 A.D. Dinga Wonanke
A.D. Dinga Wonanke Poppy Bennett
Poppy Bennett Matthew A. Addicoat
Matthew A. Addicoat- Department of Chemistry and Forensics, Nottingham Trent University, Nottingham, United Kingdom
Metal-organic frameworks, MOFs, offer an effective template for polymerisation of polymers with precisely controlled structures within the sub-nanometre scales. However, synthetic difficulties such as monomer infiltration, detailed understanding of polymerisation mechanisms within the MOF nanochannels and the mechanism for removing the MOF template post polymerisation have prevented wide scale implementation of polymerisation in MOFs. This is partly due to the significant lack in understanding of the energetic and atomic-scale intermolecular interactions between the monomers and the MOFs. Consequently in this study, we explore the interaction of varied concentration of styrene, and 3,4-ethylenedioxythiophene (EDOT), at the surface and in the nanochannel of Zn2(1,4-ndc)2 (dabco), where 1,4-ndc = 1,4-naphthalenedicarboxylate and dabco = 1,4-diazabicyclo[2.2.2]octane. Our results showed that the interactions between monomers are stronger in the nanochannels than at the surfaces of the MOF. Moreover, the MOF-monomer interactions are strongest in the nanochannels and increase with the number of monomers. However, as the number of monomers increases, the monomers turn to bind more strongly at the surface leading to a potential agglomeration of the monomers at the surface.
1 Introduction
In recent years, the interest and effort in the synthesis, characterisation, functionalisation, modelling, and designing of novel nanoporous materials have gained a massive insurgence (Materazzi et al., 2008). This owes mostly to the fact that the properties of these materials are not only dependent on how atoms are arranged within their crystals, but also on the size and shape of their pores as well as on their specific surface area. For these reasons, nanoporous materials are heavily investigated for application in gas storage, sieving, filtration, extraction, separation, sensors, drug delivery, and electrochemical energy storage and catalysis (Bastani et al., 2013; Ma et al., 2014; Zhang et al., 2014; Forest et al., 2015; Zhang et al., 2015; Wilkerson and Ramesh, 2016; Fu et al., 2017; Rafiee and Shahebrahimi, 2017).
Hitherto, the most rapidly growing and investigated classes of nanoporous materials that hold potentials for an almost limitless range of applications are known as metal-organic frameworks, MOFs (Li et al., 1999; Yaghi et al., 2000; Yaghi et al., 2003; Mueller et al., 2006). MOFs are organic-inorganic hybrid crystalline porous materials that are formed by covalently binding metal ions or clusters, also known as secondary binding units, SBUs, with organic ligands, also known as linkers, in a variety of 2- and 3-dimensional nets or topologies (Furukawa et al., 2013; Butova et al., 2016). In general, they are materials with typical low mass densities, high internal surface area and large pore volumes. Consequently, they are exploited for several applications including (but not limited to) gas storage, filtration, extraction, separation, sensors, drug delivery, electrochemical energy storage, and catalysis (Furukawa et al., 2013; Ricco et al., 2016; Pettinari et al., 2017).
Moreover, the well-defined porous network and relatively high internal surface area have opened up new avenues for the use of MOFs as a template for various chemical reactions to obtain specific regio- and stereoisomers (Uemura et al., 2005; Liu et al., 2008; Bhakta et al., 2009; Canivet et al., 2011; Distefano et al., 2013; Lee et al., 2015; Chen et al., 2016; Ding et al., 2016; Wang et al., 2017; Mochizuki et al., 2018; Anan et al., 2019; Rivera-Torrente et al., 2019; Schmidt, 2019). Amongst these potential chemical reactions, polymerisation in MOFs has gained significant scientific interest. This is primarily because the highly designable features of MOFs result in nanochannels that can be applied as a tailor-made polymerisation system to obtain highly controlled polymer structures with long-range order. Furthermore, since the MOFs act only as a scaffold for reactions, conventional polymerisation methods can be easily employed, with little or no modification, provided that the reagents and reaction conditions do not destroy the crystal structures of the MOFs (Mochizuki et al., 2018).
So far, polymerisations in MOFs have been used to effectively control polymer molecular weight distribution, stereo-regularity (tacticity), reaction sites, and copolymer sequence (Uemura et al., 2008; Uemura et al., 2009). Consequently, this provides an attractive avenue for not only the precision synthesis of novel polymer materials but also for exploring specific properties of polymer confinement. A comprehensive review describing the state-of-the-art of polymerisation in MOFs was recently published (Schmidt, 2019).
Despite the advantages resulting from polymerisation in MOFs, there is a significant lack in conceptual understanding of how to effectively control these reactions, which consequently restrains their wide-scale application. (Uemura et al., 2009). Firstly, there is still an enormous synthetic challenge on how to effectively infiltrate the monomers in the confinement of the pores before polymerisation. Secondly, there is only very little understanding of the MOF nanochannel polymerisation mechanism, the initiation process and the propagation process. Thirdly, little is known on how the monomer interacts with the MOF framework during polymerisation as well as the mechanism for removing the MOF template (Wang et al., 2017).
