 Jessica M. Rimsza
Jessica M. Rimsza Reese E. Jones2
Reese E. Jones2- 1Sandia National Laboratories, Geochemistry Department, Albuquerque, NM, United States
- 2Sandia National Laboratories, Mechanics of Materials Department, Livermore, CA, United States
Glassy silicates are substantially weaker when in contact with aqueous electrolyte solutions than in vacuum due to chemical interactions with preexisting cracks. To investigate this silicate weakening phenomenon, classical molecular dynamics (MD) simulations of silica fracture were performed using the bond-order based, reactive force field ReaxFF. Four different environmental conditions were investigated: vacuum, water, and two salt solutions (1M NaCl, 1M NaOH) that form relatively acidic and basic solutions, respectively. Any aqueous environment weakens the silica, with NaOH additions resulting in the largest decreases in the effective fracture toughness (eKIC) of silica or the loading rate at which the fracture begins to propagate. The basic solution leads to higher surface deprotonation, narrower radius of curvature of the crack tip, and greater weakening of the silica, compared with the more acidic environment. The results from the two different electrolyte solutions correspond to phenomena observed in experiments and provide a unique atomistic insight into how anions alter the chemical-mechanical fracture response of silica.
Introduction
The environmental impact of salt solutions on fracture has been well-established in both crystalline (Atkinson and Meredith, 1981; Dove, 1995) and glassy materials (Wiederhorn and Johnson, 1973). The subcritical fracture velocity in silicate glasses increases when in contact with both high and low pH solutions, with acidic conditions resulting in less extreme changes (Wiederhorn and Johnson, 1973). This has been attributed to two different mechanisms that cause subcritical fracture growth, as outlined by Dove (1995). The first reaction mechanism is facilitated by a water molecule, which forms a temporary hydrogen bond with the siloxane (Si-O-Si) oxygen and weakens the Si-O linkage until the bond is broken (Equation 1). This is often referred to as the acidic site, because an hydrogen from the water molecule is transferred to the oxygen in the siloxane bond during rupture (White et al., 1987). The result is the formation of two surface silanol groups through consumption of a water molecule:
where > and < indicate surface bound species. The second pathway is facilitated by an OH− ion that forms a temporary over-coordinated silicon until the Si-O bond is sufficiently stretched for Si-O bond breakage (Equation 2). This allows for the electrons to transfer from the siloxane to an adjacent oxygen, weakening the siloxane bond (White et al., 1987).
The result is the formation of one neutral (Si-OH) and one deprotonated (Si-O−) silanol group. Computationally, significant research has been performed to distinguish different mechanisms for silanol bond breakage in the presence of water (Rimola and Ugliengo, 2008; Morrow et al., 2009; Kubicki et al., 2012; Zapol et al., 2013; Kagan et al., 2014; Lowe et al., 2015; Rimsza and Du, 2015; Rimsza J. M. et al., 2016). The exact intermediates and their relative stabilities are still under debate, offering the explanation that both mechanisms may be occurring or, more generally, that a variety of reaction pathways are available (Du and Rimsza, 2017).
In both experimental (Wiederhorn and Johnson, 1973) and computational (Zapol et al., 2013) studies the response of silica to electrolytes is pH dependent. Reactions between the glass and water at the crack tip can result in localized pH's in the fracture volume, due to hydroxyl (-OH) exchange between the solution and the glass (Wiederhorn and Johnson, 1973). Therefore, when comparing different solutions, the concentration of OH− needs to be considered. Dove hypothesized, in the context of transition state theory, that the increased crack velocity in basic conditions is due to increased attempt frequency facilitated by the ability of the OH− ion to diffuse to the crack tip (Dove, 1995). This effect is assumed to dominate the small change in reaction enthalpy for the water-mediated (Dove, 1995; Equation 1) vs. the hydroxide-mediated (Dove, 1995; Equation 2) reaction. Additionally, White et al. (1987) hypothesized that the cations with OH− in their first solvation shell could keep OH− from accessing the crack tip, thereby altering the fracture mechanism. The confined geometry of a crack tip is also unusual, with the water or electrolyte solution becoming increasingly confined as the solution enters the wedge-like fracture volume. This causes challenges in analysis, since crack tips represent a small percent of the total volume of a system, and are distinctly different from more traditionally studied confined geometries, such as slit pores (Bonnaud et al., 2010; Ho et al., 2011; Bourg and Steefel, 2012).
Because crack tips are a small fraction of macroscopic systems and are difficult to analyze experimentally, simulations have been used to investigate fracture properties of silica in the presence of water. Until recently, the application of molecular dynamics (MD) simulations has been limited because few force fields have the functionality to simulate proton transport in water (Mahadevan and Garofalini, 2007) and even fewer have relevant functionality with electrolyte solutions. At the same time, applications of more accurate electronic structure methods such as density functional theory (DFT) have had difficulty simulating the large number of atoms required to encompass the farther reaching effects of fracture, such as the nanometer sized process zone (Célarié et al., 2007; Rimsza et al., 2017b). Recently, the development and subsequent parameterization of the bond order, reactive force field ReaxFF (Fogarty et al., 2010; van Duin et al., 2013; Yeon and van Duin, 2015) has allowed for investigations of electrolyte-silica interactions. Building from recent work on simulation of sodium hydroxide adsorption on silica surfaces (Rimsza et al., 2018b), as well as silica fracture in vacuum (Rimsza et al., 2017b) and in the presence of water (Rimsza et al., 2018a), we are now able to investigate how electrolyte solutions impact silica fracture with sufficient accuracy to make mechanistic statements on the role of different salt solutions. This is, to the best of our knowledge, the first study to investigate the impact of coupled stress and chemical reactivity on silica fracture in an electrolyte solution using reactive MD simulations. This closes a critical gap in our understanding of silica fracture mechanisms, that until now was beyond the scope of both experimental and computational methods.
Here, simulations of silica fracture are performed with four different environmental conditions: vacuum, water, and two different electrolyte solutions (1M NaCl, 1M NaOH). The systems are analyzed for structural features, including surface configurations, solution composition, and connectivity, as well as mechanical properties such as effective fracture toughness (eKIC). This provides evidence for differences in fracture mechanisms between NaCl and NaOH solutions, and how anions alter chemical-mechanical fracture of silicates.
Methods
Reactive Molecular Dynamic Force Field (ReaxFF)
Investigating the chemical-mechanical of silica requires a force field which both correctly estimates the mechanical properties of silica and the water/electrolyte interaction with the surface. Very few MD force fields exhibit both capabilities. These include one (Mahadevan and Garofalini, 2007) which is an adaptation of a dissociative water force field (Guillot and Guissani, 2001) and the other, a bond-order based force field (ReaxFF) (Van Duin et al., 2003; Fogarty et al., 2010; Pitman and Van Duin, 2012; Yeon and van Duin, 2015). ReaxFF allows for the dissociation and creation of chemical bonds and redistribution of charges through environmentally dependent charge equilibrium methods. Previous studies have used the Yeon (Yeon and van Duin, 2015) and the Pitman (Pitman and Van Duin, 2012) parametrizations of ReaxFF to investigate silica surface energies (Rimsza et al., 2017a; Yu et al., 2018) and the Fogarty (Fogarty et al., 2010) parameterization to calculate mechanical properties (Yu et al., 2016). For this work, the Yeon version of ReaxFF was selected because it is a reparametrization of the Fogarty version that improves the energetics of water-surface interactions (Rimsza J. M. et al., 2016), which are critical to chemical-mechanical fracture. Additionally, our previous investigations of silica fracture toughness (KIC) process zone size, and radius of curvature (Rimsza et al., 2017b) have been consistent with experiment. Some concerns have been raised surrounding the over estimation of bulk modulus of α-quartz, reported as 602 GPa by Yu et al. (2018). Elastic constants for the Yeon ReaxFF parametrization were calculated as E = 76.1 ± 0.3 GPa, K = 55.2 ± 5.2 GPa, μ = 21.8 ± 0.8 GPa, and ν = 0.32 ± 0.02 for the elastic modulus, bulk modulus, shear modulus, and Poisson's ratio, respectively. Reported error is from variation in strain rates. These values do not compare exactly with experimental results (E = 73.1 GPa, K = 36.7 GPa, μ = 31.2 GPa, and ν = 0.17) (Varshneya, 2013) but are within an order of magnitude. The elastic constant that is of primary concern is E, since the loading methodology (discussed below) is focused on mode I or pure tension. The overestimation of G in the Yeon parametrization (55.2 ± 5.2 GPa from simulation and 36.7 GPa from experiment) would be a larger concern for studies which focused on the analysis of silica in shear and compression. All simulations were performed using the LAMMPS MD code with the USER-REAXC package with the Yeon and van Duin Si/O/H ReaxFF parameterization (Aktulga et al., 2012; Yeon and van Duin, 2015) as the force field (included in Supporting Information).
