 Yusuke Kuramochi*†
Yusuke Kuramochi*† Osamu Ishitani*
Osamu Ishitani*- Department of Chemistry, Graduate School of Science and Engineering, Tokyo Institute of Technology, Tokyo, Japan
A cyclometalated iridium(III) complex having 2-(pyren-1-yl)-4-methylquinoline ligands [Ir(pyr)] has a strong absorption band in the visible region (ε444nm = 67,000 M−1 cm−1) but does not act as a photosensitizer for photochemical reduction reactions in the presence of triethylamine as an electron donor. Here, 1,3-dimethyl-2-(o-hydroxyphenyl)-2,3-dihydro-1H-benzo[d]imidazole (BI(OH)H) was used instead of the amine, demonstrating that BI(OH)H efficiently quenched the excited state of Ir(pyr) and can undergo the photochemical carbon dioxide (CO2) reduction catalyzed by trans(Cl)-Ru(dmb)(CO)2Cl2 (dmb = 4,4′-dimethyl-2,2′-bipyridine, Ru) to produce formate as the main product. We also synthesized a binuclear complex combining Ir(pyr) and Ru via an ethylene bridge and investigated its photochemical CO2 reduction activity in the presence of BI(OH)H.
Introduction
Today, the consumption of fossil resources releases a tremendous amount of carbon dioxide (CO2), which has had a serious impact on global climate change. The reduction in fossil resources in the future will induce shortages in both energy and carbon sources. To resolve these serious problems, the development of alternative energy systems that produce reduced volumes of CO2 by using solar light as an energy source is desirable. To utilize a wider range of visible light from the sun, nature-inspired artificial Z-scheme systems have been developed by using semiconductors modified with metal complexes (Sato et al., 2011; Sekizawa et al., 2013; Kuriki et al., 2016, 2017; Sahara et al., 2016; Kumagai et al., 2017). Some metal complex photocatalytic systems that consist of a photosensitizer (PS) and a catalyst (CAT) can selectively induce CO2 reduction and suppress hydrogen (H2) evolution. These systems require a sacrificial electron donor due to the relatively low oxidation power of the PS unit in the excited state (Yamazaki et al., 2015; Tamaki and Ishitani, 2017; Kuramochi et al., 2018a) Step-by-step excitation of both the semiconductor and the metal complex produces an electron with high reducing power and a hole with high oxidizing power, allowing for CO2 reduction by weaker electron donors such as methanol (Figure 1; Sekizawa et al., 2013).
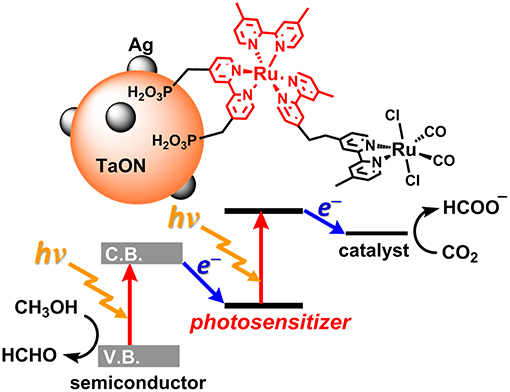
Figure 1. The artificial Z-scheme system for CO2 (carbon dioxide) reduction, consisting of a semiconductor (TaON) and a Ru(II) binuclear complex (RuRu') (Sekizawa et al., 2013).
Ru(II) tris-diimine complexes [Ru(N∧N)3]2+ have been frequently used as the PS unit of supramolecular photocatalysts, which have strong absorption in the visible region, a long lifetime of the 3MLCT excited state, and a stable one-electron reduced state. However, [Ru(N∧N)3]2+ has a problem in that one of the N∧N ligands is relatively easily released during the photocatalytic reaction to give [Ru(N∧N)2(Solvent)2]2+-type complexes that work as catalysts for CO2 reduction (Lehn and Ziessel, 1990; Yamazaki et al., 2015; Kuramochi et al., 2018a). In addition, a PS unit with stronger absorption in the visible region compared to [Ru(N∧N)3]2+ should be more favorable for constructing new photocatalytic systems.
In recent years, cyclometalated iridium(III) complexes, such as Ir(ppy)3 (ppy = 2-phenylpyridine) and [Ir(ppy)2(N∧N)]+, have been used as PSs in various photocatalytic reactions, such as H2 evolution (Goldsmith et al., 2005; Lowry and Bernhard, 2006), CO2 reduction (Thoi et al., 2013; Bonin et al., 2014; Chen et al., 2015; Rao et al., 2017, 2018), and organic synthesis (Figure 2; Prier et al., 2013; Schultz and Yoon, 2014; Shaw et al., 2016), even though the absorption by Ir(ppy)3 and [Ir(ppy)2(N∧N)]+ is relatively weak in the visible region. We already reported that [Ir(piq)2(dmb)]+ (piq = 1-phenylisoquinoline, dmb = 4,4′-dimethyl-2,2′-bipyridine), which has a stronger absorption in the visible region (ε444nm = 7,800 M−1 cm−1) than Ir(ppy)3, acts as a PS for CO2 reduction without forming decomposed species that catalyze CO2 reduction (Figure 2; Kuramochi and Ishitani, 2016). The intense absorption at the longer wavelength allowed for the selective excitation of the PS without exciting the CAT, such as fac-Re(dmb)(CO)3Br (dmb = 4,4′-dimethyl-2,2′-bipyridine). In addition, the supramolecular photocatalyst, where [Ir(piq)2(BL)]+ (BL = bridging ligand) is connected with fac-Re(BL)(CO)3Br, works as a better photocatalyst for CO2 reduction compared to the mixed system of the corresponding mononuclear complexes, i.e., [Ir(piq)2(dmb)]+ and fac-Re(dmb)(CO)3Br. Although [Ir(piq)2(dmb)]+ has a stronger absorption in the visible region compared to Ir(ppy)3 and the advantages over [Ru(N∧N)3]2+ as mentioned above, its absorption in the visible region is weaker than that of [Ru(N∧N)3]2+. It has been reported that several Ir(III) complexes that have stronger absorption bands in the visible region than [Ru(N∧N)3]2+ can act as PSs for H2 evolution (Takizawa et al., 2012, 2014, 2018; Fan et al., 2014). Fan et al. reported that an Ir(III) complex with 2-(pyren-1-yl)-4-methylquinoline ligands [Ir(pyr), Figure 2] showed a very strong absorption band in the visible region, ε(450 nm) > 60,000 M−1 cm−1. Unfortunately, it could not photocatalyze H2 evolution using K2PtCl4 as CAT in the presence of triethylamine (TEA). This inactivity was explained by the lack of photoinduced electron transfer from TEA to the excited state of Ir(pyr), as the reduction potential of the excited state of Ir(pyr) is less than the oxidation potential of TEA (Fan et al., 2014). We reported that the Ru(II)–Ru(II) supramolecular photocatalyst can selectively reduce CO2 to formic acid (HCOOH) by using 1,3-dimethyl-2-(o-hydroxyphenyl)-2,3-dihydro-1H-benzo[d]imidazole (BI(OH)H) as an electron donor (ED) with a high turnover number (TONHCOOH) and a high quantum yield (ΦHCOOH; Tamaki et al., 2015). This photocatalytic reaction does not proceed in the absence of BI(OH)H even if triethanolamine (TEOA), which has a similar oxidation potential to TEA, is used. This is because BI(OH)H has a much stronger reducing power (E = +0.02 V vs. Ag/AgNO3) (Hasegawa et al., 2006; Elgrishi et al., 2017; Kuramochi et al., 2018a) than TEA (E = +0.67 V vs. Ag/AgNO3) (Yamazaki et al., 2015; Elgrishi et al., 2017).
