 Aditya Menon
Aditya Menon James A. Thompson-Colón
James A. Thompson-Colón Newell R. Washburn
Newell R. Washburn- 1Department of Materials Science and Engineering, Carnegie Mellon University, Pittsburgh, PA, United States
- 2Covestro LLC, Pittsburgh, PA, United States
- 3Department of Chemistry and Department of Biomedical Engineering, Carnegie Mellon University, Pittsburgh, PA, United States
Polyurethanes are a broad class of material that finds application in coatings, foams, and solid elastomers. The urethane chemistry allows a diversity of monomers to be used, and prediction of mechanical properties, which are determined by complex interplay between monomer chemistry and chain architecture, is an unresolved challenge. Urethanes are based on aromatic or cyclic isocyanates and linear or branched polyols, and polymerization results in linear chains for bifunctional monomers or branched chains for multifunctional monomers. Strong intermolecular interactions between aromatic groups result in the formation of hard-segment domains that generate physical crosslinks between disorganized rubbery domains and anchor the material microstructure, contributing to resistance to deformation. Here, a general hierarchical machine learning (HML) model for predicting the stress-at-break, strain-at-break, and Tan δ for thermoplastic and thermoset polyurethanes is presented. The algorithm was trained on a library of 18 polymers with different diisocyanates, bifunctional or trifunctional polyols, and NCO:OH index. HML reduces data requirements through robust embedding of domain knowledge and surrogate data in a middle layer that bridges input variables (composition) and output responses (mechanical properties). In this work, the middle layer included information on overall polymer composition, predictions of chain architecture derived from Monte Carlo simulations of polymerization, information on interchain interactions from empirically derived molecular potentials and shifts in infrared (IR) spectroscopy absorbances. The HML predictions are shown to be more accurate than those from a random forest model directly relating composition and properties, suggesting that embedding domain knowledge provides significant advantages in predicting the properties of complex material systems based on small datasets.
Introduction
Polyurethanes are ubiquitous materials found in coatings, foams, and solid elastomers (Oertel, 1994; Engels et al., 2013). Prototypical polyurethanes are formed through step-growth polymerization of an aromatic diisocyanate and an aliphatic diol, resulting in the formation of a material having aggregated aromatic hard segments bridged by rubbery segments. This microstructure is the basis for the remarkable mechanical properties of polyurethanes characterized by primarily elastic behavior and large values of ultimate elongation. However, modern polyurethanes are based on a highly diverse family of monomers that provide control over the number of reactive isocyanate or alcohol groups, which allow the preparation of linear of branched materials, varying monomer chemistries, which tune the interactions between hard segments and soft segments, and control over the NCO:OH index, which controls the degree of polymerization. Developing a model to predict the mechanical properties of these materials based on all these compositional variables is an unresolved challenge.
There are many analytical approaches to predicting properties of polymers based on their linear or crosslinked structure. Viscoelastic and rheological properties of linear polymers have been predicted by tube models (Milner and McLeish, 1998; Pattamaprom et al., 2000; van Ruymbeke et al., 2002, 2005) where reptation theory has been extended with contour length fluctuations and constraint release mechanisms, leading to successful predictions especially with low molecular weight polymers. For crosslinked polymers, mechanical properties have been predicted using group interaction modeling (GIM) (Foreman et al., 2008) where a mean-field potential is calculated from cohesive energy and other molar constants derived using a group-additivity approach based on each component in the repeating unit. Another model for thermo-mechanical behavior prediction uses a molecular-modeling approach (Shenogina et al., 2012) whereas Eom et al show the effect of native topology on mechanical strength of crosslinked polymer chains (Eom et al., 2003).
With the advent of machine learning in many traditional scientific disciplines and the Materials Genome Initiative, there have also been many data-driven, ML-based approaches for prediction of polymer properties (de Pablo et al., 2014; Agrawal and Choudhary, 2016). Viscoelastic properties have been modeled using a multi scale computational framework on inverse Boltzmann method (Li et al., 2012) and specific properties (mechanical, thermal, optical, and electrical) have been trained on microscopic, mesoscopic, and macroscopic structures from polymer databases available online using artificial neural networks (Roy et al., 2006). Recently, Kim et al. (2018) have developed a polymer informatics platform which trains machine learning models of a dataset of high throughput DFT calculations and experimental data from the polymer literature.
Since most of the ML based approaches rely on large datasets, Hierarchical Machine Learning (HML) was developed on small experimental datasets to predict properties of complex material systems utilizing an intermediate layer between the desired responses and system variables (Menon et al., 2017). These intermediate variables are based on latent physicochemical factors from domain knowledge pertaining to the material system. This methodology was validated on a system of dispersant dosed concentrated MgO suspension, which acted as a non-setting model of cement. Building upon previous work, HML was successfully utilized to designed a superplasticizer tailored specifically for metakaolin-portland blend cement blends (Menon et al., 2018).
In this work, HML was applied to a system of linear and crosslinked polyurethanes modeling mechanical responses: stress-at-break, strain-at-break, and Tan δ with system variables which are polymer structure, molecular weights and densities of the reactants (diisocyanates, polyols), chain length of polyol and isocyanate: alcohol (NCO:OH) index. Intermediate variables utilized for predicting mechanical responses were chosen to simplistically represent intermolecular, interchain and crosslinking behaviors in the system. The input dataset of 18 synthesized polymers was split into a training set of 14 and test set of 4 data points. A model was developed on the training set and validated against the test set. Finally, a comparison of the HML algorithm was performed with a random forest (RF) model (Breiman, 2001; Pedregosa et al., 2011) that directly predicted mechanical properties based on composition using the same training set.
Training Dataset
Materials
The oligomers and monomers used to build the training set, poly(tetramethylene ether) glycol(PTMEG) (Mn = 1,000), polycaprolactone triol (PCL) (Mn = 900), toluene-2,4-diisocyanate (TDI), hexamethylene diisocyanate (HDI), isophorone diisocyanate(IPDI), were purchased from Sigma-Aldrich, as well as the catalyst used for the polymerization reaction, dibutyltin dilaurate (DBDTL). Dichloromethane was acquired from EMD Millipore. All of the materials were used as received.
