 Xiaorui Liu†
Xiaorui Liu† Chunmei Chen†
Chunmei Chen† Yinyu ZhengMi ZhangQingyi Tong
Yinyu ZhengMi ZhangQingyi Tong Junjun Liu
Junjun Liu Qun ZhouJianping WangZengwei LuoHucheng Zhu*
Qun ZhouJianping WangZengwei LuoHucheng Zhu* Yonghui Zhang*
Yonghui Zhang*- Hubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
(±)-Peniorthoesters A and B (±1 and ±2), two pairs of unprecedented spiro-orthoester enantiomers with a 1,4,6-trioxaspiro[4. 5]decane-7-one unit, were obtained from Penicillium minioluteum. Their structures were determined by spectroscopic methods, X-ray diffraction analyses, and ECD calculations. (±)-Peniorthoesters A and B are the first examples of spiro-orthoester enantiomers, and they represent the first spiro-orthoesters originating from fungi. All compounds showed potential inhibitory activities comparable to dexamethasone against NO production with IC50 values ranging from 14.2 to 34.5 μM.
Introduction
Orthoesters, a special functional group characterized by three alkoxy groups attached to a single carbon atom, are unusual structural subunits in natural products (Liao et al., 2009). Natural occurring orthoesters include several major types, such as daphnane diterpenoid orthoesters (He et al., 2002), limonoid orthoesters (Roy and Saraf, 2006), steroid orthoesters (Steyn and van Heerden, 1998), and coumarinoid orthoesters (Santana et al., 2004). In our previous study on the plant Wikstroemia chamaedaphne, three new daphnane type diterpenoids with orthoester group were isolated (Guo et al., 2015). A literature investigation revealed that most of these orthoesters originate from plants, and only a few originate from fungi, such as novofumigatonin, a meroterpenoid orthoester from Aspergillus novofumigatus (Rank et al., 2008). As a special class of natural products, orthoesters have attracted great attention due to their diverse structures and biological properties (Liao et al., 2009; Bourjot et al., 2014; Li et al., 2015; Liu et al., 2017).
Fungi have historically played an important role in drug discovery. The genus Penicillium has been shown to be a rich source of structurally unique and biologically active secondary metabolites (Meng et al., 2016; Sun et al., 2016; Luo et al., 2017) and many metabolites from Penicillium are clinically used drugs with penicillin as a representative compound. Previous studies on the secondary metabolites of Penicillium minioluteum have resulted in the identification of scores of bioactive metabolites, including isomeric furanones with cytotoxic activities against HeLa cell lines (Tang et al., 2015), sesquiterpene-conjugated amino acids with cytotoxic activities against HepG2 cells (Ngokpol et al., 2015), and hybrid polyketide-terpenoids (Iida et al., 2008). This fungus was also used to produce clovane derivatives, which are the raw materials for the synthesis of rumphellclovane A (Gontijo de Souza et al., 2012), and an enzyme from this fungus was used in the bioconversion (Kmiecik and Zymanczyk-Duda, 2017). During our ongoing search for structurally unique and biologically interesting constituents from fungi (Zhu et al., 2015; Chen et al., 2017; Zhou et al., 2017), P. minioluteum, obtained from China General Micro-biological Culture Collection Center (CGMCC), was phytochemically investigated, and two pairs of new orthoesters (Figure 1, compounds ±1 and ±2) were isolated along with their biosynthetic intermediate, sclerotinin A (3) (Xiao et al., 2009). The structures and absolute configurations of (±)-1 and (±)-2 were determined by a combination of spectroscopic methods, X-ray diffraction analyses, and ECD calculations. (±)-Peniorthoesters A (±1) and B (±2) are the first examples of spiro-orthoester enantiomers and represent the first spiro-orthoesters originating from fungi.
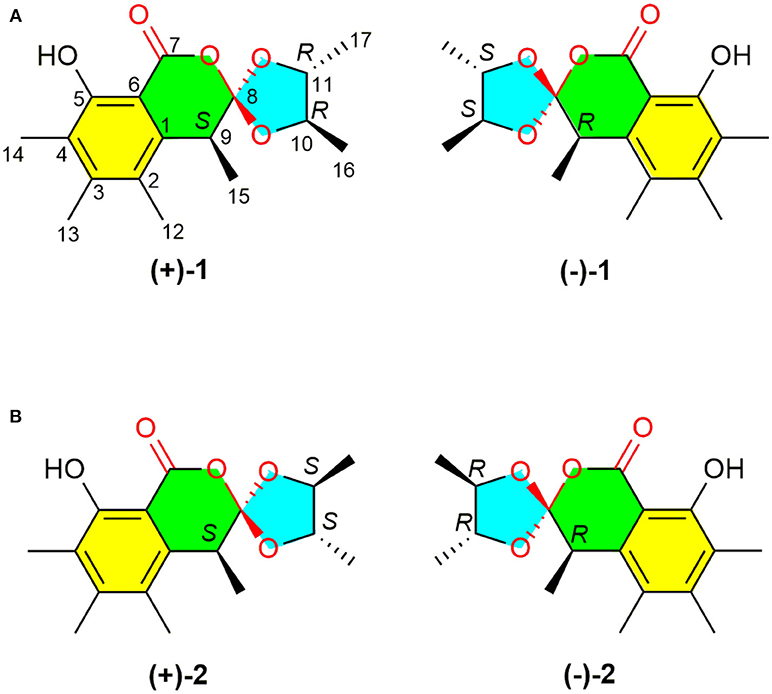
Figure 1. Structures of (±)-peniorthoesters A (±1) and B (±2).
