 Hanhui Li1*
Hanhui Li1* Jingxu Yao
Jingxu Yao- 1Guizhou University of Commerce, Guiyang, China
- 2Wuhan University of Technology, Wuhan, China
- 3Shandong University of Arts, Shandong, Jinan, China
- 4Huzhou University, Huzhou, Zhejiang, China
Introduction: The advancement of digitized material design has revolutionized the field of materials science by integrating computational modeling, machine learning, and high-throughput simulations. Traditional material discovery heavily relies on iterative physical experiments, which are often resource-intensive and time-consuming. Recent developments in high-throughput computing offer an efficient alternative by enabling large-scale simulations and data-driven predictions of material properties. However, conventional predictive models frequently suffer from limited generalization, inadequate incorporation of domain knowledge, and inefficient optimization of material structures.
Methods: To address these limitations, we propose a novel framework that combines physics-informed machine learning with generative optimization for material design and performance prediction. Our approach consists of three major components: a graph-embedded material property prediction model that integrates multi-modal data for structure–property mapping, a generative model for structure exploration using reinforcement learning, and a physics-guided constraint mechanism that ensures realistic and reliable material designs.
Results: By embedding domain-specific priors into a deep learning framework, our method significantly improves prediction accuracy while maintaining physical interpretability. Extensive experiments demonstrate that our approach outperforms state-of-the-art models in both predictive performance and optimization efficiency.
Discussion: These findings highlight the potential of digitized design methodologies to accelerate the discovery of novel materials with desired properties and to drive next-generation material innovation.
1 Introduction
The design and performance prediction of materials have always been critical challenges in materials science and engineering. Traditional experimental approaches are not only time-consuming and expensive but also often limited by the complexity of material properties and interactions Zhou et al. (2020). With the advent of computational methods, researchers have increasingly relied on simulation-based approaches to accelerate material discovery. However, these conventional simulations still struggle with efficiency, particularly when dealing with high-dimensional material spaces Angelopoulos et al. (2023). The emergence of high-throughput computing (HTC) has provided a new paradigm by enabling rapid evaluation of vast material libraries. Not only does HTC facilitate large-scale material screening, but it also enhances predictive modeling by leveraging extensive datasets Shen and Kwok (2023). The integration of HTC with data-driven methodologies has further optimized performance predictions, making it possible to identify novel materials with desirable properties efficiently. This shift towards digitized material design, combining computational power with intelligent algorithms, is transforming the field by reducing the reliance on trial-and-error experimentation and promoting data-driven innovation Wen and Li (2023).
To address the limitations of purely experimental methods, early computational material design approaches focused on symbolic AI and knowledge-based models. These methods relied on explicit rule-based representations of material properties, utilizing domain knowledge and expert-defined relationships to guide material discovery Ren et al. (2024). Expert systems and first-principles simulations, such as density functional theory (DFT), played a crucial role in predicting electronic structures and material behaviors. These models were interpretable, allowing researchers to derive fundamental insights into material interactions Li et al. (2023). However, their effectiveness was constrained by the complexity of material systems, as manually encoding all relevant physical principles and interactions proved increasingly difficult Yin et al. (2023). Knowledge-based models lacked adaptability when dealing with novel materials that deviated from established scientific understanding. As a result, these traditional approaches, while foundational, struggled with scalability and flexibility in handling high-throughput material discovery Yu et al. (2023).
To overcome the limitations of symbolic AI, researchers turned to data-driven and machine learning (ML)-based methods, which leveraged statistical patterns in material datasets rather than explicit rule-based encoding Durairaj and Mohan (2022). ML models, such as support vector machines, decision trees, and Gaussian processes, allowed for efficient material property predictions based on training data from experiments and simulations. One significant advantage of ML-based approaches was their ability to interpolate within known material spaces, offering accurate predictions without requiring explicit physical formulations Chandra et al. (2021). HTC-enabled data generation expanded the applicability of these models by providing large-scale datasets for training, thereby improving generalization. However, these approaches also had drawbacks, particularly in their reliance on high-quality labeled data Fan et al. (2021). The interpretability of ML models remained a challenge, as many predictive models acted as “black boxes,” limiting their usefulness for fundamental scientific insights. Moreover, traditional ML techniques often struggled with extrapolation beyond known data distributions, making them less effective for discovering entirely novel materials Hou et al. (2022).
To further enhance the predictive capabilities of ML-based models, deep learning (DL) and pretrained models have emerged as powerful tools in digitized material design. Unlike traditional ML approaches, deep neural networks can automatically extract complex hierarchical features from large-scale material datasets, enabling more accurate and scalable predictions Lindemann et al. (2021). The adoption of graph neural networks (GNNs), convolutional neural networks (CNNs), and transformers has revolutionized material informatics by capturing intricate structure-property relationships. Pretrained models, trained on extensive HTC-generated datasets, offer significant advantages by transferring learned knowledge to new material discovery tasks Dudukcu et al. (2022). This transfer learning capability significantly reduces the need for large labeled datasets, making deep learning particularly valuable for high-throughput screening. Generative models, such as variational autoencoders (VAEs) and generative adversarial networks (GANs), have been utilized to propose novel material candidates, further accelerating the design process Amalou et al. (2022). Despite these advancements, challenges remain in ensuring the generalizability and robustness of deep learning models, particularly in predicting out-of-distribution materials. The computational cost associated with training large-scale deep networks is another critical issue, necessitating the development of more efficient architectures and hybrid approaches that integrate physical principles with data-driven learning Xiao et al. (2021).
Building on the limitations of existing approaches, we propose a hybrid HTC-driven framework that integrates deep learning with physics-based simulations to achieve more accurate and interpretable material design. By leveraging HTC-generated large-scale datasets, our method addresses the data scarcity issue faced by ML models while maintaining the physical rigor of knowledge-based approaches. This hybrid framework combines symbolic AI for fundamental insights, machine learning for pattern recognition, and deep learning for automated feature extraction, creating a comprehensive and scalable material discovery pipeline. Our approach incorporates uncertainty quantification techniques to enhance the reliability of predictions, ensuring robust generalization to novel materials. Through the seamless integration of high-throughput simulations with advanced AI methodologies, our framework paves the way for a new era of digitized material design, enabling faster, more efficient, and scientifically grounded material discovery.
Our key contributions can be outlined as below.
• Our method integrates symbolic AI, machine learning, and deep learning, combining physical interpretability with data-driven efficiency to improve material prediction accuracy.
• The proposed framework supports multi-scale material modeling, effectively handling diverse materials across different domains while ensuring high throughput and adaptability.
• By incorporating uncertainty quantification and high-throughput computing, our approach significantly improves predictive confidence, leading to more successful experimental validation and real-world material applications.
2 Related work
2.1 High-throughput computing in materials design
High-throughput computing (HTC) has revolutionized materials design by enabling the rapid screening and discovery of novel materials with desired properties. This computational approach leverages the power of parallel processing to perform extensive first-principles calculations, thereby accelerating the identification of promising candidates for various applications. By automating and scaling computational workflows, HTC facilitates the efficient exploration of vast chemical and structural spaces, which is essential for the development of advanced materials Wang et al. (2021b). One of the fundamental aspects of HTC in materials design is its reliance on first-principles calculations, particularly those based on density functional theory (DFT). These calculations provide accurate predictions of material properties such as electronic structure, stability, and reactivity without the need for empirical parameters. By systematically varying compositional and structural parameters, HTC enables the construction of comprehensive databases that can be mined for materials with optimal characteristics Xu et al. (2020). For instance, the Materials Project has utilized HTC to compute the properties of thousands of inorganic compounds, thereby providing a valuable resource for researchers seeking materials with specific functionalities. The integration of HTC with data techniques further enhances its utility in materials design. By analyzing large datasets generated from high-throughput calculations, researchers can identify patterns and correlations that inform the design of new materials. This approach has been successfully applied to the discovery of materials for energy storage, catalysis, and electronic applications. For example, in the context of lithium-ion batteries, HTC has been employed to screen potential electrode materials by evaluating their voltage profiles, stability, and capacity, leading to the identification of novel compounds with superior performance. Moreover, HTC facilitates the exploration of complex materials systems, such as high-entropy alloys and complex oxides, where the combinatorial space is vast. By systematically sampling different compositions and structures, HTC enables the identification of stable phases and the prediction of their properties, thereby guiding experimental efforts Karevan and Suykens (2020). This approach reduces the reliance on trial-and-error experimentation, making the materials discovery process more efficient and cost-effective. The development of robust computational workflows is crucial for the successful implementation of HTC in materials design. These workflows automate the process of structure generation, property calculation, and data analysis, ensuring consistency and reproducibility. Advanced workflow management systems have been developed to handle the complexities associated with large-scale computations, including error handling, data storage, and resource allocation. For example, the mkite platform offers a distributed computing environment tailored for high-throughput materials simulations, enabling researchers to efficiently manage and execute large-scale computational studies.
