 Seyed Mahmoud Hosseini1
Seyed Mahmoud Hosseini1 Rashed Rihani2
Rashed Rihani2 Benjamin Batchelor3
Benjamin Batchelor3 Allison M. Stiller2
Allison M. Stiller2 Joseph J. Pancrazio2Walter E. Voit2,3,4
Joseph J. Pancrazio2Walter E. Voit2,3,4 Melanie Ecker2,3,4*
Melanie Ecker2,3,4*- 1Department of Chemistry and Biochemistry, The University of Texas at Dallas, Dallas, TX, United States
- 2Department of Bioengineering, The University of Texas at Dallas, Dallas, TX, United States
- 3Center for Engineering Innovation, The University of Texas at Dallas, Dallas, TX, United States
- 4Department of Materials Science and Engineering, The University of Texas at Dallas, Dallas, TX, United States
Candidate materials for next generation neural recording electrodes include shape memory polymers (SMPs). These materials have the capability to undergo softening after insertion in the body, and therefore reduce the mismatch in modulus that usually exists between the device and the tissue. Current SMP formulations, which have shown promise for neural implants, contain ester groups within the main chain of the polymer and are therefore prone to hydrolytic decomposition under physiological conditions over periods of 11–13 months in vivo, thus limiting the utility for chronic applications. Ester free polymers are stable in harsh condition (PBS at 75°C or NaOH at 37°C) and accelerated aging results suggest that ester free SMPs are projected to be stable under physiological condition for at least 7 years. In addition, the ester free SMP is compatible with microfabrication processes needed for device fabrication. Furthermore, they demonstrate in vitro biocompatibility as demonstrated by high levels of cell viability from ISO 10993 testing.
Introduction
Shape memory polymers (SMPs) are an emerging class of materials. Their capability to restore their original shape after being deformed is outstanding (Dietsch and Tong, 2007; Liu et al., 2007; Mather et al., 2009; Lendlein, 2010; Hu et al., 2012b; Hager et al., 2015) and these materials have found utility in a variety of potential applications, including airspace, (Ishizawa et al., 2003; Barrett et al., 2006; Rory Barrett et al., 2006; Yanju and Jinsong, 2010; Yanju et al., 2014) anti-counterfeiting technology, (Pretsch et al., 2012; Ecker and Pretsch, 2013, 2014a,b) textile industry, (Matilla, 2006; Hu and Chen, 2010; Hu et al., 2012a) and as medical devices (Feninat et al., 2002; Buckley et al., 2006; Baer et al., 2007; Kulshrestha and Mahapatro, 2008; Lendlein and Behl, 2008; Baudis et al., 2014; Wang et al., 2017). The triggers for SMPs to recover to their permanent shape are diverse and include direct heating above the transition temperature of the polymer, indirect heating through electric or magnetic activation, and less commonly, chemical modification including plasticization. The plasticization of the SMP with solvent molecules, e.g., water, leads to a lowering of the glass transition temperature (Tg) of the polymer due to swelling and the resulting increased free volume of the polymer chains (Immergut and Mark, 1965; Singhal et al., 2013). When the Tg of a polymer is above room temperature (RT) before, but below after plasticization, the SMP may recover its permanent shape upon immersion in aqueous solutions at ambient temperatures. At the same time, the polymer changes from a glassy (stiff) materials to a rubbery (soft) material. This behavior of SMPs is what is of interest for the development of (self)-softening SMP substrates for bioelectronics devices. Especially for cortical neural interfaces, there is a need for devices which are stiff enough during the implantation to enable a sufficient penetration of the tissue with least possible trauma. However, after implantation, materials that soften to a modulus that is much closer to the tissue modulus offer the promise of decreasing the foreign body response and micromotion effects. Previously, the Voit Lab has presented a new generation of neural implants comprising of softening thiol-ene/acrylate polymers used as substrates (Ware et al., 2013, 2014; Do et al., 2017; Ecker et al., 2017; Simon et al., 2017). These devices work well for acute experiments and for chronic experiments on the order of 1–3 months. At least one factor limiting the long term stability of current SMP devices may be the fact that these polymers contain ester groups in their backbone which are vulnerable to hydrolytic degradation under moist conditions. For future applications, including translation to the clinic, it is necessary to have substrate materials with increased durability under in vivo conditions to enable chronic experiments over the course of years (Ryu and Shenoy, 2009; Rubehn and Stieglitz, 2010; Takmakov et al., 2015; Teo et al., 2016; Lecomte et al., 2017; ASTM, 2018), Reit et al. (2015) have demonstrated, that the use of ester free thiol-monomers significantly increases the durability of thiol-ene networks while it still allows to tune the glass transition temperature and cross-link density.
Here, we present a thiol-ene SMP formulation that is chemically and structurally similar to the most recent ones, but does not contain any ester groups (Do et al., 2017; Ecker et al., 2017; Simon et al., 2017; Garcia-Sandoval et al., 2018; Shoffstall et al., 2018). We have optimized the synthesis of a new monomer and tailored the polymer composition to have similar in vivo softening capabilities as previous reported SMPs. Dynamic mechanical analysis (DMA) of the hydrolytically stable SMP revealed that the SMP has a glass transition temperature above body temperature when dry, but below body temperature after being soaked in phosphate buffered saline (PBS). Thus, the novel SMP is also able to soften under physiological conditions to a modulus that is much closer to the tissue. To verify the improved stability of the new material against hydrolysis, we have performed accelerated aging tests in PBS at elevated temperatures (75°C) for 8 weeks and in one molar sodium hydroxide (NaOH) solution at body temperature over the course of 4 weeks. Weight loss and mechanical properties were determined and compared to a SMP composition that contains ester groups in the main chain. Our results demonstrate that the ester free SMP remained stable over the course of the study whereas the ester-containing counterpart lost about 15% of mass after aging in PBS and even 39% after aging in NaOH. We have also demonstrated that the new softening polymer is biocompatible, can be sterilized, and is compatible with microfabrication methods. That makes this polymer an ideal substrate candidate for future neural implants.
Materials and Methods
Synthesis of 1,3,5-tris(3-mercaptopropyl)-1,3,5-triazinane-2,4,6-trione (TTTSH)
Trithiol monomer TTTSH was synthesized following previously reported method (Lundberg et al., 2010) with minor modifications (Scheme 1). Briefly, 30 g (120.4 mmol) 1, 3, 5-triallyl-1, 3, 5-triazine-2, 4, 6-trione (TATATO), 82.40 g (1,080 mmol) thioacetic acid, and 1.98 g (12.04 mmol) 2, 2′-azobis (2-methylpropionnitrile) (AIBN) were placed in a 500 mL three-neck round-bottom flask which was equipped with condenser and nitrogen inlet. Afterward, the reaction mixture was stirred at 65°C for 24 h under a nitrogen atmosphere. Excess thioacetic acid was removed by reduced pressure and then was reacted with methanol (100 ml) and concentrated hydrochloric acid (50 ml) at 65°C for 36 h to cleave the thioester bond. After cooling down to room temperature, water was added (300 ml) and extracted for three times with methylene chloride (300 ml). The organic mixture was washed with sodium hydrogen carbonate solution (5%), dried over MgSO4, and concentrated with reduced pressure. After purification by column chromatography with gradient hexane: ethyl acetate mixtures 1:0 to 1:4 yellowish viscous liquid was obtained.
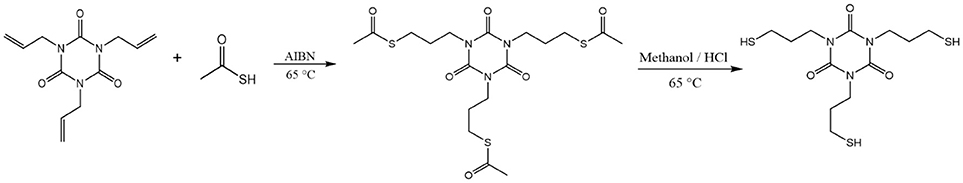
Scheme 1. Reaction scheme of the TTT-SH synthesis.