Materials and Methods
General Experimental Procedures
Optical rotations were determined in acetonitrile and dichloromethane on a PerkinElmer 341 polarimeter. UV spectra were obtained on Varian Cary 50 spectrometer. ECD spectra were obtained with a JASCO J-810 spectrometer. IR spectra were acquired on a Bruker Vertex 70 instrument. NMR spectra were recorded on Bruker AM-400 spectrometers, and the 1H and 13C NMR chemical shifts were referenced to the solvent or solvent impurity peaks for CD3Cl at δH 7.26 and δC 77.20. HRESIMS data were acquired in the positive-ion mode on a Thermo Fisher LC-LTQ-Orbitrap XL spectrometer. The crystallographic experiments were performed on a Bruker APEX DUO diffractometer equipped with graphite-monochromated Cu Kα radiation (λ = 1.541 78 Å). Semi-preparative HPLC was carried out on an instrument consisting of an Agilent 1,220 controller, an Agilent 1,220 pump, an Agilent UV detector, with a reversed-phased C18 column (5 μm, 10 × 250 mm, Welch Ultimate XB-C18), an Ultimate SiO2 column (5 μm, 10 × 250 mm, Welch Materials, Inc.), and a Chiralpak IC column (5 μm, 4.6 × 250 mm, Daicel Chiral Technologies Co., Ltd., China). Chromatography coloums (CC) were performed on silica gel (200–300 mesh; Qingdao Marine Chemical, Inc., Qingdao, China), Sephadex LH-20 (40–70 μm, Amersham Pharmacia Biotech AB, Uppsala, Sweden), and ODS (50 μm, Merck, Germany). Thin-layer chromatographies (TLC) was performed with RP-C18 F254 plates (Merck, Germany) and silica gel 60 F254 (Yantai Chemical Industry Research Institute).
Fungal Material and Fermentation
The strain in this work was bought from China General Micro-biological Culture Collection Center (CGMCC). A voucher Specimen was preserved in the herbarium of Huazhong University of Science and Technology, China. The fungal strain was cultured on potato dextrose agar (PDA) at 28°C for 8 days to prepare the seed culture. Then the strain was inoculated into 200 Erlenmeyer flasks (1 L each) which had previously been sterilized by autoclaving. Each flask contained 250 g rice and 200 mL distilled water. The flasks were incubated at 20°C for 26 days.
Extraction and Isolation
The fermented rice substrate was extracted six times in 95% aqueous EtOH at room temperature. The 95% aqueous EtOH extracts were concentrated under vacuum to afford a residue (1.5 kg). The residue was suspended in H2O and successively partitioned with petroleum ether and EtOAc. The EtOAc partition fraction (630.0 g) was subjected to a silica gel chromatograph column (CC) using petroleum ether–EtOAc and EtOAc–MeOH gradient elution to give five fractions. Fraction 2 (20.0 g) was chromatographed on C18 reversed phase (RP-18) silica gel CC (gradient elution of MeOH–H2O, 20:80–100:0) to give seven subfractions, named Fr. 2A−2G. Fr.2F was successively separated via Sephadex LH-20 CC (CH2Cl2-MeOH, 1:1) and further purified by RP-C18 HPLC to afford compounds 1 and 2 (MeCN–H2O, 45:55, 3.5 ml/min, 1 at tR 53.2 min, 6.0 mg; 2 at tR 56.7 min, 5.1 mg). Subsequently, the separation of 1 by chiral HPLC using a Daicel IC column eluting with isopropanol–n-hexane (5:95) afforded (+)-1 (tR 28.0 min, 2.1 mg) and (–)-1 (tR 30.1 min, 2.0 mg). The enantiomers (+)-2 (tR 8.2 min, 1.6 mg) and (–)-2 (tR 11.1 min, 2.7 mg) were also obtained by chiral HPLC using a Daicel IC column eluting with isopropanol–n-hexane (10:90).
Compounds (±)-1: white powder; [α] ±0 (c 0.4, MeCN); UV (MeCN) λmax (log ε) 216 (4.50), 259 (4.23), 329 (4.04) nm; IR (KBr) νmax 3,435, 2,982, 2,936, 1,661, 1,612, 1,456, 1,420, 1,383, 1,341, 921, 883, 809, 753 cm−1; 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100 MHz) data, see Table 1; HRESIMS m/z 307.1538 [M + H]+ (calcd for C17H23O5, 307.1545).
(+)-1: white amorphous powder; [α +37 (c 0.1, CH2Cl2); ECD (MeCN) 229 (Δε +1.63), 254 (Δε +4.75), 325 (Δε −0.99) nm.
(–)-1: white amorphous powder; [α −36 (c 0.1, CH2Cl2); ECD (MeCN) 229 (Δε −1.66), 254 (Δε −4.34), 325 (Δε +1.23) nm.
Compounds (±)-2: white powder; [α ±0 (c 0.3, MeCN); UV (MeCN) λmax (log ε) 216 (4.60), 259 (3.99), 329 (3.71) nm; IR (KBr) νmax 3,435, 2,980, 2,932, 1,669, 1,612, 1,456, 1,421, 1,379, 1,339, 920, 881, 810, 763 cm−1; 1H NMR (CDCl3, 400 MHz) and 13C NMR (CDCl3, 100MHz) data, see Table 1; HRESIMS m/z 307.1534 [M + H]+ (calcd for C17H23O5, 307.1545).