Polymer Synthesis
The training set consisted of 18 samples, all of which were prepared by reacting a bifunctional diisocyanate with either a bifunctional or trifunctional polyol at NCO:OH indices of 1.0, 1.2, or 1.5, as shown in Supplementary Table 1. The reactions were carried out at room temperature in 8 ml of dichloromethane as a solvent under the presence of DBTDL as a catalyst. Films were cast from the synthesized polymers and were left to dry at room temperature for 24 h and then again dried in a vacuum oven for 24 h at 60°C to remove any residual solvent.
Measurements
Stress-at-break and strain-at-break were measured for all polymers in a universal testing machine (Instron). Gage width, parallel section width and thickness for each sample tested were 12 mm, 4 mm, and 2mm, respectively. Tan δ for all polymers was measured via a frequency sweep in a Discovery HR-2 rheometer (TA instruments) and the value at 1 Hz was used as the characteristic system response in this study. FT-IR analysis was performed on 2 mm thick film specimens in a Frontier Spectrometer (PerkinElmer) in the standard wavenumber range (4000–700 cm−1).
Monte Carlo Modeling
Monte Carlo simulations of step growth polymerization were performed in R (R Core Team, 2018). We focused on parameters which would provide insight into the crosslinking tendency of polymer chains due to polyfunctionality in either the polyisocyanates or the polyol reactants. These parameters were determined through a Monte Carlo simulation of a spatially homogeneous chemical ensemble of monomers; for these simulations we used 200,000 monomers. The algorithm performs an event-based stochastic process analogous to the approach described by Mikes and Dusek (1982), then repeats the stochastic process until all of the limiting reactive group are consumed, which in our simulations was the OH group. For the recipes with NCO:OH index of 1, the simulation is at this point complete. For the recipes with index 1.2 and 1.5 we further simulate for moisture cure. Moisture cure refers to the reaction of some of the remaining unreacted—NCO react with ambient moisture to form to—NH2 with further reactions with—NCO to form urea bonds. In these cases, once all the -OH is consumed, the appropriate amount of water is added to the ensemble and the stochastic process is continued until no more bonds can be formed. The simulation provides parameters that describe the connectivity for all monomers in the post-gel thermoset. For our analysis, we identify and quantify three type of molecular configurations, shown in Figure 1: Sol-which are the oligomers that are not part of the infinite network formed during polymerization, Elastic link—which are the effective links formed between the reactive groups and form part of the crosslinked core gel component and Dangler—which are the pendant groups that are attached to the infinite network and are also known as tethered plasticizer and ineffective links. The parameters which are relevant are the effective crosslinks per kg of polymer (nEff) calculated using Miller and Macosko's recursive method (Miller and Macosko, 1976), the average molecular weight of the elastic links weighed as a percent of total polymer weight (elastic_link_mtw), the average molecular weight of the core gel component weighed as a percent of total polymer weight (Mtw), the percentage of sol present in the synthesized polymer (sol_pctWgt) and the percentage of core gel component in the synthesized polymer (Core_pctWgt).
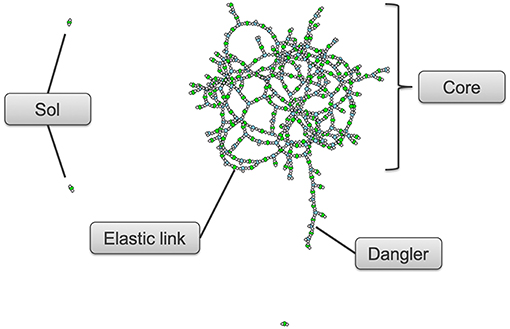
Figure 1. Schematic of sol, danglers, elastic links, and core gel components modeled over 300 monomers using Monte Carlo simulations.
HML Modeling
HML modeling was performed in Python (Rossum, 1995) using the Scikit learn library (Pedregosa et al., 2011) for machine learning estimators. HML modeling has been used with multiple systems now; in all of these systems, the top layer will represent a complex system response that has to be either predicted or optimized with respect to a bottom layer which consists of simple experimentally tunable variables. However, there is an intermediate middle layer which consists of physical or chemical factors parameterized from the variables in bottom layer through surrogate physical measurements and existing physical/chemical relationships pertaining to the specific material system. The algorithm can be better understood with the scheme shown in Figure 2.
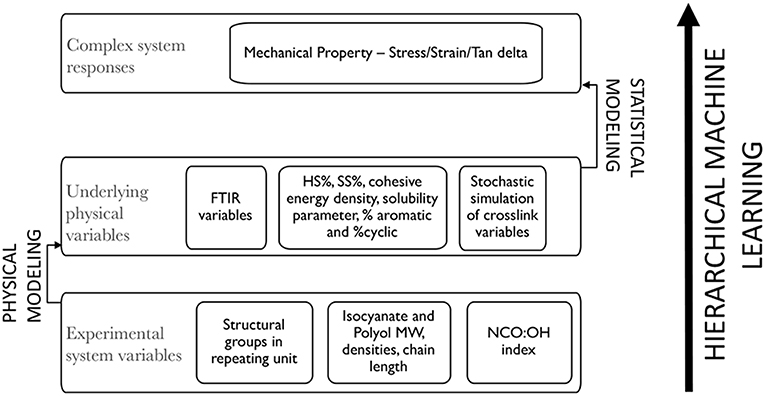
Figure 2. Schematic depicting the three layers in the Hierarchical Machine Learning approach.
The bottom layer of input or experimental variables in the model for PU consisted of the repeating unit in the synthesized polymer split into chemical structural units per kg of polymer, molecular weights and densities of the diisocyanates and polyols, the NCO:OH indices and the estimated chain length of polyols, assuming a PDI of 1. The chemical structures of the diisocyanates and polyols used in the training set are depicted in Figure 3. Each of the 18 polymers synthesized from these reactants have been categorized into a vector of structural units per kg of polymer (c, ch, ch2, ch3, c6h6, co, nh, o, nh2) calculated from the groups present in the reactants—the diisocyanates and polyols, the NCO:OH index, chain length of polyols and the total weight of the polymer synthesized.