Fabrication of Polymers
1,3,5-Triallyl- 1,3,5-triazine-2,4,6(1H,3H,5H)-trione (TATATO), Trimethylolpropane tris(3-mercaptopropionate) (TMTMP), and 2,2-Dimethoxy-2-phenylacetophenone (DMPA) were purchased from Sigma Aldrich, whereas Tris [2-(3-mercaptopropionyloxy) ethyl] isocyanurate (TMICN) was purchased from Evans Chemicals, and 1,10-Decanedithiol (DDT) from TCI Chemicals. All the chemicals were used as received without further purification. TTTSH was synthesized as described under section Synthesis of 1,3,5-tris(3-mercaptopropyl)-1,3,5-triazinane-2,4,6-trione (TTTSH). Two thiol-ene SMP compositions were prepared, an ester-free (SMP-A) and an ester containing (SMP-B) formulation, each consisting of stoichiometric quantities of thiol to alkene functionalities. Exact mole fractions are: TTTSH/DDT-TATATO = 0.3/0.2–0.5 (SMP-A) and TMTMP/TMICN-TATATO = 0.45/0.05–0.5 (SMP-B) (Figure 1). A total of 0.1 wt% DMPA of total monomer weight was dissolved in the solution for the initiation of the photopolymerization of the monomer solution. The vial was covered in aluminum foil to prevent incident light from contacting the monomer solution and kept at room temperature. Without exposing the solution to light, the vial was mixed thoroughly by planetary speed mixing.
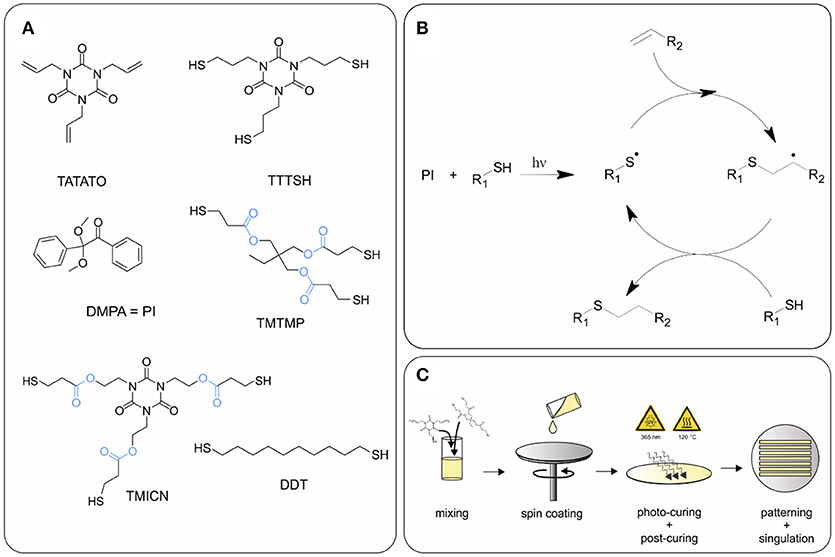
Figure 1. (A) Chemical structures of monomers and photoinitiator (PI) used for synthesis of polymers SMP-A and SMP-B. Ester groups are highlighted in blue, (B) reaction mechanism of thiol-ene click reaction, and (C) schematic of polymer fabrication.
The polymer solutions were spin cast on 75 × 50 mm glass microscope slides using a Laurell WS-650-8B spin coater. Spin speed was 350 rpm and time was 45 s for SMP-A and 600 rpm and 25 s for SMP-B in order to achieve thicknesses of about 30 μm, respectively. Polymerization was performed at ambient temperature using an UVP CL-1000 crosslinking chamber with five overhead 365 nm UV bulbs for 60 min under air. Cured samples were then placed in a vacuum oven at 120°C and 5 inHg (~16.9 kPa) for 24 h to further complete network conversion.
Test devices were either fabricated using a CO2 laser or were fabricated in the UT Dallas Class 10,000 cleanroom facility. The above SMP-on-glass substrates were used as the starting substrates in the cleanroom. Low temperature silicon nitride (using PlasmaTherm-790 PECVD) was deposited to act as a hard mask for the following plasma etching processes in which the device outline/shape was patterned. Adjacently, the nitride hard mask was etched away in the 1:10 HF dip. For some SMP samples top 4 μm thick crust was etched away in oxygen plasma (Technics RIE). In a final step, the test devices having dimensions of 4.5 × 50 mm × 30 μm were delaminated from the glass slide by soaking in water.
Accelerated Aging
SMP-A and B were subjected to two different accelerated aging scenarios; test samples were either immersed in 20 ml phosphate buffered saline (PBS) at 75°C or in 1 M sodium hydroxide (NaOH) solution at 37°C, respectively. A number of N = 3 samples were removed after 7, 14, 21, and 28 days immersion in NaOH and after 7, 14, 21, 28, 35, 42, 49, and 56 days after immersion in PBS. After removal, the samples were rinsed in de-ionized water before they were dried with a lint-free cloth. The accelerated aging in PBS follows the Arrhenius equation under the conservative assumption for biomedical polymers (Hemmerich, 1998) that Q10 = 2:
where tAA is the accelerated aging time, tRT the real aging time, TAA temperature for accelerated aging, TRT temperature for real time aging and Q10 the temperature coefficient. According to the Q10 temperature coefficient, which is a derivation of the Arrhenius equation, 1 week in PBS at 75°C is equal to 14 weeks at 37°C.
Weight and Thickness Loss
The dry weight of all samples was determined with 0.01 mg precision before and after aging. The weight loss was calculated according the following equation:
where m0 is the weight of the neat samples and mT is the weight of the samples after aging. A Marathon micrometer with 0.001 mm precision was applied to determine thickness of samples before and after the aging study. The mean value and standard deviation was calculated for N = 3 samples per aging time.
Dynamic Mechanical Analysis (DMA)
DMA was performed using a TA Instruments RSA-G2 Solids Analyzer with the immersion system in tension mode in order to quantify the storage modulus E' and tan δ of dry or in PBS soaked samples. All measurements were performed on rectangular samples as received after the clean room processing or CO2 laser cutting, having a width of 4.5 ± 0.1 mm and thicknesses of 30 ± 3 μm. The following parameters were selected: clamping distance of 15 mm, a preload force of 0.1 N, a frequency of 1 Hz, and a deformation amplitude of 0.275% strain. Dry experiments were run from 10 to 100°C or from 20 to 120°C using a heating rate of 2°C min−1. Soaking experiments were performed using the immersion system of the RSA-G2 filled with PBS. The first step (the soaking) included the heating from room temperature to 37°C followed by isothermal oscillating for 60 or 120 min. The second step comprised first cooling down to the start temperature with a rate of 3 C min−1 followed by heating from 10 to 80°C applying a heating rate of 2°C min−1. All measurements were performed on three independent specimens in order to gather statistical results. Graphics show representative measurements only.
Termogravimetric Analysis (TGA)
A Mettler Toledo TGA/DSC 1 was used to perform Thermal Gravimetric Analysis on N = 3 samples before and after accelerated aging. The polymer samples were heated from 25 to 700°C at a heating rate of 20°C/min and flow of 50 ml/min nitrogen gas. Samples were approximately 5 mg each.
Cytotoxicity Test
Cytotoxicity assays were carried out as previously described (Black et al., 2018) and in accordance with the International Organization for Standards (ISO) protocol “10993-5: Biological evaluation of medical devices”(ISO, 2008). Briefly, 50% and 100% concentration shape-memory polymer (SMP-A) extract was evaluated against Tygon-F-4040-Lubricant Tubing extract (positive control) and cell medium (negative control). Material extracts which reduced normalized cell viability percentages below 70% were considered cytotoxic in accordance with the ISO protocol (ISO, 2008).