(+)-2: white amorphous powder; [α +30 (c 0.1, CH2Cl2); ECD (MeCN) 224 (Δε +2.37), 258 (Δε +4.64), 319 (Δε −0.84) nm.
(–)-2: white amorphous powder; [α −30 (c 0.1, CH2Cl2); ECD (MeCN) 224 (Δε −2.42), 258 (Δε −6.58), 319 (Δε +1.60) nm.
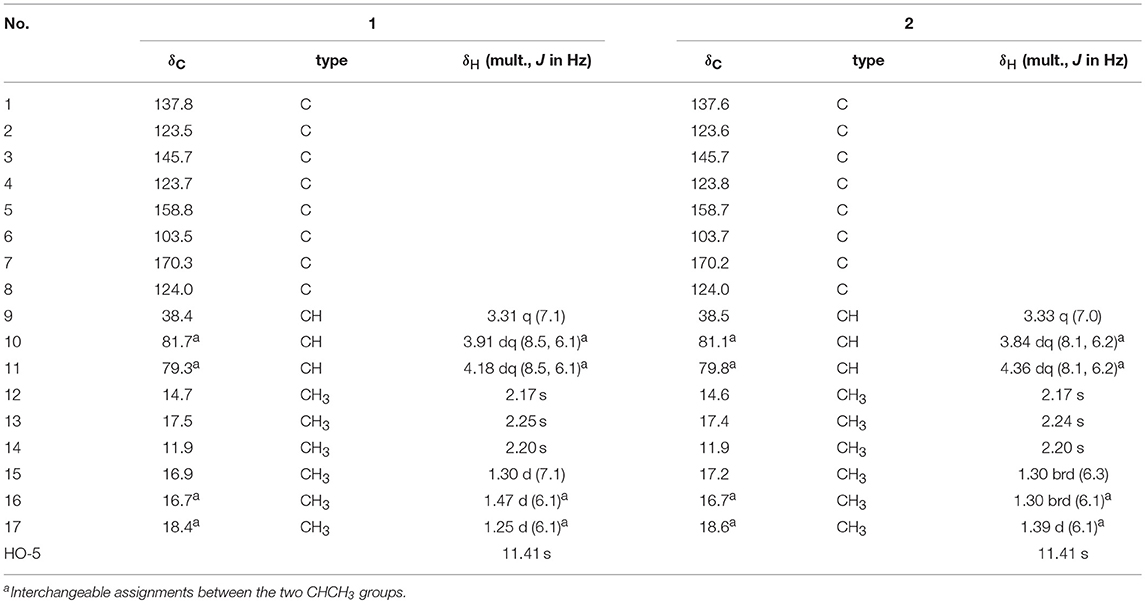
Table 1. 1H and 13C NMR Spectroscopic Data for 1 and 2 (in CDCl3).
Computational Details
The crystal structure of 9R,10S,11S-1, and 9R,10R,11R-2 were optimized at the B3LYP/6-31G(d) level in acetonitrile with the IEFPCM solvation model using Gaussian 09 program. The harmonic vibrational frequencies were calculated to confirm the stability of the optimized structure. The electronic circular dichroism (ECD) spectrum were calculated using the TDDFT methodology at the LC-wPBE/6-311++G(d,p) level with acetonitrile as solvent by the IEFPCM solvation model implemented in Gaussian 09 program. The ECD spectra was simulated using a Gaussian function with a bandwidth σ of 0.3 eV. The UV correction was applied to generate the final spectra (Zhu, 2015).
Single-Crystal X-ray Diffraction Analysis and Crystallographic Data
Crystallographic data of compound 1 (CCDC 1840165): C17H22O5, M = 306.34, monoclinic, T = 297(2) K, λ = 1.54178 Å, colorless platelet (crystallized from distilled water at room temperature), size 0.12 × 0.10 × 0.10 mm3, a = 11.7937(4) Å, b = 32.5593(12) Å, c = 8.1659(3) Å, α = 90.00°, β = 91.95(2)°, γ = 90.00°, V = 3,133.84(19) Å3, space group P21/c, Z = 8, Dc = 1.299 g/cm3, μ(CuKα) = 0.782 mm−1, F(000) = 1312, 48082 reflections and 5,729 independent reflections (Rint = 0.0528) were collected in the θ range of 2.71° ≤ θ ≤ 69.99° with index ranges of h(−14/14), k(−39/39), l(−9/9), completeness θmax = 98%, data/restraints/parameters 5,729/0/412. Largest difference peak and hole = 0.257 and −0.184 e Å−3. The final R1 values were 0.0489 (I > 2σ(I)). The final wR(F2) values were 0.1364 (I > 2σ(I)). The final R1 values were 0.0521 (all data). The final wR(F2) values were 0.1381 (all data). The goodness of fit on F2 was 1.045.
Crystallographic data of compound 2 (CCDC 1840166): C17H22O5, M = 306.34, monoclinic, T = 297(2) K, λ = 1.54178 Å, colorless platelet (crystallized from distilled water at room temperature), size 0.12 × 0.10 × 0.10 mm3, a = 7.4709(2) Å, b = 8.9377(12) Å, c = 13.5720(3) Å, α = 92.43°, β = 100.25(2)°, γ = 113.76°, V = 809.60(19) Å3, space group P-1, Z = 2, Dc = 1.257 g/cm3, μ(CuKα) = 0.757 mm−1, F(000) = 328, 14,855 reflections and 2,814 independent reflections (Rint = 0.0374) were collected in the θ range of 5.45° ≤ θ ≤ 70.86° with index ranges of h(−8/7), k(−10/10), l(−16/16), completeness θmax = 95%, data/restraints/parameters 2,814/0/207. Largest difference peak and hole = 0.232 and −0.241 e Å−3. The final R1 values were 0.0588 (I > 2σ(I)). The final wR(F2) values were 0.1750 (I > 2σ(I)). The final R1 values were 0.0647 (all data). The final wR(F2) values were 0.1855 (all data). The goodness of fit on F2 was 1.076.