Silica Structure Model
To investigate crack propagation in silica, thin, quasi-2D simulation cells (140 × 140 × 28 Å) were created through a melt-and-quench procedure. Three replicas of the silica glass were generated to enable statistical averaging. The initial cristobalite configurations contained 38,400 atoms in a 2:1 oxygen to silicon ratio. These systems were heated to 4,000 K, then held for 100 ps to melt the crystal structure under periodic boundary conditions. They were then cooled from 4,000 to 300 K at a rate of 5 K/ps, which is reported to accurately replicate bond distance and angles in glassy silica (Lane, 2015). Densities were controlled during cooling at the measured density of silica, 2.2 g/cm3 (Mozzi and Warren, 1969), through a canonical (NVT) ensemble which controls the number (N) of atoms, volume (V), and temperature (T) of the simulation with a damping coefficient of 100 time steps and a time-step of 0.5 fs. After the systems had reached 300 K, the density fluctuated in simulations that alternated isothermal-isobaric dynamics with energy minimization to achieve low energy configurations. Final silica structures had a density of 2.187 ± 0.001 g/cm3.
First a slit crack has been introduced into the silica on a half-plane by deleting bonds between atoms on opposite sides of the half-plane, then the crack was mechanically loaded by displacement of far-field atoms in a region outside a cylinder centered on the crack tip (Figure 1). This displacement field was taken from the linear elastic fracture mechanics (LEFM) continuum solution for a semi-infinite slit crack in mode I (tension) loading that is spatially varying and parameterized by KI (Lawn and Wilshaw, 1993, Equation 2.11). Prior to loading, the in-plane (x and y) boundary conditions were changed to non-periodic since periodicity is lost during loading of the system (Figure 1). Via the prescribed displacement field, a pre-existing slit crack on a halfplane was first opened to a KI of 0.05 MPa√m. Alternating minimizations and low temperature NVT relaxations were then performed to reach an equilibrium structure. Subsequently, the systems were loaded by incrementally increasing KI by 0.071 MPa√m. While holding the periphery fixed to enforce the prescribed mode I displacement, the interior region of atoms was integrated at 300 K with a 0.1 fs time step for 5 ps with NVT dynamics at each mechanical loading step. The effect of different relaxation times between each loading step was previously tested for values of 2.5, 5, and 10 ps (Rimsza et al., 2018a). It was found that changing the relaxation time did not significantly alter the eKIC values. Therefore, it appears that variation in the glassy silica structure has a greater impact on the mechanical response than the relaxation time used between loading steps. Through 100 loading steps the system was fractured with a final loading state of KI = 0.65 MPa√m. During loading, four different chemical environments (vacuum, water, 1M NaCl, and 1M NaOH) were introduced in separate simulations to investigate the role of different aqueous solution compositions on fracture properties.
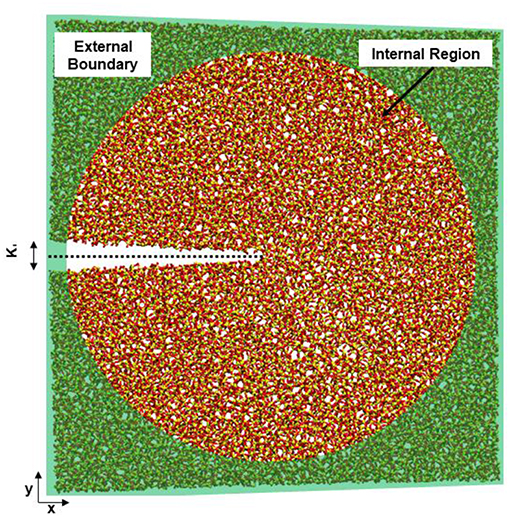
Figure 1. Schematic of the quasi-2D silica system with a slit crack, oxygen atoms are red and silicon atoms are yellow. The half plane where bonds are severed to form a slit crack is denoted by a dashed line. The boundary region (radius 3.2 nm), where the motion of the atoms is prescribed, is shaded green. In the internal cylindrical region, the atoms are free to relax to a minimum energy configuration while atoms in the boundary are fixed to the displacement prescribed by the mode I loading. The axis of the cylinder is out-of-plane and the thickness of the system is 2.8 nm.
We use effective fracture toughness (eKIC) in this paper, which is different than the KIC used in classical fracture mechanics. Classically, the KIC is the point at which a pre-existing fracture converts from subcritical to critical behavior. Due to our small system size, a single fracture event, in which a few bonds break at the fracture tip, results in fracture velocities on the order of 10 m/s. These high fracture velocities are indicative of a critical behavior, but several subsequent events would need to occur to produce an experimentally measurable change in fracture length. Therefore, we are defining the loading at which the first fracture event occurs as eKIC to highlight that it is (1) defined differently than the KIC in classical mechanics and (2) is likely to be an underestimation of the experimentally-derived KIC values, which do not have the atomistic level accuracy exhibited in these simulations. Also, as we will see, eKIC is impacted by the chemical environment unlike the strict definition of KIC defined as an intrinsic material property in fracture mechanics text books (Lawn and Wilshaw, 1993).
For the vacuum systems, no water was introduced, and the internal surfaces stay unhydroxylated during the simulation. When aqueous solutions (water, 1M NaCl, 1M NaOH) are introduced it is necessary to add water to the fracture volume as the crack opens, so that water is available to wet the surface as the system is loaded. A grand canonical Monte Carlo (GCMC) method was used to add water into the crack during the simulation. The GCMC input used a chemical potential of −250 kcal/mol, corresponding to a target density of liquid water, and 10,000 attempts were made to insert water molecules at the beginning of every loading step. To avoid inserting water directly into the glass matrix, a modified GCMC method was used that limited insertion to within 3 Å of an existing water molecule. To maintain stable NaCl and NaOH concentrations, one NaCl or NaOH was added following the addition of water. Each NaCl or NaOH was added as a molecule at the widest point of the fracture, rather than inserted near the crack tip, to emulate the diffusion of ions from the bulk fluid. A reflective wall at the crack opening was used to keep the solution inside the fracture without adding external pressure.
After insertion of the water and NaCl/NaOH molecules, 5 ps of NVT dynamics at 300 K was performed to allow for equilibration of the system. The relaxation time for water-silica interactions was investigated by performing several fracture simulations with shorter (2.5 ps) and longer (10 ps) periods between loading steps. Previous studies have found that a range of relaxation times resulted in overlapping silica energy curves and minimal differences in the results (Rimsza et al., 2018a), indicating that variation in glass structure dominate differences in fracture due to simulated relaxation times.
Due to the high variation in the glassy silica structure, we used 12 replicas of the pre-cracked systems in each of the four loading conditions (48 simulations in total). These 12 replicas were constructed by rotating the three original silica systems by 0, 90,180, and 270° in plane and then introducing slit cracks on the -x half-plane. The results from these 12 systems are reported as the average and standard error unless otherwise noted.
As part of the analysis of the systems, the radius of curvature of the crack tip was calculated by fitting the locations of the surface atoms to a second order polynomial (y = f(x) = ax2 + bx + c) and then obtaining the crack tip radius (ρ) from the formula (Howard, 1988):
The radius of curvature has been previously calculated for silica fracture in MD simulations using this method (Rimsza et al., 2017b).
The coordination environment of the Na+ ion was analyzed to identify how the salt solution was altering the silica surface. The coordination was calculated by using interatomic distances with cut-offs: Si-O (2.3 Å), H-O (1.25 Å), Na-O (2.20 Å), O-O (1.5 Å), and H-H (1.0Å). Cut-off values were selected based on previous investigations of Na+ adsorption on silica surfaces from NaOH solutions using the ReaxFF force fields (Rimsza et al., 2018b). See Rimsza et al. (2018b) for the pair distribution functions used for cut-off selection and further discussion.
Results
Solution and Surface Chemistry
During fracture the energy balance is controlled by the energetic cost associated with creating new surfaces, namely the surface energy, as described through the Griffith criterion (Lawn and Wilshaw, 1993). Therefore, a detailed understanding of the surface chemistry as the fluid infiltrates the fracture and reacts with freshly fractured surface is critical to identify the mechanisms that are responsible for environmentally-assisted fracture. Previous work has been done on the interactions between electrolyte solutions and silica surfaces (Ho et al., 2011; DelloStritto et al., 2016; Pfeiffer-Laplaud and Gaigeot, 2016); however, these have not typically included the application of external loading. A stress field can alter the structure of the silica by introducing voids, stretching bonds and ring structures (Muralidharan et al., 2005), and generally increasing the reactivity of the silica matrix (Rimsza J. M. et al., 2016).
