federated learning at unprecedented scale unlocks benefits in QSAR without compromising proprietary information. J. Chem. Inf. Model.64 (7), 2331–2344. 10.1021/acs.jcim.3c00799
44
Hoofnagle J. H. Serrano J. Knoben J. E. Navarro V. J. (2013). LiverTox: a website on drug-induced liver injury. Hepatology57 (3), 873–874. 10.1002/hep.26175
45
ICH (2020). S2(R1) genotoxicity testing and data interpretation for pharmaceuticals intended for human use. Available online at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/s2r1-genotoxicity-testing-and-data-interpretation-pharmaceuticals-intended-human-use.
46
ICH (2022). S1B(R1) addendum to S1B testing for carcinogenicity of pharmaceuticals. Available online at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/s1br1-addendum-s1b-testing-carcinogenicity-pharmaceuticals.
47
ICH (2023). M7(R2) assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk. Available online at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-m7r2-assessment-and-control-dna-reactive-mutagenic-impurities-pharmaceuticals.
48
Jiang C. S. Zhao P. P. Li W. H. Tang Y. Liu G. X. (2020). In silico prediction of chemical neurotoxicity using machine learning. Toxicol. Res.9 (3), 164–172. 10.1093/toxres/tfaa016
49
Jiang D. J. Wu Z. X. Hsieh C. Y. Chen G. Y. Liao B. Wang Z. et al (2021). Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models. J. Cheminformatics13 (1), 12. 10.1186/s13321-020-00479-8
50
Karim A. Lee M. Balle T. Sattar A. (2021). CardioTox net: a robust predictor for hERG channel blockade based on deep learning meta-feature ensembles. J. Cheminformatics13 (1), 60. 10.1186/s13321-021-00541-z
51
Kim H. Park M. Lee I. Nam H. (2022). BayeshERG: a robust, reliable and interpretable deep learning model for predicting hERG channel blockers. Briefings Bioinforma.23 (4), bbac211. 10.1093/bib/bbac211
52
Kim S. Thiessen P. A. Bolton E. E. Chen J. Fu G. Gindulyte A. et al (2016). PubChem substance and compound databases. Nucleic Acids Res.44 (D1), D1202–D1213. 10.1093/nar/gkv951
53
Kocot-Kepska M. Zajaczkowska R. Mika J. Wordliczek J. Dobrogowski J. Przeklasa-Muszynska A. (2021). Peripheral mechanisms of neuropathic pain-the role of neuronal and non-neuronal interactions and their implications for topical treatment of neuropathic pain. Pharmaceuticals14 (2), 77. 10.3390/ph14020077
54
Konda L. S. K. P. S. K. Kristam R. (2019). hERG liability classification models using machine learning techniques. Comput. Toxicol.12, 100089. 10.1016/j.comtox.2019.100089
55
Kosnik M. B. Strickland J. D. Marvel S. W. Wallis D. J. Wallace K. Richard A. M. et al (2020). Concentration-response evaluation of ToxCast compounds for multivariate activity patterns of neural network function. Archives Toxicol.94 (2), 469–484. 10.1007/s00204-019-02636-x
56
Kotsampasakou E. Ecker G. F. (2017). Predicting drug-induced cholestasis with the help of hepatic transporters—an in silico modeling approach. J. Chem. Inf. Model.57 (3), 608–615. 10.1021/acs.jcim.6b00518
57
Kragstrup T. W. Glintborg B. Svensson A. L. McMaster C. Robinson P. C. Deleuran B. et al (2022). Waiting for JAK inhibitor safety data. Rmd Open8 (1), e002236. 10.1136/rmdopen-2022-002236
58
Krewski D. Acosta D. Andersen M. Anderson H. Bailar J. C. Boekelheide K. et al (2010). Toxicity testing in the 21st century: a vision and a strategy. J. Toxicol. Environ. Health-Part B-Critical Rev.13 (2-4), 51–138. 10.1080/10937404.2010.483176
59
Kuhn M. Letunic I. Jensen L. J. Bork P. (2016). The SIDER database of drugs and side effects. Nucleic Acids Res.44 (D1), D1075–D1079. 10.1093/nar/gkv1075
60
Lagunin A. Filimonov D. Zakharov A. Xie W. Huang Y. Zhu F. C. et al (2009). Computer-aided prediction of rodent carcinogenicity by PASS and CISOC-psct. Qsar and Comb. Sci.28 (8), 806–810. 10.1002/qsar.200860192
61