Determination of No Production
RAW 264.7 cells were obtained from the Boster Biological Technology Co., Ltd (Wuhan, China) and maintained in DMEM containing 10% heat-inactived fetal bovine serum (FBS) (Gibco BRL Co, Grand Island, NY, United States) at 37°C in humidified incubator containing 5% CO2. All tested compounds were dissolved in DMSO (the final concentration of DMSO was <0.25% in assay). RAW 264.7 cells were seeded into 48-well plates (1 × 105cells/well) for 24 h and then pretreated with different concentrations (1–40 μM) of test compounds. After being incubated for 3 h, the cells were stimulated with 100 ng/ml LPS (final concentration) for another 24 h. Dexamethasone was used as the positive control in the experiment. NO content in the supernatant was measured using Griess reagent. The absorbance at 540 nm was measured on a microplate reader. All the experiments were performed in three independent replicates.
Cytotoxic Activity
Cell lines were cultured in RPMI-1640 or DMEM medium (HyClone, USA), supplemented with 10% fetal bovine serum (HyClone, USA) at 37°C in a humidified atmosphere with 5% CO2. For cell viability assay, cells were plated into 96-well plates in 50 μl of medium and then compounds were serially diluted in medium and delivered to the cells as 2 × solutions in 50 μl of medium. After 48 h, cell viability was detected by a CCK-8 Kit (Dojindo,Japan) according to the instruction. Growth relative to untreated cells was calculated (positive control, anticancer drug VP16), and this data was used for the dose-response curve, the IC50 (50% inhibiting concentration) of compounds to each cell lines were calculated by SPSS.
Results and Discussions
Compound 1 was isolated as a white powder. Its UV spectrum exhibited absorption maxima at 216 and 260 nm. Its IR spectrum indicated the presence of an OH functionality (3,435 cm−1), a conjugated carbonyl group (1,661 cm−1), and an aromatic ring (1,612 and 1,456 cm−1). The molecular formula of 1 was determined to be C17H22O5 by HRESIMS with an [M + H]+ ion peak at m/z 307.1538 (calcd for C17H23O5, 307.1545), implying seven degrees of unsaturation. The 1H NMR spectroscopic data of 1 (Table 1) revealed the presence of two oxygenated methines [δH 4.18 (1H, dq, J = 8.5, 6.1 Hz, H-11) and 3.91 (1H, dq, J = 8.5, 6.1 Hz, H-10)], one sp3 methine [δH 3.31, 1H, q, J = 7.1 Hz, H-9], and six methyl groups [δH 1.47 (d, J = 6.1 Hz, H3-16), 1.30 (d, J = 7.1 Hz, H3-15), 1.25 (d, J = 6.1 Hz, H3-17), 2.17 (s, H3-12), 2.20 (s, H3-14), and 2.25 (s, H3-13)]. The 13C NMR spectrum of 1 exhibited signals assignable to a conjugated carbonyl (δC 170.3), a hexa-substituted benzene ring [δC 158.8, 145.7, 137.8, 123.7, 123.5, and 103.5], one oxygenated quaternary carbon (δC 124.0), six methyl groups and three methines (including two oxygenated ones). The above analyses confirmed the presence of an ester carbonyl group and a hexa-substituted benzene ring, which account for five degrees of unsaturation, indicating the presence of two additional rings. With the aid of the HSQC spectrum, all protons were unambiguously assigned to their respective carbons.
The planar structure of 1 was elucidated on the basis of 1H–1H COSY and HMBC experiments (Figure 2). The HMBC spectrum of 1 displayed correlations from H3-14 to C-3, C-4, and C-5; from H3-13 to C-2, C-3, and C-4; from H3-12 to C-1, C-2, and C-3; and from H-9 to C-1, and C-6, which together with the HMBC correlations from the OH to C-4, C-5, and C-6 constructed the hexa-substituted benzene ring. In addition, two spin systems of H3-17/H-11/H-10/H3-16 and H-9/H3-15 were established from the 1H–1H COSY spectrum. Therefore, the HMBC correlations from H-9 to C-1, C-6, and C-8 and from H3-15 to C-1, C-8, and C-9 suggested the C-15/C-9/C-8 fragment was connected to the benzene ring via C-9. Moreover, the ester carbonyl (δC 170.3) was connected to C-6 based on the chemical shifts of C-6 (δC 103.5), C-1 (δC 137.8), and C-3 (δC 145.7). Combined with the chemical shifts of C-10 (δC 81.7) and C-11 (δC 79.3), the C-17/C-11/C-10/C-16 fragment was proposed to be a 2,3-butanediol unit, which should be linked with C-8 and form a 4,5-dimethyl-1,3-dioxolane moiety. Finally, a lactone ring was proposed between C-7 and C-8 to satisfy the above deduced tricyclic ring system as well as the chemical shift of C-8 (δC 124.0). This planar structure satisfied all of the correlations observed in the 2D NMR spectra and the chemical shifts in the 1D NMR spectra.