Material extracts were made by soaking 3 cm2/ml of positive control and SMP A in Dulbecco's Modified Eagle Medium (DMEM) at 37°C, 5% CO2, and 95% relative humidity for 24 h in a polystyrene, glass-bottom 24 well plate (Greiner Bio-One, Austria). NCTC clone 929 fibroblasts (ATCC, USA) were routinely sub-cultured and seeded in a separate 24 well plate at a density of 100,000 cells per well in complete cell medium (DMEM with 10% horse serum) and allowed to incubate at 37°C, 5% CO2, and 95% relative humidity for 24 h until a semi-confluent monolayer of cells was formed. Cell media was replaced with the respective material extract for 24 h before being stained using a LIVE/DEAD Cytotoxicity kit for mammalian cells (Thermo Fisher, L3324) using manufacturer protocol. Briefly, Cells were washed three times with sterile PBS and incubated at 37°C with 2 μM Calcein AM (CaAM) and 4 μM Ethidium Homodimer (EthD-1) for 15 min. CaAM dye stained the cytoplasm of live cells while EthD-1 stained the nucleus of apoptotic cells. 2 × 2 field stitched fluorescent images (10x objective) were taken in each well using an inverted microscope (Nikon Ti eclipse).
Live/dead cell counts were quantified using ImageJ (NIH). Briefly, images were treated with a 2.0 Gaussian Blur then automatically counted based on local intensity maxima. Further analysis using a MATLAB program identified cells that exhibited both live and dead stains based on cell-to-cell proximity through and were removed from the live count. Cell viability percentage was defined as the ratio of live cells to the total number of cells. Cell viability percentages reported were normalized to the negative control.
Results
Synthesis of Ester Free Monomer
Trifunctional thiol (TTTSH) was synthesized by radical addition reaction between TATATO and thioacetic acid followed by hydrolysis in acidic media. Column chromatography was applied to remove byproducts. Yield: 30 g (71%). 1H NMR, 13C NMR and FTIR, support the successful synthesis (detailed plots are shown in SI, Supplementary Figures 1–3). 1H NMR [600 MHz, CDCl3, δ (ppm)]: 3.98 (t, J = 7 Hz, 6H, -N-CH2-CH2-), 2.54 (dt, J = 7 Hz/ J = 8 Hz, -CH2-SH), 1.94 (quintet, J = 7 Hz, 6H, -CH2-CH2-CH2-), 1.52 (t, 3H, J = 8 Hz, -SH).13C NMR [150 MHz, CDCl3, δ (ppm)]: 149.03(–C = O), 41.84 (–NCH2–), 31.87 (–CH2–), 21.95 (–CH2SH). FT-IR (cm−1): 2962, 2933, 2854, 2566 (νS−H), 1671 (νC = O), 1454, 1423, 1373, 1334, 1288, 1230, 759.
Softening Effect on Pristine SMPs
Our aim was to synthesize an ester free SMP formulation with similar softening properties as previously used ester-containing thiol-ene and thiol-ene/acrylate polymer compositions. In order to mimic the effect of body fluids on mechanical properties of the polymer SMP-A, dynamic mechanical analysis was performed in dry and soaked conditions (Figure 2A). The glass transition temperature (Tg) and storage modulus (E') in the glassy and rubbery state was tuned to be similar to the previously used SMP-B (Figure 2B). Soaked conditions were achieved by immersing the polymers in phosphate buffered saline (PBS) at 37°C and monitoring the storage modulus loss until the modulus no longer decreases. We determined that the composition consisting of TTTSH/DDT-TATATO = 0.3/0.2–0.5 had comparable values. Soaking in PBS led to a modulus decrease for SMP-A from 1,020 ± 67 MPa at room temperature (23°C) to 22.1 ± 0.3 MPa after 20 min at 37°C, while the modulus of SMP-B dropped from 2,187 ± 98 MPa to 28.8 ± 0.4 MPa. After soaking in PBS, the peaks of loss modulus and tan (delta) which show the glass transition temperature of both SMPs decreased by 12–14°C compared to the dry values (Figure 2). For SMP-A, Tg dropped from 46.3 ± 0.7 to 34.6 ± 0.8°C and for the SMP-B, fell from 47.8 ± 0.4 to 33.0 ± 0.6°C.
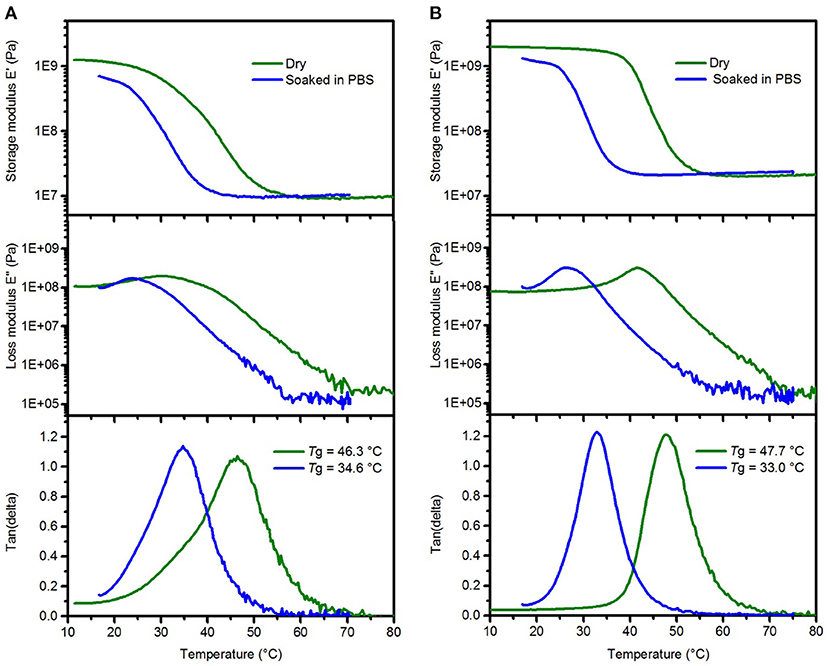
Figure 2. Representative DMA data of SMP-A (A) and SMP-B (B) showing the changes in the storage modulus (top), loss modulus (center), and tan delta (bottom) after softening in PBS, respectively.
Accelerating Aging Test in PBS at 75°C
Chronic implantable bioelectronic devices must consist of stable substrate materials in biological environment to enable operation for many years in vivo (Lyu and Untereker, 2009). To compare the hydrolytic stability of the sample's network in physiological conditions, aging in PBS was performed. In order to accelerate the aging process, the temperature was increased to 75°C. The effect of the aging conditions on mass change (Figure 3) and viscoelastic behavior (Figure 4) of the samples were then assessed. As shown in Figure 3A, the SMP-B was stable for nearly 4 weeks, but thereafter began to continually lose mass until the test was stopped. After 8 weeks at elevated temperature, SMP-B lost 14.7 ± 0.9% of its original mass. On the other hand, SMP-A exhibited no weight loss. A similar trend is seen in Figure 3B, where SMP-A displayed no change in thickness over the testing period, whereas SMP-B began to thin after 5 weeks. At the end of 8 weeks SMP-B lost 9.8 ± 1.6% of its original thickness.
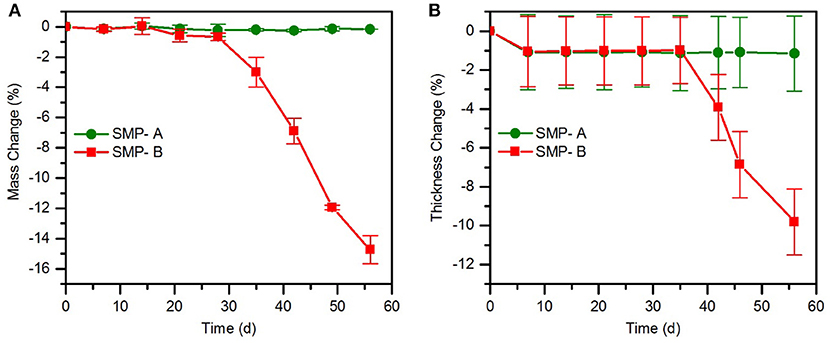
Figure 3. Mass change (A) and thickness change (B) of SMP-A and SMP-B after various aging times in PBS at 75°C.