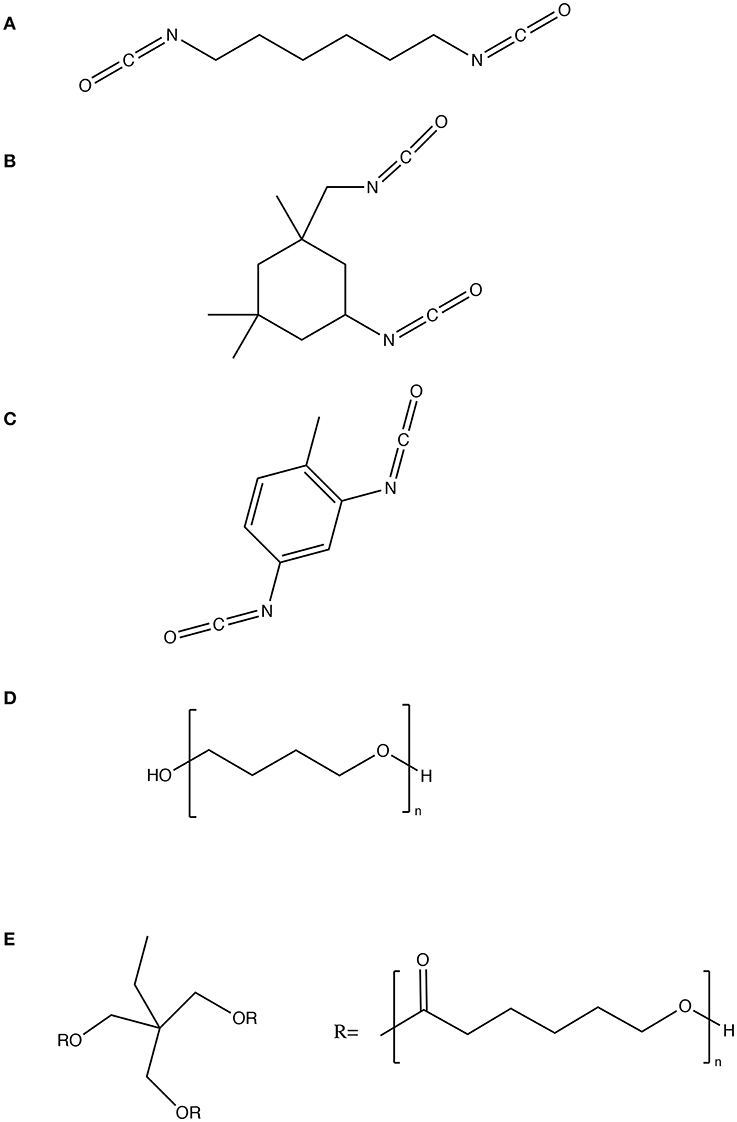
Figure 3. Chemical structures of reactants used in the training set (A) hexamethylene diisocyanate (B) isophorone diisocyanate (C) toluene diisocyanate (D) PTMEG 1000 and (E) polycaprolactone 900.
The middle layer or the intermediate physical/chemical variables have been grouped into three categories: the first will probe into the intermolecular interactions and absorption characteristics of the synthesized polymer molecule through FT-IR spectroscopy (Siesler, 1980). The presence of spectral features in specific regions of the spectrum is indicative of certain functional groups which have vibrational modes with large displacements and are minimally affected by the presence of other functional groups or atoms (Griffiths and de Haseth, 2007). For our study on polyurethanes, the wavenumbers exhibited by CO and NH groups as well as the ratio of absorbance values of NH to CO groups for each sample, are of particular importance and will provide better correlation to mechanical responses. It has been observed before that the CO stretching vibration and NH stretching vibration show different wavenumbers depending on the degree of H-bonding as well as crosslinking occurring due to trifunctional hydroxyl group polyol which significantly impact mechanical properties of such polyurethanes (Tsai et al., 1998). It is expected that a particular amount or degree of these factors, h-bonding and crosslinking, which results in the optimal mechanical response suited to a particular end-user application. The degree of H-bonding is also influenced through the symmetry of chemical structure of the reactants and presence of even/odd number of atoms (Caracciolo et al., 2009), making these variables highly significant for model prediction. These variables have been parameterized with respect to all the bottom-layer variables using a Gaussian regression-based framework using the scikit learn—ML library in Python (Williams and Rasmussen, 1996; Pedregosa et al., 2011). The FT-IR variables were then recalculated using the predict function from the Gaussian process models to be used with the top-layer variables.
The second set of middle layer variables consists of intermolecular chain interactions and properties pertaining to polyurethane polymer system. Hard segment (HS%) and soft segment (SS%) were easily calculated by mass of diisocyanate and polyols with respect to the total mass of the polymer. Similarly, % aromatic and % cyclic (non-aromatic) nature was calculated based on mass of respective structural units with respect to total mass of polymer. The solubility parameter and cohesive energy density (CED) was calculated using molar attraction constant, molar volume and cohesive energy of the polymer repeating unit using a group additivity-based approach on the structural units present in the bottom layer. The values for group contributions to the molar attraction constant, molar volume and cohesive energy are easily available in literature and have been extensively used before, for other polymer systems.
The third set of middle layer variables were the predictions of chain architecture from Monte Carlo simulations as described in the section above. Based on existing literature, these three categories of variables sufficiently model the main forces and interactions that govern the mechanical behavior of polyurethanes—the microstructure consisting of soft and hard domains which control permanent deformation, high modulus and tensile strength, hydrogen bonding between neighboring polymer chains control the elasticity as well as strain deformation behavior whereas simulation of chemical crosslinking addresses the mechanical behavior due to network formation.
Finally, the top layer, which consists of system responses (stress-at-break, strain-at-break, and Tan δ) have been modeled with the middle-layer variables using a random forest regression-based model from the scikit-ML library in Python. Random forest regression is an ensemble learning technique based on multiple decision trees learned from the provided variables. One of the advantages of a random forest model is its use of bagging or bootstrap aggregation where each decision tree is modeled on a subset of the input set but by drawing samples with replacement the subset has the same size as the original input set. Then, averaging is performed on all the decision trees to improve the prediction accuracy and to control overfitting. The number of decision trees used in our training set is equal to 100 and the max depth of trees was unrestricted since modeling was performed on a sparse dataset and are not concerned about memory consumption or computational efficiency, thus leading to better predictive power for the model. The estimator used from scikit -ML library is a “RandomForestRegressor” with the following attributes: bootstrap = True, criterion = “mse,” max_depth = None, max_features = “auto,” max_leaf_nodes = None, min_impurity_decrease = 0.0, min_impurity_split = None, min_samples_leaf = 1, min_samples_split = 2, min_weight_fraction_leaf = 0.0, n_estimators = 100, n_jobs = 4, oob_score = False, random_state = 0, verbose = 0, warm_start = False. Each of these attributes are well described at the scikit learn website for random forest regression:
https://scikit-learn.org/stable/modules/generated/sklearn.ensemble.RandomForestRegressor.html
and the code is available with Supplementary Files.