Lanevskij K. Didziapetris R. Sazonovas A. (2022). Physicochemical QSAR analysis of hERG inhibition revisited: towards a quantitative potency prediction. J. Computer-Aided Mol. Des.36 (12), 837–849. 10.1007/s10822-022-00483-0
62
Lee D. Yoo S. (2025). hERGAT: predicting hERG blockers using graph attention mechanism through atom- and molecule-level interaction analyses. J. Cheminformatics17 (1), 11. 10.1186/s13321-025-00957-x
63
Lee S. Yoo S. (2024). InterDILI: interpretable prediction of drug-induced liver injury through permutation feature importance and attention mechanism. J. Cheminformatics16 (1), 1. 10.1186/s13321-023-00796-8
64
Legradi J. B. Di Paolo C. Kraak M. H. S. van der Geest H. G. Schymanski E. L. Williams A. J. et al (2018). An ecotoxicological view on neurotoxicity assessment. Environ. Sci. Eur.30, 46–34. 10.1186/s12302-018-0173-x
65
Li Q. Y. Jorgensen F. S. Oprea T. Brunak S. Taboureau O. (2008). hERG classification model based on a combination of support vector machine method and GRIND descriptors. Mol. Pharm.5 (1), 117–127. 10.1021/mp700124e
66
Li S. M. Zhang L. Feng H. W. Meng J. H. Xie D. Yi L. W. et al (2021). MutagenPred-GCNNs: a graph convolutional neural network-based classification model for mutagenicity prediction with data-driven molecular fingerprints. Interdiscip. Sciences-Computational Life Sci.13 (1), 25–33. 10.1007/s12539-020-00407-2
67
Li T. Tong W. D. Roberts R. Liu Z. C. Thakkar S. (2021). DeepCarc: deep learning-powered carcinogenicity prediction using model-level representation. Front. Artif. Intell.4, 757780. 10.3389/frai.2021.757780
68
Liew C. Y. Lim Y. C. Yap C. W. (2011). Mixed learning algorithms and features ensemble in hepatotoxicity prediction. J. Computer-Aided Mol. Des.25 (9), 855–871. 10.1007/s10822-011-9468-3
69
Liu A. N. K. Seal S. Yang H. B. Bender A. (2023). Using chemical and biological data to predict drug toxicity. Slas Discov.28 (3), 53–64. 10.1016/j.slasd.2022.12.003
70
Liu L. L. Zhang L. Feng H. W. Li S. M. Liu M. Zhao J. et al (2021). Prediction of the blood-brain barrier (BBB) permeability of chemicals based on machine-learning and ensemble methods. Chem. Res. Toxicol.34 (6), 1456–1467. 10.1021/acs.chemrestox.0c00343
71
Liu M. Zhang L. Li S. M. Yang T. Z. Liu L. L. Zhao J. et al (2020). Prediction of hERG potassium channel blockage using ensemble learning methods and molecular fingerprints. Toxicol. Lett.332, 88–96. 10.1016/j.toxlet.2020.07.003
72
Liu T. Q. Lin Y. M. Wen X. Jorissen R. N. Gilson M. K. (2007). BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res.35, D198–D201. 10.1093/nar/gkl999
73
Liu Y. Ding S. Zhou S. Fan W. Tan Q. (2024). Moleculargpt: open large language model (llm) for few-shot molecular property prediction. arXiv:2406.12950
74
Lundberg S. M. Lee S. I. (2017). “A unified approach to interpreting model predictions,” in NIPS'17: Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach California USA, December 4 - 9, 2017, 4768–4777.)
75
Mak K. K. Pichika M. R. (2019). Artificial intelligence in drug development: present status and future prospects. Drug Discov. Today24 (3), 773–780. 10.1016/j.drudis.2018.11.014
76
Martínez M. J. Sabando M. V. Soto A. J. Roca C. Requena-Triguero C. Campillo N. E. et al (2022). Multitask deep neural networks for ames mutagenicity prediction. J. Chem. Inf. Model.62 (24), 6342–6351. 10.1021/acs.jcim.2c00532
77
Mastropietro A. Pasculli G. Feldmann C. Rodríguez-Pérez R. Bajorath J. (2022). EdgeSHAPer: bond-centric Shapley value-based explanation method for graph neural networks. Iscience25 (10), 105043. 10.1016/j.isci.2022.105043
78
Mazumdar B. Sarma P. K. D. Mahanta H. J. Sastry G. N. (2023). Machine learning based dynamic consensus model for predicting blood-brain barrier permeability. Comput. Biol. Med.160, 106984. 10.1016/j.compbiomed.2023.106984
79
McNaught A. D. Wilkinson S. (2025). 'toxicity' in IUPAC compendium of chemical terminology. 5th ed.IUPAC: International Union of Pure and Applied Chemistry.