In addition to traditional first-principles approaches such as density functional theory (DFT), several alternative paradigms have recently gained traction in the high-throughput computing community. Among these, interatomic potentials—empirically derived functions describing interactions between atoms—have long been used for large-scale simulations, albeit with limitations in generalizability and transferability. More recently, machine learning-based potentials have emerged as powerful surrogates for ab initio methods. These include models like the Moment Tensor Potential (MTP), Gaussian Approximation Potentials (GAP), Deep Potential Molecular Dynamics (DeePMD), and graph-based neural potentials such as NequIP Zheng and Chen (2021). These frameworks are trained on DFT-calculated data but can perform orders of magnitude faster while retaining high fidelity. In addition, “universal potentials” have been developed to generalize across different material classes, further increasing their applicability in exploratory studies. Such advances in interatomic modeling offer significant speed and scalability advantages, enabling high-throughput workflows to simulate complex phenomena like phase stability, defect formation, and diffusion with greater computational efficiency Prifling et al. (2021). The incorporation of these potentials into HTC pipelines complements traditional DFT-based approaches and broadens the scope of feasible investigations in modern digitized materials discovery.
2.2 Machine learning for performance prediction
The integration of machine learning (ML) techniques into materials science has significantly enhanced the ability to predict material performance, thereby accelerating the discovery and optimization of new materials. By learning from existing data, ML models can identify complex patterns and relationships that are not easily discernible through traditional methods, enabling accurate predictions of material properties and behaviors Wang et al. (2024). One of the primary applications of ML in materials science is the prediction of properties based on compositional and structural features. By training models on datasets obtained from experiments or high-throughput computations, researchers can develop predictive models that estimate properties such as band gaps, elastic moduli, and thermal conductivities. For instance, supervised learning algorithms have been employed to predict the formation energies of inorganic compounds, facilitating the identification of thermodynamically stable materials Altan and Karasu (2021). Similarly, ML models have been used to predict the photovoltaic efficiencies of organic molecules, guiding the design of more efficient solar cell materials. Feature engineering plays a crucial role in the success of ML models for performance prediction. By selecting appropriate descriptors that capture the underlying physics and chemistry of materials, researchers can improve the accuracy and interpretability of the models. Descriptors such as atomic radii, electronegativities, and coordination numbers have been utilized to represent materials in a form suitable for ML algorithms Wen et al. (2021). Advancements in automatic feature selection and representation learning, including the use of graph-based methods, have further enhanced the capability of ML models to handle complex materials systems. The combination of ML with high-throughput computing has led to the development of hybrid approaches for materials discovery. In such frameworks, ML models are trained on data generated from high-throughput calculations and subsequently used to predict properties of unexplored materials, thereby reducing the computational burden Moskolaï et al. (2021). This approach has been applied to the discovery of thermoelectric materials, where ML models trained on computational data have identified promising candidates with high figures of merit. Unsupervised learning techniques, such as clustering and dimensionality reduction, have also been employed to explore materials datasets. These methods can reveal hidden structures in data, classify materials into categories with similar properties, and identify outliers that may exhibit novel behaviors. For example, clustering algorithms have been used to group materials based on their electronic structures, aiding in the systematic exploration of materials for electronic applications Morid et al. (2021). The integration of ML into materials science also extends to the development of inverse design strategies, where desired properties are specified, and the corresponding material structures are predicted. Generative models, such as variational autoencoders and generative adversarial networks, have been utilized to propose new materials with target properties, thereby shifting the paradigm from trial-and-error experimentation to rational design Zhao et al. (2022). Machine learning has emerged as a powerful tool for predicting material performance, offering the ability to rapidly and accurately estimate properties based on existing data. The synergy between ML and high-throughput computing holds great promise for accelerating materials discovery and optimization, ultimately leading to the development of advanced materials for a wide range of applications.
2.3 Data-driven materials informatics
Data-driven materials informatics has emerged as a transformative approach within materials science, fundamentally altering traditional research paradigms. Rather than relying exclusively on experimental trial-and-error, this methodology employs advanced data analysis techniques to systematically harness information from extensive materials datasets. As data availability in materials science grows exponentially—spurred by improvements in experimental techniques, computational simulations, and open-access databases—the need to effectively manage and interpret this information becomes increasingly critical Wang et al. (2021a). One of the core elements that distinguishes data-driven informatics from traditional approaches is its capacity to identify previously hidden correlations within complex, high-dimensional datasets. Through sophisticated statistical techniques, machine learning algorithms, and pattern recognition, this approach can rapidly uncover intricate relationships between structural features, processing conditions, and resulting material properties Widiputra et al. (2021). Such insights facilitate a deeper, more predictive understanding of how specific material configurations influence performance, streamlining the discovery process significantly. The incorporation of high-throughput computing (HTC) technologies further enhances the efficacy of data-driven informatics by accelerating data generation and analysis. HTC methods allow researchers to systematically generate massive amounts of computational data, covering a broad spectrum of possible material configurations. When combined with advanced analytics, this approach significantly reduces the time required to screen and identify promising new materials, offering substantial advantages over conventional, experimentally intensive techniques Yang and Wang (2021). Machine learning and deep learning techniques play a pivotal role in data-driven materials informatics, particularly through their ability to model highly nonlinear relationships within large datasets. Unlike traditional modeling methods that depend heavily on predefined rules or simplistic empirical correlations, machine learning models—such as neural networks, support vector machines, and ensemble methods—learn complex interactions directly from the data. This flexibility enables accurate predictions across diverse materials systems, especially when combined with careful feature engineering and domain-specific knowledge Ruan et al. (2021). Recent advances in deep learning, notably graph neural networks (GNNs) and convolutional neural networks (CNNs), have particularly enhanced the capability to capture structural and compositional information inherent in material datasets. These advanced architectures effectively represent materials as complex interconnected networks, capturing atomic-level interactions and higher-order structural motifs. By doing so, they significantly improve the accuracy of property predictions and enable more robust generalization to novel, unexplored regions of the materials landscape Kim and King (2020). Another essential dimension of data-driven informatics involves generative modeling and inverse design methodologies. Leveraging techniques such as variational autoencoders (VAEs) and generative adversarial networks (GANs), researchers can systematically propose entirely novel materials with tailored characteristics. This strategy shifts the paradigm from reactive discovery—where experiments or computations test predefined materials—to proactive creation, where desirable properties dictate structural exploration. Coupled with reinforcement learning and physics-informed constraints, generative approaches are instrumental in guiding the search toward feasible, high-performance material solutions Bachmann et al. (2022). Data-driven materials informatics represents a profound shift toward computational and algorithmically informed material design, providing powerful new tools that integrate data analytics, computational simulations, machine learning, and generative modeling. This approach not only accelerates material discovery but also delivers deeper scientific insights, paving the way for more systematic, rational, and efficient development of advanced materials tailored explicitly for targeted applications.
3 Methods
3.1 Overview
The field of Digitized Material Design has gained significant attention in recent years due to its potential to revolutionize material discovery, manufacturing, and performance optimization. Traditional material design relies on iterative physical experiments, which are costly and time-consuming. With advancements in computational modeling, machine learning, and high-throughput simulations, digitized approaches enable a more efficient and systematic exploration of material properties and structures.
This section provides an overview of our proposed method for digitized material design. Our approach consists of three major components: a formalized problem definition and mathematical representation of material properties; a novel computational model for material property prediction and structure optimization; and a domain-specific strategy that integrates physics-informed priors and data-driven methodologies, ensuring accuracy and generalizability. In Section 3.2, we introduce the fundamental concepts and notations necessary for describing the digitized material design problem. We formulate the relationship between material structures, properties, and their digital representations using a mathematical framework that bridges computational models and real-world materials. We also provide an overview of relevant theoretical foundations, including multi-scale modeling, graph-based material representations, and statistical learning approaches used to capture the complex interactions governing material behavior. Building upon this foundation, in Section 3.3, we present our proposed model, which leverages deep learning architectures and physics-based simulations to predict material properties from digital representations. Our model is designed to incorporate multi-modal data sources, enabling it to learn complex structure-property relationships with high accuracy. Unlike conventional approaches that rely solely on empirical data, our model integrates domain knowledge through hybrid modeling techniques, combining data-driven learning with fundamental physical principles. In Section 3.4, we describe our strategic approach to optimizing material design. Our methodology employs a combination of generative modeling, inverse design techniques, and reinforcement learning to iteratively refine material candidates. By leveraging uncertainty quantification and active learning, our approach ensures that the model efficiently explores the material design space while maintaining robustness and interpretability. We introduce a novel evaluation metric that balances predictive accuracy and computational efficiency, allowing for scalable deployment in practical applications. Our method for digitized material design provides a unified framework that integrates computational modeling, machine learning, and domain-specific knowledge. By systematically structuring the material discovery process, our approach significantly enhances the efficiency of material development, reduces experimental costs, and accelerates the transition from theoretical design to real-world applications.