Results and Discussion
Figure 4 shows the mechanical responses for the training set. In general, Tan δ and strain-at-break are expected to decrease and stress-at-break is expected to increase with increasing NCO:OH index associated with the transition from rubbery toward a glassy state (Petrović et al., 2002; Levine et al., 2012) due to the presence of increasing urea linkages in the polymer network from moisture cure of excess NCO content. Furthermore, similar trends are expected with a general replacement of a bifunctional polyol with a higher functionality polyol, owing to crosslinking and network formation (Dušek and Dušková-Smrčková, 2000). Crosslinking also occurs either physically through hydrogen bonding between hard urethane segments or chemically through allophanate linkages due to excess NCO content during the polymerization reaction (Kontou et al., 1990). We see that even though some of our samples show expected behavior, others behave differently in either different response metrics or in all of them. In Figure 5, it can be observed that there is poor correlation between the various responses under a wide range of attained measurements. The diagonal grid represents the one-dimensional spread of values for single responses whereas the top right section represents the scatter plot correlation between response pairs and the bottom grid represents the two-dimensional spread as well as density of values for the response pairs. This suggests that there are multiple factors controlling these responses that may be competing.
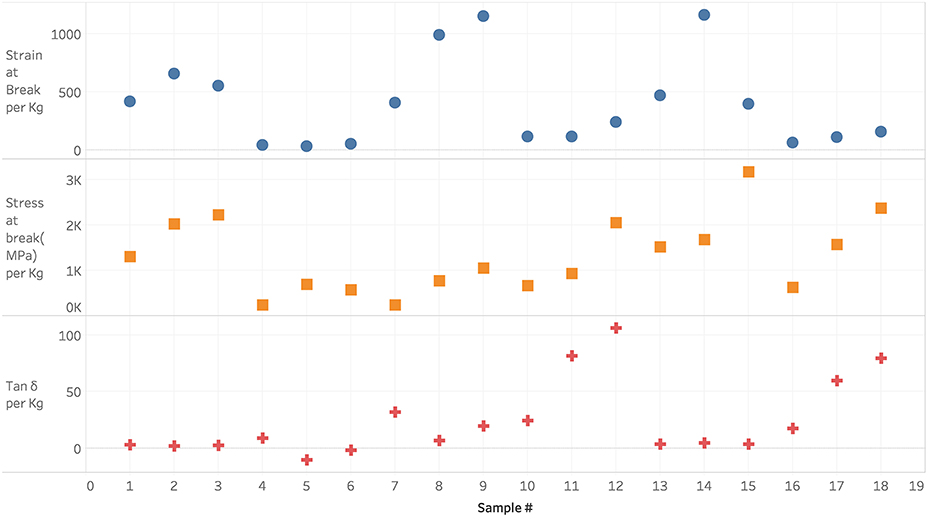
Figure 4. Mechanical responses measured for the training set of 18 polymers.
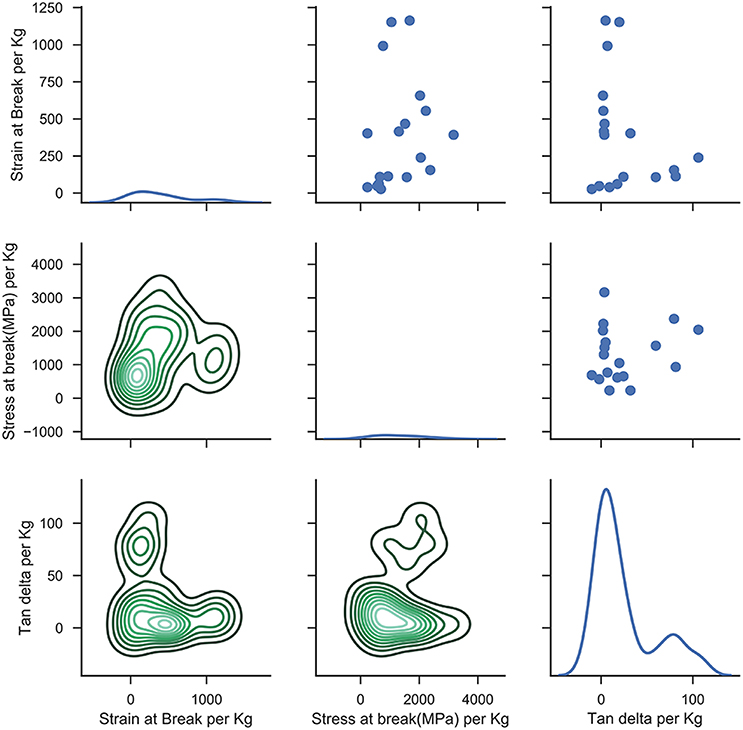
Figure 5. Lattice plot depicting the relation between various mechanical properties.
In order to deconvolute the relationship between these mechanical responses and the variables in the bottom layer of the model, the middle layer variables of our algorithm were parameterized in terms of the variables in bottom layer. Here, CO wavenumber, NH wavenumber and the ratio of NH absorbance per CO absorbance were modeled with a Gaussian Process regression using a train/test split to ensure accuracy and predictive capability. The train and test scores for the three variables are shown in Table 1. The IR values were regenerated from the learned GP model to be used further in the next training step.

Table 1. Training and test scores for GP model between FTIR variables and bottom layer variables.
The solubility parameter and the cohesive energy density for the polymers were calculated using group contribution methods (Van Krevelen and Te Nijenhuis, 2009) using the equation (1) for solubility parameter
where Fi is the molar attraction contribution and Vm, i is the molar volume contribution for the ith structural unit in the bottom layer and the equation (2) for cohesive energy density is
where Ecoh, i is the cohesive energy contribution for the ith structural unit in the bottom layer. HS%, SS%, nEff, elastic_link_mtw, Mtw, sol_pctWgt, and core_pctWgt were calculated and simulated as mentioned in the previous section.