The first evidence of polymerisation in MOFs was from the pioneering work of Uemura and co-workers on the radical polymerisation of styrene (Uemura et al., 2005). In this study, the styrene monomer was shown to fully infiltrate the nanochannel by immersing the MOF in the liquid monomer, while excess styrene at the external surface was removed by subjecting the host crystals to reduced pressure. The result from powder X-ray diffraction studies showed that the newly synthesised polymer was fully encapsulated in the nanochannel of the MOF. In a recent study, (Wang et al., 2017), Wang and co-workers performed an oxidative polymerisation of 3,4-ethylenedioxythiophene, EDOT, in a MOF. This time, the authors encountered a significant challenge in fully infiltrating the monomer into the MOF nanochannels and results from this study showed an agglomeration of monomers at the surface of the MOF.
An in-depth understanding of the energetic and atomic-scale intermolecular interactions between the monomers both at the surface and in the nanochannels of the pores would be a significant step towards understanding how to fully control these reactions. Consequently in this study, we explore the interaction of styrene and EDOT, at the surface and in the nanochannel of the MOF, Zn2(1,4-ndc)2 (dabco), where 1,4-ndc = 1,4-naphthalenedicarboxylate and dabco = 1,4-diazabicyclo [2.2.2]octane. Zn2(1,4-ndc)2 (dabco), hereafter referred to as ZnPW-NDC MOF, is a MOF possessing a zinc paddlewheel building block on which the naphthalenedicarboxylate linkers are joined to form two-dimensional square grids, which are pillared by the dabco ligands as shown in Figure 1 (Klein et al., 2012).
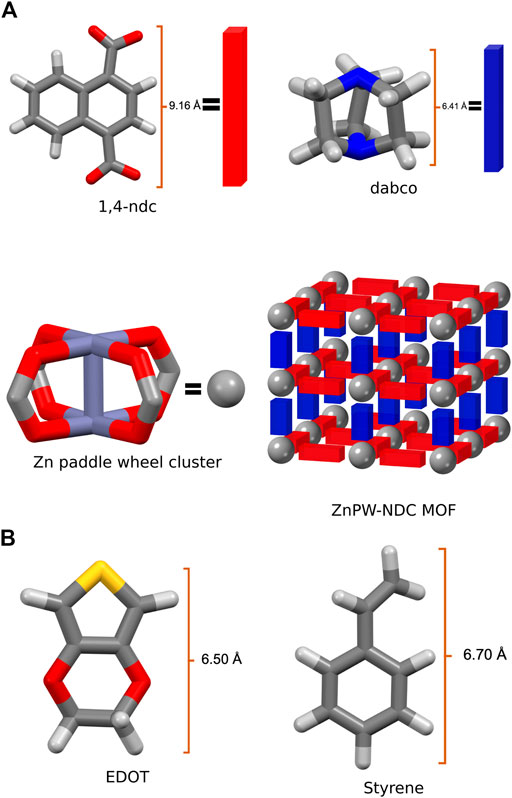
FIGURE 1. (A) the secondary building units of Zn2 (1,4-ndc)2 (dabco), represented as a net, where the organic linkers constitute the edges and the metal cluster the nodes, (B) Illustration of monomers EDOT and Styrene.
2 Methods
To fully explore the intermolecular interactions between the monomers and the ZnPW-NDC MOF, 100 ZnPW-NDC MOF
EMOF⊃monomers is the ground state optimised energy of the MOF
A molecular dynamics, MD, simulation was then performed on all the optimal MOF
All computational data corresponding to detailed energetics, intermolecular interactions and MD trajectories can be freely downloaded from http://doi.org/10.5281/zenodo.4382475.
3 Results and Discussions
3.1 Binding Energy Analysis
The binding energies for the most stable MOF
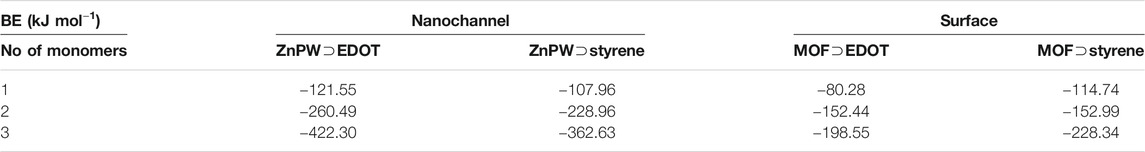
TABLE 1. UFF-Zenorg binding energies for the most stable MOF
In all cases, the strength of this intermolecular interaction increases with the number of monomers. The interactions are generally observed to be stronger in the nanochannels with ZnPW
3.2 Pairwise Root-Mean-Square Deviation
A pairwise root-mean-square deviation (RMSD) was performed to provide a visual inspection of how each structure changes over time. In the pairwise RMSD, we compute the RMSD of each snapshot in the trajectory with respect to all the other snapshots. The RMSD along the diagonals have values of zero, which correspond to the RMSD of a snapshot with itself. Low RMSD values at the off-diagonal regions correspond to snapshots whose structures are similar to the reference snapshot, while higher values correspond to dissimilar structures. Consequently, occupation of a given state can be observed as blocks of similar RMSDs along the diagonal. The pairwise RMSD for all the MD trajectories are presented in Figures 2, 3, wherein the snapshots are converted into picoseconds, ps and RMSD presented in Ångstrom, Å.