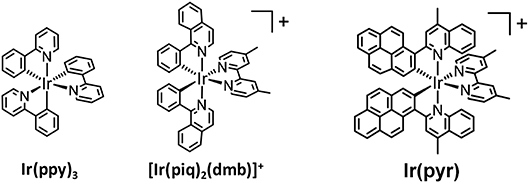
Figure 2. Structures of iridium(III) complexes.
Herein, we report the successful use of Ir(pyr) as a PS for CO2 reduction by using BI(OH)H as ED and trans(Cl)-Ru(dmb)(CO)2Cl2 (Ru) as CAT. We also synthesized a supramolecular photocatalyst from Ir(pyr) (Ir(pyr)–Ru; Scheme 1) and investigated its photocatalytic activity for CO2 reduction.
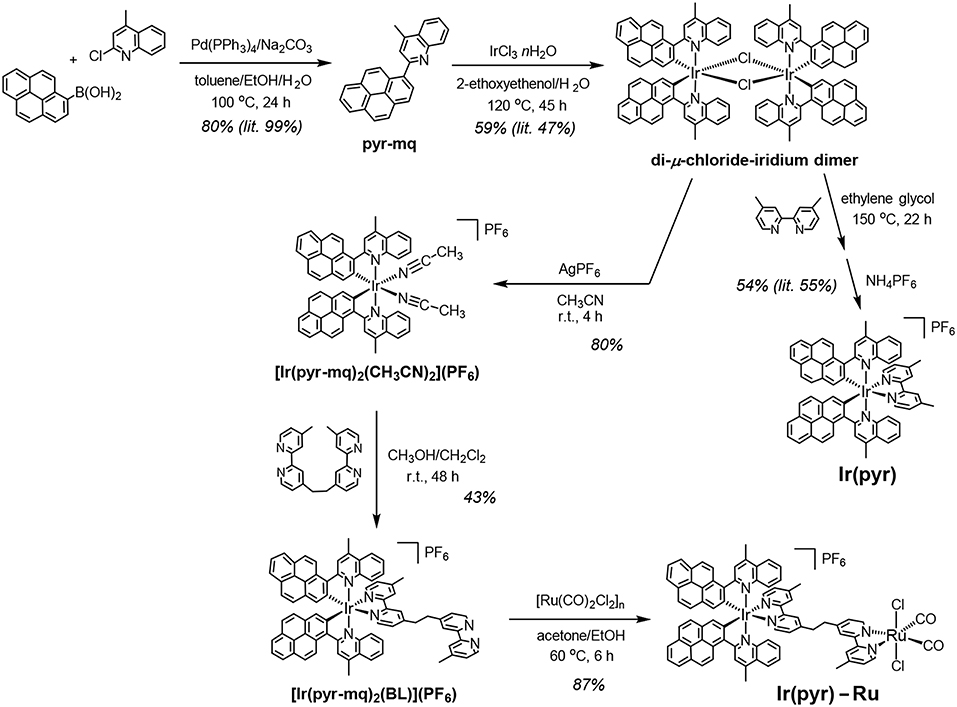
Scheme 1. Synthetic routes to Ir(pyr)–Ru and Ir(pyr).
Results and Discussion
Synthesis of the Binuclear Complex, Ir(pyr)–Ru
Ir(pyr)–Ru was synthesized according to Scheme 1. The di-μ-chloride-iridium dimer (Fan et al., 2014) was reacted with AgPF6 in acetonitrile to give the mononuclear acetonitrile–iridium complex. This complex was reacted with 1,2-bis(4′-methyl-[2,2′-bipyridin]-4-yl)ethane (BL), and the crude product was isolated using a silica gel column giving [Ir(pyr-mq)2(BL)](PF6) in 43% yield, based on the acetonitrile–iridium complex. This was reacted with [Ru(CO)2Cl2]n, which was pretreated by refluxing in the solvent (Kuramochi et al., 2015), giving the desired binuclear complex Ir(pyr)–Ru as a PF6 salt in 87% yield.