After generating the set of middle layer variables, a random forest regression model was fitted between the latter and the mechanical responses (stress-at-break, strain-at-break, and Tan δ), and the train/test scores for all responses are shown in Table 2. The feature importance values from the RF model are shown in Figure 6, the predicted vs. test values are shown in Figure 7 and the ensemble averaged trees are shown in Figure 8.
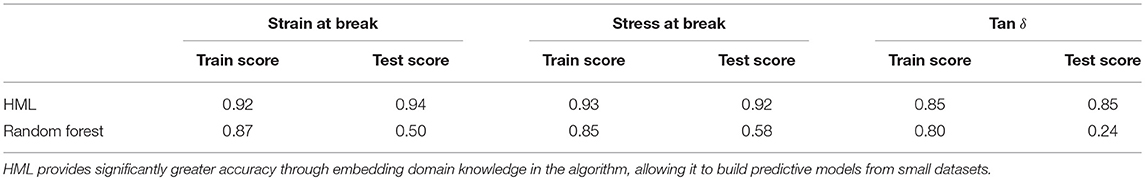
Table 2. Training and test scores for HML and Random Forest modeling of mechanical properties as a function of composition.
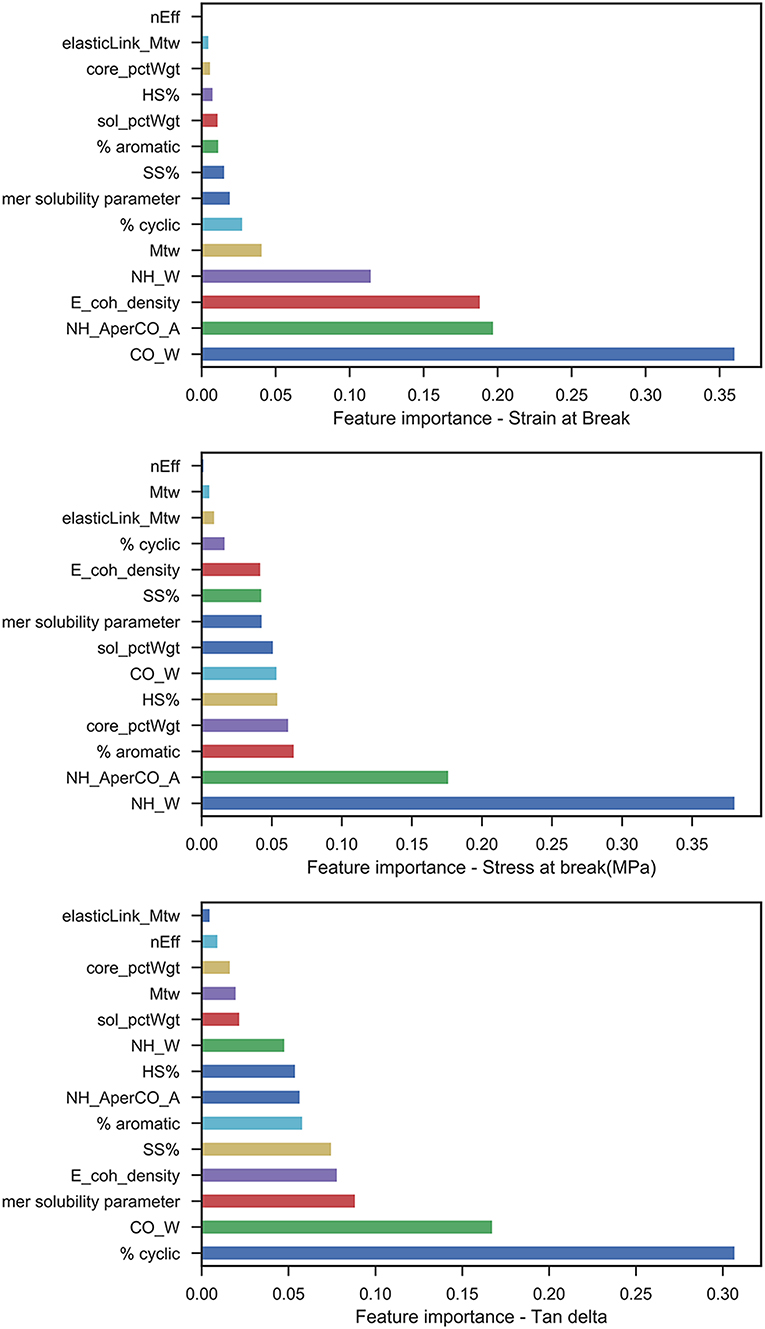
Figure 6. Feature importance plots from the trained Random Forest model for each of the mechanical responses measured.
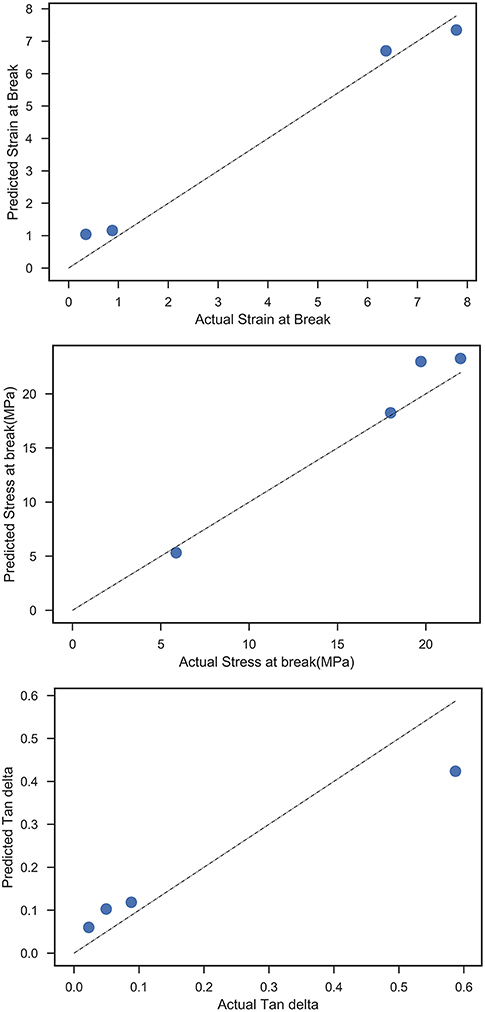
Figure 7. Predicted vs. actual values for all mechanical responses from the HML model.