Here we identify four different surface species. The most dominant surface species when the surface is contact with an aqueous solution is a silanol group, with a dangling bond terminated by one hydrogen (Si-OH). Several other surface species are present including protonated silanols (Si-OH2), deprotonated silanols (Si-O−), and protonated siloxane oxygen atoms (Si-OH-Si). In this work the crack surface is dominated by either Si-OH or Si-O−, with concentrations of Si-OH2 and Si-OH-Si being <0.06 #/nm2.
The concentration of silanol groups on silica surfaces varies, but is reported as 4.0–5.0 #/nm2 (Zhuravlev, 2000) and represents the total number of surface sites including neutral, deprotonated, and protonated silanol groups. We analyze all the available surface sites for protonation to determine how the silica surface has relaxed in the presence of the different solution compositions. Fewer surface sites would suggest that more silanol groups have recombined, forming Si-O-Si from Si-OH and Si-O− sites. In our simulated silica, the initial concentration of available sites for formation of Si-OH groups is ~3.05–3.5 #/nm2 (Figure 2A). The slight differences in surface site concentrate at 0.15 MPa√m is due to initial relaxation of the system before loading is applied. The silica surfaces exposed to salt solutions exhibit more sites (Si-OH, Si-O−) than those in pure water, suggesting that these surfaces have higher concentrations of broken Si-O bonds.
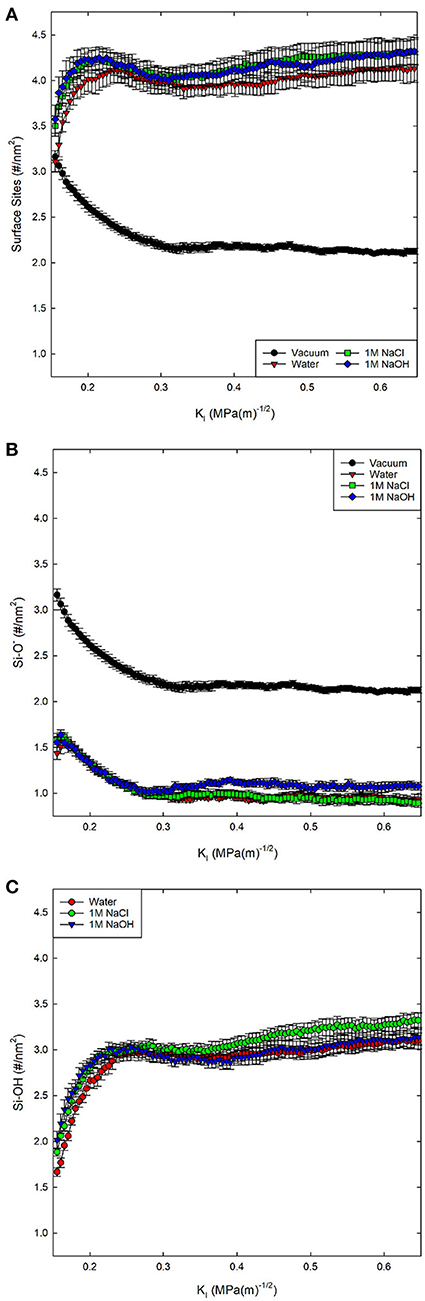
Figure 2. Total concentration of (A) surface sites (Si-OH, Si-O−, Si-OH2) on the fracture surfaces of silica in four environmental conditions, (B) the concentration of Si-O−, and (C) Si-OH on the surface. No Si-OH sites are present under vacuum conditions. KI indexes the far-field mechanical loading and fracture occurs at a KI of ~0.25 MPa-m−1/2.
By separating the types of sites (Figures 2B,C), it becomes apparent that the surface species formed in 1M NaCl solution are more likely to be Si-OH in contrast to those formed in the 1M NaOH solution. The concentration of surface sites (Si-O−, Si-OH, Si-OH2) and level of relaxation has been explicitly tied to the KIC of the material (Lawn and Wilshaw, 1993) through the surface energy and elastic modulus.
The surface structure is therefore influenced by solution composition. These variations can be simulated when a reactive force field is used (compared with traditional MD force fields that do not allow for molecular disassociation). In these simulations the bonding states of atoms can change and the introduction of a NaOH or NaCl molecule into solution does not guarantee that the molecules will remain intact as the simulation evolves. Investigations of NaOH solution wetting a silica surface with a reactive force field (Rimsza et al., 2018b) noted the development of many different aqueous species including OH−, H3O+, H2, and O2. Here, the solution composition is tracked over time to look at the development of these species.
In other work, the steric effects of solution infiltration in fractures focused on what, if any, impact occurred with changing crack tip accessibility (Jones et al., 2018), identifying that the size of the molecules was less important than the reactivity in altering the fracture properties of the material. Salt solutions are known to alter the silica surface due to adsorption (Dove and Craven, 2005). In the present context, the addition of OH− creates a basic solution that increases the dissolution rates of glass (Du and Rimsza, 2017), while the addition of Cl− has a less significant effect on the pH of the solution. Studies of the dissolution rate of glasses have shown the rate increases in both acidic and basic conditions, but is slower in acidic conditions (Alexander et al., 1954). In Figure 3, the OH− and H3O+ concentrations influencing pH in the simulations are reported as molar concentrations based on the number of water molecules in the fracture volume. Clearly, the most rapid change occurs during the initial loading, before the crack propagates, where OH− decreases rapidly and H3O+ increases slightly. The formation of a steady-state concentration occurs at ~0.25–0.3 MPa√m, due to a balance of the rate of water infiltration and addition of NaCl or NaOH molecules as the fracture is loaded.
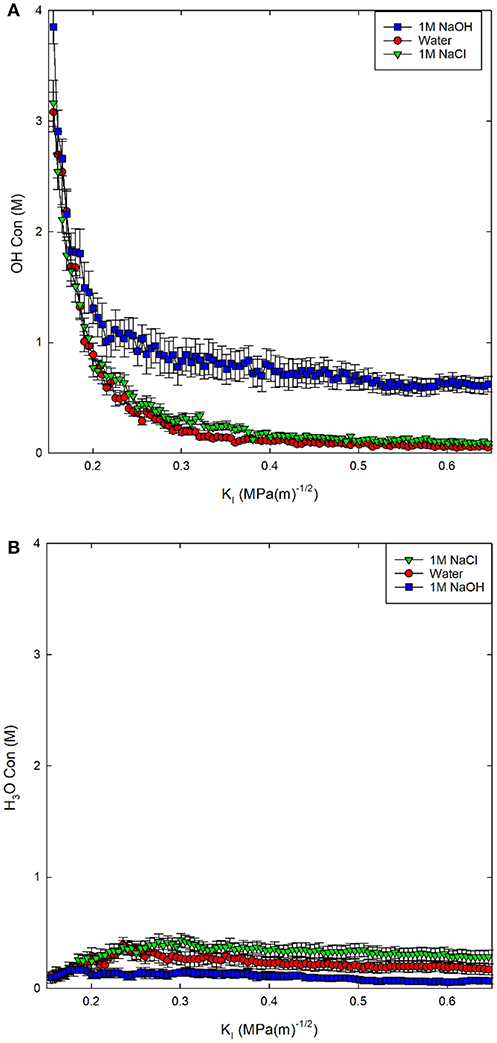
Figure 3. (A) OH− concentration in solution and (B) H3O+ concentration in solution reported as a molarity (M) for three environmental conditions.
The addition of the NaOH molecules results in a relatively steady OH− concentration of 1M (Figure 3A). The concentration of OH− drops more dramatically to <0.25 M for both the 1M NaCl and pure water solutions. The addition of NaCl molecules results in a higher concentration of both OH− and H3O+ in solution compared to pure water; however, both solutions become slightly acidic, with higher concentrations of H3O+ than OH−. The concentration of H3O+ increases with decreasing pH: 1M NaOH < water < 1M NaCl, as expected. The deviation from neutral pH indicates that in both salt solutions, silica dissolution should be higher than in pure water, increasing the chemical impact on chemical-mechanical subcritical fracture growth.