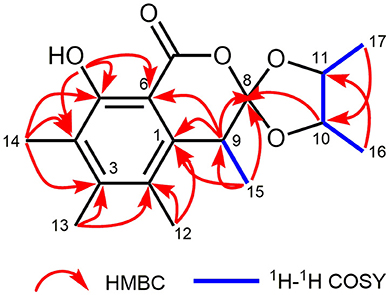
Figure 2. Key 1H−1H COSY and HMBC correlations of 1.
A NOESY experiment was performed on 1, but no interactions useful for determining the relative configuration were observed. Unfortunately, the relative configuration of H-10 and H-11 could also not be determined from their coupling constants because they were located on a five-membered ring. To confidently assign the configuration of 1, we tried to crystallize it so we could use X-ray single-crystal analysis. After a number of attempts, a high-quality single-crystal of 1 was finally obtained from a mixture of methyl alcohol and water. The X-ray crystallography data (CCDC 1840165) obtained with Cu Kα radiation confirmed the structure of 1 (Figure 3). However, because it has a centrosymmetric monoclinic space group of chiral P21/c, indicating the crystal is racemic, the absolute configuration of 1 could not be determined. After analyses by using various types of chiral columns, the racemic nature of this solution was further confirmed by the presence of two peaks in the HPLC chromatogram acquired using a chiral Daicel IC column (Figure 4). Finally, compounds (+)-1 and (–)-1 were successfully obtained, and they showed specific rotations with opposite signs {(+)-1: [α37 (c 0.1, CH2Cl2); (–)-1: [α-36 (c 0.1, CH2Cl2)}. In addition, the ECD spectra of (+)-1 and (–)-1 displayed similar signal intensities but mirror-image Cotton effects (Figure 5).
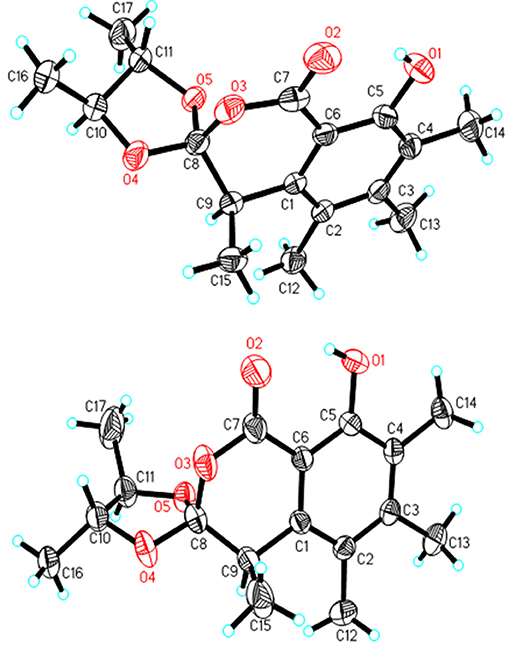
Figure 3. X-ray structures of 1 and 2.
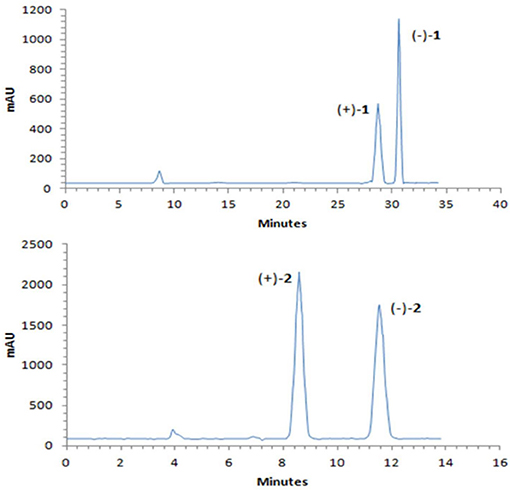
Figure 4. Chiral HPLC separation profiles of (±)-1 and (±)-2.
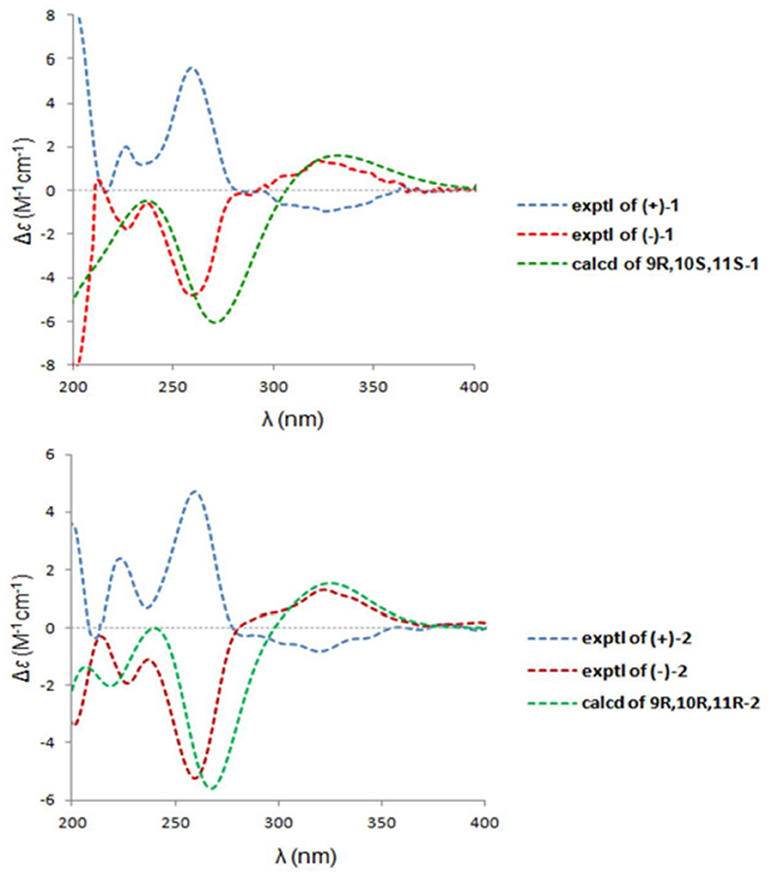
Figure 5. The experimental ECD spectra of (±)-1 and (±)-2 and the cal-culated ECD curves.