Photophysical and Electrochemical Properties
Fan et al. reported the photophysical and electrochemical properties of Ir(pyr) in dichloromethane or tetrahydrofuran (Fan et al., 2014). Since these solvents are less polar than the solvents suitable for CO2 reduction, such as N,N-dimethylacetamide (DMA), we measured the photophysical and electrochemical properties of Ir(pyr) in DMA (Kuramochi et al., 2014). Figure 3 shows the ultraviolet–visible (UV–vis) absorption spectrum of Ir(pyr) in DMA, which shows a much stronger absorption in the visible region (ε444nm = 67,000 M−1 cm−1) compared to [Ir(piq)2(dmb)]+ and [Ru(dmb)3]2+. According to the time-dependent density functional theory (TD-DFT) calculation of the UV–vis spectrum of Ir(pyr) in DMA (Figure 4, red bars), the strong absorption band at 444 nm is due to the transitions from the highest occupied molecular orbital (HOMO)-1 to the lowest unoccupied molecular orbital (LUMO)+2 and from HOMO to LUMO+1, which correspond to the π−π* transitions of the pyrene moieties. The absorption at a wavelength >500 nm is assigned to the transition from HOMO-1 to LUMO and corresponds to the transition from the interligand transition from the dmb to the pyrene moieties and might include some contribution from the singlet–triplet transitions, as described in the literature (Fan et al., 2014). The emission spectrum of Ir(pyr) in DMA is shown in Figure 4, and the Franck–Condon line-shape analysis (Figure S1) gave a 0–0 band energy gap of 14,500 cm−1 for Ir(pyr), which is lower than that for [Ir(piq)2(dmb)]+ (16,950 cm−1) (Kuramochi and Ishitani, 2016).
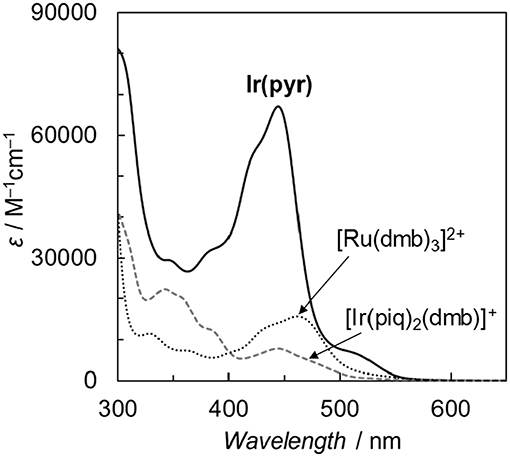
Figure 3. Comparison of the ultraviolet–visible (UV–vis) absorption spectra of Ir(pyr), [Ir(piq)2(dmb)](PF6), and [Ru(dmb)3](PF6)2 in N,N-dimethylacetamide (DMA) at 298 K.
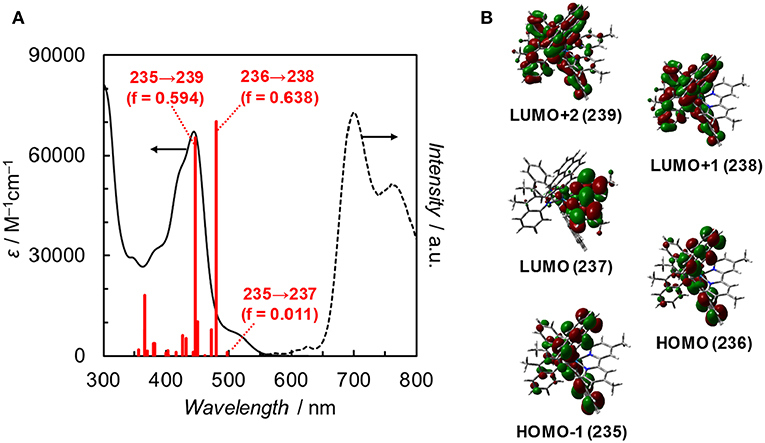
Figure 4. (A) UV–vis absorption spectrum (solid line), TDDFT theoretical excitations (red bars, >350 nm, f: oscillator strength), and emission spectrum (dotted line) of Ir(pyr) in DMA at 298 K. (B) Frontier orbitals of Ir(pyr). Calculation method: B3LYP/LANL2DZ(Ir)/6-31G(d,p) (H, C, N) levels by using polarizable continuum model (PCM) with the default for DMA, isovalue = 0.02.
The redox potentials of Ir(pyr) in DMA were obtained by cyclic voltammetry (CV; Figure S2) and differential pulse voltammetry (DPV) and are summarized in Table 1 together with those of [Ir(piq)2(dmb)]+ and Ru (Kuramochi and Ishitani, 2016; Kuramochi et al., 2018b). The oxidation waves of the Ir complexes were measured in acetonitrile due to its wide-potential window. The CV showed three each of reversible cathodic and irreversible anodic waves, indicating that Ir(pyr) is stable against reduction but relatively unstable to oxidation on the CV timescale. According to the DFT calculation, the LUMO mainly distributes across the dmb ligand of Ir(pyr) (Figure 4). Thus, it is expected that the first electron is injected into the dmb ligand, which benefits the electron transfer from the one-electron reduced species of Ir(pyr) to the Ru moiety. This property has been previously observed in [Ir(piq)2(dmb)]+ (Kuramochi and Ishitani, 2016).
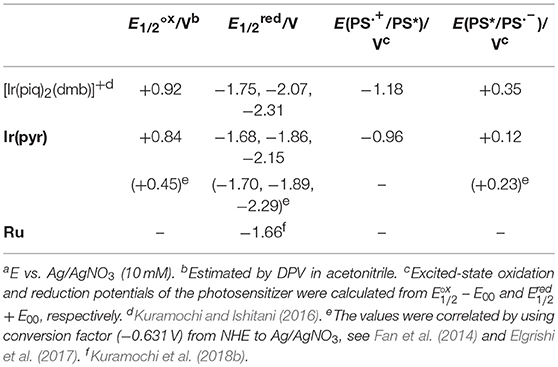
Table 1. Electrochemical data in DMA (dichloromethane or THF) at 298 Ka.
Emission Quenching by Electron Donors
The emission intensity of Ir(pyr)–Ru was similar to that of Ir(pyr), suggesting that oxidative quenching of the excited state of the Ir unit by the Ru unit does not proceed in Ir(pyr)–Ru. This is reasonable because the oxidative quenching process is endothermic; the oxidation potential of the excited state of Ir(pyr) is much more positive (−0.96 V; Table 1) than the reduction potential of Ru (−1.66 V).