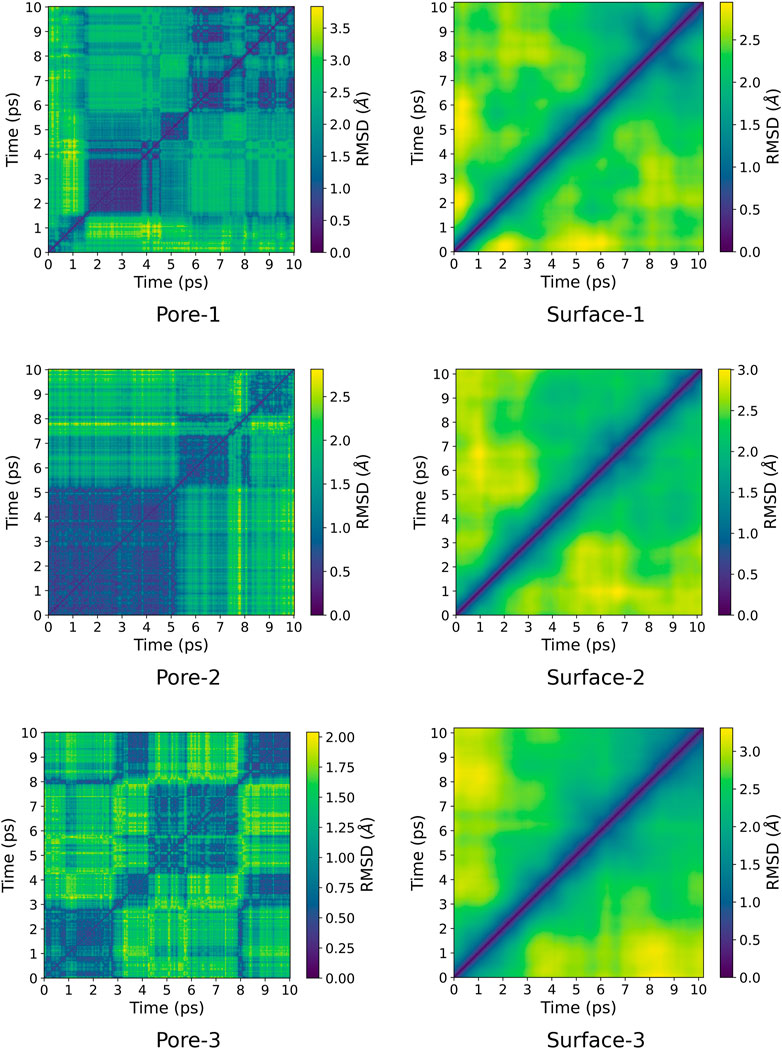
FIGURE 2. Pairwise RMSD plot for ZnPW
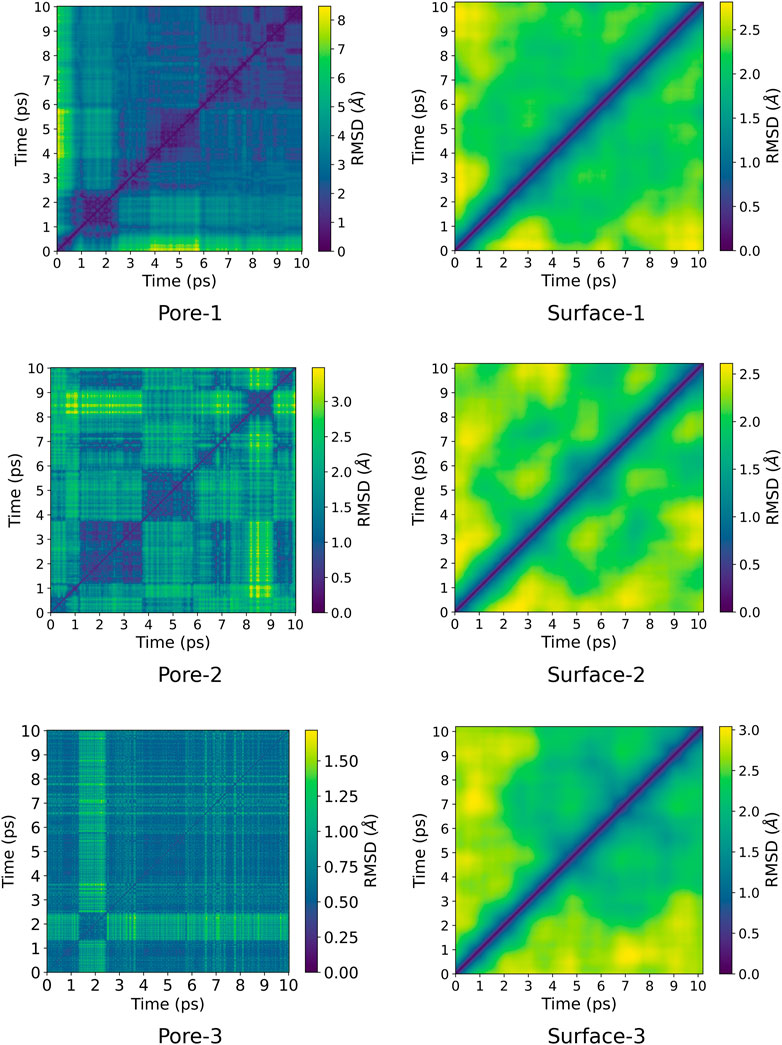
FIGURE 3. Pairwise RMSD plot for ZnPW
The RMSD for every point in Figures 2, 3 were computed using the formula in Eq. 2 and the python script can freely be downloaded (https://github.com/bafgreat/Pairwise-RMSD).