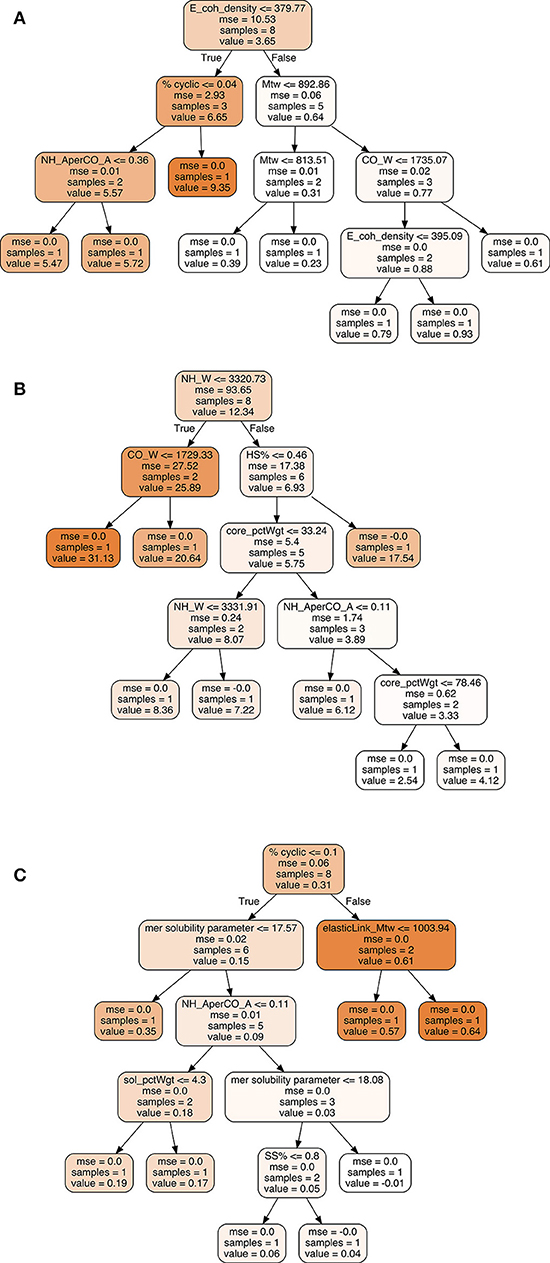
Figure 8. Ensemble averaged decision tree from Random Forest model trained to predict (A) strain-at-break (B) stress-at-break, and (C) Tan δ.
Interestingly, the most important features for prediction of strain-at-break from the trained model were CO wavenumber, NH absorbance per CO absorbance, cohesive energy density, and NH wavenumber. This could be due to the vibrational shift in the CO and NH bands, which relate to the chemical environment obtained with different reactant combinations (polyols and diisocyanates) whereas the ratio of carbonyl peak absorbance to amide peak absorbance may indicate the effect of NCO:OH index. The shift in frequencies is an indication of hydrogen bonding strength between polymer chains, primarily due to interactions between CO and NH groups. Strong hydrogen bonding between the groups will make the bond within the carbonyl and NH groups weaker, for e.g., HDI and PTMEG 1000 exhibit strong hydrogen bonding, thus higher strain at break as the CO wavenumber is ~1,683 cm−1 and NH wavenumber is ~3,318 cm−1. Similarly, between HDI and PCL 900, weaker hydrogen bonding reduces strain at break where the CO wavenumber is ~1,732 cm−1 and NH wavenumber is ~3,380 cm−1. Cohesive energy density is a proxy for intermolecular forces within polymer chains, and as such strongly links to forces required for mechanical deformation of a polymeric material. CED is also notable as a property prediction tool for calculating relative strain at failure for similarly networked chains i.e., under presence of moderate chemical crosslinking, CED can improve toughness of chains during large strain deformation (Safranski and Gall, 2008). Other parameters in our model such as % cyclic and Mtw (average molar weight of core gel component as a percentage of total polymer weight) also have smaller yet influencing behavior on strain.
Apart from the FTIR derived variables, the model for stress shows a reliance on hard segment %, core_pctWgt (percentage of core gel component) and % aromatic behavior. This makes sense as the hard segment in a polyurethane is the load bearing component under mechanical deformation and as a result, induces most of the elastic response in the system. At higher HS%, urethane—urethane hydrogen bonding in particular is also increased. The core gel component represents the crosslinked structure in a polyurethane which again corresponds to mechanical strength and load bearing nature of the polymer. It is interesting that the model identified % aromatic behavior as an important feature: it impacts mechanical strength due to much more efficient hydrogen bonding and pi-stacking between aromatic groups in neighboring chains. Aromatic groups in isocyanates account for stiffer chains and result in a higher melting point polyurethane as well.
Tan δ represents the damping behavior in the mechanical performance of a viscoelastic polymer, i.e., the ratio of plastic behavior to elastic behavior. Thus, it was not surprising to observe both hard segment % and soft segment % as important features, however % cyclic behavior has an interestingly significant impact on Tan δ. Even though cyclic groups correspond to stiffness and rigidity, they contribute less than aromatic groups due to the possibility of configurational isomerism as well as non-planar structures. This might explain the ability of these groups to absorb more energy while mechanical stress is applied and provide a good balance between elastic and plastic performance. Other crucial features which were identified in the model were the CO wavenumber, cohesive energy density, and mer solubility parameter.
Each random forest model shown in Figure 8 represents an averaged decision tree from an ensemble of decision trees, for each mechanical response. The random forest classifier from Python scikit learn uses bootstrap aggregating in which multiple decision trees are modeled on subsets of training data, chosen randomly with replacement. Each predictor or feature is learned and is split for values based on a mean squared error reduction scheme, which is continued until all the data is split till the last node. Bootstrap aggregation is an excellent stochastic method of avoiding overfitting in the trained model and reduces variance in result without increasing the bias of the model. In random forests, this allows for an out-of-bag (OOB) error estimate to measure prediction error of a trained decision tree on the subset of data not used in that tree, thus negating the need of an independent validation dataset.