Emission quenching of Ir(pyr) by 1-benzyl-1,4-dihydronicotinamide (BNAH) was inefficient: Stern–Volmer constant (KSV) = 13 M−1 in DMA. Assuming that the emission lifetime is 3.1 μs (Fan et al., 2014), the quenching rate constant (kq) was estimated to be 4.2 × 106 M−1 s−1. When a stronger electron donor, BI(OH)H (Hasegawa et al., 2006; Tamaki et al., 2015), was used, the emission of Ir(pyr) was more efficiently quenched (Figure 5); KSV reached 3,000 M−1 in DMA/TEOA (5:1 v/v), and kq was 9.7 × 108 M−1 s−1, which is close to the diffusion-controlled rate constant (Tamaki et al., 2013). The KSV of Ir(pyr)–Ru by BI(OH)H was 2,800 M−1, which is similar to that of Ir(pyr). In previous work by Fan et al. (2014), Ir(pyr) did not work as a PS for H2 evolution because the emission from Ir(pyr) was not quenched by TEA. This emission is not quenched by TEOA as well because TEOA has a similar oxidation potential to TEA. Conversely, BI(OH)H significantly quenches the emission from Ir(pyr), indicating efficient electron transfer from BI(OH)H to the excited Ir(pyr).
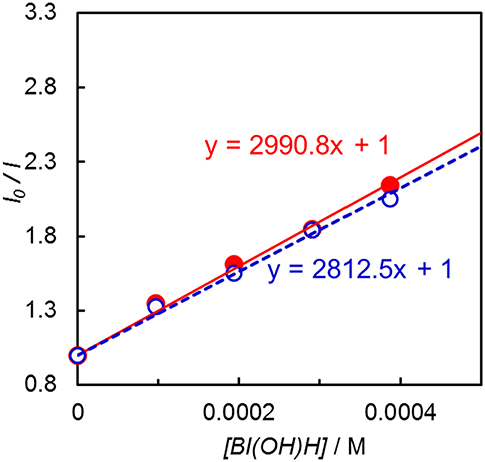
Figure 5. Emission quenching of the excited Ir(pyr) (closed red circle) and Ir(pyr)–Ru (open blue circle) by BI(OH)H in DMA/TEOA (5:1 v/v) at 298 K.
Photocatalytic CO2 Reduction
DMA-TEOA (5:1 v/v) mixed solutions containing both Ir(pyr) and Ru or Ir(pyr)–Ru (0.05 mM) as the photocatalysts and BI(OH)H as the ED were irradiated at λex > 480 nm under a CO2 atmosphere. In both cases, HCOOH was mainly detected with small amounts of CO and H2. Figure 6 shows the time profiles of product formation during the photocatalytic reaction. Blank experiments in the absence of the Ru catalyst produced trace amounts of CO (3.9 μmol) and formate (7.6 μmol) after 24 h. From the Stern–Volmer constants, the quenching efficiencies of the excited states of Ir(pyr) and Ir(pyr)–Ru by 0.1 M BI(OH)H were estimated as ηq > 99%, indicating that the excited states of Ir(pyr) and Ir(pyr)–Ru were almost completely quenched by BI(OH)H under these reaction conditions. Figure 6 shows that Ir(pyr) does work as a PS for CO2 reduction when using BI(OH)H. The time profiles for the mixture of Ir(pyr) and Ru showed a linear increase reaching a TONHCOOH = ~2,000 during 24-h irradiation, indicating that Ir(pyr) has a high durability during photocatalytic CO2 reduction. Although Ir(pyr)–Ru also worked as a photocatalyst for CO2 reduction, it showed a lower activity compared to the mixture of Ir(pyr) and Ru. While the initial formation rates of the products were similar, the reaction stopped after just 5 h of irradiation in the case of Ir(pyr)–Ru. Because the reaction solution of Ir(pyr)–Ru was decolorized during the photocatalytic reaction, the low activity of Ir(pyr)–Ru would result from its low durability. The decoloration was also observed in irradiation experiments of [Ir(piq)2(dmb)]+ and ED without the CAT because of hydrogenation of the ligands in [Ir(piq)2(dmb)]+ (Kuramochi and Ishitani, 2016). Thus, it is also expected that the decoloration of Ir(pyr)–Ru is caused by hydrogenation of the Ir unit. In Ir(pyr)–Ru, the accumulated electron(s) might be stabilized because of electron hopping between the Ir and Ru units, which might be enhanced by the hydrogenation of the Ir unit.
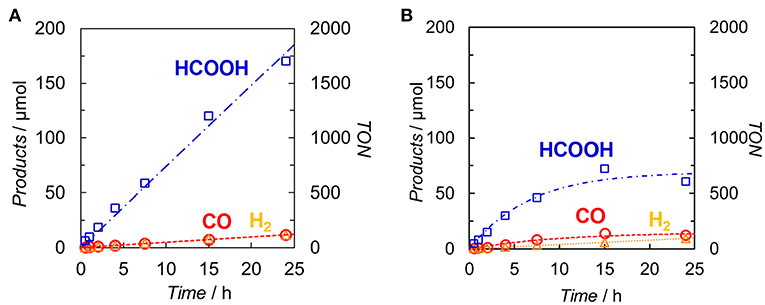
Figure 6. Time dependences of the products during the photo-irradiation of CO2-saturated DMA/TEOA (5:1 v/v, 2.0 ml) solutions containing (A) a mixed system of Ir(pyr) (0.05 mM) and Ru (0.05 mM) or (B) Ir(pyr)–Ru (0.05 mM) in the presence of BI(OH)H (0.1 M): CO (◦), HCOOH (□) and H2 (Δ). A 500-W high-pressure Hg lamp was used for the irradiation (λ > 480 nm).