Here,
The pairwise RMSD for the nano-channels of both ZnPW
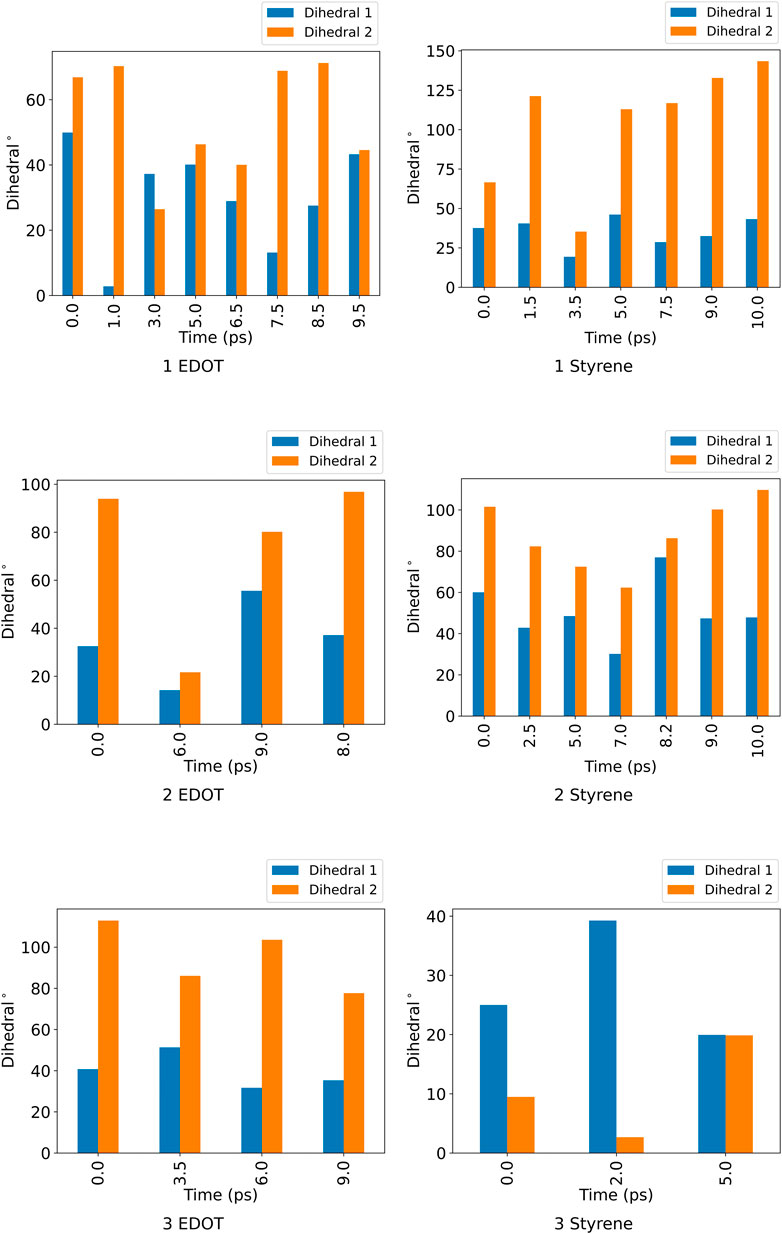
FIGURE 4. Dihedral angles for the two pairs positionally distinct ndc linker in the unit cell for the distinct conformational states of ZnPW
For the ZnPW
At the surface, the transitions from one conformational state to the other are far less distinct. Here, the snapshots show more dissimilarity across the trajectory, indicative of less favourable interactions for which the systems may want to explore for a longer time scale. In the presence of three monomers, there are few regions of similar RMSD at larger time scale.
3.3 Radial Distribution Analysis
A radial distribution function (RDF) was computed for all the MD trajectories to analyse the ZnPW-NDC MOF
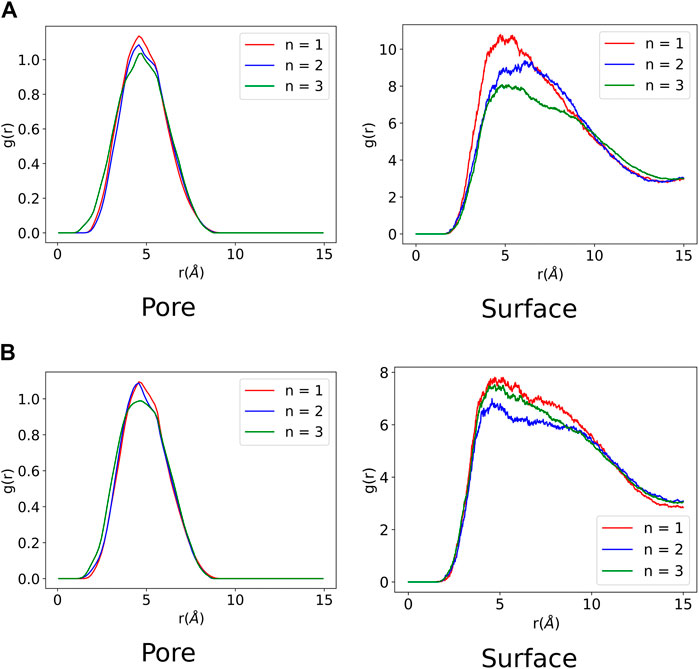
FIGURE 5. Radial distribution function of ZnPW
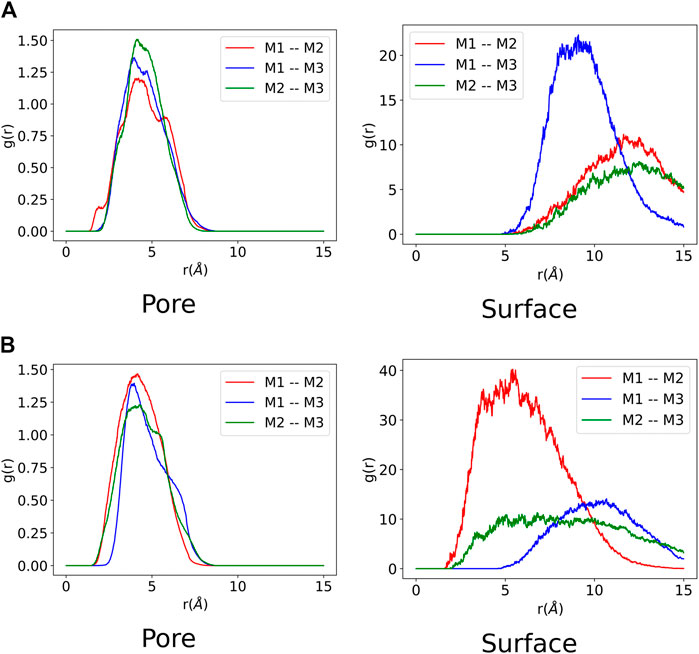
FIGURE 6. Monomer-monomer radial distribution function from each molecular dynamic trajectory composed of three monomers. Each monomer is labelled
It can be observed from Figure 5 that all the monomers have favourable intermolecular interactions with the ZnPW both at the surface and in the nanochannels, which are within the Van der Waals intermolecular range (r < 3.5 Å). However for EDOT, it can be observed from Figure 5A that as the number of monomers increases to three, the number of interactions below 2Å increases both at the surface and in the nanochannel, represented by the 3 green peaks. A similar observation is seen for the surface interaction of ZnPW
The monomer-monomer interactions for systems containing three monomers are presented in Figure 6. It can be observed for both ZnPW-NDC MOF
3.4 Contact Analysis
A contact analysis was computed for MOF
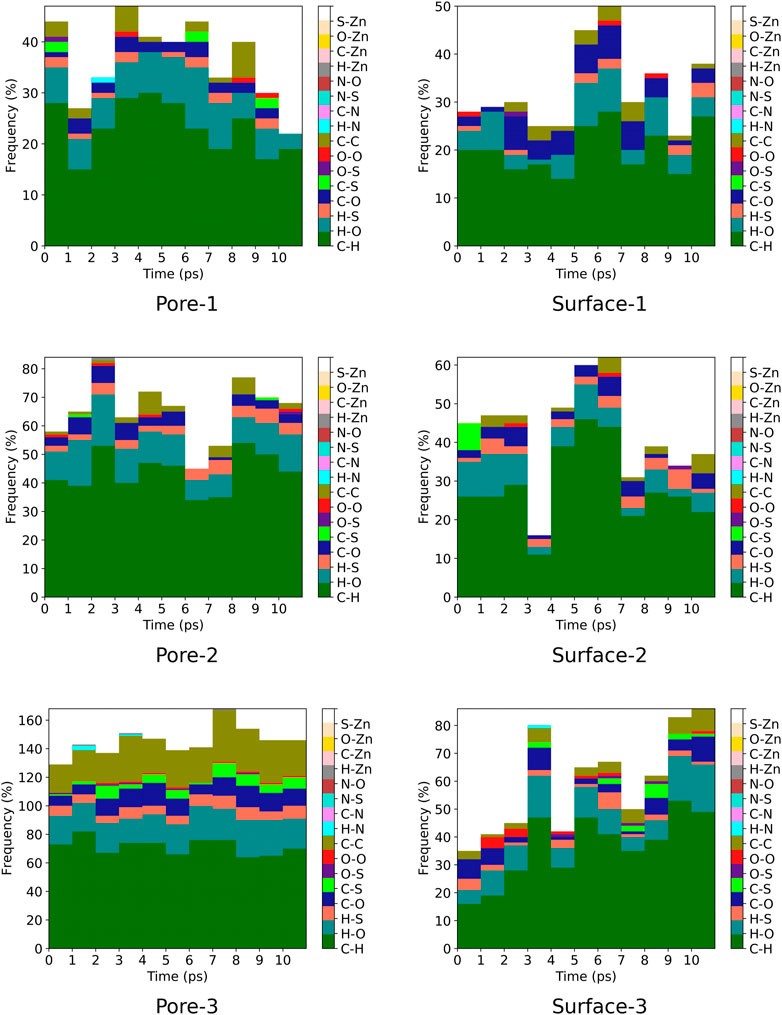
FIGURE 7. Frequency of ZnPW
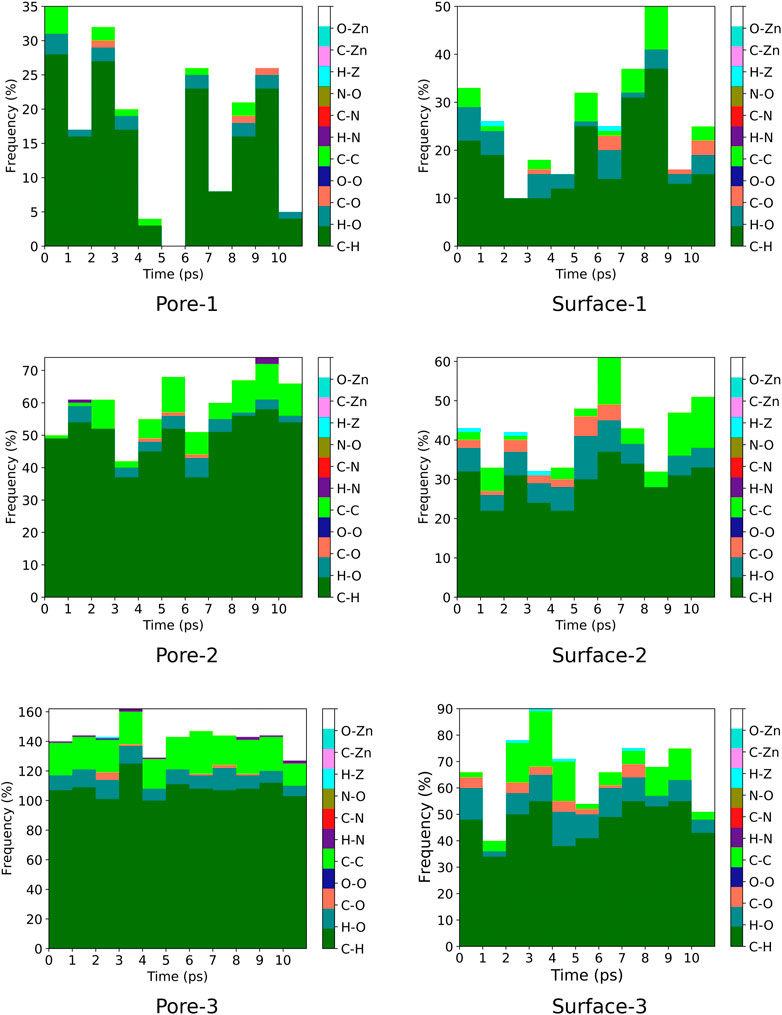
FIGURE 8. Frequency of ZnPW
For the nanochannel and surface interaction of both monomers with the ZnPW-NDC MOF, the C-H, H-O and C-C are the most common interactions. These correspond to strong hydrogen-bond and hydrophobic interactions occurring between the monomers and the ZnPW. The number of these interactions are observed to increase with the concentration of monomers. In ZnPW-NDC MOF
4 Conclusion
In this study, we investigated the intermolecular interactions of two monomers, styrene and EDOT, with ZnPW-NDC MOF metal organic framework. It was observed for both monomers that the ZnPW-NDC MOF
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. All computational data corresponding to detailed energetics, intermolecular interactions and MD trajectories can be freely downloaded from http://doi.org/10.5281/zenodo.4382475. Further inquiries can be directed to the corresponding author.