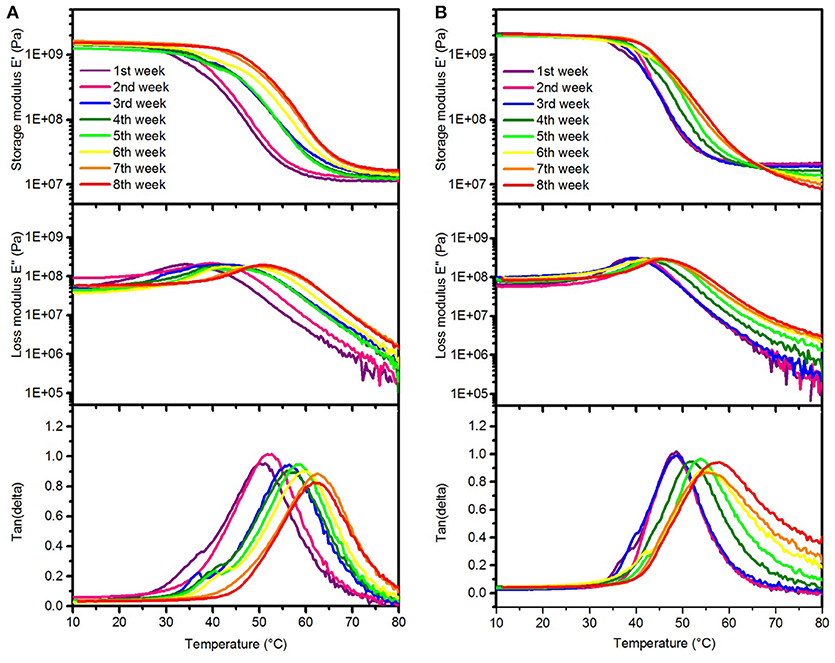
Figure 4. Representative DMA measurements of SMP-A (A) and SMP-B (B) showing changes in the storage modulus (top), loss modulus (center), and tan delta (bottom) after various aging times in PBS at 75°C, respectively.
The DMA data of the SMPs aged in PBS indicates that Tg increased each week, which can be seen in the peak shifts of tan delta and the loss modulus, respectively. As seen in Figure 4, the Tgfor both SMP-A and SMP-B displayed higher glass transition temperatures after soaking in PBS at 75°C (shifting from 47 to~61°C). Figure 4B indicates that rubbery modulus of SMP-B decreases gradually and tan delta peak is getting wider and asymmetric with increasing aging time. On the other hand SMP-A does not show any changes in the shape of graphs.
Accelerated Aging Test in 1 M NaOH Solution
The effect of harsh conditions (1.0 M NaOH, 37°C) on our two different polymers (SMP-A and B) was investigated. Mass and thickness changes (Figure 5) and mechanical properties (Figure 6) of the polymers were compared to the initial pristine and dry polymers. Figure 5A shows the change in polymer mass over time. Over the course of the 4 weeks investigation, SMP-A had no appreciable loss of mass, whereas SMP-B showed remarkable mass change during 4 weeks with a final mass loss of 38.7 ± 0.3%. In addition to the weight loss study, the thickness of each polymer was monitored during the aging. Figure 5B indicates significant thickness loss (42.1 ± 1.7%) for SMP-B over the course of the aging test, while SMP-A was stable.
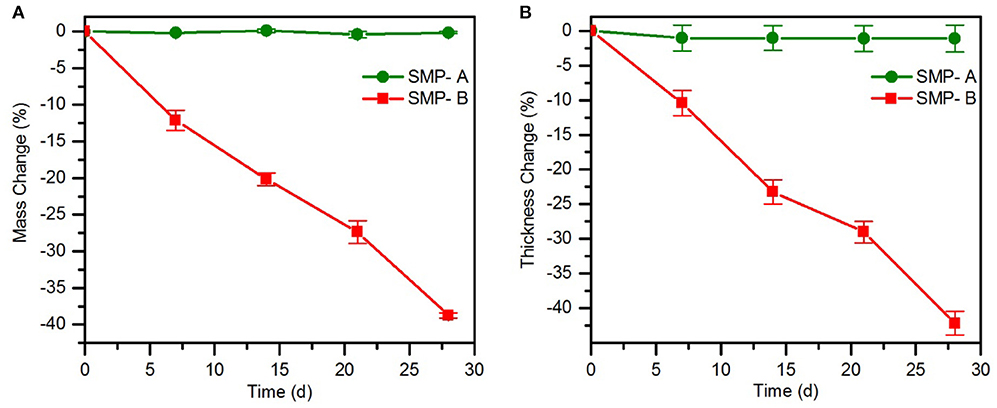
Figure 5. (A) Mass change of SMP-A and SMP-B in 1 M NaOH with (B) corresponding thickness loss.
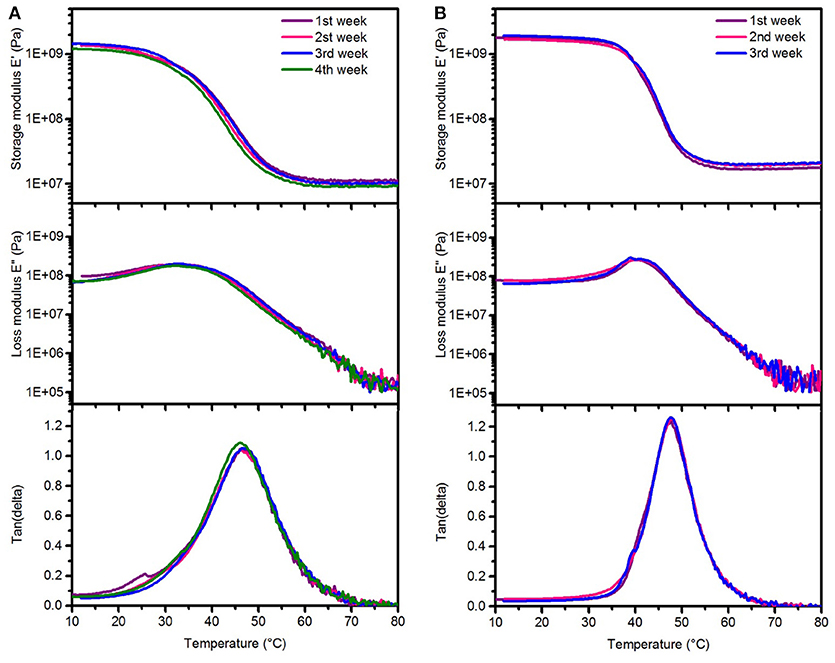
Figure 6. Representative dynamic mechanical analysis after aging test in 1 M NaOH solution showing changes in the storage modulus (top), loss modulus (center), and tan delta (bottom) after various aging times at 37°C (A) SMP-A (B) SMP-B.