Figure 7 illustrates the time profiles of the products during the irradiation of CO2-saturated DMA/TEOA (5:1 v/v, 2.0 ml) solutions containing Ir(pyr) or [Ru(dmb)3]2+ as PS in the presence of trans-Ru(bpy)(CO)2Cl2 (bpy = 2,2-bipyridine) and BI(OH)H as CAT and ED, respectively. The initial formation rate of HCOOH in the system using Ir(pyr) (Figure 7A) is slower than that using [Ru(dmb)3]2+ (Figure 7B), although Ir(pyr) has more intense absorption band at >480 nm than [Ru(dmb)3]2+ (Figure 3). In Figure 7, trans-Ru(bpy)(CO)2Cl2 is used instead of Ru. Although trans-Ru(bpy)(CO)2Cl2 has a much less negative reduction potential (−1.51 V vs. Ag/AgNO3; Kuramochi et al., 2015) than Ru (−1.66 V vs. Ag/AgNO3), the initial formation rate of HCOOH in the system using trans-Ru(bpy)(CO)2Cl2 (Figure 7A) is similar to that using Ru (Figure 6A), suggesting that the electron transfer process from the one-electron reduced Ir(pyr) to CAT is not the rate-determining step and does not significantly affect the reaction rate. Considering that the emission of Ir(pyr) is almost completely quenched, the slow initial formation rate of HCOOH in Ir(pyr) would result from the competitive back-electron transfer process soon after the electron transfer from BI(OH)H to the excited state of Ir(pyr) in the solvent cage (Kavarnos, 1993; Nakada et al., 2015).
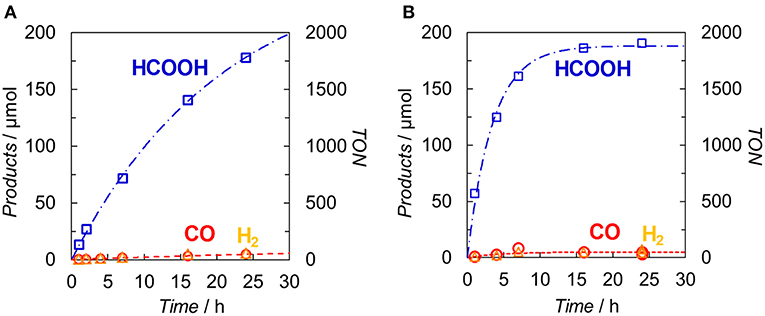
Figure 7. Time dependences of the products during the photo-irradiation of CO2-saturated DMA/TEOA (5:1 v/v, 2.0 ml) solutions containing (A) Ir(pyr) (0.05 mM) or (B) [Ru(dmb)3]2+ (0.05 mM) in the presence of trans-Ru(bpy)(CO)2Cl2 (0.05 mM) and BI(OH)H (0.1 M): CO (◦), HCOOH (□) and H2 (Δ). A 500-W high-pressure Hg lamp was used for the irradiation (λ > 480 nm).
Conclusion
Photocatalytic CO2 reduction using Ir(pyr) as PS, which has a strong absorption in the visible region, proceeded efficiently for more than 1 day when a suitable electron donor, BI(OH)H, and Ru were used as CAT. A new supramolecular photocatalyst, Ir(pyr)–Ru, was successfully synthesized, which exhibited a similar reaction rate during the initial stage of CO2 reduction to that of the mixed system, but the durability of Ir(pyr)–Ru was lower than that of the mixed system. While Ir(pyr) showed high durability in the mixed system, the initial formation rate of HCOOH tended to be slower than that of the catalytic system using [Ru(dmb)3]2+ as PS, which is possibly due to the faster back-electron transfer from the reduced Ir(pyr) to the oxidized BI(OH)H.
Experimental Section
General Procedure
All chemicals and solvents were of commercial reagent quality and were used without further purification unless otherwise stated. DMA was dried over molecular sieves of size 4 Å and distilled under reduced pressure. TEOA was distilled under reduced pressure. Tetraethylammonium tetrafluoroborate was dried in vacuo at 100°C overnight before use. [Ir(piq)2(BL)](PF6) (Kuramochi and Ishitani, 2016), BNAH (Mauzerall and Westheimer, 1955), BI(OH)H (Hasegawa et al., 2005; Zhu et al., 2008), Ru (Anderson et al., 1995), and BL (Sun et al., 1997) were synthesized according to literature procedures. 1H NMR spectra were recorded on an AL400 NMR spectrometer. IR spectra were measured in dichloromethane on a JASCO FT/IR-610 spectrometer. Electrospray ionization–mass spectroscopy (ESI-MS) was undertaken using a SHIMADZU LCMS-2010A system with acetonitrile as a mobile phase. UV–vis absorption spectra were recorded with a JASCO V-670 instrument. Emission spectra were measured at 25°C under an Ar atmosphere using a JASCO FP-8600 spectrofluorometer with correlation for the detector sensitivity. Emission quenching experiments were performed in DMA or DMA/TEOA (5:1 v/v) solutions containing a complex and several different concentrations of BNAH or BI(OH)H.
Emission Spectral Fitting
Double-mode Franck–Condon band shape analysis was used to fit the emission spectra. The spectral fittings were carried out according to the following equation (Caspar et al., 1984) using the Wavemetrics Igor software.
I(ν) is the relative emission intensity at frequency ν. E00 is the energy gap between the zeroth vibrational levels in the ground and excited states, n1 and n2 are the vibrational quantum numbers of the high- and low-frequency vibrational modes, respectively, S1 and S2 are the Huang–Rhys factors, and ν1/2 is the half-width at half-maximum (fwhm) of the individual vibronic band. The 0–0 band energy gaps between the lowest excited state and the ground state were obtained from the emission spectral fitting (Figure S1).
[Ir(pyr-mq)2(CH3CN)2](PF6)
[(pyr-mq)2Ir-μ-Cl]2 (100 mg, 5.4 × 10−5 mol), AgPF4 (31 mg, 1.2 × 10−4 mol), and acetonitrile (10 ml) were placed in a 50-ml flask. The mixture was stirred for 4 h at room temperature. The resulting suspension was filtered through Celite pad to remove AgCl. The filtrate was concentrated to ca. 1 ml, and the product was precipitated by the addition of diethyl ether (10 ml). After cooling the suspension for 30 min at 0°C, the product was collected by filtration, giving 96 mg (80%) of the titled compound as a dark orange solid: 1H NMR (CDCl3, 400 MHz) δ 9.18 (brs, 2H), 8.73 (d, J = 8.8 Hz, 2H), 8.56 (s, 2H), 8.23 (d, J = 7.2 Hz, 2H), 8.15–7.95 (m, 8H), 7.85–7.70 (m, 6H), 7.27 (m, 2H), 6.40 (brs, 2H), 3.23 (s, 6H), 2.22 (s, 6H, CH3CN).