Author Contributions
MAA devised the study. PB and LC ran the calculations. ADDW performed md calculations, did analysis and wrote the manuscript.
Funding
MA thanks the EPSRC for a New Investigator award, EP/S015868/1. We also acknowledge HPC resources on THOMAS via membership of the UK's HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/P020194).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.716294/full#supplementary-material
References
Addicoat, M. A., Coupry, D. E., and Heine, T. (2014). AuToGraFS: Automatic Topological Generator for Framework Structures. J. Phys. Chem. A 118 (40), 9607–9614. doi:10.1021/jp507643v
Addicoat, M. A., Fukuoka, S., Page, A. J., and Irle, S. (2013). Stochastic Structure Determination for Conformationally Flexible Heterogenous Molecular Clusters: Application to Ionic Liquids. J. Comput. Chem. 34 (30), 2591–2600. doi:10.1002/jcc.23420
Anan, S., Mochizuki, Y., Kokado, K., and Sada, K. (2019). Step-growth Copolymerization between an Immobilized Monomer and a mobile Monomer in Metal-Organic Frameworks. Angew. Chem. Int. Edition 58 (24), 8018–8023. doi:10.1002/anie.201901308
Bastani, D., Esmaeili, N., and Asadollahi, M. (2013). Polymeric Mixed Matrix Membranes Containing Zeolites as a Filler for Gas Separation Applications: A Review. J. Ind. Eng. Chem. 19 (2), 375–393. doi:10.1016/j.jiec.2012.09.019
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., and Haak, J. R. (1984). Molecular Dynamics with Coupling to an External bath. J. Chem. Phys. 81 (8), 3684–3690. doi:10.1063/1.448118
Bhakta, R. K., Herberg, J. L., Jacobs, B., Highley, A., Behrens, R., Ockwig, N. W., et al. (2009). Metal-organic Frameworks as Templates for Nanoscale Naalh4. J. Am. Chem. Soc. 131 (37), 13198–13199. doi:10.1021/ja904431x
Butova, V. V., Soldatov, M. A., Guda, A. A., Lomachenko, K. A., and Lamberti, C. (2016). Metal-organic Frameworks: Structure, Properties, Methods of Synthesis and Characterization. Russ. Chem. Rev. 85, 280–307. doi:10.1070/rcr4554
Canivet, J., Aguado, S., Daniel, C., and Farrusseng, D. (2011). Engineering the Environment of a Catalytic Metal-Organic Framework by Postsynthetic Hydrophobization. ChemCatChem. 3 (4), 675–678. doi:10.1002/cctc.201000386
Chen, T., Huo, P., Hou, J.-L., Xu, J., Zhu, Q.-Y., and Dai, J. (2016). Confinement Effects of Metal-Organic Framework on the Formation of Charge-Transfer Tetrathiafulvalene Dimers. Inorg. Chem. 55 (24), 12758–12765. doi:10.1021/acs.inorgchem.6b02062
Ding, B., Wang, J., Chang, Z., Xu, G., Hao, X., Shen, L., et al. (2016). Self-sacrificial Template-Directed Synthesis of Metal-Organic Framework-Derived Porous Carbon for Energy-Storage Devices. ChemElectroChem. 3 (4), 668–674. doi:10.1002/celc.201500536