Dynamic mechanical analysis (DMA) was performed to investigate the stability of thermomechanical properties of both SMPs after aging in NaOH (Figure 6). It can be seen that there are no changes in the profiles of the storage/loss moduli, and tan delta. We could not measure the thermomechanical properties of SMP-B after 4 weeks of aging in NaOH, since the polymers were already too degraded and the specimen were brittle and ruptured (see Supplementary Figure 4 in SI). These findings are in line with the weight loss and thickness measurements, which also revealed drastic changes. Thermal gravimetric analysis (TGA) was performed in order to compare thermal stability of materials before and after the aging study in NaOH and PBS (Figure 7). It can be seen in Figure 7A that SMP-A (top) was not affected by the aging study in both media and degradation temperature and mass loss rate did not change. However, SMP-B (bottom) displays a slightly lower onset in the decomposition temperature after 4 weeks in NaOH (from 320 to 300°C) and even larger shift (from 320 to 250°C) after 8 weeks in PBS media. Figure 7B displays a more detailed view on the aging effects of 75°C PBS on SMP-B. The onset of the decomposition temperature shifts downward about 8–10°C per week. In addition, the mass loss rate pattern changes over time, in detail the shoulder at higher temperature diminishes.
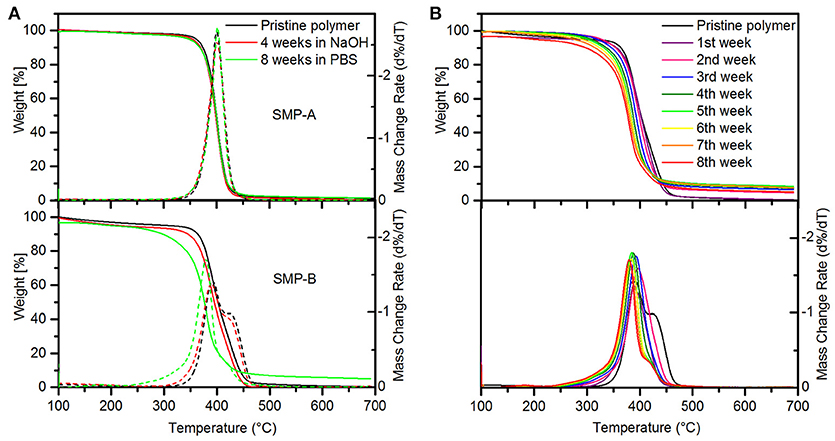
Figure 7. Thermal gravimetric analysis of SMPs. (A) Showing weight loss and mass change rate of SMP-A (top) and SMP-B (bottom) after 4 weeks in 1 M NaOH solution at 37°C (red line) and 8 weeks in PBS at 75°C (green line). (B) Weight loss (top) and mass change rate (bottom) of SMP-B in PBS over 8 weeks.
Cytotoxicity Test
To evaluate the cytotoxicity of SMP-A in vitro, we carried out live/dead assays based on material extract treatments in accordance with ISO protocol 10993-5. After fibroblasts were incubated for 24 h in the material extract, cell viability percentages were calculated and normalized to the negative control (Figure 8). SMP A at 50 and 100% concentrations had normalized viability percentages of 97.8 ± 0.8% (mean ± SEM, n = 6) and 93.6 ± 1% (mean ± SEM, n = 6), respectively. The positive control had a significantly lower viability percentage of 21.8 ± 4.7% (mean ± SEM, n = 6) (Figure 8B). Normalized viability percentages for SMP A at both 50 and 100% concentrations were both above the 70% threshold and deemed non-cytotoxic in accordance with ISO protocol 10993-5.(ISO, 2008) The in vitro cytotoxicity of SMP-B was evaluated in a different study (Black et al., 2018) and is therefore not shown detailed herein. The other study revealed that SMP-B (which was named FS SMP in the other study) had normalized viability percentages of 99.1 ± 0.7 % in the case of NCTC fibroblasts.
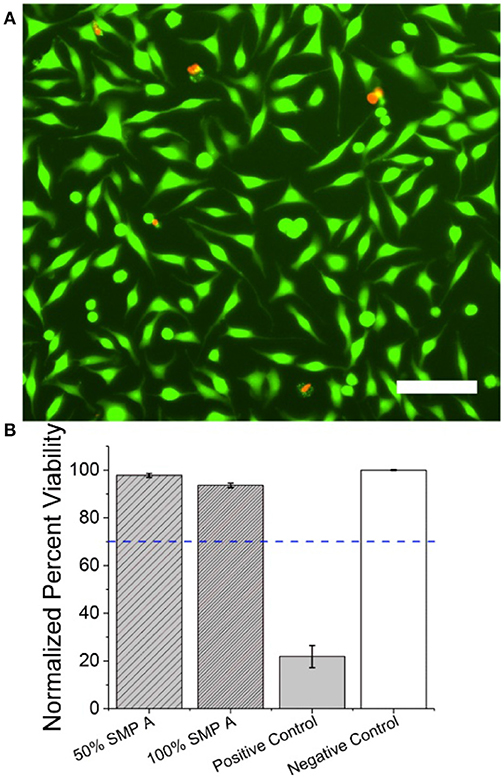
Figure 8. SMP-A Cytotoxicity assays using NCTC fibroblasts (A) Fluorescent image of NCTC fibroblasts stained with CaAM (green) and EthD-1 (red). Green cells represent live cells while red cells represent apoptotic cells. Scale bar represents 100 μ m (B) Mean normalized cell viability percentage of SMP A at 50 and 100% concentrations, positive control, and negative control. Blue dotted line represents the 70% threshold based on ISO protocols (ISO, 2008) needed to pass for non-cytotoxic materials.
Discussion
The focus of this study was to compare the stability of two types of SMPs that are applicable for flexible bioelectronic devices. Finding the appropriate substrate for the use in self-softening bioelectronic devices requires an understanding of the chemical structure and composition of the monomers for fine-tuning of the glass transition temperature of the final polymer. Glass transition (Tg) is critical to designing polymeric substrates for softening bioelectronic devices, since the Tg must be higher than body temperature prior to insertion for easy handling. After insertion however, a Tg higher than body temperature may cause an inflammatory response because the polymer is still in its glassy (stiff) state. Both SMP-A and SMP-B were synthesized utilizing a thiol-click polymerization mechanism and the glass transitions were tuned to be between 42 and 46°C in dry conditions and around 34°C when immersed in PBS. The mechanical properties of the SMPs change in the aqueous environments due to the plasticization effect of water molecules on polymer films. The storage modulus E′ decreased significantly after 25 and 10 min immersion in PBS at 37°C, for SMP-A and SMP-B, respectively. Therefore, the glass transition in dry condition is high enough for handling during insertion and low enough to minimize an inflammatory response under physiological conditions. After finding the proper composition for both SMPs, the polymers were evaluated for long-term stability in two different media; PBS to mimic the aqueous environment in vivo at elevated temperature (75°C) and a harsher alkaline solution (NaOH) at 37°C. Afterward, the weight loss and thermomechanical properties of the SMPs were investigated.
The weight loss and thickness loss data of polymers in PBS (Figure 3) indicates that SMP-B is stable until the fourth week but thereafter begins to continually lose weight and thickness up to 15 and 10%, respectively. According to equation 1, aging test at 75°C for 1 week is equivalent to 14 weeks at body temperature. Therefore, SMP-B is expected to start to degrade after about 56 weeks (14 months) under physiological conditions. While this time span might be long enough for many chronic and sub-chronic studies on animals, which usually last 3 months to a year, it would not be sufficient for long-term chronic applications, which may take several years to decades. In contrast, SMP-A data shows that ester-free polymers are stable under these conditions until the end of this study, which was 8 weeks (projected to ~26 months under physiological conditions) without any signs of degradation. Harsh conditions (1 M NaOH) were used to further accelerate aging, which will be discussed later.