[Ir(pyr-mq)2(BL)](PF6)
The bridging ligand (BL, 80 mg, 2.2 × 10−4 mol), dichloromethane (40 ml), and methanol (20 ml) were placed in a 100-ml flask, and the system was purged with argon gas. A solution of [Ir(pyr-mq)2(CH3CN)2](PF6) (80 mg, 7.3 × 10−5 mol) in dichloromethane (10 ml) and methanol (5 ml) was then added dropwise at room temperature. The reaction mixture was stirred at room temperature. The reaction progress was monitored with ESI-MS. After stirring for 48 h, the resulting solution was evaporated. The residue was purified with a silica gel column (1 to 2 vol% methanol in dichloromethane). The second red band eluted with 2 vol% methanol was collected and evaporated to dryness, giving 42 mg (43% based on the iridium precursor) of the titled complex as a dark red solid: ESI-MS m/z: 1,243 ([M–PF]+). 1H NMR (CDCl3, 400 MHz) δ 8.94 (brs, 1H), 8.92 (brs, 1H), 8.60 (d, J = 2.8 Hz, 2H), 8.51 (d, J = 5.2 Hz, 1H), 8.45 (d, J = 5.2 Hz, 1H), 8.24–8.12 (m, 8H), 8.02 (s, 1H), 8.00 (s, 1H), 7.91–7.76 (m, 8H), 7.58 (s, 2H), 7.52–7.38 (m, 5H), 7.35 (m, 1H), 7.25–7.21 (m, 1H), 7.09 (d, J = 4.4 Hz, 1H), 6.99–6.88 (m, 3H), 6.77 (m, 1H), 3.13–3.00 (m, 4H), 2.97 (s, 3H), 2.95 (s, 3H), 2.45 (s, 3H), 2.42 (s, 3H).
Ir(pyr)–Ru
An acetone/ethenol (1:2 v/v) mixed solution (6 ml) containing [Ru(CO)2Cl2]n (9.9 mg, 4.3 × 10−5/n mol) was refluxed for 1 h, and then [Ir(pyr-mq)2(BL)](PF6) (30 mg, 2.2 × 10−5 mol) was added to it. The reaction mixture was heated to 60°C and stirred for 4.5 h under Ar atmosphere. As the reaction proceeded, the starting red solution became a red suspension. The resulting solid was filtered and washed with ethanol. The solid was dissolved in dichloromethane (ca. 2 ml) and filtered to remove insoluble materials. The solution was evaporated to afford 30 mg (87%) of the titiled compound as a dark red solid. ESI-MS m/z: 1,471 ([M–PF]+). FT-IR νCO/cm−1: 1,992, 2,058. Anal. calcd (%) for C78H54Cl2F6IrN6O2PRu· 3H2O: C, 56.08; N, 5.03; H, 3.62. Found (%): C, 55.88; N, 4.81; H, 3.21. 1H NMR (CDCl3, 400 MHz) δ 8.97–8.88 (m, 4H), 8.74 (brs, 1H), 8.65–8.62 (m, 3H), 8.42 (brs, 1H), 8.30 (brs, 1H), 8.25–8.21 (m, 2H), 8.18–8.15 (m, 2H), 8.03–8.01 (m, 2H), 7.96–7.79 (m, 8H), 7.61 (s, 1H), 7.56 (s, 1H), 7.53–7.49 (m, 2H), 7.45–7.40 (m, 4H), 7.36 (m, 1H), 7.28–7.24 (m, 1H), 7.09 (d, J = 5.6 Hz, 1H), 6.98–6.88 (m, 3H), 3.05–2.94 (m, 4H), 2.99 (s, 3H), 2.98 (s, 3H), 2.61 (s, 3H), 2.47 (s, 3H).
Photocatalytic CO2 Reduction
DMA-TEOA (2 ml; 5:1 v/v) solutions containing a mixture of PS and CAT or the supramolecular Ir(pyr)–Ru complex and BI(OH)H were bubbled with CO2 for 30 min. Photo-irradiations were carried out in 11-mL Pyrex tubes (i.d. = 8 mm) with light at λ > 480 nm using a 500-W high-pressure Hg lamp combined with a K2CrO4 solution filter (30% w/w, optical path length: 1 cm) using a merry-go-round apparatus. The reaction temperature was maintained at 25°C using an IWAKI constant-temperature system (CTS-134A). The gaseous reaction products (CO and H2) were quantified with GC-TCD (GL Science GC323), and the product (formate) in the solutions was analyzed with a capillary electrophoresis system (Otuka Electronics Co.CAPI-3300I).
Computational Methods
DFT calculations were carried out using the Gaussian 09 package of programs (Frisch et al., 2009). Each structure was fully optimized using the B3LYP functional using the 6-31G(d,p) basis set for all atoms except Ir and the standard double-ζ type LANL2DZ basis set with the effective core potential of Hay–Wadt for Ir. The calculation was carried out by using the polarizable continuum model (PCM) with default parameter for DMA. The stationary points were verified using the vibrational analysis.
Author Contributions
YK concieved the research and conducted experiments. OI directed the project and co-wrote the paper.