The absolute configurations of the two enantiomers of (±)-1 were further determined by comparing their experimental ECD spectra with those predicted by time-dependent density functional theory (TDDFT) calculations at the B3LYP/6-31G(d) level. As shown in Figure 5, the calculated ECD curve of 9R,10S,11S-1 displayed good agreement with the experimental curve of (–)-1. Therefore, the absolute configurations of (+)-1 and (–)-1 were elucidated as 9S,10R,11R and 9R,10S,11S, respectively.
Compound 2, obtained as a white powder, possesses the same molecular formula (C17H22O5) as that of compound 1 based on its HRESIMS data with an [M + H]+ ion peak at m/z 307.1534 (calcd for C17H23O5, 307.1545). A detailed comparison of its NMR spectroscopic data with those of 1 indicated that the main differences between 1 and 2 were tiny changes in the chemical shifts of C-10 and C-11 as well as their protons [δC 81.1 (C-10), 79.8 (C-11); δH 3.84 (1H, dq, J = 8.1, 6.2 Hz, H-10), 4.36 (1H, dq, J = 8.1, 6.2 Hz, H-11) in 2; δC 81.7 (C-10), 79.3(C-11); δH 3.91 (1H, dq, J = 8.5, 6.1 Hz, H-10), 4.18 (1H, dq, J = 8.5, 6.1 Hz, H-11) in 1]. These findings, combined with the 2D NMR data, implied that 2 has the same planar structure as 1, and it should be a stereoisomer of 1. The planar structure of 2 was further confirmed by analyses of its 1H–1H COSY and HMBC spectra. Unfortunately, the NOESY experiment of 2 also did not show any NOESY correlations useful in for the elucidation of the relative configuration of compound 2.
Similarly, after many attempts, we finally determined the relative configuration of compound 2 by X-ray crystallography analysis with Cu Kα radiation (Figure 3, CCDC 1840166). This single-crystal is triclinic with space group of chiral P-1, also indicating it is racemic. Compound 2 was then separated into a pair of enantiomers by a method similar to what was used for compound 1 (Figure 4), and the enantiomers showed opposite optical rotations {(+)-2: [α30 (c 0.1, CH2Cl2); (–)-2: [α-30 (c 0.1, CH2Cl2)} and mirror image ECD curves (Figure 5). The absolute configurations of the two enantiomers of 2 were further determined by ECD calculations. As shown in Figure 5, the calculated ECD curve of 9R,10R,11R-2 closely resembled the experimental curve of (–)-2, and the absolute configurations of (+)-2 and (–)-2 were elucidated as 9S,10S,11S and 9R,10R,11R, respectively.
To the best of our knowledge, (±)-1 and (±)-2 are the first examples of spiro-orthoester enantiomers with an unusual 1,4,6-trioxaspiro[4.5]decane-7-one unit, and they represent the first spiro-orthoesters originating from fungi. The proposed biosynthetic pathway of 1 and 2 was outlined in Figure 6. First, the condensation of acetyl-CoA and four molecules of malonyl-CoA by a polyketide synthase formed i, which underwent cycloaddition and methylations to form precursor sclerotinin A (3). Then, sclerotinin A underwent isomerization and hydrolytic cleavage to afford iv, which further formed vi by a H+ mediated double bond isomerization. After that, intermediate vii was produced by an aldol condensation, which further generated the key intermediates viii and x via an esterification reaction with 2R,3R-butanediol and 2S,3S-butanediol (Ji et al., 2011), respectively. Finally, compounds (±)-1 and (±)-2 were formed via condensation and lactonization reactions.
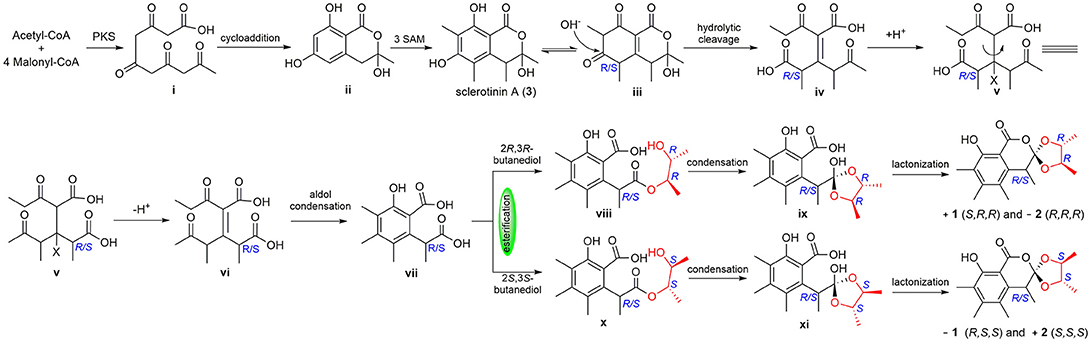
Figure 6. Proposed biosynthetic pathway of 1 and 2.