Electrolyte Chemistry and Adsorption
For the salt solutions, the silica surface interacts not just with water, but also with dissolved Na+ and either Cl− (1M NaCl) or OH− (1M NaOH) ions. Both salt solutions change the concentrations of surface species (Si-O−, Si-OH, Si-OH2) when compared with water; this can be partially attributed to adsorption of Na+ to maintain local charge balance. In a previous study, we used ReaxFF (Rimsza et al., 2018b) to investigate Na+ adsorption environments with OH− counter ions and identified several adsorption structures with Na+ ions coordinated by a mixture of water, silanols, and siloxane oxygen atoms. In this work, eight distinct Na+ structures are found in the NaCl and NaOH solutions. Different Na+ structures are identified by the coordinating oxygen species, which include the oxygen atoms in water molecules, silanol groups, and siloxane groups. The most common species is a free Na+ ion coordinated exclusively by water molecules (Figure 4A). These cations have at least one water molecule between them and the silica surface, and account for ~75% of the Na+ ions in the 1M NaCl simulations compared to only 61.5% in the 1M NaOH simulations (Table 1). The next three structures are inner-sphere monodentate (Figure 4B), bidentante (Figure 4C), and tridentate (not pictured), which include Na+ coordinated by water molecules and by one, two, or three Si-OH groups, respectively. These types of configurations (including the tridentate) were also observed on flat surfaces at 1M NaOH concentrations (Rimsza et al., 2018b). In contrast, the coordination of Na+ by Si-O− species did not occur on flat surfaces at the same NaOH concentration, where Si-O− was more commonly coordinated with either neighboring silanol groups or water (Rimsza et al., 2018b). However, in the fracture simulations, coordination of Na+ by Si-O− does occur and is more common than the tridentate species (Table 1). We hypothesize that the confining conditions at the crack tip results in increased Si-O− interactions with Na+ (Figure 4D) compared with flat surfaces. The final three configurations include Na+ that is at least partially coordinated by siloxane oxygen species. The first is a buried species (Figure 4F), which is a Na+ coordinated only by siloxane oxygen and indicates the Na+ has been entirely incorporated into the silica structure. Na+ diffusing into bulk silica occurs readily due to the open structure of the silica and Na+ is a common component of industrial glasses (Smit et al., 1978; Varshneya, 2013). The next is an adsorbed species (Figure 4E), defined as a Na+ ion coordinated by siloxane and water oxygen, but no silanol. Finally, there is a mixed coordination structure at the surface with Na+ ions coordinated by water, silanol, and siloxane oxygen atoms. Overall, the higher free and lower adsorbed Na+ ion concentrations in the 1M NaCl simulations may be leading to the higher Si-OH surface concentration and the lower deprotonation that is seen on the surface.
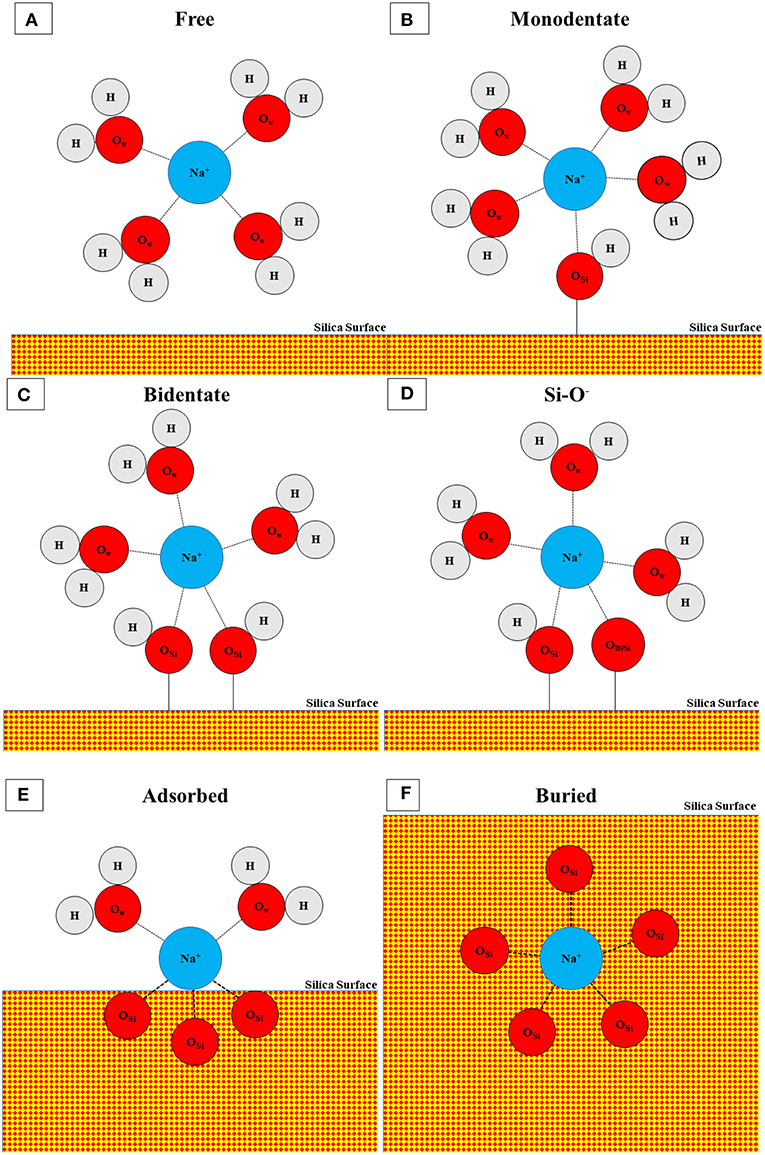
Figure 4. Schematic of coordination structures for Na+ including (A) free, (B) monodentate, (C) bidentate, (D) Si-O−, (E) adsorbed, and (F) buried. Tridentate and mixed configurations are not pictured. Atoms are represented with circles and bonds by connecting lines. Oxygen in water (OW), oxygen in silicon (OSi), and oxygen in deprotonated silanol (ODPSi) are distinguished by subscript and silica is denoted with red-yellow checks. Not to scale.
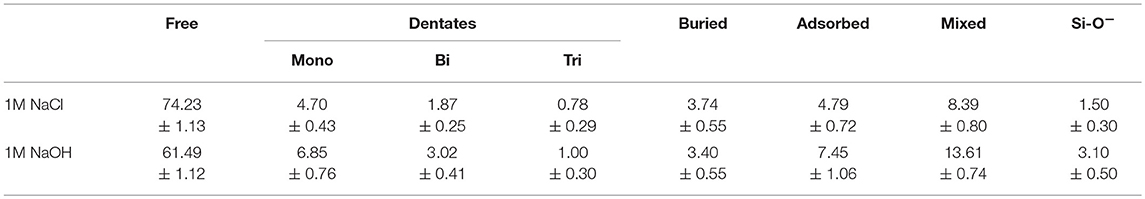
Table 1. Distribution (%) of Na+ coordination environments in 1M NaCl and 1M NaOH solutions.
In addition to the coordination environment, the location of the Na+ ions can vary inside the fracture geometry. Several hypotheses on the role of pH on the fracture of silica suggest that the accessibility of the crack tip by OH− is the primary means of enhancing fracture in basic solutions (White et al., 1987); therefore, understanding how the confined geometry of the crack tip alters the fracture properties is critical. Snapshots of the fracture geometry and the infiltrating fluid (Figure 5) provide some geometric insight into the crack tip accessibility by solution species. For silica fracture in water, the crack tip is primarily filled with complete water molecules along with a few H3O+; the OH− ions present do not migrate to the crack tip, in contrast to those in NaOH and NaCl solutions. In the NaCl solution, the fracture volume is filled with both Na+ and Cl− ions, along with some OH− and H3O+ ions. Therefore, it appears that the presence of Na+ with a Cl− counter ion may allow for easier migration toward the crack tip. In the breakdown of the different coordination environments (Supporting Information, Table S2) there is some limited coordination of the Na+ ion by Cl−, with an average of 0.52 ± 0.02 Cl− ions coordinating each free Na+ ion in the first solvation shell. In the NaOH system, the Na+ ions have limited diffusion into the fracture volume, so that most of the aqueous species near the crack tip are either surface coordinated OH− or free H3O+ molecules. NaCl and NaOH solutions result in different fracture properties (discussed below), which is unexpected considering that both are promoting the diffusion of OH− to the crack tip and potentially encouraging the hydroxide–mediated reaction (Equation 2). It is hypothesized that the hydration shells of the Na+ and Cl− ions are affecting the OH−/H3O+/H2O-silica interactions, altering the energy barriers or the frequency of interactions (White et al., 1987) and the presence of different counter ions at the crack tip alters the activity of the OH−.