3.2 Preliminaries
In this section, we establish the mathematical framework for Digitized Material Design by formulating the problem in a structured manner. We introduce key notations, define the relationships between material structures and properties, and outline the computational representations used to describe digitized materials. This formalization provides the foundation for our proposed model and optimization strategy.
A material can be characterized by its structure, properties, and processing conditions. We represent a material as a tuple (Equation 1):
where
The structural representation
where
The properties of a material,
where
For computational modeling, we assume that the material properties can be parameterized by a vector
where
In digitized material design, structures and properties are often represented using graph-based models or neural descriptors. We consider a graph-based representation where a material structure is modeled as a weighted graph (Equation 5):
where
The transition from a structural representation to a property prediction model is typically governed by differential equations. A common approach is to use a partial differential equation (PDE) formulation (Equation 6):
where
Given a dataset
where
Material design involves solving an inverse problem: given a target property
where
3.3 Graph-embedded material property prediction model (GEM-PPM)
In this section, we propose the Graph-Embedded Material Property Prediction Model (GEM-PPM), which is designed to accurately learn relationships between material structures and properties from digitized representations (As shown in Figure 1). The proposed model highlights three key innovations.
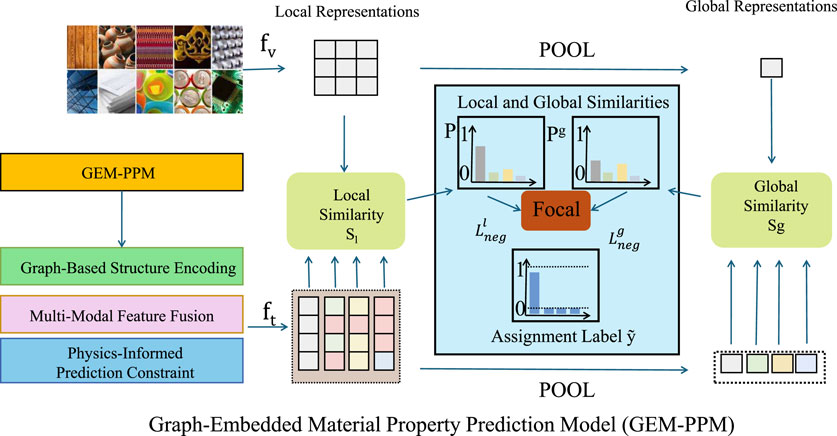
Figure 1. Architecture of the proposed Graph-Embedded Material Property Prediction Model (GEM-PPM). The model takes digitized material representations as input and encodes them into structural graphs, which are processed through a graph neural network to extract both local and global structural features. These structural embeddings are fused with external condition embeddings—such as temperature or synthesis parameters—through a multi-modal fusion module that includes concatenation, gating, and bilinear interaction. The resulting feature representations are used to compute local and global similarities with visual descriptors, followed by pooling and assignment to guide contrastive alignment. A focal loss is applied to emphasize informative negative pairs, and a physics-informed constraint is introduced to enforce consistency with governing physical laws during training. This unified framework enables accurate and physically meaningful material property prediction from structure–condition pairs.
3.3.1 Graph-based structure encoding
To effectively encode and utilize the intricate structural characteristics of materials, we represent each material as a weighted graph defined as (Equation 9):
where
where
where the pooling function can be a simple mean, sum, or a more complex attention-based readout mechanism. In some cases, to incorporate edge-level contributions more explicitly, a joint aggregation of edge and node embeddings is also considered (Equation 12):
where
where
3.3.2 Multi-Modal Feature Fusion
To enhance the expressiveness and predictive power of the model, we introduce a multi-modal feature fusion mechanism that integrates structural representations of materials with various external factors such as temperature, pressure, and synthesis conditions. These external conditions often play a critical role in determining the physical and chemical behavior of materials. The structural representation, denoted as
where
where
where
where
3.3.3 Physics-informed prediction constraint
To ensure physically consistent predictions in data-driven modeling, especially for problems governed by well-established physical laws, it is crucial to incorporate domain-specific physical constraints directly into the learning objective. Physics-Informed Machine Learning (PIML) achieves this by embedding the governing equations, such as conservation laws, constitutive relations, or equilibrium conditions, into the model training process through a regularization loss (As shown in Figure 2). The physics-based loss component can be defined as follows (Equation 19):
where
where
with
To ensure energy consistency, another constraint often considered is the principle of minimum potential energy, represented by (Equation 23):
where
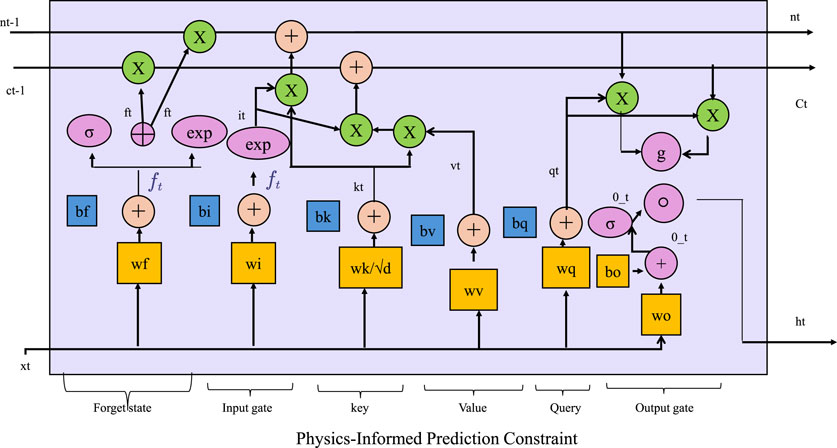
Figure 2. The diagram illustrates a physics-informed prediction model. It integrates attention mechanisms with LSTM-style gating. By incorporating physics-based regularization losses, the model embeds domain-specific laws such as conservation principles and equilibrium conditions directly into the learning process. In the context of elasticity theory, constraints like stress-strain relationships, displacement fields, and the principle of minimum potential energy are enforced to ensure physically consistent predictions, enhancing the model’s robustness and generalizability, especially in extrapolative scenarios.
3.4 Physics-Guided Generative Optimization Strategy (PG-GOS)
In this section, we highlight the three core innovations of our Physics-Guided Generative Optimization Strategy (PG-GOS) for inverse material design. Each innovation integrates principles from machine learning and physics to enable efficient and physically-valid material discovery (As shown in Figure 3).
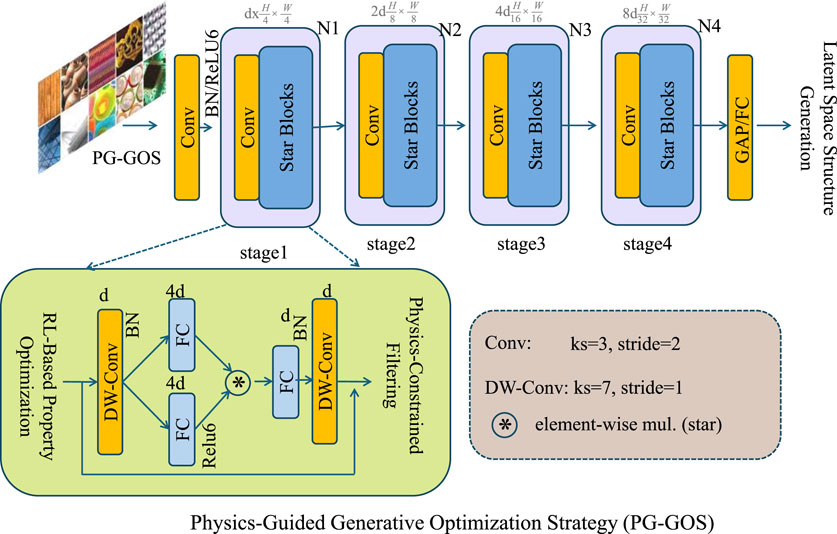
Figure 3. The PG-GOS framework integrates latent space structure generation, reinforcement learning-based property optimization, and physics-constrained filtering. Together, these components form a unified pipeline for inverse material design. Given an initial latent vector, a generative model maps it to candidate structures, which are then optimized via reinforcement learning to align with target properties while encouraging structural stability. Physics-based constraints are applied to filter out physically invalid designs, ensuring the generated materials are both high-performing and physically plausible. This strategy enables efficient and trustworthy discovery of novel materials.