80
Mulliner D. Schmidt F. Stolte M. Spirkl H. P. Czich A. Amberg A. (2016). Computational models for human and animal hepatotoxicity with a global application scope. Chem. Res. Toxicol.29 (5), 757–767. 10.1021/acs.chemrestox.5b00465
81
Munawar S. Vandenberg J. I. Jabeen I. (2019). Molecular docking guided grid-independent descriptor analysis to probe the impact of water molecules on conformational changes of hERG inhibitors in drug trapping phenomenon. Int. J. Mol. Sci.20 (14), 3385. 10.3390/ijms20143385
82
Myung Y. de Sa A. G. C. Ascher D. B. (2024). Deep-PK: deep learning for small molecule pharmacokinetic and toxicity prediction. Nucleic Acids Res.52 (W1), W469–W475. 10.1093/nar/gkae254
83
Negami T. Araki M. Okuno Y. Terada T. (2019). Calculation of absolute binding free energies between the hERG channel and structurally diverse drugs. Sci. Rep.9 (1), 16586. 10.1038/s41598-019-53120-6
84
Nohmi T. (2018). Thresholds of genotoxic and non-genotoxic carcinogens. Toxicol. Res.34 (4), 281–290. 10.5487/Tr.2018.34.4.281
85
NRC (1994). Science and judgment in risk assessment. National research Council (US) Committee on risk Assessment of hazardous air pollutants. Washington (DC): National Academies Press US. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK208246/.
86
OECD (2014). “Guidance document on the validation of (quantitative) structure-activity relationship [(Q)SAR] models,” in OECD series on testing and assessment. Paris.
87
Pang X. D. He X. J. Yang Y. Wang L. Sun Y. Z. Cao H. M. et al (2025). NeuTox 2.0: a hybrid deep learning architecture for screening potential neurotoxicity of chemicals based on multimodal feature fusion. Environ. Int.195, 109244. 10.1016/j.envint.2024.109244
88
Papamokos G. Silins I. (2016). Combining QSAR modeling and text-mining techniques to link chemical structures and carcinogenic modes of action. Front. Pharmacol.7, 284. 10.3389/fphar.2016.00284
89
Pei Q. Z. Zhang W. Zhu J. H. Wu K. H. Gao K. Y. Wu L. J. et al (2023). “BioT5: enriching cross-modal integration in biology with chemical knowledge and natural language associations,” in 2023 Conference on Empirical Methods in Natural Language Processing, Emnlp 2023, Singapore, December 6–10, 2023, 1102–1123.
90
Pognan F. Beilmann M. Boonen H. C. M. Czich A. Dear G. Hewitt P. et al (2023). The evolving role of investigative toxicology in the pharmaceutical industry. Nat. Rev. Drug Discov.22 (4), 317–335. 10.1038/s41573-022-00633-x
91
Quinton A. Latry P. Biour M. (1993). Hepatox(C) - a database about hepatotoxic drugs. Gastroenterologie Clinique Biol.17 (5bis), H116–H120.