DMA data (Figure 4) reveals that the glass transition temperature of both polymers shifts to a higher temperature with increasing aging time, which is indicated by shifted loss modulus and tan delta peaks. In order to investigate whether this effect is due to the PBS or due to the relatively high temperature (this effect was not seen upon aging on NaOH at 37°C), additional experiments were performed. We aged SMP-A samples in de-ionized (DI) water at 75°C for up to 3 weeks, respectively, before DMA measurements were conducted and compared to the aging study in PBS at the same temperature. DMA data (shown in SI, Supplementary Figure 5) revealed that the samples aged in water showed similar shifting in Tg, which shows that the elevated temperature causes annealing effects. We do not expect to see such effects at body temperature. In addition, the DMA data shows that after 4 weeks of aging test in PBS, the rubbery storage modulus started to decrease and tan delta got wider which means the SMP-B started to lose the crosslink density. Since accelerating aging test was performed at 75°C (above the glass transition of the polymer), the mobility of polymer chains and diffusion of water molecules were increased. Therefore, the water migration into the polymer was faster than the reaction rate of the hydrolysis, which leads to bulk degradation (Lyu and Untereker, 2009). TGA data (Figure 7B) also confirms that at higher temperature water molecules can diffuse into the polymer structure and hydrolize ester functional groups. That causes a loss in the crosslink density of polymer. With that, the thermal stability of the polymer was reduced as shown by decreased decomposition temperatures. The diminishing shoulder at higher temperatures can be attributed to the fact that the polymer network got already broken down by hydrolysis. Fragments of the hydrolyzed TMICN and TMTMP monomers have already left the polymeric network and therefore do no longer contribute to the thermal decomposition profile of the polymer. To further investigate the degradation of the polymers, gas chromatography-mass spectroscopy (GS-MS) was applied on the PBS solution after aging. GC-MS data (shown in SI, Supplementary Figure 7) indicates that tris(hydroxymethyl)propane was released from SMP-B. In contrast, however, SMP-A did not reveal any degradation products in the PBS after aging. That confirms our assumption that the ester-containing SMP-B undergoes hydrolysis since tris(hydroxymethyl)propane is a fragment of the monomer TMTMP, which used for the synthesis of this polymer (Supplementary Scheme 1).
Figure 5 shows the weight loss and thickness loss of SMP-A and SMP-B in NaOH solution. According to the graphs, SMP-A, which does not contain ester groups, is completely stable and the weight and thickness did not change over the course of the aging study. On the other hand, ester-group containing SMP-B started to hydrolize which leads to a 38% and 42% decrease in weight and thickness, respectively after 4 weeks. It was seen that SMP-B had a maximum weight loss of 15% after 8 weeks in PBS at 75°C, which was approximately the same as for aging in NaOH for 9 days. Therefore, we assume that the aging in NaOH at 37°C is roughly six times faster than in PBS at 75°C. Based on the equation 1, 1 week in PBS at 75°C is equal to 14 weeks at 37°C. Taking both considerations into account, we estimate that 4 weeks degradation in NaOH at 37°C is equal to 24 weeks at 75°C. Therefore, it could be concluded that SMP-A is projected to be completely stable for at least 7 years under physiological conditions.
The DMA data of SMP-A and SMP-B after aging in NaOH (Figure 6) indicates that Tg, loss and storage moduli of SMP-A are completely conserved, while SMP-B degrades too rapidly for data collection after 4 weeks. All of the SMP-B samples tore apart during testing before obtaining a glass transition temperature (see Supplementary Figure 4 in SI). Since salts and ions have low solubility in polymer chains, hydroxide ions did not diffuse into the polymer structure and hydrolysis took place on the surface (Lyu and Untereker, 2009). Therefore, erosion of the polymer occurred from the outside to the inside of the film. Another finding was, that in contrast to the aging in PBS, the rubbery modulus of SMP-B did not decrease. That indicates that there are no changes in the polymeric network and the crosslink density. These findings support the hypothesis that aging in NaOH follows surface erosion rather than bulk degradation. We have also noticed, that the aging at 37°C was not affecting the Tg of the polymers, which indicates that no annealing took place at this temperature. Additionally, we have performed ATR-FTIR of polymers SMP-A and SMP-B before and after both aging scenarios (Supplementary Figure 8 in SI). SMP-A shows as expected no changes in surface chemistries after both aging studies. SMP-B however has shown only minor changes after 4 weeks in NaOH, wheras it shows more distinct changes after aging for 8 weeks in PBS. It can be seen for example a broad peak appearing between 2,500 and 3,500 cm−1, which can be assigned to the hydroxyl (OH) and carboxyl (OH) streching vibration signatures, which is due to hydrolized polymer fragments. These measurements support the bulk vs. surface erosion theory further. Even if ATR is a surface method, the penetration depth of the IR beam is between 0.5 and 5 μm into the bulk of the sample. Therefore, the bulk degraded sample (PBS aged) shows a higher number of degraded moieties per volume measured.
Thermogravimetric analysis graph (Figure 7) displays SMP data before and after aging in NaOH. Figure 7A indicates that SMP-A is completely stable, while for SMP-B the onset of decomposition shifts about 30–40°C toward lower temperatures after aging. In general, polymers with decomposition temperatures higher than ~300°C are favorable for the fabrication of bioelectronic devices. The micro-fabrication of such devices uses processes such as photolithography, metal deposition, reactive ion etching, and chemical etching.
It should be noted that long term implants are not only subjected to hydrolytic degradation in an in vivo environment. Takmakov et al. (2015) pointed out that hydrolytic degradation alone may not adequately capture the aggressive chemical environment that is created by activated immune cells, which release digestive enzymes and reactive oxygen species (ROS). They have developed an in vitro system to simulate degradation of neural implants that can occur in a stressful environment by using hydrogen peroxide to mimic the effect of ROS generation during the brain's injury response. While this presents an important part of real live conditions, the focus of the present study is on the hydrolytic degradation only, because we wanted to demonstrate the improved durability of the ester free SMP-A against the ester containing SMP-B. In future studies however, we will perform experiments to evaluate the durability of thiol-ene and thiol-ene/acrylate formulations against oxidative species such as hydrogen peroxide. We do not expect to see any differences between ester containing and ester free versions because the ester groups are reported in literature to be not susceptible for oxidation (Lyu and Untereker, 2009). The sulfide groups however, may undergo oxidation to sulfoxides and sulfones using 30% aqueous hydrogen peroxide (Jeyakumar and Chand, 2006; Gregori et al., 2008).
The ultimate goal is to utilize the hydrolytically stable SMP-A as a substrate for neural interfaces and therefore have an alternative to the currently used softeing SMP versions (Simon et al., 2017; Garcia-Sandoval et al., 2018). To validate that the polymer is compatible with microfabrication processes, SMP-A was spin-coated on silicon wafers and subjected to photolithography as previously decribed (Ecker et al., 2017). Briefly, the polymer was covered by a silicon nitride (SiN) hard mask before it was coated with positive photoresist. Afterward, a photomask was used to pattern the pacifier probe through the photoresist by exposure to UV light. Next, the probe was subjected to dry etch with SF6 to remove unpatterned hard mask. Subsequently, oxygen plasma was applied to remove excess polymer and patterned photoresist. Finally, the sample was etched with hydrofluoric acid to remove SiN that was on the SMP (Garcia-Sandoval et al., 2018). The micro-fabricated pacifier probes were also used for the in vitro cytotoxicity test.
Another important aspect is that materials for biomedical applications should be biocompatible and sterilizable. The materials need to be able to show a desirable performance with respect to a specific medical treatment, without any unwanted effects on the tissues (Johnson and Shiraishi, 2014; Huang et al., 2017). One of the tests for evaluating prospective biocompatibility of biomedical devices before clinical survey is in vitro cytotoxicity (Johnson and Shiraishi, 2014). To investigate the potential toxicity without affecting the mechanical or chemical properties of the polymer, cytotoxicity tests were performed on pacifier probes by incubating fibroblasts with extract of SMP-A for 24 h. Figure 8 confirms that the extract of the polymer at both concentrations is reliable and the polymer is expected to be safe for biomedical applications. We have seen, that SMP-B produces some water soluble byproducts after hydrolysis. These byproducts may undergo some deleterious reactions including oxidation in vivo. Therefore, the degraded polymer may not prove to be as biocompatible as the original polymer. Additional studies, such as functional neurotoxicity or cytotoxicity assays using primary cortical neurons, (Charkhkar et al., 2014) need to be performed to further investigate potential harm of degradation product. On the other hand, SMP-A sustained even in harsh conditions without any signs of degradation and therefore is less of a biocompatibility concern. Biomedical devices need to be sterilized properly for in vivo studies. To inquire the effect of sterilization on mechanical properties, SMP-A was subjected to sterilization with ethylene oxide (EtO) as previously described (Ecker et al., 2017). Since the sterilization with EtO was performed at a low temperature, it has negligible effects on the mechanical properties of SMPs. DMA data (shown in SI, Supplementary Figure 6) before and after sterilization shows that storage modulus and glass transition did not change remarkably.