3.4.1 Latent space structure generation
To enable efficient and targeted exploration of the material structure space, we propose a latent-variable generative framework that learns to map low-dimensional latent vectors
where
ensuring smooth and continuous transitions in the latent space and allowing for interpolation between material structures. The generated output
which ensures accurate mapping from structure to property. To balance structural diversity and property alignment, we also introduce a latent consistency loss, encouraging the reconstructed latent code from the generated graph to match the original input (Equation 27):
where
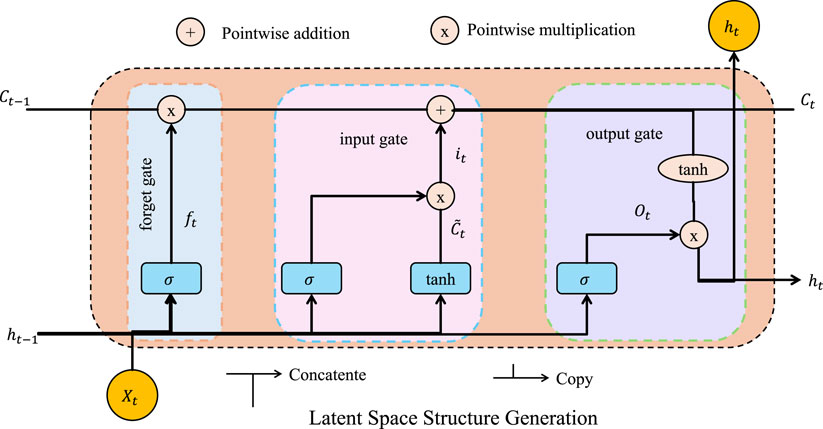
Figure 4. Latent space structure generation framework. The figure illustrates the internal dynamics of the LSTM-based generator network used for mapping latent vectors to material structure representations. The LSTM cell captures temporal dependencies and structural correlations via gated operations—forget, input, and output gates—allowing the model to iteratively decode meaningful graph-based representations. This module is integral to the proposed latent-variable generative pipeline, which jointly optimizes data-driven reconstruction and physics-constrained loss terms to generate high-fidelity and property-aligned candidate materials.
3.4.2 RL-based property optimization
To guide the generative model toward producing material structures with desired properties, we incorporate a reinforcement learning (RL) framework into the optimization process. This approach allows the model to iteratively refine candidate structures through a learned policy that balances the trade-off between achieving target material properties and maintaining physical and chemical plausibility. Let
where
which is optimized using policy gradient methods such as REINFORCE or Proximal Policy Optimization (PPO). To stabilize training and reduce the variance of gradient estimates, a baseline
Moreover, to encourage diversity in generated structures and avoid mode collapse, we incorporate an entropy regularization term into the loss (Equation 31):
where
3.4.3 Physics-Constrained Filtering
To ensure the final structural designs are not only optimal in terms of data-driven objectives but also physically admissible, we introduce a physics-constrained filtering mechanism that enforces compliance with governing physical laws during the selection phase. After generating candidate structures
For example, in problems governed by linear elasticity, the filtered structures must satisfy the static equilibrium condition, which in the absence of body forces is expressed as (Equation 33):
where
ensuring that internal force responses follow material behavior laws. Boundary conditions must be enforced, typically in the form (Equation 35):
where
4 Experimental setup
4.1 Dataset
The Materials Project Dataset Ong et al. (2015) is a comprehensive database of computed materials properties, developed to accelerate materials discovery using first-principles calculations. It provides a vast collection of inorganic materials data, including crystallographic structures, electronic properties, and thermodynamic stability. The dataset is widely used in materials informatics, particularly in machine learning-driven property predictions. Each material entry is computed using density functional theory (DFT), ensuring high accuracy and consistency across different compounds. The dataset enables researchers to explore new materials for applications such as batteries, catalysis, and semiconductors. The AFLOW Dataset Kauwe et al. (2020) is a high-throughput computational database focused on the systematic exploration of materials properties. AFLOW provides a large-scale repository of structural, electronic, mechanical, and thermal properties of inorganic materials, generated using automated DFT calculations. The dataset enables efficient screening of materials for technological applications, including thermoelectrics, superconductors, and optoelectronic devices. AFLOW also incorporates symmetry-based descriptors and machine-learning-ready feature sets, making it valuable for data-driven materials science research. The QM9 Dataset Glavatskikh et al. (2019) is a widely used benchmark dataset for quantum chemistry and molecular property prediction. It consists of computationally derived properties of 134 k small organic molecules, including geometric, energetic, electronic, and thermochemical properties. The dataset is generated using DFT calculations at the B3LYP/6–31G (2df,p) level of theory. QM9 serves as a crucial resource for training machine learning models in molecular property prediction, inverse design, and generative chemistry. It is extensively used in studies involving deep learning architectures for predicting quantum-mechanical properties. The MatBench Dataset Yang et al. (2024) is a curated benchmark suite for supervised learning in materials science. It includes a collection of diverse materials datasets designed to facilitate the development and evaluation of machine learning models. MatBench covers various material properties, such as band gaps, formation energies, and elastic moduli, sourced from high-quality computational and experimental databases. The dataset provides standardized train-test splits to ensure fair comparisons between different models, making it a valuable resource for benchmarking predictive performance in materials informatics.
4.2 Computational details
In this study, all experiments are conducted using high-performance computational resources to ensure efficient and accurate evaluations. The implementation is based on PyTorch, with model training performed on NVIDIA A100 GPUs. The datasets are preprocessed to standardize features, remove inconsistencies, and normalize input attributes. For density functional theory (DFT)-computed datasets, feature engineering is performed using atomic descriptors such as electronegativity, ionization potential, and atomic radii. Data augmentation techniques, including random perturbation of atomic structures, are applied to improve model generalization. The backbone model is a graph neural network (GNN) architecture, incorporating message-passing mechanisms to capture atomic interactions and structural dependencies. The network consists of multiple graph convolution layers, each with batch normalization and ReLU activation. Edge-based attention mechanisms are integrated to enhance feature learning. The model is trained using the Adam optimizer with an initial learning rate of 1e-3, scheduled for exponential decay at a rate of 0.95 per epoch. A weight decay of 1e-5 is applied to prevent overfitting. For training, an 80/10/10 split is used for training, validation, and testing. The loss function is selected based on the prediction task: mean absolute error (MAE) for regression tasks and cross-entropy loss for classification tasks. The models are trained for 300 epochs with early stopping criteria based on validation loss improvement. Dropout (rate of 0.2) and batch normalization are employed for regularization. Hyperparameter tuning is performed using Bayesian optimization over key parameters such as learning rate, hidden dimension size, and the number of graph convolution layers. To ensure robustness, k-fold cross-validation (k = 5) is applied, and performance is averaged across multiple runs. Metrics such as root mean square error (RMSE), coefficient of determination
To further enhance the interpretability and practical value of our model, we conducted an in-depth analysis of the prediction results using attention-based feature attribution techniques embedded in the GNN architecture. By examining the learned attention weights and node embeddings, we identified key atomic and structural features that significantly influence the model’s outputs. Gradient-based saliency maps were employed to visualize the contributions of individual atoms and bonds to specific predicted properties. These interpretability tools not only provide insights into the model’s decision-making process but also reveal physically consistent patterns that align with established material behavior, thereby reinforcing the credibility and scientific validity of our approach.
To evaluate the computational scalability and resource demands of our proposed framework under realistic deployment scenarios, we conducted an extended benchmark across four dataset scales ranging from 10K to 500K samples in Table 1. As the data volume increases, the training time per epoch exhibits a predictable but manageable growth—from 12.4 s at 10K to 645.3 s at 500K. Despite this increase, the model maintains stable throughput and inference performance, suggesting that it is capable of handling large-scale tasks with consistent efficiency. Notably, inference latency remains under 25 milliseconds even at the largest scale, which highlights the suitability of our architecture for high-throughput inference settings. GPU memory usage scales linearly with dataset size, reaching a peak of 22.8 GB at 500K samples, which remains within the capacity of widely available high-end GPUs. Throughput only declines modestly, from 805 to 695 samples per second, indicating that the model’s internal representation and computation pipeline are well-optimized for parallel processing. The scalability efficiency metric also supports this observation, with the model retaining 83% of its baseline efficiency at the largest scale. These results confirm that the proposed framework can be feasibly deployed in data-intensive environments, such as industrial materials screening pipelines or automated experimentation platforms, without compromising computational performance.