Funding
This work was supported by JST CREST (grant number JPMJCR13L1) and the Strategic International Collaborative Research Program (SICORP) of JST.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00259/full#supplementary-material
References
Anderson, P. A., Deacon, G. B., Haarmann, K. H., Keene, F. R., Meyer, T. J., Reitsma, D. A., et al. (1995). Designed synthesis of mononuclear Tris(heteroleptic) ruthenium complexes containing bidentate polypyridyl ligands. Inorg. Chem. 34, 6145–6157. doi: 10.1021/ic00128a028
Bonin, J., Robert, M., and Routier, M. (2014). Selective and efficient photocatalytic CO2 reduction to CO using visible light and an iron-based homogeneous catalyst. J. Am. Chem. Soc. 136, 16768–16771. doi: 10.1021/ja510290t
Caspar, J. V., Westmoreland, T. D., Allen, G. H., Bradley, P. G., Meyer, T. J., and Woodruff, W. H. (1984). Molecular and electronic structure in the metal-to-ligand charge-transfer excited states of d6 transition-metal complexes in solution. J. Am. Chem. Soc. 106, 3492–3500. doi: 10.1021/ja00324a017
Chen, L., Guo, Z., Wei, X.-G., Gallenkamp, C., Bonin, J., Anxolabéhère-Mallart, E., et al. (2015). Molecular catalysis of the electrochemical and photochemical reduction of CO2 with earth-abundant metal complexes. selective production of CO vs HCOOH by switching of the metal center. J. Am. Chem. Soc. 137, 10918–10921. doi: 10.1021/jacs.5b06535
Elgrishi, N., Chambers, M. B., Wang, X., and Fontecave, M. (2017). Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 46, 761–796. doi: 10.1039/C5CS00391A
Fan, S., Zong, X., Shaw, P. E., Wang, X., Geng, Y., Smith, A. R., et al. (2014). Energetic requirements of iridium(III) complex based photosensitisers in photocatalytic hydrogen generation. Phys. Chem. Chem. Phys. 16, 21577–21585. doi: 10.1039/C4CP02997F
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09, Revision D.01. Wallingford, CT.
Goldsmith, J. I., Hudson, W. R., Lowry, M. S., Anderson, T. H., and Bernhard, S. (2005). Discovery and high-throughput screening of heteroleptic iridium complexes for photoinduced hydrogen production. J. Am. Chem. Soc. 127, 7502–7510. doi: 10.1021/ja0427101
Hasegawa, E., Seida, T., Chiba, N., Takahashi, T., and Ikeda, H. (2005). Contrastive photoreduction pathways of benzophenones governed by regiospecific deprotonation of imidazoline radical cations and additive effects. J. Org. Chem. 70, 9632–9635. doi: 10.1021/jo0514220
Hasegawa, E., Takizawa, S., Seida, T., Yamaguchi, A., Yamaguchi, N., Chiba, N., et al. (2006). Photoinduced electron-transfer systems consisting of electron-donating pyrenes or anthracenes and benzimidazolines for reductive transformation of carbonyl compounds. Tetrahedron 62, 6581–6588. doi: 10.1016/j.tet.2006.03.061
Kumagai, H., Sahara, G., Maeda, K., Higashi, M., Abe, R., and Ishitani, O. (2017). Hybrid photocathode consisting of a CuGaO2 p-type semiconductor and a Ru(II)–Re(I) supramolecular photocatalyst: non-biased visible-light-driven CO2 reduction with water oxidation. Chem. Sci. 8, 4242–4249. doi: 10.1039/C7SC00940B
Kuramochi, Y., Fukaya, K., Yoshida, M., and Ishida, H. (2015). trans-(Cl)-[Ru(5,5′-diamide-2,2′-bipyridine)(CO)2Cl2]: synthesis, structure, and photocatalytic CO2 reduction activity. Chem. Eur. J. 21, 10049–10060. doi: 10.1002/chem.201500782
Kuramochi, Y., and Ishitani, O. (2016). Iridium(III) 1-phenylisoquinoline complexes as a photosensitizer for photocatalytic CO2 reduction: a mixed system with a Re(I) catalyst and a supramolecular photocatalyst. Inorg. Chem. 55, 5702–5709. doi: 10.1021/acs.inorgchem.6b00777
Kuramochi, Y., Ishitani, O., and Ishida, H. (2018a). Reaction mechanisms of catalytic photochemical CO2 reduction using Re(I) and Ru(II) complexes. Coord. Chem. Rev. 373, 333–356. doi: 10.1016/j.ccr.2017.11.023
Kuramochi, Y., Itabashi, J., Toyama, M., and Ishida, H. (2018b). Photochemical CO2 reduction catalyzed by trans(Cl)-[Ru(2,2′-bipyridine)(CO)2Cl2] bearing two methyl groups at 4,4′-, 5,5′- or 6,6′-positions in the ligand. ChemPhotoChem 2, 314–322. doi: 10.1002/cptc.201700201
Kuramochi, Y., Kamiya, M., and Ishida, H. (2014). Photocatalytic CO2 reduction in N,N-dimethylacetamide/water as an alternative solvent system. Inorg. Chem. 53, 3326–3332. doi: 10.1021/ic500050q
Kuriki, R., Matsunaga, H., Nakashima, T., Wada, K., Yamakata, A., Ishitani, O., et al. (2016). Nature-inspired, highly durable CO2 reduction system consisting of a binuclear ruthenium(II) complex and an organic semiconductor using visible light. J. Am. Chem. Soc. 138, 5159–5170. doi: 10.1021/jacs.6b01997
Kuriki, R., Yamamoto, M., Higuchi, K., Yamamoto, Y., Akatsuka, M., Lu, D., et al. (2017). Robust binding between carbon nitride nanosheets and a binuclear ruthenium (II) complex enabling durable, selective CO2 reduction under visible light in Aqueous solution. Angew. Chem. Int. Ed. 56, 4867–4871. doi: 10.1002/anie.201701627
Lehn, J.-M., and Ziessel, R. (1990). Photochemical reduction of carbon dioxide to formate catalyzed by 2,2t-bipyridine- or 1,10-phenanthroline-ruthenium(II) complexes. J. Organomet. Chem. 382, 157–173. doi: 10.1016/0022-328X(90)85224-M
Lowry, M. S., and Bernhard, S. (2006). Synthetically tailored excited states: phosphorescent, cyclometalated iridium(III) complexes and their applications. Chem. Eur. J. 12, 7970–7977. doi: 10.1002/chem.200600618