Compounds (±)-1 and (±)-2 were tested for their inhibitory activities against NO production in lipopolysaccharide (LPS)-induced RAW264.7 cells. The results revealed that (+)-1, (–)-1, (+)-2, and (–)-2 exhibited potential inhibitory activities with IC50 values of 34.5, 29.6, 23.5, and 15.2 μM, respectively (Table 2). Interestingly, for both pairs of enantiomers, the levorotatory compounds (–1 and –2) showed better inhibitory effects than the dextrorotatory compounds (+1 and +2). Moreover, both (+)-2 and (–)-2 showed better inhibitory effects than those of (+)-1 and (–)-1 as well as the positive control, dexamethasone. We also tested cytotoxicity of these compounds, but even at the concentration of 40 μM, none of them showed cytotoxicity activity.

Table 2. Inhibition of LPS-Induced NO Production.
Conclusion
In conclusion, two pairs of new spiro-orthoester enantiomers, (±)-peniorthoesters A and B (±1 and ±2), were isolated from P. minioluteum. These compounds, characterized by an unexpected 1,4,6-trioxaspi-ro[4.5]decane-7-one unit, are the first examples of spiro-orthoester enantiomers, and they represent the first spiro-orthoesters originating from fungi. All of them showed potential inhibitory activities against NO production in activated macrophages with IC50 values ranging from 14.2 to 34.5 μM, which are comparable to the positive control, dexamethasone. Their highly functionalized structures and promising biological activities will attract considerable attention from the pharmacological, synthetic, and biosynthetic communities.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
XL and CC conducted the main experiments and wrote the manuscript. YiZ, MZ, and QZ carried out bioassays. JL did the ECD calculations. JW and ZL analyzed the spectroscopic data. HZ and YoZ initiated and oversaw all research. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by the Program for Changjiang Scholars of Ministry of Education of the People's Republic of China (No. T2016088); the National Natural Science Foundation for Distinguished Young Scholars (No. 81725021); Innovative Research Groups of the National Natural Science Foundation of China (81721005); the Academic Frontier Youth Team of HUST; and the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College (HUST). We thank the Analytical and Testing Center at Huazhong University of Science and Technology for assistance in the acquisition of the ECD, UV, and IR spectra.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00605/full#supplementary-material
The Supplementary Material includes Full NMR, HRESIMS, UV, and IR spectra of 1 and 2; detailed of the ECD calculations of 1 and 2; X-ray data of 1 and 2 (PDF); and crystallographic data (CIF).
References
Bourjot, M., Leyssen, P., Neyts, J., Dumontet, V., and Litaudon, M. (2014). Trigocherrierin A, a potent inhibitor of chikungunya virus replication. Molecules 19, 3617–3627. doi: 10.3390/molecules19033617
Chen, K., Zhang, X., Sun, W., Liu, J., Yang, J., Chen, C., et al. (2017). Manginoids A–G: seven monoterpene–shikimate-conjugated meroterpenoids with a spiro ring system from Guignardia mangiferae. Org. Lett. 19, 5956–5959. doi: 10.1021/acs.orglett.7b02955
Gontijo de Souza, G., Oliveira, T. S., Takahashi, J. A., Collado, I. G., Macias-Sanchez, A. J., and Hernandez-Galan, R. (2012). Biotransformation of clovane derivatives. Whole cell fungi mediated domino synthesis of rumphellclovane A. Org. Biomol. Chem. 10, 3315–3320. doi: 10.1039/C2OB07114B
Guo, J., Tian, J., Yao, G., Zhu, H., Xue, Y., Luo, Z., et al. (2015). Three new 1α-alkyldaphnane-type diterpenoids from the ?ower buds of Wikstroemia chamaedaphne. Fitoterapia 106, 242–246. doi: 10.1016/j.fitote.2015.09.017
He, W., Cik, M., Appendino, G., Puyvelde, L. V., Leysen, J. E., and De Kimpe, N. (2002). Daphnane-type deterpene orthoesters and their biological activities. Mini. Rev. Med. Chem. 2, 185–200. doi: 10.2174/1389557024605492
Iida, M., Ooi, T., Kito, K., Yoshida, S., Kanoh, K., Shizuri, Y., et al. (2008). Three New polyketide–terpenoid hybrids from Penicillium sp. Org. Lett. 10, 845–848. doi: 10.1021/ol7029867
Ji, X. J., Huang, H., and Ouyang, P. K. (2011). Microbial 2,3-butanediol production: a state-of-the-art review. Biotechnol. Adv. 29, 351–364. doi: 10.1016/j.biotechadv.2011.01.007
Kmiecik, N., and Zymanczyk-Duda, E. (2017). Enantio convergent biotransformation of O,O-dimethyl-4-oxoazetidin-2-ylphosphonate using fungal cells of Penicillium minioluteum and purified enzymes. Bioorg. Chem. 71, 81–85. doi: 10.1016/j.bioorg.2017.01.014