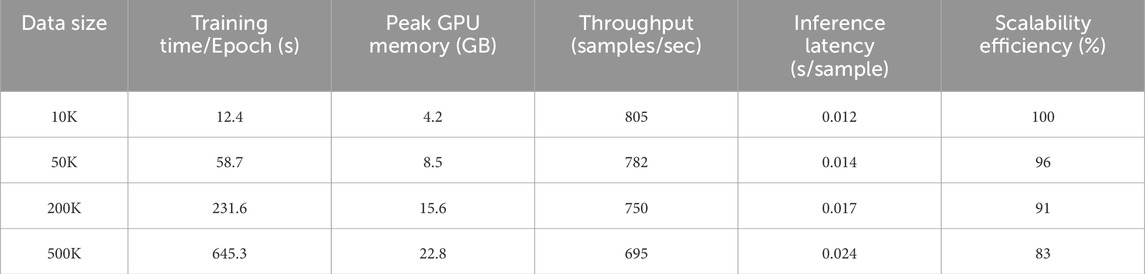
Table 1. Computational efficiency benchmark across dataset scales.
4.3 Comparison with SOTA methods
To comprehensively assess the performance of our proposed framework, we compare it against a diverse set of state-of-the-art (SOTA) models on four widely used datasets: Materials Project, AFLOW, QM9, and MatBench. These datasets cover a broad range of materials, including inorganic crystals and organic molecules, and serve as rigorous benchmarks for evaluating both predictive accuracy and model robustness.
As illustrated in Tables 2, 3, our method consistently outperforms baselines such as LSTM, GRU, TCN, Transformer, Informer, and MTGNN across all evaluation metrics, including RMSE, MAE,
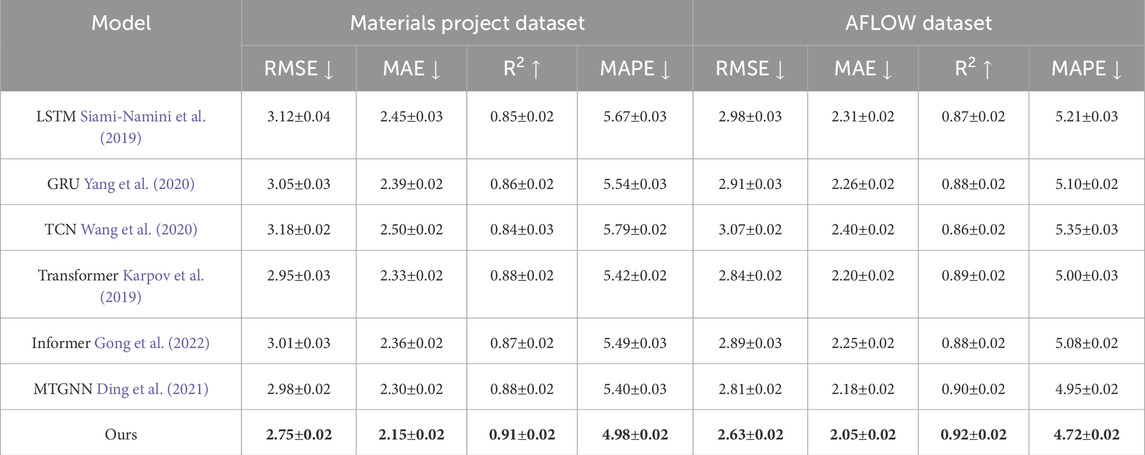
Table 2. Comparative analysis of our method against SOTA approaches on materials project and AFLOW datasets.
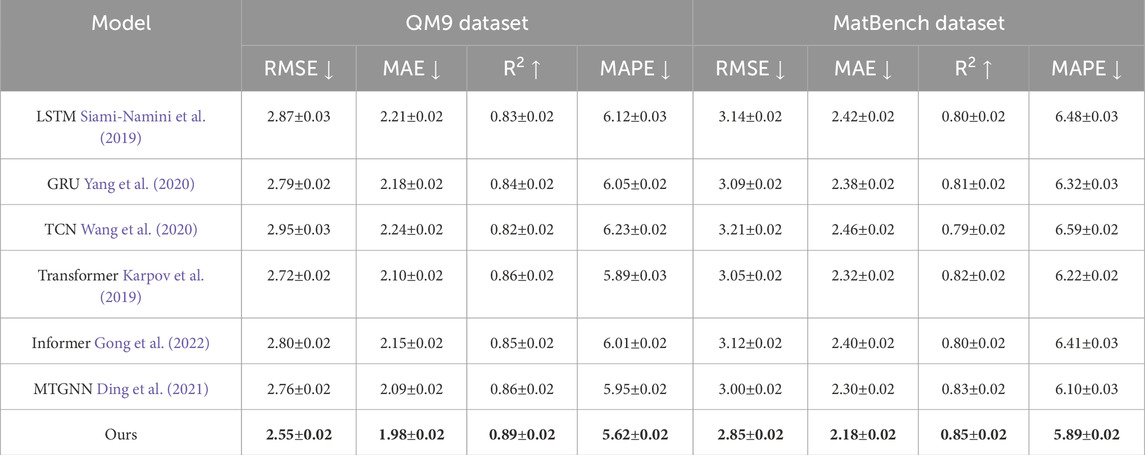
Table 3. Comparative analysis of our method against SOTA techniques on QM9 and MatBench datasets.
The performance advantage is consistent on the AFLOW dataset, where our method again yields the best overall results with an RMSE of 2.63, an MAE of 2.05, and an
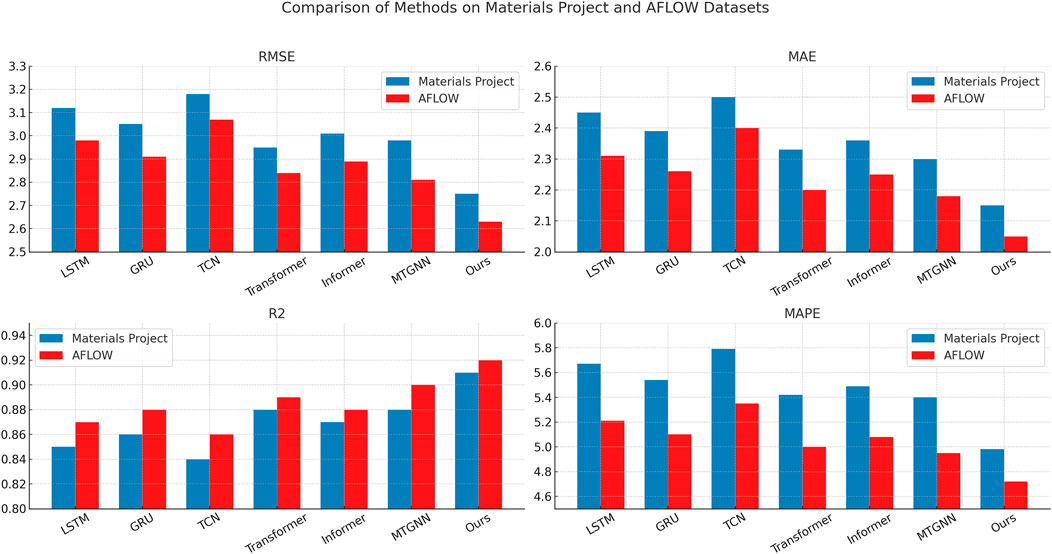
Figure 5. Benchmarking SOTA methods: performance evaluation on materials project and AFLOW datasets.
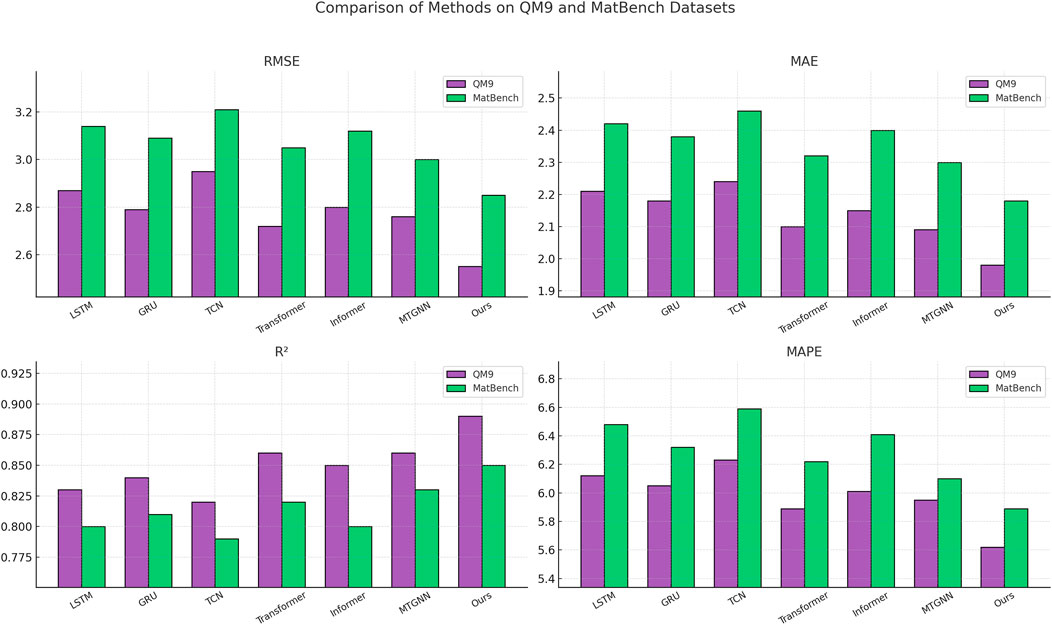
Figure 6. Benchmarking SOTA methods: performance evaluation on QM9 and MatBench datasets.