Distefano, G., Suzuki, H., Tsujimoto, M., Isoda, S., Bracco, S., Comotti, A., et al. (2013). Highly Ordered Alignment of a Vinyl Polymer by Host-Guest Cross-Polymerization. Nat. Chem. 5, 335–341. doi:10.1038/nchem.1576
Forest, C., Chaumont, P., Cassagnau, P., Swoboda, B., and Sonntag, P. (2015). Polymer Nano-Foams for Insulating Applications Prepared from Co2 Foaming. Prog. Polym. Sci. 41, 122–145. doi:10.1016/j.progpolymsci.2014.07.001
Fu, Y., Qu, Z., and Zhou, L. (2017). Prediction of the Effective thermal Conductivity of Aerogel Nano-Porous Materials. Energ. Proced. 105, 4769–4775. doi:10.1016/j.egypro.2017.03.938
Furukawa, H., Cordova, K. E., O’Keeffe, M., and Yaghi, O. M., (2013), The Chemistry and Applications of Metal-Organic Frameworks, Science 341. 1230444.doi:10.1126/science.1230444
Gowers, R. J., Linke, M., Barnoud, J., Tyler, J., Reddy, E., Melo, M. N., et al. (2016). MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations, in Proceedings of the 15th Python in Science Conference (Sebastian Benthall and Scott Rostrup, eds.), pp. 98 – 105.
Hourahine, B., Aradi, B., Blum, V., Bonafé, F., Buccheri, A., Camacho, C., et al. (2020). Dftb+, a Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations. J. Chem. Phys. 152 (12), 124101. doi:10.1063/1.5143190
Klein, N., Hoffmann, H. C., Cadiau, A., Getzschmann, J., Lohe, M. R., Paasch, S., et al. (2012). “Structural Flexibility and Intrinsic Dynamics in the M2(2,6-Ndc)2(dabco) (M = Ni, Cu, Co, Zn) Metal–Organic Frameworks. J. Mater. Chem. 22, 10303–10312. doi:10.1039/c2jm15601f
Lee, M., Shin, S. M., Jeong, N., and Thallapally, P. K. (2015). Chiral Environment of Catalytic Sites in the Chiral Metal-Organic Frameworks. Dalton Trans. 44, 9349–9352. doi:10.1039/c5dt01322d
Levine, B. G., Stone, J. E., and Kohlmeyer, A. (2011). Fast Analysis of Molecular Dynamics Trajectories with Graphics Processing Units-Radial Distribution Function Histogramming. J. Comput. Phys. 230 (9), 3556–3569. doi:10.1016/j.jcp.2011.01.048
Li, H., Eddaoudi, M., O'Keeffe, M., and Yaghi, O. M. (1999). Design and Synthesis of an Exceptionally Stable and Highly Porous Metal-Organic Framework. Nature 402, 276–279. doi:10.1038/46248
Liu, B., Shioyama, H., Akita, T., and Xu, Q. (2008). Metal-organic Framework as a Template for Porous Carbon Synthesis. J. Am. Chem. Soc. 130 (16), 5390–5391. doi:10.1021/ja7106146
Ma, J., Sanchez, J. P., Wu, K., Couples, G. D., and Jiang, Z. (2014). A Pore Network Model for Simulating Non-ideal Gas Flow in Micro- and Nano-Porous Materials. Fuel 116, 498–508. doi:10.1016/j.fuel.2013.08.041
Martyna, G. J., Tuckerman, M. E., Tobias, D. J., and Klein, M. L. (1996). Explicit Reversible Integrators for Extended Systems Dynamics. Mol. Phys. 87 (5), 1117–1157. doi:10.1080/00268979600100761