Mauzerall, D., and Westheimer, F. (1955). 1-benzyldihydronicotinamide—a model for reduced DPN. J. Am. Chem. Soc. 77, 2261–2264. doi: 10.1021/ja01613a070
Nakada, A., Koike, K., Nakashima, T., Morimoto, T., and Ishitani, O. (2015). Photocatalytic CO2 reduction to formic acid using a Ru(II)–Re(I) supramolecular complex in an aqueous solution. Inorg. Chem. 54, 1800–1807. doi: 10.1021/ic502707t
Prier, C. K., Rankic, D. A., and MacMillan, D. W. C. (2013). Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363. doi: 10.1021/cr300503r
Rao, H., Bonin, J., and Rober, M. (2018). Toward visible-light photochemical CO2-to-CH4 conversion in aqueous solutions using sensitized molecular catalysis. J. Phys. Chem. C 122, 13834–13839. doi: 10.1021/acs.jpcc.8b00950
Rao, H., Schmidt, L. C., Bonin, J., and Robert, M. (2017). Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 548, 74–77. doi: 10.1038/nature23016
Sahara, G., Kumagai, H., Maeda, K., Kaeffer, N., Artero, V., Higashi, M., et al. (2016). Photoelectrochemical reduction of CO2 coupled to water oxidation using a photocathode with a Ru(II)–Re(I) complex photocatalyst and a CoOx/TaON photoanode. J. Am. Chem. Soc. 138, 14152–14158. doi: 10.1021/jacs.6b09212
Sato, S., Arai, T., Morikawa, T., Uemura, K., Suzuki, T. M., Tanaka, H., et al. (2011). Selective CO2 conversion to formate conjugated with H2O oxidation utilizing semiconductor/complex hybrid photocatalysts. J. Am. Chem. Soc. 133, 15240–15243. doi: 10.1021/ja204881d
Schultz, D. M., and Yoon, T. P. (2014). Solar synthesis: prospects in visible light photocatalysis. Science 343:1239176. doi: 10.1126/science.1239176
Sekizawa, K., Maeda, K., Domen, K., Koike, K., and Ishitani, O. (2013). Artificial Z-scheme constructed with a supramolecular metal complex and semiconductor for the photocatalytic reduction of CO2. J. Am. Chem. Soc. 135, 4596–4599. doi: 10.1021/ja311541a
Shaw, M. H., Twilton, J., and MacMillan, D. W. (2016). Photoredox catalysis in organic chemistry. J. Org. Chem. 81, 6898–6926. doi: 10.1021/acs.joc.6b01449
Sun, L., Berglund, H., Davydov, R., Norrby, T., Hammarstroem, L., Korall, P., et al. (1997). Binuclear ruthenium–manganese complexes as simple artificial models for photosystem II in green plants. J. Am. Chem. Soc. 119, 6996–7004. doi: 10.1021/ja962511k
Takizawa, S., Kano, R., Ikuta, N., and Murata, S. (2018). An anionic iridium(III) complex as a visible-light absorbing photosensitizer. Dalton Trans. 47, 11041–11046. doi: 10.1039/C8DT02477D
Takizawa, S., Pérez-Bolívar, C., Anzenbacher, P. Jr., and Murata, S. (2012). Cationic iridium complexes coordinated with coumarin dyes – sensitizers for visible-light-driven hydrogen generation. Eur. J. Inorg. Chem. 2012, 3975–3979. doi: 10.1002/ejic.201200474
Takizawa, S., Shimada, K., Sato, Y., and Murata, S. (2014). Controlling the excited state and photosensitizing property of a 2-(2-Pyridyl)benzo[b]thiophene-based cationic iridium complex through simple chemical modification. Inorg. Chem. 53, 2983–2995. doi: 10.1021/ic402778x
Tamaki, Y., and Ishitani, O. (2017). Supramolecular photocatalysts for the reduction of CO2. ACS Catal. 7, 3394–3409. doi: 10.1021/acscatal.7b00440
Tamaki, Y., Koike, K., and Ishitani, O. (2015). Highly efficient, selective, and durable photocatalytic system for CO2 reduction to formic acid. Chem. Sci. 6, 7213–7221. doi: 10.1039/C5SC02018B
Tamaki, Y., Koike, K., Morimoto, T., and Ishitani, O. (2013). Substantial improvement in the efficiency and durability of a photocatalyst for carbon dioxide reduction using a benzoimidazole derivative as an electron donor. J. Catal. 304, 22–28. doi: 10.1016/j.jcat.2013.04.002
Thoi, V. S., Kornienko, N., Margarit, C. G., Yang, P., and Chang, C. J. (2013). Visible-light photoredox catalysis: selective reduction of carbon dioxide to carbon monoxide by a nickel N-heterocyclic carbene-isoquinoline complex. J. Am. Chem. Soc. 135, 14413–14424. doi: 10.1021/ja4074003
Yamazaki, Y., Takeda, H., and Ishitani, O. (2015). Photocatalytic reduction of CO2 using metal complexes. J. Photochem. Photobiol. C 2015, 106–137. doi: 10.1016/j.jphotochemrev.2015.09.001
Zhu, X.-Q., Zhang, M.-T., Yu, A., Wang, C.-H., and Cheng, J.-P. (2008). Hydride, hydrogen atom, proton, and electron transfer driving forces of various five-membered heterocyclic organic hydrides and their reaction intermediates in acetonitrile. J. Am. Chem. Soc. 130, 2501–2516. doi: 10.1021/ja075523m
Keywords: strong visible-light absorption, metal complex, photocatalyst, electron donor, rhenium, CO2 reduction, photosensitizer, iridium
Citation: Kuramochi Y and Ishitani O (2019) An Ir(III) Complex Photosensitizer With Strong Visible Light Absorption for Photocatalytic CO2 Reduction. Front. Chem. 7:259. doi: 10.3389/fchem.2019.00259
Received: 31 January 2019; Accepted: 01 April 2019;
Published: 01 May 2019.
Edited by:
Hitoshi Ishida, Kitasato University, JapanCopyright © 2019 Kuramochi and Ishitani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yusuke Kuramochi, a3VyYW1vY2hpeUBycy50dXMuYWMuanA=
Osamu Ishitani, aXNoaXRhbmlAY2hlbS50aXRlY2guYWMuanA=
†Present Address: Yusuke Kuramochi, Department of Chemistry, Faculty of Science Division II, Tokyo University of Science, Tokyo, Japan