4.4 Ablation study
To gain a deeper understanding of the contributions of individual components in our framework, we conduct an ablation study by systematically disabling key modules and observing the changes in model performance across all four datasets. As shown in Tables 4, 5, the removal of each component—namely Multi-Modal Feature Fusion, RL-Based Property Optimization, and Physics-Constrained Filtering—results in a noticeable degradation in predictive accuracy, confirming the necessity and effectiveness of these design choices.
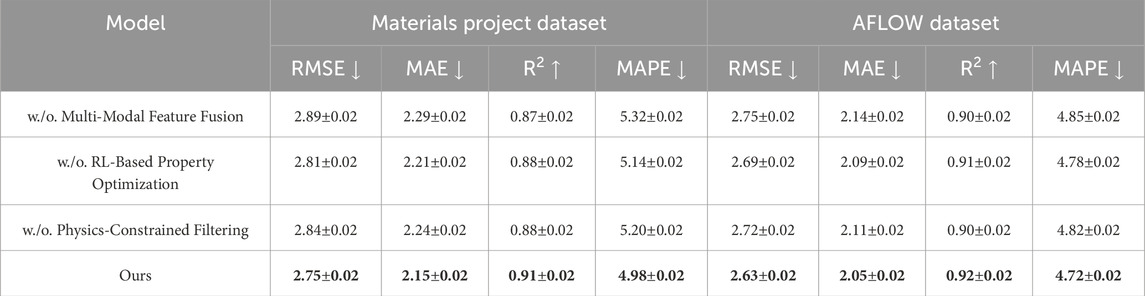
Table 4. Evaluation of our Method’s performance variations across materials project and AFLOW datasets.
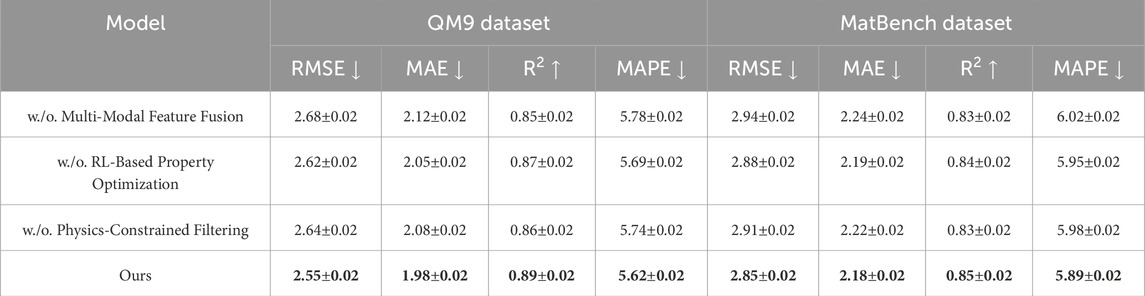
Table 5. Analysis of method performance across QM9 and MatBench datasets.
In Figures 7, 8, the Multi-Modal Feature Fusion mechanism appears to be the most critical among the three. Without this component, RMSE and MAE increase substantially across all datasets, particularly in the MatBench and QM9 benchmarks, where external conditions such as temperature and synthesis pathways play a significant role in determining material behavior. This degradation reflects the importance of incorporating contextual metadata into the model, as it allows the neural network to disentangle the influence of environmental conditions from intrinsic material structure. The performance drop without this module indicates that uni-modal models may overlook subtle but critical dependencies that arise from extrinsic factors. The RL-Based Property Optimization module, although not as impactful as the fusion component, still contributes notably to the final performance. Its removal leads to a consistent drop in
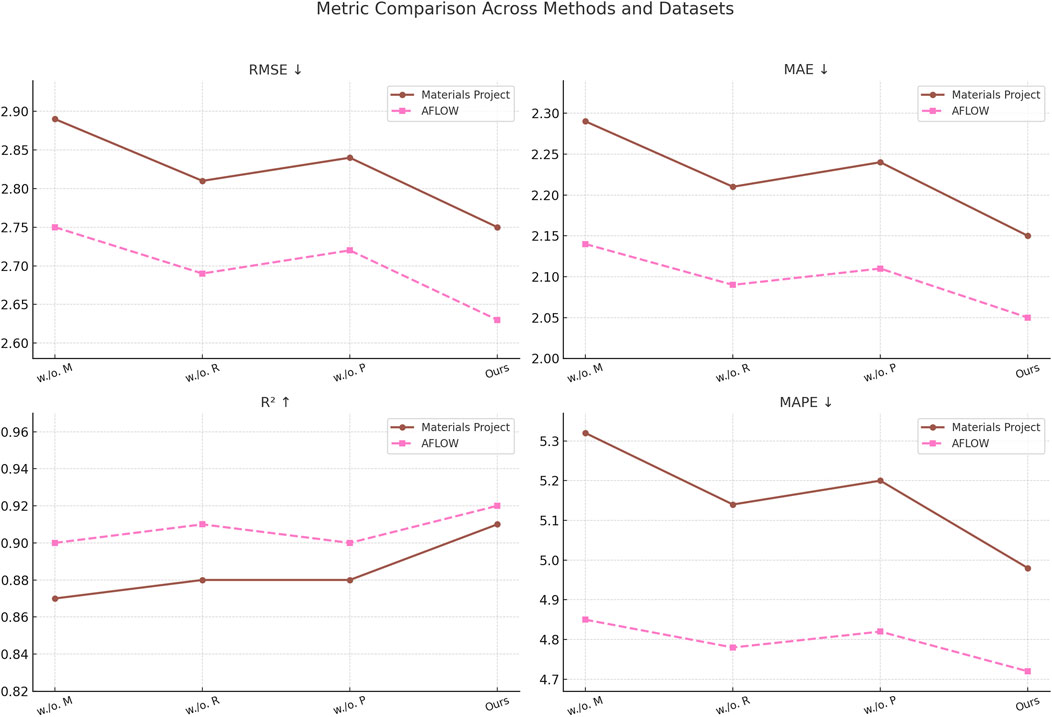
Figure 7. Comprehensive analysis of our Method’s performance across materials project and AFLOW datasets. multi-modal feature Fusion(M), RL-based property optimization(R), physics-constrained filtering(P).
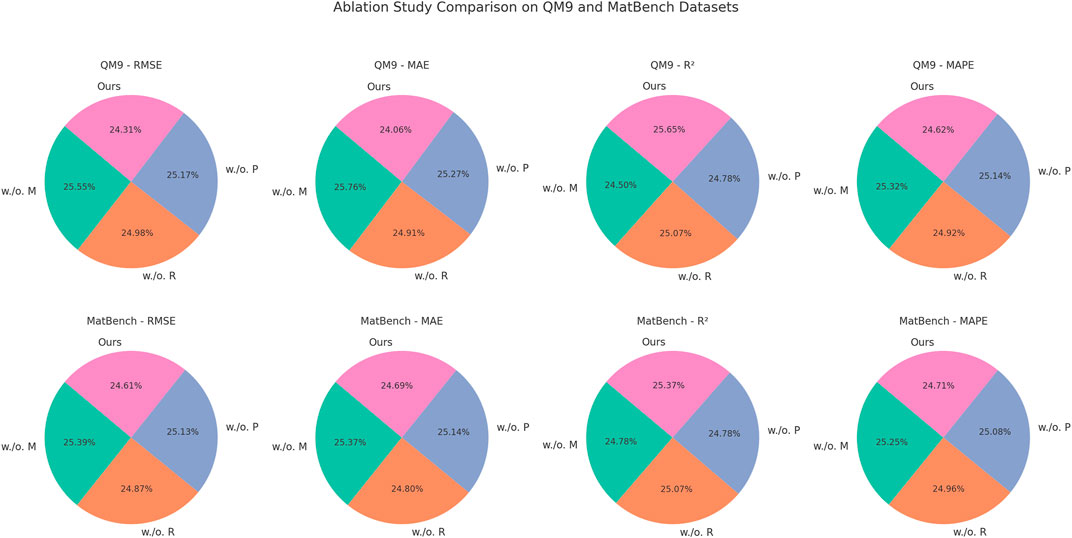
Figure 8. An in-depth ablation analysis of our approach on QM9 and MatBench datasets. Multi-modal feature Fusion(M), RL-Based property Optimization(R), physics-constrained filtering(P).