Conclusion
Ester free (SMP-A) and ester containing (SMP-B) polymers with comparable thermomechanical properties and softening capabilities were prepared and their stability in vitro was evaluated. Accelerated aging tests were performed on both polymers and their stability was compared. According to the test results, ester free polymers are projected to be stable for at least 7 years in the biological environment, whereas the ester containing polymer should be stable for approximately 1 year. Therefore, the new ester-free polymer (SMP-A) is a good candidate for future devices. It was stable under the tested conditions and is therefore much more reliable and robust than SMP-B, but still biocompatible. Furthermore, fabrication of pacifier probes demonstrated that SMP-A was stable in cleanroom processes and conserved its mechanical properties. Next steps will include the fabrication of fully functional neural interfaces and their testing in vitro as well as in vivo.
Author Contributions
SH and ME: Conceptualization; SH, RR, BB, and AS: Methodology; SH, RR, BB, and ME: Formal Analysis; SH, RR, and BB: Data Curation; SH and ME: Writing–Original Draft Preparation; SH and ME: Writing–Review and Editing; SH, RR, and BB: Visualization; ME: Supervision; ME: Project Administration; WV and JP: Resources; JP and WV: Funding Acquisition; All authors approved the final version to be published and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Center for Engineering Innovation and in parts by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award No. W81XWH-15-1-0607. Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. Additionally, the contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. The authors want to thank Dr. Taylor Ware for allowing us to use the environmental DMA.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmats.2018.00066/full#supplementary-material
References
ASTM (2018). Standard Guide for Accelerated Aging of Sterile Barrier Systems for Medical Devices. ASTM F1980–07.
Baer, G., Wilson, T. S., Matthews, D. L., Maitland, D. (2007). Shape-memory behavior of thermally stimulated polyurethane for medical applications. J. Appl. Polym. Sci. 103, 3882–3892. doi: 10.1002/app.25567
Barrett, R., Francis, W., Abrahamson, E., Lake, M. S., and Scherbarth, M. (2006). “Qualification of elastic memory composite hinges for spaceflight applications,” in 47th AIAA/ASME/ASCE/AHS/ASC Structures, Structural Dynamics, and Materials Conference (Newport, RI). doi: 10.2514/6.2006-2039
Baudis, S., Behl, M., and Lendlein, A. (2014). Smart polymers for biomedical applications. Macromol. Chem. Phys. 215, 2399–2402. doi: 10.1002/macp.201400561
Black, B. J., Ecker, M., Stiller, A., Rihani, R., Danda, V. R., Reed I., et al. (2018). In vitro compatibility testing of thiol-ene/acrylate-based shape memory polymers for use in implantable neural interfaces. J. Biomed. Mater. Res. A doi: 10.1002/jbm.a.36478. [Epub ahead of print].
Buckley, P. R., McKinley, G. H., Wilson, T. S., Small, W., Benett W.J, Bearinger J.P., et al. (2006). Inductively heated shape memory polymer for the magnetic actuation of medical devices. IEEE Transac. Biomed. Eng. 53, 2075–2083. doi: 10.1109/TBME.2006.877113
Charkhkar, H., Frewin, C., Nezafati, M., Knaack, G. L., Peixoto, N., Saddow S. E., et al. (2014). Use of cortical neuronal networks for in vitro material biocompatibility testing. Biosensors Bioelectron. 53, 316–323. doi: 10.1016/j.bios.2013.10.002
Dietsch, B., and Tong, T. (2007). A review - Features and benefits of shape memory polymers (SMPs). J. Adv. Mater. 39, 3–12. Available online at: http://apps.webofknowledge.com/full_record.do?product=WOS&search_mode=GeneralSearch&qid=8&SID=8BSUhmnqWxNvZmIqxlx&page=1&doc=1
Do, D. H., Ecker, M., and Voit, W. E. (2017). Characterization of a Thiol-Ene/acrylate-based polymer for neuroprosthetic implants. ACS Omega 2, 4604–4611. doi: 10.1021/acsomega.7b00834
Ecker, M., Danda, V., Shoffstall, A. J., Mahmood, S. F., Joshi-Imre, A., Frewin C. L., et al. (2017). Sterilization of Thiol-ene/Acrylate based shape memory polymers for biomedical applications. Macromol. Mater. Eng. 302:1600331. doi: 10.1002/mame.201600331
Ecker, M., and Pretsch, T. (2013). Durability of switchable QR code carriers under hydrolytic and photolytic conditions. Smart Mater. Struct. 22:094005. doi: 10.1088/0964-1726/22/9/094005
Ecker, M., and Pretsch, T. (2014a). Multifunctional poly(ester urethane) laminates with encoded information. RSC Adv. 4, 286–292. doi: 10.1039/C3RA45651J
Ecker, M., and Pretsch, T. (2014b). Novel design approaches for multifunctional information carriers. RSC Adv. 4, 46680–46688. doi: 10.1039/C4RA08977D
Feninat, F. E., Laroche, G., Fiset, M., and Mantovani, D. (2002). Shape Memory materials for biomedical applications. Adv. Eng. Mater. 4, 91–104. doi: 10.1002/1527-2648(200203)4:3<91::AID-ADEM91>3.0.CO;2-B
Garcia-Sandoval, A., Pal, A., Mishra, A. M., Sherman, S., Parikh, A., Joshi-Imre A., et al. (2018). Chronic softening spinal cord stimulation arrays. J. Neural Eng. 15:045002. doi: 10.1088/1741-2552/aab90d
Gregori, F., Nobili, I., Bigi, F., Maggi, R., Predieri, G., and Sartori, G. (2008). Selective oxidation of sulfides to sulfoxides and sulfones using 30% aqueous hydrogen peroxide and silica-vanadia catalyst. J. Mol. Catal. A Chem. 286, 124–7. doi: 10.1016/j.molcata.2008.02.004
Hager, M. D., Bode, S., Weber, C., and Schubert, U. S. (2015). Shape memory polymers: past, present and future developments. Prog. Polym. Sci. 49–50, 3–33. doi: 10.1016/j.progpolymsci.2015.04.002
Hemmerich, K. J. (1998). General aging theory and simplified protocol for accelerated aging of medical devices. Medi. Plastic Biomater. 5, 16–23.
Hu, J., and Chen, S. (2010). A review of actively moving polymers in textile applications. J. Mater. Chem. 20, 3346–3355. doi: 10.1039/b922872a
Hu, J., Meng, H., Li, G., and Ibekwe, S. I. (2012a). A review of stimuli-responsive polymers for smart textile applications. Smart Mater. Struct. 21:053001. doi: 10.1088/0964-1726/21/5/053001
Hu, J., Zhu, Y., Huang, H., and Lu, J. (2012b). Recent advances in shape memory polymers: structure, mechanism, functionality, modeling and applications. Prog. Polym. Sci. 37, 1720–1763. doi: 10.1016/j.progpolymsci.2012.06.001
Huang, Y., Zhou, J., Hakamivala, A., Wu, J., Hong, Y., Borrelli, J., et al. (2017). An optical probe for detecting chondrocyte apoptosis in response to mechanical injury. Sci. Rep. 7:10906. doi: 10.1038/s41598-017-10653-y
Immergut, E. H., and Mark, H. F. (1965). Principles of plasticization. plasticization and plasticizer processes. Am. Chem. Soc. 48, 1–26. doi: 10.1021/ba-1965-0048.ch001
Ishizawa, J., Imagawa, K., Minami, S., Hayashi, S., and Miwa, N. (2003). “Research on Application of Shape Memory Polymers to Space Inflatable Systems,” in Proceeding of the 7th International Symposium on Artificial Intelligence, Robotics and Automation in Space: i-SAIRAS (Nara).