92
Rao M. Nassiri V. Alhambra C. Snoeys J. Van Goethem F. Irrechukwu O. et al (2023). AI/ML models to predict the severity of drug-induced liver injury for small molecules. Chem. Res. Toxicol.36 (7), 1129–1139. 10.1021/acs.chemrestox.3c00098
93
Regev A. (2014). Drug-induced liver injury and drug development: industry perspective. Seminars Liver Dis.34 (2), 227–239. 10.1055/s-0034-1375962
94
Reiser P. Neubert M. Eberhard A. Torresi L. Zhou C. Shao C. et al (2022). Graph neural networks for materials science and chemistry. Commun. Mater.3 (1), 93. 10.1038/s43246-022-00315-6
95
Ren N. Atyah M. Chen W. Y. Zhou C. H. (2017). The various aspects of genetic and epigenetic toxicology: testing methods and clinical applications. J. Transl. Med.15, 110. 10.1186/s12967-017-1218-4
96
Richard A. M. Huang R. L. Waidyanatha S. Shinn P. Collins B. J. Thillainadarajah I. et al (2021). The Tox21 10K compound library: collaborative chemistry advancing toxicology. Chem. Res. Toxicol.34 (2), 189–216. 10.1021/acs.chemrestox.0c00264
97
Richard A. M. Judson R. S. Houck K. A. Grulke C. M. Volarath P. Thillainadarajah I. et al (2016). ToxCast chemical landscape: paving the road to 21st century toxicology. Chem. Res. Toxicol.29 (8), 1225–1251. 10.1021/acs.chemrestox.6b00135
98
Rodríguez-Pérez R. Bajorath J. (2020). Interpretation of machine learning models using shapley values: application to compound potency and multi-target activity predictions. J. Computer-Aided Mol. Des.34 (10), 1013–1026. 10.1007/s10822-020-00314-0
99
Ryu J. Y. Lee M. Y. Lee J. H. Lee B. H. Oh K. S. (2020). DeepHIT: a deep learning framework for prediction of hERG-induced cardiotoxicity. Bioinformatics36 (10), 3049–3055. 10.1093/bioinformatics/btaa075
100
Schrenk D. (2018). What is the meaning of 'A compound is carcinogenic. Toxicol. Rep.5, 504–511. 10.1016/j.toxrep.2018.04.002
101
Schwaller P. Laino T. Gaudin T. Bolgar P. Hunter C. A. Bekas C. et al (2019). Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. Acs Central Sci.5 (9), 1572–1583. 10.1021/acscentsci.9b00576
102
Seal S. Williams D. Hosseini-Gerami L. Mahale M. Carpenter A. E. Spjuth O. et al (2024). Improved detection of drug-induced liver injury by integrating predicted and experimental data. Chem. Res. Toxicol.37 (8), 1290–1305. 10.1021/acs.chemrestox.4c00015
103
Shi Y. P. Hua Y. Q. Wang B. B. Zhang R. Q. Li X. (2022). In silico prediction and insights into the structural basis of drug induced nephrotoxicity. Front. Pharmacol.12, 793332. 10.3389/fphar.2021.793332
104
Shinada N. K. Koyama N. Ikemori M. Nishioka T. Hitaoka S. Hakura A. et al (2022). Optimizing machine-learning models for mutagenicity prediction through better feature selection. Mutagenesis37 (3-4), 191–202. 10.1093/mutage/geac010
105
Storchi L. Cruciani G. Cross S. (2023). DeepGRID: deep learning using GRID descriptors for BBB prediction. J. Chem. Inf. Model.63 (17), 5496–5512. 10.1021/acs.jcim.3c00768
106
Tang Q. Nie F. L. Zhao Q. Chen W. (2022). A merged molecular representation deep learning method for blood-brain barrier permeability prediction. Briefings Bioinforma.23 (5), bbac357. 10.1093/bib/bbac357
107
Thakkar S. Li T. Liu Z. C. Wu L. H. Roberts R. Tong W. D. (2020). Drug-induced liver injury severity and toxicity (DILIst): binary classification of 1279 drugs by human hepatotoxicity. Drug Discov. Today25 (1), 201–208. 10.1016/j.drudis.2019.09.022
108
Tibo A. He J. Z. Janet J. P. Nittinger E. Engkvist O. (2024). Exhaustive local chemical space exploration using a transformer model. Nat. Commun.15 (1), 7315. 10.1038/s41467-024-51672-4
109
Valentine W. M. (2020). Toxic peripheral neuropathies: agents and mechanisms. Toxicol. Pathol.48 (1), 152–173. 10.1177/0192623319854326
110