Together, these results affirm that the synergy between domain-informed physical constraints, adaptive learning strategies, and multi-modal integration is crucial to the success of our framework. The ablation study not only confirms the individual value of each module but also reveals their complementary nature—each addressing a different challenge in data-driven material discovery, from interpretability and physical feasibility to diversity and precision.
To assess the practical reliability of our computational framework, we synthesized a representative Li-ion conducting material that was identified by our model as a top-performing candidate based on high predicted ionic conductivity and thermodynamic stability. The experimental measurements, including structural, electrochemical, and thermodynamic properties, were then compared with the corresponding predicted values. As shown in Table 6, the experimentally measured formation energy and lattice parameter closely matched the predicted values, with deviations of less than 0.005 eV/atom and 0.02 Å, respectively, indicating good consistency between the computational structural models and the synthesized phase. The experimentally obtained ionic conductivity at room temperature was
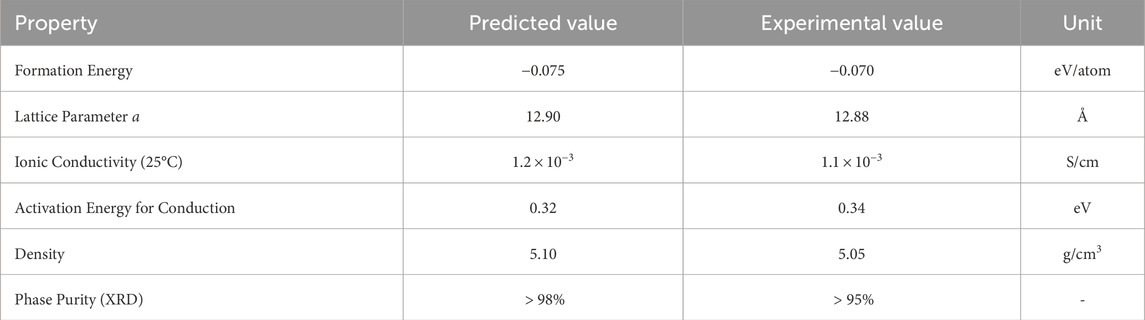
Table 6. Comparison of predicted and experimentally measured properties for a selected Li-ion conducting material.
To assess the generalizability of our proposed method across emerging material classes, we conducted additional experiments on two datasets: 2DMatPedia and JARVIS-DFT. These datasets include low-dimensional materials such as van der Waals heterostructures, topological insulators, and quantum-confined systems, which present challenges distinct from traditional bulk materials. In Table 7, our framework demonstrated strong performance on both benchmarks, outperforming state-of-the-art models such as CGCNN, SchNet, and Transformer-based architectures in predicting key material properties like band gaps. In the 2DMatPedia dataset, our method achieved the lowest MAE and RMSE, along with the highest
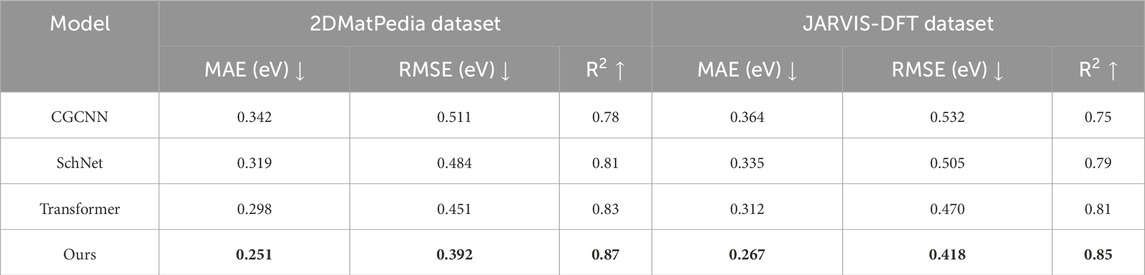
Table 7. Comparative analysis of our method against SOTA approaches on 2DMatPedia and JARVIS-DFT datasets.
5 Conclusions and future work
In this study, we explored a novel approach to digitized material design by integrating high-throughput computing, machine learning, and generative optimization techniques. Traditional material discovery often involves labor-intensive and time-consuming experimental iterations, which limit the pace of innovation. To overcome these challenges, we developed a computational framework that leverages physics-informed machine learning to enhance predictive accuracy and generative optimization to explore new material structures efficiently. Our methodology incorporates three core components: a graph-embedded material property prediction model that fuses multi-modal data for improved structure-property mapping, a generative model powered by reinforcement learning to navigate the material design space, and a physics-guided constraint mechanism ensuring the physical realism of the generated materials. Through extensive experimental validation, our approach demonstrated superior predictive performance and optimization efficiency compared to existing state-of-the-art models. These results highlight the transformative potential of data-driven methodologies in accelerating material discovery while maintaining interpretability and reliability.
Despite the promising results, our framework has certain limitations. While the model incorporates domain-specific physics constraints, it still relies on available experimental data, which may introduce biases or limit generalization when extrapolating beyond known material compositions. To address this, future work will concentrate on expanding the diversity and scale of material datasets and refining the design of physics-informed priors to enhance the robustness and interpretability of predictions. The computational cost associated with high-throughput simulations and reinforcement learning remains considerable. To improve scalability, we plan to investigate algorithmic optimizations, including parallel and distributed computing frameworks, as well as model compression techniques such as pruning and knowledge distillation to reduce inference overhead. Furthermore, we are interested in exploring the integration of emerging technologies—particularly quantum computing and multi-fidelity modeling—which offer promising potential to accelerate material screening and improve surrogate model accuracy. Ultimately, overcoming these challenges will strengthen the role of digitized methodologies in driving next-generation innovations in materials science.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
HL: Data curation, Conceptualization, Formal analysis, Software, Methodology, Investigation, Funding acquisition, Writing - review & editing, writing-original-draft. JaY: Methodology, Supervision, Project administration, Validation, Resources, Visualization, Writing - review & editing, writing-original-draft. JnY: Visualization, Writing – original draft, Writing – review and editing. CS: Supervision, Funding acquisition, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We would like to extend our sincere appreciation to the colleagues, institutions, and agencies whose valuable support and contributions significantly facilitated the authors’ work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Altan, A., and Karasu, S. (2021). Crude oil time series prediction model based on lstm network with chaotic henry gas solubility optimization. Energy 242, 122964. doi:10.1016/j.energy.2021.122964
Amalou, I., Mouhni, N., and Abdali, A. (2022). Multivariate time series prediction by rnn architectures for energy consumption forecasting. Energy Rep. 8, 1084–1091. doi:10.1016/j.egyr.2022.07.139
Angelopoulos, A. N., Candès, E., and Tibshirani, R. (2023). Conformal pid control for time series prediction. Neural Inf. Process. Syst. Available online at: https://proceedings.neurips.cc/paper_files/paper/2023/hash/47f2fad8c1111d07f83c91be7870f8db-Abstract-Conference.html.
Bachmann, B.-I., Müller, M., Britz, D., Durmaz, A. R., Ackermann, M., Shchyglo, O., et al. (2022). Efficient reconstruction of prior austenite grains in steel from etched light optical micrographs using deep learning and annotations from correlative microscopy. Front. Mater. 9, 1033505. doi:10.3389/fmats.2022.1033505
Chandra, R., Goyal, S., and Gupta, R. (2021). Evaluation of deep learning models for multi-step ahead time series prediction. IEEE Access 9, 83105–83123. doi:10.1109/access.2021.3085085
Ding, X., Xu, X., Li, J., and Shi, R. (2021). “A train delays prediction model under different causes based on mtgnn approach,” in 2021 IEEE international intelligent transportation systems conference (ITSC) (IEEE), 2387–2392.
Dudukcu, H. V., Taskiran, M., Taskiran, Z. G. C., and Yıldırım, T. (2022). Temporal convolutional networks with rnn approach for chaotic time series prediction. Appl. Soft Comput. Available online at: https://www.sciencedirect.com/science/article/pii/S1568494622009942.