Figure 5. Atomic positions of Na+, Cl−, OH−, and H3O+ molecules inside the crack tip for three different solutions (A) water, (B), 1M NaCl, and (C) 1M NaOH. Water is not shown for clarity. All 12 replicates are overlaid to sample the variety of atomic positions and are at a constant loading of 0.2 MPa√m. Atom types: Si (yellow), Na+ (blue), Cl− (green), OH− (red), H3O+ (white).
Material Chemistry
Siloxane bond strength in the glass structure varies with the number of oxygen bonds connecting two silica tetrahedra. Previous MD (Mahadevan and Garofalini, 2008) and DFT (Criscenti et al., 2006) simulations have been used to investigate the relative Si-O bond strength of different linkages to elucidate glass dissolution mechanisms as a function of glass composition. Here, the Qn (n = 0–4) descriptor is used to distinguish silicon species, where n is the number of siloxane oxygen atoms attached to a silicon. A perfectly coordinated silicon atom is bonded to exactly four siloxane oxygen atoms and, hence, is a Q4 species. Surfaces, defects, and network modifiers (e.g., Na+, Li+, Ca2+) can disrupt the glass structure, introducing silicon with lower concentrations of siloxane bonds and therefore lower Qn species. Occasionally Q5 species can also be formed, typically in low concentrations (<3%); this is a known defect in silica glass simulated with ReaxFF that has been previously reported for different silica systems (Rimsza J. et al., 2016; Rimsza and Du, 2018). Q5 species have been identified by both MD simulations (Rimsza J. et al., 2016) and nuclear magnetic resonance (NMR) characterization (Zotov and Keppler, 1998), but at lower concentrations. By looking at the change in Qn species in the structure, we can compare the relative strength of the siloxane bonds that link Qn species in the glass.
The change in Qn species from the first to the last loading step (0.15 MPa√m to 0.65 MPa√m) is provided in Figure 6 and generally, we expect higher Qn species to transition to lower Qn species during dissolution (Q4 → Q3 → Q2 → Q1 → Q0). Regardless of environment, our simulations show that there is a decrease in Q4 species. This is balanced by an increase in Q3, Q2, and Q1 species. Interestingly, a water environment results in a decrease in the Q0 species during loading, suggesting that the few Q0 species that are formed reprecipitate in pure water. Previous computational results showed that during dissolution without mechanical stress, the Q1 → Q0 conversion has the lowest activation energy (Mahadevan and Garofalini, 2008). Therefore, when dissolution occurs we would expect a decrease in Q1 species and an increase in Q0. In contrast, the Q4 → Q3 and Q3 → Q2 transitions have higher activation energies and, hence, these species should be maintained in the system under dissolution-only conditions. As Figure 6 demonstrates, Q3 and Q2 concentrations are also maintained in these chemo-mechanical fracture simulations. When water is present, Q3 and Q2 appear to be more stable than in dry conditions, consistent with predictions from dissolution-only studies.
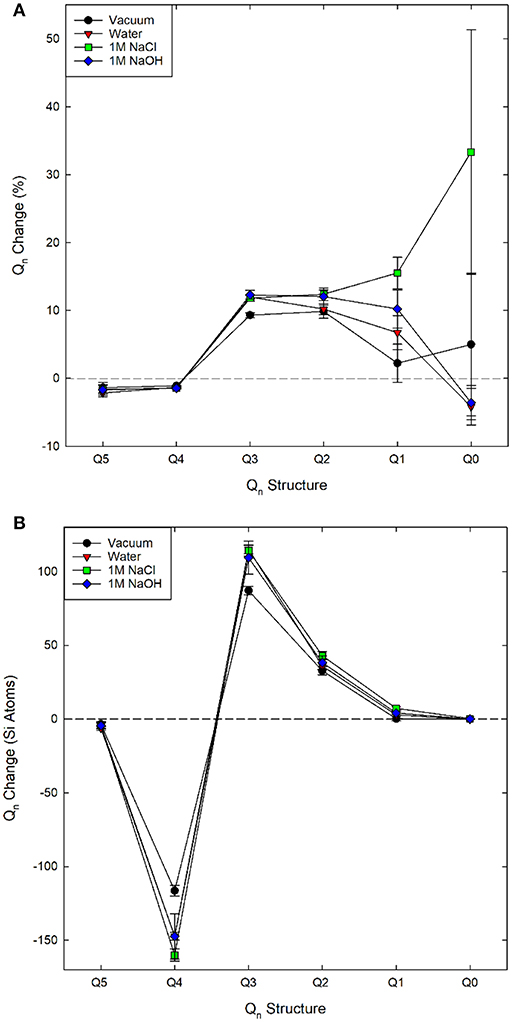
Figure 6. Change in (A) percent and (B) total Qn speciation as a function of solution chemistry over the loading range 0.5 MPa√m to 0.65 MPa√m.
The impact of solution composition is most apparent when examining the Q1 species data. Generally, there is an increase in Q1 from vacuum to water to 1M NaOH to 1M NaCl. The only simulations that exhibit silica monomers released from the surface are those with a 1M NaCl solution, and the Q0 is still in very low concentrations. In contrast, bulk glass dissolution is expected to occur most readily for the 1M NaOH solution (due to the higher pH). In the 1M NaCl solution the ~33% increase seen in Figure 6 is an average increase of total Q0 speciation of ~0.5 silicon atoms, which is large only with respect to the low initial concentration in the replica systems. Despite the low change, several previous investigations have not seen spontaneous dissolution (Dove, 1999; Kubicki et al., 2012; Zapol et al., 2013; Rimsza and Du, 2017, 2018).
Material Mechanics
It is well-established that the strength of the silica decreases in the presence of an aqueous environment, which is typically attributed to dissolution inside the fracture volume and around the crack tip. The radius of curvature of the crack is a structural parameter that is connected to the amount of local inelasticity in a brittle material, with a blunter tip being associated with more inelasticity. It has also been investigated (Rimsza et al., 2017b) for its role in identifying the level of dissolution in the fracture volume, and how this impacts the chemical-mechanical fracture. Previous work (Rimsza et al., 2018a) noted that, in the presence of water, the radius of curvature decreased, resulting in a narrow crack tip, suggesting a higher stress concentration and a lower threshold for fracture propagation. Interestingly, the simulations with a 1M NaCl solution exhibit fractures with a larger radius of curvature than those with a 1M NaOH solution or pure water (Figure 7), and are the simulations that exhibit the highest increase in Q1 species, and the only formation of simulations in which Q0 species. Based on these results, we hypothesize that the presence of a highly reactive fluid like the NaCl solution under confinement may cause rapid increases in dissolution and in the radius of a crack tip, even within the picosecond timeframe in these MD simulations.
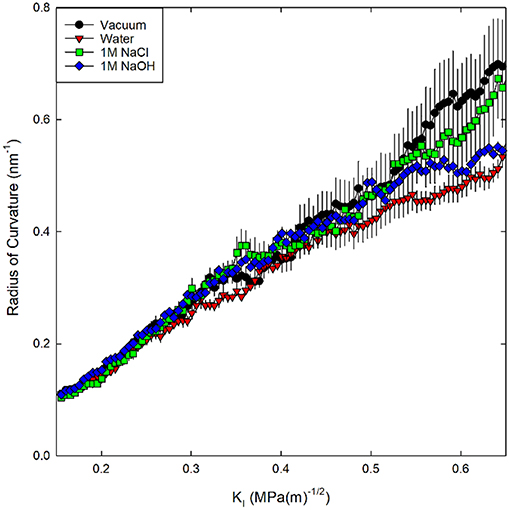
Figure 7. Radius of curvature for silica systems in four chemical environments.
Due to the metastable nature of glass, the silica network within the bulk material is somewhat inelastic and is modified when an external stress is applied. The energy associated with the structural changes in the bulk material is described by the energy dissipation (Gdiss) term in the well-known strain energy release rate (GIC) equation:
which accounts for the energetic cost of fracture propagation in terms of the surface energy (2γs) associated with new surfaces and a non-conservative process to create them (i.e., irreversible changes in bulk silica structure quantified by Gdiss). The Gdiss term is extracted from the atomistic simulations through the change in internal energy of the silica (dU) and the added surface area (dA) (Rimsza et al., 2017b).
A derivation for Equation (5) is included in Appendix I. Here, surface area was calculated through construction of a surface mesh along the crack face with a spherical probe molecule with a radius of 4.5 Å as implemented in the Ovito Visualization Tool (Stukowski, 2009). In previous investigations, the energy dissipation has been associated with the amount of inelasticity occurring around the crack tip (Rimsza et al., 2017b, 2018a). Using this description, the following trend in inelastic energy generation was calculated for the four fracture environments considered: vacuum > 1M NaOH solution > 1M NaCl solution > pure water (Figure 8). The higher Gdiss value for the 1M NaOH solution compared with the other aqueous solutions suggests that there is more unrecoverable inelastic strain in the region surrounding the crack tip.