Materazzi, S. (2008). “Coordination Compounds and Inorganics,” in Recent Advances, Techniques and Applications. Editors M. E. Brown, and P. K. Gallagher (Elsevier Science B.V.), 439–502. vol. 5 of Handbook of Thermal Analysis and Calorimetry. doi:10.1016/s1573-4374(08)80015-5
Michaud-Agrawal, N., Denning, E. J., Woolf, T. B., and Beckstein, O. (2011). Mdanalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 32 (10), 2319–2327. doi:10.1002/jcc.21787
Mochizuki, S., Kitao, T., and Uemura, T. (2018). Controlled Polymerizations Using Metal-Organic Frameworks. Chem. Commun. 54, 11843–11856. doi:10.1039/c8cc06415f
Moreira, N. H., Dolgonos, G., Aradi, B., da Rosa, A. L., and Frauenheim, T. (2009). Toward an Accurate Density-Functional Tight-Binding Description of Zinc-Containing Compounds. J. Chem. Theor. Comput. 5 (3), 605–614. doi:10.1021/ct800455a
Mueller, U., Schubert, M., Teich, F., Puetter, H., and Schierle-Arndt, K. (2006), Metal-organic Frameworks Prospective Industrial Applications, J. Mater. Chem. 16. 626–636.doi:10.1039/b511962f
Pettinari, C., Marchetti, F., Mosca, N., Tosi, G., and Drozdov, A. (2017). Application of Metal-Organic Frameworks. Polym. Int. 66 (6), 731–744. doi:10.1002/pi.5315
Rafiee, E., and Shahebrahimi, S. (2017). Organic-inorganic Hybrid Polyionic Liquid Based Polyoxometalate as Nano Porous Material for Selective Oxidation of Sulfides. J. Mol. Struct. 1139, 255–263. doi:10.1016/j.molstruc.2017.03.041
Rappe, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A., and Skiff, W. M. (1992). Uff, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 114 (25), 10024–10035. doi:10.1021/ja00051a040
Ricco, R., Pfeiffer, C., Sumida, K., Sumby, C. J., Falcaro, P., Furukawa, S., et al. (2016). “Emerging Applications of Metal–Organic Frameworks. CrystEngComm 18, 6532–6542. doi:10.1039/c6ce01030j
Rivera-Torrente, M., Pletcher, P. D., Jongkind, M. K., Nikolopoulos, N., and Weckhuysen, B. M. (2019). Ethylene Polymerization over Metal-Organic Framework Crystallites and the Influence of Linkers on Their Fracturing Process. ACS Catal. 9 (4), 3059–3069. doi:10.1021/acscatal.9b00150
Rohwer, E. J., Akbarimoosavi, M., Meckel, S. E., Liu, X., Geng, Y., Lawson Daku, L. M., et al. (2018). Dipole Moment and Polarizability of Tunable Intramolecular Charge Transfer States in Heterocyclic-Conjugated Molecular Dyads Determined by Computational and Stark Spectroscopic Study. The J. Phys. Chem. C 122 (17), 9346–9355. doi:10.1021/acs.jpcc.8b02268
Schmidt, B. V. K. J. (2019). Metal-organic Frameworks in Polymer Science:polymerization Catalysis, Polymerization Environment, and Hybrid Materials. Macromolecular Rapid Commun. 41 (1), 1900333. doi:10.1002/marc.201900333