Vamathevan J. Clark D. Czodrowski P. Dunham I. Ferran E. Lee G. et al (2019). Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov.18 (6), 463–477. 10.1038/s41573-019-0024-5
111
Venkatraman V. (2021). FP-ADMET: a compendium of fingerprint-based ADMET prediction models. J. Cheminformatics13 (1), 75. 10.1186/s13321-021-00557-5
112
Villeneuve D. L. Crump D. Garcia-Reyero N. Hecker M. Hutchinson T. H. LaLone C. A. et al (2014). Adverse outcome pathway (AOP) development I: strategies and principles. Toxicol. Sci.142 (2), 312–320. 10.1093/toxsci/kfu199
113
Walker P. A. Ryder S. Lavado A. Dilworth C. Riley R. J. (2020). The evolution of strategies to minimise the risk of human drug-induced liver injury (DILI) in drug discovery and development. Archives Toxicol.94 (8), 2559–2585. 10.1007/s00204-020-02763-w
114
Wang S. Q. Sun H. Y. Liu H. Li D. Li Y. Y. Hou T. J. (2016). ADMET evaluation in drug discovery. 16. Predicting hERG blockers by combining multiple pharmacophores and machine learning approaches. Mol. Pharm.13 (8), 2855–2866. 10.1021/acs.molpharmaceut.6b00471
115
Wang T. Y. Sun J. Q. Zhao Q. (2023). Investigating cardiotoxicity related with hERG channel blockers using molecular fingerprints and graph attention mechanism. Comput. Biol. Med.153, 106464. 10.1016/j.compbiomed.2022.106464
116
Wang Y. M. Huang M. T. Deng H. Li W. H. Wu Z. R. Tang Y. et al (2023). Identification of vital chemical information via visualization of graph neural networks. Briefings Bioinforma.24 (1), bbac577. 10.1093/bib/bbac577
117
Wellawatte G. P. Seshadri A. White A. D. (2022). Model agnostic generation of counterfactual explanations for molecules. Chem. Sci.13 (13), 3697–3705. 10.1039/d1sc05259d
118
Wen J. Zhang X. Rush E. Panickan V. A. Li X. Y. Cai T. R. et al (2023). Multimodal representation learning for predicting molecule-disease relations. Bioinformatics39 (2), btad085. 10.1093/bioinformatics/btad085
119
Wigh D. S. Goodman J. M. Lapkin A. A. (2022). A review of molecular representation in the age of machine learning. Wiley Interdiscip. Reviews-Computational Mol. Sci.12 (5), e1603. 10.1002/wcms.1603
120
Wishart D. S. Feunang Y. D. Guo A. C. Lo E. J. Marcu A. Grant J. R. et al (2018). DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res.46 (D1), D1074–D1082. 10.1093/nar/gkx1037
121
Wu L. L. Yan B. W. Han J. S. Li R. J. Xiao J. He S. et al (2023). TOXRIC: a comprehensive database of toxicological data and benchmarks. Nucleic Acids Res.51 (D1), D1432–D1445. 10.1093/nar/gkac1074
122
Wu W. X. Qian J. Y. Liang C. J. Yang J. Y. Ge G. B. Zhou Q. P. et al (2023). GeoDILI: a robust and interpretable model for drug-induced liver injury prediction using graph neural network-based molecular geometric representation. Chem. Res. Toxicol.36 (11), 1717–1730. 10.1021/acs.chemrestox.3c00199
123
Wu Z. X. Wang J. K. Du H. Y. Jiang D. J. Kang Y. Li D. et al (2023). Chemistry-intuitive explanation of graph neural networks for molecular property prediction with substructure masking. Nat. Commun.14 (1), 2585. 10.1038/s41467-023-38192-3
124
Xu C. Y. Cheng F. X. Chen L. Du Z. Li W. H. Liu G. X. et al (2012). In silico prediction of chemical ames mutagenicity. J. Chem. Inf. Model.52 (11), 2840–2847. 10.1021/ci300400a
125
Xu H. Y. Zhao Y. P. Zhang Y. X. Han J. S. Zan P. He S. et al (2024). Deep active learning with high structural discriminability for molecular mutagenicity prediction. Commun. Biol.7 (1), 1071. 10.1038/s42003-024-06758-6
126
Xu J. H. J. Henstock P. V. Dunn M. C. Smith A. R. Chabot J. R. de Graaf D. (2008). Cellular imaging predictions of clinical drug-induced liver injury. Toxicol. Sci.105 (1), 97–105. 10.1093/toxsci/kfn109
127
Xu P. C. Ji X. B. Li M. J. Lu W. C. (2023). Small data machine learning in materials science. Npj Comput. Mater.9 (1), 42. 10.1038/s41524-023-01000-z