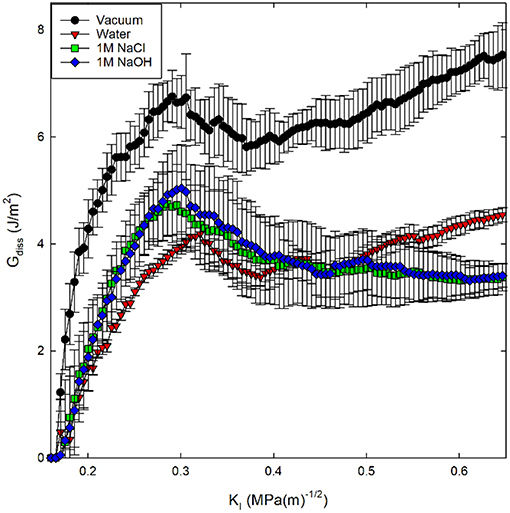
Figure 8. Energy dissipation (Gdiss) during crack loading and subsequent crack propagation for silica systems in four environmental conditions. Gdiss is calculated from Equation (5).
Identification of eKIC values for the four silica systems was based on step-wise changes in the potential energy of the silica during loading, as outlined in previous studies of silica fracture (Rimsza et al., 2017b). A sudden change in energy is accompanied by bond breakage, indicating a crack propagation event. The loading at which these crack propagation events occur are averaged over multiple simulations to estimate the eKIC for the system. The results identify that the silica systems fractured in vacuum exhibit the highest eKIC value, the fewest fracture events, and the highest GIC value (Table 2). This is consistent with experimental observations that silica is stronger in a non-reactive or dry environment (Rimsza et al., 2018a). Experiments have shown that the addition of any electrolytes that perturb the pH of the solution make the system fracture at lower loads than in neutral water (Dove, 1995). From the simulation data reported here, the eKIC values overlap, but it appears that the silica in 1M NaCl solution has a slightly higher eKIC than that in water or a 1M NaOH solution. It is hypothesized that a broader crack tip radius (Figure 7) reduces the drive for fracture propagation at these time scales.
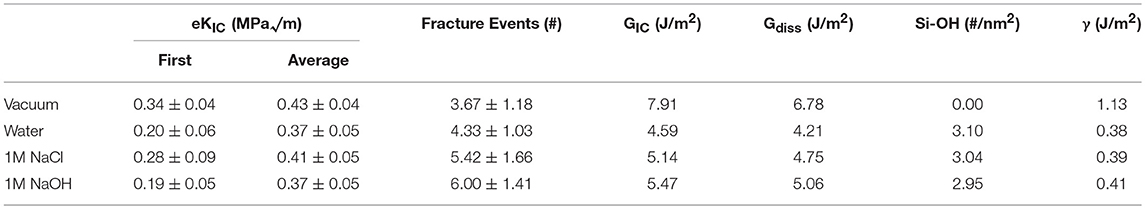
Table 2. Fracture properties of silica in four different environmental conditions (vacuum, water, 1M NaCl, 1M NaOH) as the average and standard deviation of twelve unique simulations.
Discussion
The differences in solution composition and the unique confined geometry of the crack tip generate unusual chemical environments that impact the fracture properties of silica. In this work, the confined 1M NaCl aqueous solution is slightly acidic, and the confined 1M NaOH solution is alkaline (Figure 3). The elevated OH− concentration leads to an increased number of Si-O− surface sites, which allows for the formation of Na+ adsorption complexes in the confined space that are not seen on flat surfaces (Rimsza et al., 2018b). These complexes include Na+ coordinated by Si-O− surface species and Na+ coordinated by siloxane oxygen and water molecules only (Table 1). With increasing confinement of the aqueous solution as the molecules approach the crack tip, more interactions between the Na+ ions and the surface occur, possibly made more energetically favorable due to increased surface deprotonation.
Snapshots of the fracture geometry also indicate that the solution composition influences the diffusion of electrolyte ions into the crack tip. For example, in the 1M NaCl solution the Na+ ions infiltrate the crack tip much farther than in the 1M NaOH solution (Figure 5). This observation supports the hypothesis that changing the composition of the hydration shells of Na+ and Cl− ions may alter the accessibility of the crack tip and the reactivity of the infiltrating fluid (White et al., 1987). This finding also highlights the importance of understanding how aqueous electrolyte solutions are interacting with surfaces in confined geometries, which appear to be where most critical surface reactions and stress concentrators occur.
Increased radius of curvature of the crack tip in the salt solutions provides further evidence that the crack morphology is impacted by the infiltrating fluid. The increase in the radius of the crack tip has been proposed to be a function of dissolution events (Freiman et al., 2009). This chemo-mechanical effect is apparently related to the fracture properties of silica, with the 1M NaCl solution resulting in a higher eKIC for the silica than in the 1M NaOH solution of pure water. We hypothesize that the increase in formation of Q1 and Q0 species in the 1M NaCl solution (Figure 6) is resulting in crack tip blunting (Figure 7) that dissipates energy through mechanisms other than fracture propagation. Additionally, the interaction between the Na+/Cl− ions and the aqueous OH− may be limiting the ability of the OH− to interact with the crack tip, resulting in a larger eKIC (see Figure 5 and related discussion).
Interactions between electrolyte solutions and the surface impact the formation of Q1 and Q0 species, which are a critical components of glass degradation (Guin et al., 2005; Wiederhorn et al., 2011). Dissolution has been difficult to investigate with atomistic simulations, because of the pico- or nanosecond time frames accessible by MD simulations. In this study, we used a Qn distribution analysis to determine if any of the aqueous solutions were causing local dissolution. The 1M NaCl solution caused the most dissolution events in the simulations, demonstrated by increased concentrations of Q1 and Q0 species (Figure 6). Most atomistic simulations to date have not generated Q0 species (without positioning atoms to facilitate a Q1 → Q0 conversion) (Kagan et al., 2014; Rimsza and Du, 2017, 2018). The Qn distribution analysis leads us to conclude that the silica dissolves most readily in the 1M NaCl solution. This conclusion is at odds with experimental observations that bulk glass dissolution occurs most readily in basic solutions (Alexander et al., 1954). However, previous atomistic ab initio DFT studies of reaction barrier heights have reported that the Si-O-Si bond is more easily broken with an adsorbed proton on the bridging oxygen, which would be expected to occur more readily in acidic conditions, than when one of the terminating Si has a deprotonated oxygen (Nangia and Garrison, 2008; Zapol et al., 2013); these results are also inconsistent with experimental observations on bulk glass dissolution.
This coupled chemical-mechanical fracture study from atomistic methods has highlighted several factors influencing the weakening of silica that appear to be at odds with accepted theory or chemical intuition. A ranking of proposed factors for environmentally assisted fracture is included in Table 3. From this data we can see that the environment which results in the highest number of fracture events and lowest eKIC (NaOH) does not exhibit the fastest dissolution as evidenced by the formation of Q1 and Q0 species. There does not appear to be a direct correlation between the factors: dissolution, Si-O− concentration, crack tip accessibility, or radius of curvature, and the fracture properties: eKIC or the number of fracture events. Generally, our simulations show a pure aqueous environment decreases the radius of curvature and the eKIC. Furthermore, the inclusion of either NaOH or NaCl increases the number of fracture events, silica dissolution, and the crack tip accessibility; however, separating out the impact of the two electrolytes is more complicated. We hypothesize that both the geometry of the fracture and composition of the fluid alter the reactivity of the system through:
1. Compared with flat surfaces, the nanoconfined crack tip favors different adsorbed species and concentrations and limits diffusion thereby increasing the residence time of reactive species and altering the chemical gradients in solution.
2. The presence of Na+, Cl−, or OH− ions impacts the reaction frequency between the surface and water molecules, affects the protonation and deprotonation of the surface, and exposes strained and unstrained bonds along the fracture surface and at the crack tip to the solution.

Table 3. Ranking of factors that influence environmentally assisted fracture from highest (1) to lowest (4).
Therefore, dissolution and reaction rates inside the fracture are distinct from those observed in bulk or equilibrium systems. These hypotheses will be explored through a targeted study of the details of the crack geometry. In conclusion, we propose that this investigation demonstrates that the geometry of the fracture is impacting the water and ion accessibility at the crack tip, the kinetics, and the reactions occurring at the fracture surfaces, and, results in behavior unique to fracture conditions on the nanoscale.