te Velde, G., Bickelhaupt, F. M., Baerends, E. J., Fonseca Guerra, C., van Gisbergen, S. J. A., Snijders, J. G., et al. (2001). Chemistry with Adf. J. Comput. Chem. 22 (9), 931–967. doi:10.1002/jcc.1056
Uemura, T., Kitagawa, K., Horike, S., Kawamura, T., Kitagawa, S., Mizuno, M., et al. (2005). Radical Polymerisation of Styrene in Porous Coordination Polymers. Royal Soc. Chem., 5968–5970. doi:10.1039/b508588h
Uemura, T., Ono, Y., Kitagawa, K., and Kitagawa, S. (2008). Radical Polymerization of Vinyl Monomers in Porous Coordination Polymers: Nanochannel Size Effects on Reactivity, Molecular Weight, and Stereostructure. Macromolecules 41 (1), 87–94. doi:10.1021/ma7022217
Uemura, T., Yanai, N., and Kitagawa, S. (2009). Polymerization Reactions in Porous Coordination Polymers. Chem. Soc. Rev. 38, 1228–1236. doi:10.1039/b802583p
Wang, T., Farajollahi, M., Henke, S., Zhu, T., Bajpe, S. R., Sun, S., et al. (2017). Functional Conductive Nanomaterials via Polymerisation in Nano-Channels: Pedot in a Mof. Mater. Horizon. 4, 64–71. doi:10.1039/c6mh00230g
Wilkerson, J. W., and Ramesh, K. T. (2016). A Closed-form Criterion for Dislocation Emission in Nano-Porous Materials under Arbitrary Thermomechanical Loading. J. Mech. Phys. Sol. 86, 94–116. doi:10.1016/j.jmps.2015.10.005
Yaghi, O. M., O'Keeffe, M., and Kanatzidis, M. (2000). Design of Solids from Molecular Building Blocks: Golden Opportunities for Solid State Chemistry. J. Solid State. Chem. 152 (1), 1–2. doi:10.1006/jssc.2000.8733
Yaghi, O. M., O'Keeffe, M., Ockwig, N. W., Chae, H. K., Eddaoudi, M., and Kim, J. (2003). Reticular Synthesis and the Design of New Materials. Nature 423 (6941), 705–714. doi:10.1038/nature01650
Zhang, H., Fang, W., Li, Z., and Tao, W. (2015). The Influence of Gaseous Heat Conduction to the Effective thermal Conductivity of Nano-Porous Materials. Int. Commun. Heat Mass Transfer 68, 158–161. doi:10.1016/j.icheatmasstransfer.2015.08.027
Keywords: metal-organic frameworks (MOFs), polymerisation, host-guest interaction, DFTB, molecular dynamics
Citation: Wonanke AD, Bennett P, Caldwell L and Addicoat MA (2021) Role of Host-Guest Interaction in Understanding Polymerisation in Metal-Organic Frameworks. Front. Chem. 9:716294. doi: 10.3389/fchem.2021.716294
Received: 28 May 2021; Accepted: 05 July 2021;
Published: 21 July 2021.
Edited by:
Tony D. James, University of Bath, United KingdomReviewed by:
Shenhui Li, Chinese Academy of Sciences (CAS), ChinaHiroyasu Yamaguchi, Osaka University, Japan
Xin Wu, The University of Sydney, Australia
Copyright © 2021 Wonanke, Bennett, Caldwell and Addicoat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew A. Addicoat, bWF0dGhldy5hZGRpY29hdEBudHUuYWMudWs=