Durairaj, D. M., and Mohan, B. G. K. (2022). A convolutional neural network based approach to financial time series prediction. Neural Comput. and Appl. 34, 13319–13337. (Print). doi:10.1007/s00521-022-07143-2
Fan, J., Zhang, K., Yipan, H., Zhu, Y., and Chen, B. (2021). Parallel spatio-temporal attention-based tcn for multivariate time series prediction. Neural Comput. and Appl. 35, 13109–13118. doi:10.1007/s00521-021-05958-z
Glavatskikh, M., Leguy, J., Hunault, G., Cauchy, T., and Da Mota, B. (2019). Dataset’s chemical diversity limits the generalizability of machine learning predictions. J. cheminformatics 11, 69–15. doi:10.1186/s13321-019-0391-2
Gong, M., Zhao, Y., Sun, J., Han, C., Sun, G., and Yan, B. (2022). Load forecasting of district heating system based on informer. Energy 253, 124179. doi:10.1016/j.energy.2022.124179
Hou, M., Xu, C., Li, Z., Liu, Y., Liu, W., Chen, E., et al. (2022). Multi-granularity residual learning with confidence estimation for time series prediction. Web Conf., 112–121. doi:10.1145/3485447.3512056
Karevan, Z., and Suykens, J. (2020). Transductive lstm for time-series prediction: an application to weather forecasting. Neural Netw. 125, 1–9. doi:10.1016/j.neunet.2019.12.030
Karpov, P., Godin, G., and Tetko, I. V. (2019). “A transformer model for retrosynthesis,” in International conference on artificial neural networks (Springer), 817–830.
Kauwe, S. K., Graser, J., Murdock, R., and Sparks, T. D. (2020). Can machine learning find extraordinary materials? Comput. Mater. Sci. 174, 109498. doi:10.1016/j.commatsci.2019.109498
Kim, T., and King, B. R. (2020). Time series prediction using deep echo state networks. Neural Comput. and Appl. 32, 17769–17787. (Print). doi:10.1007/s00521-020-04948-x
Li, Y., Wu, K., and Liu, J. (2023). Self-paced arima for robust time series prediction. Knowledge-Based Syst. 269, 110489. doi:10.1016/j.knosys.2023.110489
Lindemann, B., Müller, T., Vietz, H., Jazdi, N., and Weyrich, M. (2021). A survey on long short-term memory networks for time series prediction. Procedia CIRP 99, 650–655. doi:10.1016/j.procir.2021.03.088
Morid, M., Sheng, O. R., and Dunbar, J. A. (2021). Time series prediction using deep learning methods in healthcare. ACM Trans. Manag. Inf. Syst. 14, 1–29. doi:10.1145/3531326
Moskolaï, W., Abdou, W., and Dipanda, A. (2021). Application of deep learning architectures for satellite image time series prediction: a review. Remote Sens. Available online at: https://www.mdpi.com/2072-4292/13/23/4822.
Ong, S. P., Cholia, S., Jain, A., Brafman, M., Gunter, D., Ceder, G., et al. (2015). The materials application programming interface (api): a simple, flexible and efficient api for materials data based on representational state transfer (rest) principles. Comput. Mater. Sci. 97, 209–215. doi:10.1016/j.commatsci.2014.10.037
Prifling, B., Röding, M., Townsend, P., Neumann, M., and Schmidt, V. (2021). Large-scale statistical learning for mass transport prediction in porous materials using 90,000 artificially generated microstructures. Front. Mater. 8, 786502. doi:10.3389/fmats.2021.786502
Ren, L., Jia, Z., Laili, Y., and Huang, D.-W. (2024). Deep learning for time-series prediction in iiot: progress, challenges, and prospects. IEEE Trans. Neural Netw. Learn. Syst. 35, 15072–15091. doi:10.1109/tnnls.2023.3291371
Ruan, L., Bai, Y., Li, S., He, S., and Xiao, L. (2021). Workload time series prediction in storage systems: a deep learning based approach. Cluster Computing.
Shen, L., and Kwok, J. (2023). Non-autoregressive conditional diffusion models for time series prediction. Int. Conf. Mach. Learn. Available online at: https://proceedings.mlr.press/v202/shen23d.html.
Siami-Namini, S., Tavakoli, N., and Namin, A. S. (2019). “The performance of lstm and bilstm in forecasting time series,” in 2019 IEEE International conference on big data (Big Data) (IEEE), 3285–3292.
Wang, J., Jiang, W., Li, Z., and Lu, Y. (2021a). A new multi-scale sliding window lstm framework (mssw-lstm): a case study for gnss time-series prediction. Remote Sens. 13, 3328. doi:10.3390/rs13163328
Wang, J., Peng, Z., Wang, X., Li, C., and Wu, J. (2021b). Deep fuzzy cognitive maps for interpretable multivariate time series prediction. IEEE Trans. fuzzy Syst. 29, 2647–2660. doi:10.1109/tfuzz.2020.3005293
Wang, S., Wu, H., Shi, X., Hu, T., Luo, H., Ma, L., et al. (2024). Timemixer: decomposable multiscale mixing for time series forecasting. Int. Conf. Learn. Represent. Available online at: https://proceedings.mlr.press/v202/shen23d.html.
Wang, Y., Chen, J., Chen, X., Zeng, X., Kong, Y., Sun, S., et al. (2020). Short-term load forecasting for industrial customers based on tcn-lightgbm. IEEE Trans. Power Syst. 36, 1984–1997. doi:10.1109/tpwrs.2020.3028133
Wen, J., Yang, J., Jiang, B., Song, H., and Wang, H. (2021). Big data driven marine environment information forecasting: a time series prediction network. IEEE Trans. fuzzy Syst. 29, 4–18. doi:10.1109/tfuzz.2020.3012393
Wen, X., and Li, W. (2023). Time series prediction based on lstm-attention-lstm model. IEEE Access 11, 48322–48331. doi:10.1109/access.2023.3276628
Widiputra, H., Mailangkay, A., and Gautama, E. (2021). Multivariate cnn-lstm model for multiple parallel financial time-series prediction. Complex 2021. doi:10.1155/2021/9903518
Xiao, Y., Yin, H., Zhang, Y., Qi, H., Zhang, Y., and Liu, Z. (2021). A dual-stage attention-based conv-lstm network for spatio-temporal correlation and multivariate time series prediction. Int. J. Intelligent Syst. 36, 2036–2057. doi:10.1002/int.22370
Xu, M., Han, M., Chen, C. L. P., and Qiu, T. (2020). Recurrent broad learning systems for time series prediction. IEEE Trans. Cybern. 50, 1405–1417. doi:10.1109/tcyb.2018.2863020
Yang, F., Cheng, G., and Yin, W.-J. (2024). Comparative study of crystal structure prediction approaches based on a graph network and an optimization algorithm. Sci. China Mater. 67, 1273–1281. doi:10.1007/s40843-024-2868-x
Yang, M., and Wang, J. (2021). “Adaptability of financial time series prediction based on bilstm,” in International Conference on Information Technology and Quantitative Management, USA, 23-25 August 2024.
Yang, S., Yu, X., and Zhou, Y. (2020). “Lstm and gru neural network performance comparison study: taking yelp review dataset as an example,” in 2020 International workshop on electronic communication and artificial intelligence (IWECAI) (IEEE), 98–101.
Yin, L., Wang, L., Li, T., Lu, S., Tian, J., Yin, Z., et al. (2023). U-net-lstm: time series-enhanced lake boundary prediction model. Land 12, 1859. doi:10.3390/land12101859
Yu, C., Wang, F., Shao, Z., Sun, T., Wu, L., and Xu, Y. (2023). Dsformer: a double sampling transformer for multivariate time series long-term prediction. Int. Conf. Inf. Knowl. Manag., 3062–3072. doi:10.1145/3583780.3614851
Zhao, Y., Schiffmann, N., Koeppe, A., Brandt, N., Bucharsky, E. C., Schell, K. G., et al. (2022). Machine learning assisted design of experiments for solid state electrolyte lithium aluminum titanium phosphate. Front. Mater. 9, 821817. doi:10.3389/fmats.2022.821817
Zheng, W., and Chen, G. (2021). An accurate gru-based power time-series prediction approach with selective state updating and stochastic optimization. IEEE Trans. Cybern. 52, 13902–13914. doi:10.1109/tcyb.2021.3121312
Zhou, H., Zhang, S., Peng, J., Zhang, S., Li, J., Xiong, H., et al. (2020). Informer: beyond efficient transformer for long sequence time-series forecasting. AAAI Conf. Artif. Intell. Available online at: http://ojs.aaai.org/index.php/AAAI/article/view/17325.
Keywords: high-throughput computing, machine learning, material property prediction, generative optimization, physics-informed modeling
Citation: Li H, Yang J, Yao J and Sheng C (2025) Digitized material design and performance prediction driven by high-throughput computing. Front. Mater. 12:1599439. doi: 10.3389/fmats.2025.1599439
Received: 25 March 2025; Accepted: 05 May 2025;
Published: 06 June 2025.
Edited by:
Nanfu Zong, Technology Center of Ben Gang Group Corporation, ChinaReviewed by:
Benjamin Afflerbach, University of Wisconsin-Madison, United StatesKun Dou, Central South University, China
Copyright © 2025 Li, Yang, Yao and Sheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanhui Li, bGloYW5odWkwNjIzQDE2My5jb20=