Conclusion
To investigate weakening of silica in contact with aqueous electrolyte solutions, classical MD simulations of silica fracture were performed using the bond-order based, reactive force field ReaxFF. Four different environmental conditions were investigated, including vacuum, water, and two salt solutions (1M NaCl, 1M NaOH). The highlights of our findings are:
• Any aqueous condition weakens the silica, with the addition of basic NaOH further encouraging fractures to propagate, possibly due to increased interactions between ions and the silica surface.
• The 1M NaCl solution generates increased Q1 and Q0 species compared with 1M NaOH and water, altering the geometry of the crack tip and the fracture properties of the silica, in contrast to bulk glass dissolution. Additionally, the observed formation of Q0 silicon in solution is a rarely identified dissolution event in atomistic simulations and may be a function of nanoconfinement, mechanical stress, or both.
• The nanoconfinement of the crack tip alters Na+ adsorption and surface properties compared with flat surfaces, and its role in chemical-mechanical fracture should be explored further.
Overall, reactive classical MD study provides unique insight into how anions alter the chemical-mechanical fracture response of silica and indicate that critical factors for fracture at the nanoscale may differ from those that are seen in bulk systems over long time scales.
In summary, this work provides a mechanistic investigation of chemo-mechanical fracture in silica in several different aqueous solutions. By using atomistic simulations, we have shown that we can identify factors that are unique to the nanoscale environment and are difficult or impossible to separate out through current experimental techniques. This is, to the best of our knowledge, the first study of its kind to consider the mechanical and chemical impact on silica fracture in an electrolyte solution from a reactive MD simulation, which includes the formation and breakage of bonds. This study builds upon an expertise in hydroxylation (Rimsza et al., 2017a) and sodium adsorption (Rimsza et al., 2018b) on silica surfaces, atomistic mechanisms of silica fracture in vacuum (Rimsza et al., 2017b) and in aqueous environments (Rimsza et al., 2018a) and adds the complexity of electrolyte solutions. This work provides a unique atomistic perspective on chemically assisted silica fracture which has thus far been too complex to be investigated with a molecular level of detail and indicates that similar investigations of multicomponent glass compositions can be undertaken in the future as suitable potentials become available.
Author Contributions
JR performed the simulations, analyzed the data, and wrote the manuscript. LC and RJ helped design the simulations and all authors edited and discussed the manuscript prior to submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Mark Wilson (Sandia National Laboratories) is acknowledged for his technical support in the development of the grand canonical Monte Carlo (GCMC) methods used in the manuscript. This work was fully supported by the Laboratory Directed Research and Development (LDRD) program of Sandia National Laboratories. Sandia National Laboratories is a multi-mission laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC., a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmats.2019.00079/full#supplementary-material
Supporting information includes the ReaxFF force field used in this manuscript as a pdf file.
References
Aktulga, H. M., Fogarty, J. C., Pandit, S. A., and Grama, A. Y. (2012). Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques. Parall. Comput. 38, 245–259. doi: 10.1016/j.parco.2011.08.005
Alexander, G. B., Heston, W., and Iler, R. K. (1954). The solubility of amorphous silica in water. J. Phys. Chem. 58, 453–455. doi: 10.1021/j150516a002
Atkinson, B. K., and Meredith, P. G. (1981). Stress corrosion cracking of quartz: a note on the influence of chemical environment. Tectonophysics 77, T1–T11. doi: 10.1016/0040-1951(81)90157-8
Bonnaud, P., Coasne, B., and Pellenq, R. J. (2010). Molecular simulation of water confined in nanoporous silica. J. Phys. 22:284110. doi: 10.1088/0953-8984/22/28/284110
Bourg, I. C., and Steefel, C. I. (2012). Molecular dynamics simulations of water structure and diffusion in silica nanopores. J. Phys. Chem. C 116, 11556–11564. doi: 10.1021/jp301299a
Célarié, F., Ciccotti, M., and Marliere, C. (2007). Stress-enhanced ion diffusion at the vicinity of a crack tip as evidenced by atomic force microscopy in silicate glasses. J. Non Crystall. Solids 353, 51–68. doi: 10.1016/j.jnoncrysol.2006.09.034
Criscenti, L. J., Kubicki, J. D., and Brantley, S. L. (2006). Silicate glass and mineral dissolution: calculated reaction paths and activation energies for hydrolysis of a Q3 Si by H3O+ using ab initio methods. J. Phys. Chem. A 110, 198–206. doi: 10.1021/jp044360a
DelloStritto, M. J., Kubicki, J. D., and Sofo, J. O. (2016). Effect of Ions on H-bond structure and dynamics at the quartz (101)–water interface. Langmuir 32, 11353–11365. doi: 10.1021/acs.langmuir.6b01719
Dove, P. M. (1995). Geochemical controls on the kinetics of quartz fracture at subcritical tensile stresses. J. Geophys. Res. Solid Earth 100, 22349–22359. doi: 10.1029/95JB02155
Dove, P. M. (1999). The dissolution kinetics of quartz in aqueous mixed cation solutions. Geochim. Cosmochim. Acta 63, 3715–3727. doi: 10.1016/S0016-7037(99)00218-5
Dove, P. M., and Craven, C. M. (2005). Surface charge density on silica in alkali and alkaline earth chloride electrolyte solutions. Geochim. Cosmochim. Acta 69, 4963–4970. doi: 10.1016/j.gca.2005.05.006
Du, J., and Rimsza, J. M. (2017). Atomistic computer simulations of water interactions and dissolution of inorganic glasses. NPJ Mater. Degrad. 1:16. doi: 10.1038/s41529-017-0017-y
Fogarty, J. C., Aktulga, H. M., Grama, A. Y., Van Duin, A. C,, and Pandit, S. A. (2010). A reactive molecular dynamics simulation of the silica-water interface. J. Chem. Phys. 132:174704. doi: 10.1063/1.3407433
Freiman, S. W., Wiederhorn, S. M., and Mecholsky, J. J. Jr (2009). Environmentally enhanced fracture of glass: a historical perspective. J. Am. Ceramic Soc. 92, 1371–1382. doi: 10.1111/j.1551-2916.2009.03097.x
Guillot, B., and Guissani, Y. (2001). How to build a better pair potential for water. J. Chem. Phys. 114, 6720–6733. doi: 10.1063/1.1356002
Guin, J. P., Wiederhorn, S. M., and Fett, T. (2005). Crack-tip structure in soda–lime–silicate glass. J. Am. Ceramic Soc. 88, 652–659. doi: 10.1111/j.1551-2916.2005.00108.x
Ho, T. A., Argyris, D., Cole, D., and Striolo, A. (2011). Aqueous NaCl and CsCl solutions confined in crystalline slit-shaped silica nanopores of varying degree of protonation. Langmuir 28, 1256–1266. doi: 10.1021/la2036086
Jones, R., Tucker, W. C., Rimsza, J., and Crisenti, L. (2018). Atomic-scale interaction of a crack and an infiltrating fluid. Chem. Phys. Lett. X 1:100005. doi: 10.1016/j.cpletx.2018.100005
Kagan, M., Lockwood, G. K., and Garofalini, S. H. (2014). Reactive simulations of the activation barrier to dissolution of amorphous silica in water. Phys. Chem. Chem. Phys. 16, 9294–9301. doi: 10.1039/C4CP00030G
Kubicki, J. D., Sofo, J. O., Skelton, A. A., and Bandura, A. V. (2012). A new hypothesis for the dissolution mechanism of silicates. J. Phys. Chem. C 116, 17479–17491. doi: 10.1021/jp300623v
Lane, J. M. D. (2015). Cooling rate and stress relaxation in silica melts and glasses via microsecond molecular dynamics. Phys. Rev. E 92:012320. doi: 10.1103/PhysRevE.92.012320
Lawn, B., and Wilshaw, T. R. (1993). Fracture of Brittle Solids. New York, NY: Cambridge University Press. doi: 10.1017/CBO9780511623127