128
Xu Y. J. Dai Z. W. Chen F. J. Gao S. S. Pei J. F. Lai L. H. (2015). Deep learning for drug-induced liver injury. J. Chem. Inf. Model.55 (10), 2085–2093. 10.1021/acs.jcim.5b00238
129
Yan B. W. Ye X. N. Wang J. Han J. S. Wu L. L. He S. et al (2022). An algorithm framework for drug-induced liver injury prediction based on genetic algorithm and ensemble learning. Molecules27 (10), 3112. 10.3390/molecules27103112
130
Yang H. B. Lou C. F. Sun L. X. Li J. Cai Y. C. Wang Z. et al (2019). admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics35 (6), 1067–1069. 10.1093/bioinformatics/bty707
131
Yang P. C. DeMarco K. R. Aghasafari P. Jeng M. T. Dawson J. R. D. Bekker S. et al (2020). A computational pipeline to predict cardiotoxicity from the atom to the rhythm. Circulation Res.126 (8), 947–964. 10.1161/Circresaha.119.316404
132
Yang T. B. Ding X. Y. Mcmichael E. Pun F. W. Aliper A. Ren F. et al (2024). AttenhERG: a reliable and interpretable graph neural network framework for predicting hERG channel blockers. J. Cheminformatics16 (1), 143. 10.1186/s13321-024-00940-y
133
Ying R. Bourgeois D. You J. X. Zitnik M. Leskovec J. (2019). “GNNExplainer: generating explanations for graph neural networks,” in Proceedings of the 33rd Conference on Neural Information Processing Systems NeurIPS, Vancouver, Canada, 8-14 December 2019, 9240–9251.
134
Yu M.-S. Lee J. Lee Y. Cho D. Oh K.-S. Jang J. et al (2025). hERGBoost: a gradient boosting model for quantitative IC50 prediction of hERG channel blockers. Comput. Biol. Med.184, 109416. 10.1016/j.compbiomed.2024.109416
135
Zhang H. Ren J. X. Ma J. X. Ding L. (2019). Development of an in silico prediction model for chemical-induced urinary tract toxicity by using naive Bayes classifier. Mol. Divers.23 (2), 381–392. 10.1007/s11030-018-9882-8
136
Zhang S. Z. Yan Z. Y. Huang Y. Y. Liu L. H. He D. L. Wang W. et al (2022). HelixADMET: a robust and endpoint extensible ADMET system incorporating self-supervised knowledge transfer. Bioinformatics38 (13), 3444–3453. 10.1093/bioinformatics/btac342
137
Zhang Y. M. Zhao J. N. Wang Y. C. Fan Y. R. Zhu L. Yang Y. et al (2019). Prediction of hERG K+ channel blockage using deep neural networks. Chem. Biol. and Drug Des.94 (5), 1973–1985. 10.1111/cbdd.13600
138
Zhao X. Sun Y. H. Zhang R. Q. Chen Z. Y. Hua Y. Q. Zhang P. et al (2022). Machine learning modeling and insights into the structural characteristics of drug-induced neurotoxicity. J. Chem. Inf. Model.62 (23), 6035–6045. 10.1021/acs.jcim.2c01131
139
Zheng S. J. Yan X. Yang Y. D. Xu J. (2019). Identifying structure-property relationships through SMILES syntax analysis with self-attention mechanism. J. Chem. Inf. Model.59 (2), 914–923. 10.1021/acs.jcim.8b00803
140
Zhu H. Martin T. M. Ye L. Sedykh A. Young D. M. Tropsha A. (2009). Quantitative structure-activity relationship modeling of rat acute toxicity by oral exposure. Chem. Res. Toxicol.22 (12), 1913–1921. 10.1021/tx900189p
Summary
Keywords
artificial intelligence, drug discovery, toxicity, in silico methods, virtual screening
Citation
Lee H, Kim J, Kim J-W and Lee Y (2025) Recent advances in AI-based toxicity prediction for drug discovery. Front. Chem. 13:1632046. doi: 10.3389/fchem.2025.1632046
Received
20 May 2025
Accepted
18 June 2025
Published
08 July 2025
Volume
13 - 2025
Edited by
Xin Wang, Shenzhen Technology University, China
Reviewed by
Georgios Papamokos, University of Ioannina, Greece
Ashwin Dhakal, The University of Missouri, United States
Updates
Copyright
© 2025 Lee, Kim, Kim and Lee.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ji-Woon Kim, jiwoonkim@khu.ac.kr; Yoonji Lee, yoonjilee@cau.ac.kr
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.