Li, L., Knickelbein, K., Zhang, L., Wang, J., Obrinske, M., Ma, G. Z., et al. (2015). Amphiphilic sugar poly (orthoesters) as pH-responsive nanoscopic assemblies for acidity-enhanced drug delivery and cell killing. Chem. Commun. 51, 13078–13081. doi: 10.1039/C5CC04078G
Liao, G. S., Chen, H. D., and Yue, J.-M. (2009). Plant orthoesters. Chem. Rev. 109, 1092–1140. doi: 10.1021/cr0782832
Liu, F., Yang, X., Ma, J., Yang, Y., Xie, C., Tuerhong, M., et al. (2017). Nitric oxide inhibitory daphnane diterpenoids as potential anti-neuroinflammatory agents for AD from the twigs of Trigonostemon thyrsoideus. Bioorg. Chem. 75, 149–156. doi: 10.1016/j.bioorg.2017.09.007
Luo, M., Cui, Z., Huang, H., Song, X., Sun, A., Dang, Y., et al. (2017). Amino acid conjugated anthraquinones from the marine-derived fungus Penicillium sp. SCSIO sof101. J. Nat. Prod. 80, 1668–1673. doi: 10.1021/acs.jnatprod.7b00269
Meng, L. H., Wang, C. Y., Mandi, A., Li, X. M., Hu, X. Y., Kassack, M. U., et al. (2016). Three Diketopiperazine alkaloids with spirocyclic skeletons and one bisthiodiketopiperazine derivative from the mangrove-derived endophytic fungus Penicillium brocae MA-231 Org. Lett. 18, 5304–5307. doi: 10.1021/acs.orglett.6b02620
Ngokpol, S., Suwakulsiri, W., Sureram, S., Lirdprapamongkol, K., Aree, T., Wiyakrutta, S., et al. (2015). Drimane sesquiterpene-conjugated amino acids from a marine isolate of the fungus Talaromyces minioluteus (Penicillium Minioluteum). Mar. Drugs 13, 3567–3580. doi: 10.3390/md13063567
Rank, C., Phipps, R. K., Harris, P., Fristrup, P., Larsen, T. O., and Gotfredsen, C. H. (2008). N ovofumigatonin, a new orthoester meroterpenoid from Aspergillus novofumigatus. Org. Lett. 10, 401–404. doi: 10.1021/ol7026834
Roy, A., and Saraf, S. (2006). Limonoids: overview of significant bioactive triterpenes distributed in plants kingdom. Biol. Pharm. Bull. 29, 191–201. doi: 10.1248/bpb.29.191
Santana, L., Uriarte, E., Roleira, F., Milhazes, N., and Borges, F. (2004). Furocoumarins in medicinal chemistry. Synthesis, natural occurrence and biological activity. Curr. Med. Chem. 11, 3239–3261. doi: 10.2174/0929867043363721
Steyn, P. S., and van Heerden, F. R. (1998). Bufadienolides of plant and animal origin. Nat. Prod. Rep. 15, 397–413. doi: 10.1039/A815397Y
Sun, J., Zhu, Z. X., Song, Y. L., Dong, D., Zheng, J., Liu, T., et al. (2016). Nitric Oxide Inhibitory Meroterpenoids from the Fungus Penicillium purpurogenum MHZ 111. J. Nat. Prod. 79, 1415–1422. doi: 10.1021/acs.jnatprod.6b00160
Tang, H. Y., Zhang, Q., Gao, Y. Q., Zhang, A. I., and Gao, J. M. (2015). Miniolins A–C, novel isomeric furanones induced by epigenetic manipulation of Penicillium minioluteum. RSC Adv. 5, 2185–2190. doi: 10.1039/C4RA11712C
Xiao, N., Gao, J., Cai, X., and She, Z. (2009). Secondary metabolites of mangrove endophytic fungus SK5 in the South China Sea. Zhongyaocai 32, 1843–1845. doi: 10.3321/j.issn:1001-4454.2009.12.017
Zhou, P., Wu, Z., Tan, D., Yang, J., Zhou, Q., Zeng, F., et al. (2017). Atrichodermones A–C, three new secondary metabolites from the solid culture of an endophytic fungal strain, Trichoderma atroviride. Fitoterapia 123, 18–22. doi: 10.1016/j.fitote.2017.09.012
Zhu, H., Chen, C., Xue, Y., Tong, Q., Li, X. N., Chen, X., et al. (2015). Asperchalasine A, a cytochalasan dimer with an unprecedented decacyclic ring system, from Aspergillus flavipes. Angew. Chem. Int. Ed. 54, 13374–13378. doi: 10.1002/anie.201506264
Keywords: peniorthoesters, Penicillium minioluteum, spiro-orthoester, enantiomers, NO production inhibition activity
Citation: Liu X, Chen C, Zheng Y, Zhang M, Tong Q, Liu J, Zhou Q, Wang J, Luo Z, Zhu H and Zhang Y (2018) (±)-Peniorthoesters A and B, Two Pairs of Novel Spiro-Orthoester en-antiomers With an Unusual 1,4,6-Trioxaspi-ro[4.5]decane-7-One Unit From Penicillium minioluteum. Front. Chem. 6:605. doi: 10.3389/fchem.2018.00605
Received: 02 August 2018; Accepted: 26 November 2018;
Published: 07 December 2018.
Edited by:
Bin-Gui Wang, Institute of Oceanology (CAS), ChinaReviewed by:
Dipesh Dhakal, Sun Moon University, South KoreaChristophe Salome, SpiroChem AG, Switzerland
Copyright © 2018 Liu, Chen, Zheng, Zhang, Tong, Liu, Zhou, Wang, Luo, Zhu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hucheng Zhu, emh1aHVjaGVuZ0BodXN0LmVkdS5jbg==
Yonghui Zhang, emhhbmd5aEBtYWlscy50am11LmVkdS5jbg==
†These authors have contributed equally to this work