In order to evaluate the prediction efficiency on the sparseness of the training set, a standard big data approach was taken where the same random forest framework was applied on a training set containing the mechanical responses and our tunable formulation variables namely, polyol choice, diisocyanate choice, and NCO:OH index. As expected, the model converged with significantly lower test scores as shown in Table 2 and the predicted responses vs. test data can be seen in Figure 9.
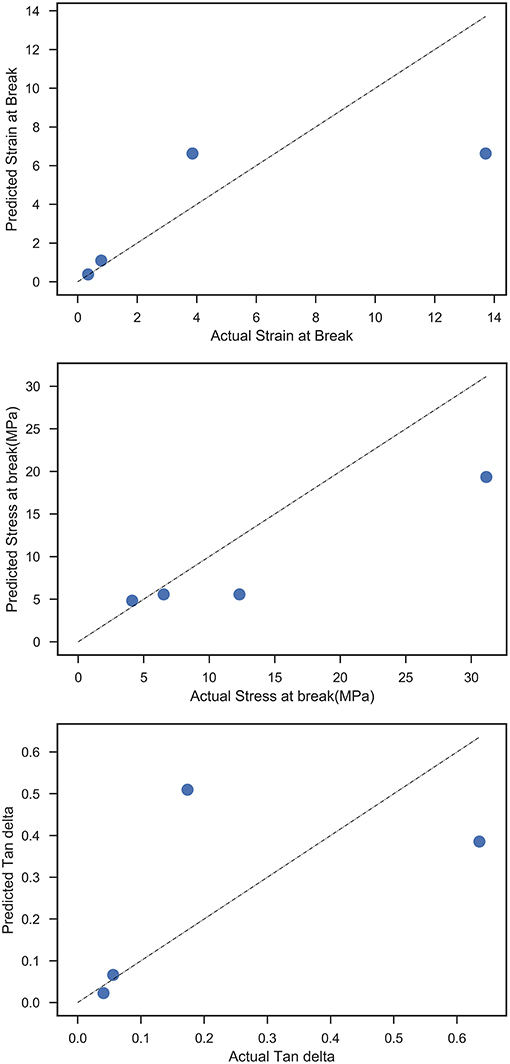
Figure 9. Predicted vs. actual values for all mechanical responses from the Random Forest model, using a black-box approach.
If compared with other approaches, this methodology mainly benefits from the planned surrogate physical and chemical measurements, and existing scientific literature to embed domain knowledge with statistical learning to strongly improve predictive capability. In traditional material industries, high throughput data for a single product family with specific end user application is hard to collect; it requires huge investment in time, effort, and cost. Thus, computational techniques relying on big data will not be beneficial for shortening the research and development cycle in such industries as shown earlier. Analytical approaches are better in some respects as they have physical laws and chemistry as underpinnings for property prediction however most of the approaches are highly complex to practically apply in an industry setting and highly sensitive to lack of required data/measurements. Design of experiments offers an accepted method for predicting structure-property relationships for small datasets, yet it does not provide insight into how the underlying forces interact with each other to achieve a specific system response since it is purely statistical in nature and may not accurately predict synergies between variables. HML aims to learn from the shortcomings as well as advantages of the previous mentioned approaches by utilizing and building upon the existing scientific domain expertise with much lesser measurements, providing a tool for not only property prediction but also to elucidate upon the nature of physical and chemical interactions that shape a system response.
Conclusion
Using HML algorithm, mechanical responses of a training set of polyurethanes were as a function of monomer chemistry, index, and chain architecture. The accuracy was compared against a random forest model and it was found that HML produced significantly better predictions of the test data. This was attributed to integration of an intermediate layer of variables comprising domain knowledge based physicochemical factors which significantly improved the model relating experimental formulation variables and mechanical responses of the cured elastomers. Some of the advantages of this approach are (a) the possibility of modeling categorical and qualitative responses of polyurethane products to formulation and processing variables and (b) predicting the properties of novel monomers, such as bio based materials. In future work, we intend to model such responses and also test our model by substituting polyols and diisocyanates to further investigate the predictive nature and capability of the HML algorithm on polymer systems.
Author Contributions
AM (CMU) conducted experiments, performed machine-learning modeling, and wrote parts of the manuscript. JT-C (Covestro, LLC) performed Monte Carlo simulations and wrote parts of the manuscript. NW (CMU) oversaw all aspects of the research and wrote parts of the manuscript.
Conflict of Interest Statement
JT-C is employed by Covestro, LLC and NW has started a company to explore commercial applications of the algorithm described here and declares a potential conflict of interest. NW owns Ansatz AI, LLC. AM declares no competing financial interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmats.2019.00087/full#supplementary-material
References
Agrawal, A., and Choudhary, A. (2016). Perspective: materials informatics and big data: realization of the “fourth paradigm” of science in materials science. APL Mater. 4:053208. doi: 10.1063/1.4946894
Caracciolo, P. C., Buffa, F., and Abraham, G. A. (2009). Effect of the hard segment chemistry and structure on the thermal and mechanical properties of novel biomedical segmented poly(esterurethanes). J. Mater. Sci. Mater. Med. 20, 145–155. doi: 10.1007/s10856-008-3561-8
de Pablo, J. J., Jones, B., Kovacs, C. L., Ozolins, V., and Ramirez, A. P. (2014). The materials genome initiative, the interplay of experiment, theory and computation. Curr. Opin. Solid State Mater. Sci. 18, 99–117. doi: 10.1016/j.cossms.2014.02.003
Dušek, K., and Dušková-Smrčková, M. (2000). Network structure formation during crosslinking of organic coating systems. Prog. Polym. Sci. 25, 1215–1260. doi: 10.1016/S0079-6700(00)00028-9
Engels, H.-W., Pirkl, H.-G., Albers, R., Albach, R. W., Krause, J., Hoffmann, A., et al. (2013). Polyurethanes: versatile materials and sustainable problem solvers for today's challenges. Angew. Chemie Int. Ed. 52, 9422–9441. doi: 10.1002/anie.201302766
Eom, K., Li, P.-C., Makarov, D. E., and Rodin, G. J. (2003). Relationship between the mechanical properties and topology of cross-linked polymer molecules: parallel strands maximize the strength of model polymers and protein domains. J. Phys. Chem. B 107, 8730–8733. doi: 10.1021/jp035178x
Foreman, J. P., Porter, D., Behzadi, S., and Jones, F. R. (2008). A model for the prediction of structure-property relations in cross-linked polymers. Polymer 49, 5588–5595. doi: 10.1016/j.polymer.2008.09.034
Griffiths, P. R., and de Haseth, J. A. (2007). Fourier Transform Infrared Spectrometry. Hoboken, NJ: John Wiley & Sons, Inc.