ISO (2008). ISO 10993-5 Biological Valuation of Medical Devices, Part 5: Tests for in vitro Cytotoxicity. Geneva: International Organization for Standardization.
Jeyakumar, K., and Chand, D. K. (2006). Selective oxidation of sulfides to sulfoxides and sulfones at room temperature using H2O2 and a Mo(VI) salt as catalyst. Tetrahedron Lett. 47, 4573–4576. doi: 10.1016/j.tetlet.2006.04.153
Johnson, A. and Shiraishi, T. (2014). “Biocompatibility of precious metals for medical applications,” in Precious Metals for Biomedical Applications, eds N. Baltzer and T. Copponnex (Woodhead Publishing), 37–55. doi: 10.1533/9780857099051.1.37
Kulshrestha, A. S., and Mahapatro, A. (2008). Polymers for biomedical applications. Polym. Biomed. Appl. 977, 1–7. doi: 10.1021/bk-2008-0977.ch001
Lecomte, A., Degache, A., Descamps, E., Dahan, L., and Bergaud, C. (2017). In vitro and in vivo biostability assessment of chronically-implanted Parylene C neural sensors. Sensors Actuators B Chemical 251(Suppl. C):1001–8. doi: 10.1016/j.snb.2017.05.057
Lendlein, A., and Behl, M. (2008). “Shape-memory polymers for biomedical applications,” in CIMTEC 2008 - Proceedings of the 3rd International Conference on Smart Materials, Structures and Systems - Smart Materials and Micro/Nanosystems (Acireale), 96–102.
Liu, C., Qin, H., and Mather, P. T. (2007). Review of progress in shape-memory polymers. J. Mater. Chem. 17, 1543–1558. doi: 10.1039/b615954k
Lundberg, P., Bruin, A., Klijnstra, J. W., Nystrom, A. M., Johansson M., Malkoch, M., et al. (2010). Poly(ethylene glycol)-based thiol-ene hydrogel coatings-curing chemistry, aqueous stability, and potential marine antifouling applications. ACS Appl. Mater. Interfaces 2, 903–912. doi: 10.1021/am900875g
Lyu, S., and Untereker, D. (2009). Degradability of polymers for implantable biomedical devices. Int. J. Mol. Sci. 10:4033. doi: 10.3390/ijms10094033
Mather, P. T., Luo, X., and Rousseau, I. A. (2009). Shape memory polymer research. Annu. Rev. Mater. Res. 39, 445–471. doi: 10.1146/annurev-matsci-082908-145419
Matilla, H. R. (2006). Intelligent Textiles and Clothing. Cambridge, UK: Woodhead Publishing in Textiles.
Pretsch, T., Ecker, M., Schildhauer, M., and Maskos, M. (2012). Switchable information carriers based on shape memory polymer. J. Mater. Chem. 22, 7757–7766. doi: 10.1039/c2jm16204k
Reit, R., Zamorano, D., Parker, S., Simon, D., Lund, B., Voit, W., et al. (2015). Hydrolytically stable thiol–ene networks for flexible bioelectronics. ACS Appl. Mater. Interfaces 7, 28673–28681. doi: 10.1021/acsami.5b10593
Rory Barrett, W. F., Erik, A., Mark Lake, S., and Mark, S. (2006). “Qualification of elastic memory composite hinges for spaceflight applications,”in 47nd IAA/ASME/ASCE/AHS/ASC SDM Conference. (Newport, RI: AIAA-2006-2039).
Rubehn, B., and Stieglitz, T. (2010). In vitro evaluation of the long-term stability of polyimide as a material for neural implants. Biomaterials 31, 3449–3458. doi: 10.1016/j.biomaterials.2010.01.053
Ryu, S. I., and Shenoy, K. V. (2009). Human cortical prostheses: lost in translation? Neurosurg. Focus 27:E5. doi: 10.3171/2009.4.FOCUS0987
Shoffstall, A. J., Srinivasan, S., Willis, M., Stiller, A. M., Ecker, M., Voit W. E., et al. (2018). A mosquito inspired strategy to implant microprobes into the brain. Sci. Rep. 8:122. doi: 10.1038/s41598-017-18522-4
Simon, D. M., Charkhkar, H. C., St John, Rajendran, S., Kang, T., Reit, R., et al. (2017). Design and demonstration of an intracortical probe technology with tunable modulus. J. Biomed. Mater. Res. A 105, 159–168. doi: 10.1002/jbm.a.35896
Singhal, P., Boyle, A., Brooks, M. L., Infanger, S., Letts, S., Small W., et al. (2013). Controlling the actuation rate of low-density shape-memory polymer foams in water. Macromol. Chem. Phys. 214, 1204–1214. doi: 10.1002/macp.201200342
Takmakov, P., Ruda, K., Scott Phillips, K., Isayeva, I. S., Krauthamer, V., and Welle C. G., (2015). Rapid evaluation of the durability of cortical neural implants using accelerated aging with reactive oxygen species. J. Neural Eng. 12:026003. doi: 10.1088/1741-2560/12/2/026003
Teo, A. J. T., Mishra, A., Park, I., Kim, J., Park, T., and Yoon, J. (2016). Polymeric biomaterials for medical implants and devices. ACS Biomater. Sci. Eng. 2, 454–472. doi: 10.1021/acsbiomaterials.5b00429
Wang, K., Strandman, S., and Zhu, X. X. (2017). A mini review: shape memory polymers for biomedical applications. Front. Chem. Sci. Eng. 11, 143–153. doi: 10.1007/s11705-017-1632-4
Ware, T., Simon, D., Hearon, K., Kang, T. H., Maitland, D., et al. (2013). Thiol-click chemistries for responsive neural interfaces. Macromol. Biosci. 13, 1640–1647. doi: 10.1002/mabi.201300272
Ware, T., Simon, D., Liu, C., Musa, T., Vasudevan, S., Sloan, A., et al. (2014). Thiol-ene/acrylate substrates for softening intracortical electrodes. J. Biomed. Mater. Res. B Appl. Biomater. 102, 1–11. doi: 10.1002/jbmb.32946
Yanju, L., Haiyang, D., Liwu, L., and Jinsong, L. (2014). Shape memory polymers and their composites in aerospace applications: a review. Smart Mater. Struct. 23:23001. doi: 10.1088/0964-1726/23/2/023001
Keywords: neural interfaces, softening behavior, accelerated aging, hydrolytic stable, shape memory polymer, thiol-ene degradation, chronic viable polymer
Citation: Hosseini SM, Rihani R, Batchelor B, Stiller AM, Pancrazio JJ, Voit WE and Ecker M (2018) Softening Shape Memory Polymer Substrates for Bioelectronic Devices With Improved Hydrolytic Stability. Front. Mater. 5:66. doi: 10.3389/fmats.2018.00066
Received: 09 August 2018; Accepted: 24 October 2018;
Published: 15 November 2018.
Edited by:
Ulrich G. Hofmann, Universitätsklinikum Freiburg, GermanyReviewed by:
Ahmed El-Fiqi, Dankook University, South KoreaAjay Devidas Padsalgikar, St. Jude Medical, United States
Copyright © 2018 Hosseini, Rihani, Batchelor, Stiller, Pancrazio, Voit and Ecker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melanie Ecker, bWVsYW5pZS5lY2tlckB1dGRhbGxhcy5lZHU=