Lowe, B. M., Skylaris, C. -K., and Green, N. G. (2015). Acid-base dissociation mechanisms and energetics at the silica–water interface: an activationless process. J. Colloid Interface Sci. 451, 231–244. doi: 10.1016/j.jcis.2015.01.094
Mahadevan, T., and Garofalini, S. (2007). Dissociative water potential for molecular dynamics simulations. J. Phys. Chem. B 111, 8919–8927. doi: 10.1021/jp072530o
Mahadevan, T., and Garofalini, S. (2008). Dissociative chemisorption of water onto silica surfaces and formation of hydronium ions. J. Phys. Chem. C 112, 1507–1515. doi: 10.1021/jp076936c
Morrow, C. P., Nangia, S., and Garrison, B. J. (2009). Ab initio investigation of dissolution mechanisms in aluminosilicate minerals. J. Phys. Chem. A 113, 1343–1352. doi: 10.1021/jp8079099
Mozzi, R., and Warren, B. (1969). The structure of vitreous silica. J. Appl. Crystallogr. 2, 164–172. doi: 10.1107/S0021889869006868
Muralidharan, K., Simmons, J., Deymier, P., and Runge, K. (2005). Molecular dynamics studies of brittle fracture in vitreous silica: review and recent progress. J. Non Crystall. Solids 351, 1532–1542. doi: 10.1016/j.jnoncrysol.2005.03.026
Nangia, S., and Garrison, B. J. (2008). Reaction rates and dissolution mechanisms of quartz as a function of pH. J. Phys. Chem. A 112, 2027–2033. doi: 10.1021/jp076243w
Pfeiffer-Laplaud, M., and Gaigeot, M. -P. (2016). Adsorption of singly charged ions at the hydroxylated (0001) α-quartz/water interface. J. Phys. Chem. C 120, 4866–4880. doi: 10.1021/acs.jpcc.5b10947
Pitman, M. C., and Van Duin, A. C. (2012). Dynamics of confined reactive water in smectite clay–zeolite composites. J. Am. Chem. Soc. 134, 3042–3053. doi: 10.1021/ja208894m
Rimola, A., and Ugliengo, P. (2008). A quantum mechanical study of the reactivity of (SiO) 2-defective silica surfaces. J. Chem. Phys. 128:204702. doi: 10.1063/1.2929827
Rimsza, J., Deng, L., and Du, J. (2016). Molecular dynamics simulations of nanoporous organosilicate glasses using Reactive Force Field (ReaxFF). J. Non Crystall. Solids 431, 103–111. doi: 10.1016/j.jnoncrysol.2015.04.031
Rimsza, J., and Du, J. (2015). Ab initio molecular dynamics simulations of the hydroxylation of nanoporous silica. J. Am. Ceramic Soc. 98, 3748–3757. doi: 10.1111/jace.13731
Rimsza, J., and Du, J. (2017). Interfacial structure and evolution of the water–silica gel system by reactive force-field-based molecular dynamics simulations. J. Phys. Chem. C 121, 11534–11543. doi: 10.1021/acs.jpcc.7b02734
Rimsza, J., and Du, J. (2018). Nanoporous silica gel structures and evolution from reactive force field-based molecular dynamics simulations. NPJ Mater. Degrad. 2:18. doi: 10.1038/s41529-018-0039-0
Rimsza, J., Jones, R., and Criscenti, L. (2017a). Surface structure and stability of partially hydroxylated silica surfaces. Langmuir 33, 3882–3891. doi: 10.1021/acs.langmuir.7b00041
Rimsza, J., Jones, R. E., and Criscenti, L. J. (2017b). Crack propagation in silica from reactive classical molecular dynamics simulations. J. Am. Ceramic Soc. 101, 1488–1499. doi: 10.1111/jace.15292
Rimsza, J. M., Jones, R. E., and Criscenti, L. J. (2018a). Chemical effects on subcritical fracture in silica from molecular dynamics simulations. J. Geophys. Res. Solid Earth 123, 9341–9354. doi: 10.1029/2018JB016120
Rimsza, J. M., Jones, R. E., and Criscenti, L. J. (2018b). Interaction of NaOH solutions with silica surfaces. J. Colloid Interface Sci. 516, 128–137. doi: 10.1016/j.jcis.2018.01.049
Rimsza, J. M., Yeon, J., van Duin, A. C., and Du, J. (2016). Water interactions with nanoporous silica: comparison of ReaxFF and ab Initio based molecular dynamics simulations. J. Phys. Chem. C 120, 24803–24816. doi: 10.1021/acs.jpcc.6b07939
Smit, W., Holten, C., Stein, H., De Goeij, J., and Theelen, H. (1978). A radiotracer determination of the adsorption of sodium ion in the compact part of the double layer of vitreous silica. J. Colloid Interface Sci. 63, 120–128. doi: 10.1016/0021-9797(78)90042-5
Stukowski, A. (2009). Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simulat. Mater. Sci. Eng. 18:015012. doi: 10.1088/0965-0393/18/1/015012
Van Duin, A. C., Strachan, A., Stewman, S., Zhang, Q., Xu, X., and Goddard, W. A. (2003). ReaxFFSiO reactive force field for silicon and silicon oxide systems. J. Phys. Chem. A 107, 3803–3811. doi: 10.1021/jp0276303
van Duin, A. C., Zou, C., Joshi, K., Bryantsev, V., and Goddard, W. A. (2013). A ReaxFF reactive force-field for proton transfer reactions in bulk water and its applications to heterogeneous catalysis. Computat. Catal. 14, 223–243. doi: 10.1039/9781849734905-00223
White, G. S., Freiman, S. W., Wiederhorn, S. M., and Coyle, T. D. (1987). Effects of counterions on crack growth in vitreous silica. J. Am. Ceramic Soc. 70, 891–895. doi: 10.1111/j.1151-2916.1987.tb04912.x
Wiederhorn, S., and Johnson, H. (1973). Effect of electrolyte pH on crack propagation in glass. J. Am. Ceramic Soc. 56, 192–197. doi: 10.1111/j.1151-2916.1973.tb12454.x
Wiederhorn, S. M., Guin, J.-P., and Fett, T. (2011). The use of atomic force microscopy to study crack tips in glass. Metallurg. Mater. Trans. A 42, 267–278. doi: 10.1007/s11661-010-0411-3
Yeon, J., and van Duin, A. C. (2015). ReaxFF molecular dynamics simulations of hydroxylation kinetics for amorphous and nano-silica structure, and its relations with atomic strain energy. J. Phys. Chem. C 120, 305–317. doi: 10.1021/acs.jpcc.5b09784
Yu, Y., Krishnan, N. A., Smedskjaer, M. M., Sant, G., and Bauchy, M. (2018). The hydrophilic-to-hydrophobic transition in glassy silica is driven by the atomic topology of its surface. J. Chem. Phys. 148:074503. doi: 10.1063/1.5010934
Yu, Y., Wang, B., Wang, M., Sant, G., and Bauchy, M. (2016). Revisiting silica with ReaxFF: Towards improved predictions of glass structure and properties via reactive molecular dynamics. J. Non Crystall. Solids 443, 148–154. doi: 10.1016/j.jnoncrysol.2016.03.026
Zapol, P., He, H., Kwon, K. D., and Criscenti, L. J. (2013). First-principles study of hydrolysis reaction barriers in a sodium borosilicate glass. Int. J. Appl. Glass Sci. 4, 395–407. doi: 10.1111/ijag.12052
Zhuravlev, L. (2000). The surface chemistry of amorphous silica. Zhuravlev Model Colloids Surfaces A Physicochem. Eng. Aspects 173, 1–38. doi: 10.1016/S0927-7757(00)00556-2
Zotov, N., and Keppler, H. (1998). The structure of sodium tetrasilicate glass from neutron diffraction, reverse Monte Carlo simulations and Raman spectroscopy. Phys. Chem. Min. 25, 259–267. doi: 10.1007/s002690050112
Appendix 1
The Griffiths strain energy release rate (G) for a crack in an elastic material is given by
where UE is the elastic potential energy, WL is the external work, and A the fracture surface area. In this case the fracture toughness is associated with the critical value of G, GIC = 2γS, which is equal to the energetic cost of creating new surfaces, i.e., twice the surface energy γS. To account for energy dissipated during fracture, a dissipation energy term (Gdiss) is typically added:
Using that the internal energy of the system (U) is the elastic potential energy (UE) minus the external work due to loading (WL) plus the surface energy (US):
and assuming Gdiss and γs are constant, we arrive at
After equating the change of surface energy dUs = 2γsdA, we obtain
Keywords: fracture, silica, molecular dynamics simulation, dissolution, electrolytes
Citation: Rimsza JM, Jones RE and Criscenti LJ (2019) Mechanisms of Silica Fracture in Aqueous Electrolyte Solutions. Front. Mater. 6:79. doi: 10.3389/fmats.2019.00079
Received: 26 November 2018; Accepted: 05 April 2019;
Published: 24 April 2019.
Edited by:
Lothar Wondraczek, Friedrich Schiller University Jena, GermanyReviewed by:
Joachim Deubener, Clausthal University of Technology, GermanyMathieu Bauchy, University of California, Los Angeles, United States
Copyright © 2019 Rimsza, Jones and Criscenti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica M. Rimsza, anJpbXN6YUBzYW5kaWEuZ292