Kim, C., Chandrasekaran, A., Huan, T. D., Das, D., and Ramprasad, R. (2018). Polymer genome: a data-powered polymer informatics platform for property predictions. J. Phys. Chem. C 122, 17575–17585. doi: 10.1021/acs.jpcc.8b02913
Kontou, E., Spathis, G., Niaounakis, M., and Kefalas, V. (1990). Physical and chemical cross-linking effects in polyurethane elastomers. Colloid Polym. Sci. 268, 636–644. doi: 10.1007/BF01410405
Levine, F., Escarsega, J., and La Scala, J. (2012). Effect of isocyanate to hydroxyl index on the properties of clear polyurethane films. Prog. Org. Coatings 74, 572–581. doi: 10.1016/j.porgcoat.2012.02.004
Li, Y., Tang, S., Abberton, B. C., Kröger, M., Burkhart, C., Jiang, B., et al. (2012). A predictive multiscale computational framework for viscoelastic properties of linear polymers. Polymer 53, 5935–5952. doi: 10.1016/j.polymer.2012.09.055
Menon, A., Childs, C. M., Poczós, B., Washburn, N. R., and Kurtis, K. E. (2018). Molecular engineering of superplasticizers for metakaolin-portland cement blends with hierarchical machine learning. Adv. Theory Simulations 2:1800164. doi: 10.1002/adts.201800164
Menon, A., Gupta, C., Perkins, K. M., DeCost, B. L., Budwal, N., Rios, R. T., et al. (2017). Elucidating multi-physics interactions in suspensions for the design of polymeric dispersants: a hierarchical machine learning approach. Mol. Syst. Des. Eng. 2, 263–273. doi: 10.1039/C7ME00027H
Mikes, J., and Dusek, K. (1982). Simulation of polymer network formation by the Monte Carlo method. Macromolecules 15, 93–99. doi: 10.1021/ma00229a018
Miller, D. R., and Macosko, C. W. (1976). A New Derivation of Post Gel Properties of Network Polymers. Macromolecules 9, 206–211. doi: 10.1021/ma60050a004
Milner, S. T., and McLeish, T. C. B. (1998). Reptation and contour-length fluctuations in melts of linear polymers. Phys. Rev. Lett. 81, 725–728. doi: 10.1103/PhysRevLett.81.725
Oertel, G. (1994). Polyurethane Handbook: Chemistry- Raw Materials-Processing-Application-Properties. Second Edition. New York, NY: Hanser Publishers.
Pattamaprom, C., Larson, R. G., and Van Dyke, T. J. (2000). Quantitative predictions of linear viscoelastic rheological properties of entangled polymers. Rheol. Acta 39, 517–531. doi: 10.1007/s003970000104
Pedregosa, F., Varoquaux, G., Gramfort, A., Michel, V., Thirion, B., Grisel, O., et al. (2011). Scikit-learn: machine learning in {p}ython. J. Mach. Learn. Res. 12, 2825–2830.
Petrović, Z. S., Zhang, W., Zlatanić, A., Lava, C. C., and Ilavský, M. (2002). Effect of OH/NCO molar ratio on properties of soy-based polyurethane networks. J. Polym. Environ. 10, 5–12. doi: 10.1023/A:1021009821007
R Core Team (2018). R: A Language and Environment for Statistical Computing. Available online at: http://www.r-project.org/ (accessed February 1, 2019).
Rossum, G. (1995). Python Reference Manual. Amsterdam: CWI (Centre for Mathematics and Computer Science).
Roy, N. K., Potter, W. D., and Landau, D. P. (2006). Polymer property prediction and optimization using neural networks. IEEE Trans. Neural Netw. 17, 1001–1014. doi: 10.1109/TNN.2006.875981
Safranski, D. L., and Gall, K. (2008). Effect of chemical structure and crosslinking density on the thermo-mechanical properties and toughness of (meth)acrylate shape memory polymer networks. Polymer 49, 4446–4455. doi: 10.1016/j.polymer.2008.07.060
Shenogina, N. B., Tsige, M., Patnaik, S. S., and Mukhopadhyay, S. M. (2012). Molecular modeling approach to prediction of thermo-mechanical behavior of thermoset polymer networks. Macromolecules 45, 5307–5315. doi: 10.1021/ma3007587
Siesler, H. W. (1980). Fourier transform infrared (ftir) spectroscopy in polymer research. J. Mol. Struct. 59, 15–37. doi: 10.1016/0022-2860(80)85063-0
Tsai, Y.-M., Yu, T.-L., and Tseng, Y.-H. (1998). Physical properties of crosslinked polyurethane. Polym. Int. 47, 445–450. doi: 10.1002/(SICI)1097-0126(199812)47:4 <445::AID-PI82>3.0.CO;2-B
Van Krevelen, D. W., and Te Nijenhuis, K. (2009). Properties of Polymers. Amsterdam: Elsevier. doi: 10.1016/B978-0-08-054819-7.X0001-5
van Ruymbeke, E., Keunings, R., and Bailly, C. (2005). Prediction of linear viscoelastic properties for polydisperse mixtures of entangled star and linear polymers: modified tube-based model and comparison with experimental results. J. Nonnewton. Fluid Mech. 128, 7–22. doi: 10.1016/j.jnnfm.2005.01.006
van Ruymbeke, E., Keunings, R., Stéphenne, V., Hagenaars, A., and Bailly, C. (2002). Evaluation of reptation models for predicting the linear viscoelastic properties of entangled linear polymers. Macromolecules 35, 2689–2699. doi: 10.1021/ma011271c
Keywords: polyurethane, machine learning, structure-property relationship, property prediction, tunability
Citation: Menon A, Thompson-Colón JA and Washburn NR (2019) Hierarchical Machine Learning Model for Mechanical Property Predictions of Polyurethane Elastomers From Small Datasets. Front. Mater. 6:87. doi: 10.3389/fmats.2019.00087
Received: 05 February 2019; Accepted: 08 April 2019;
Published: 08 May 2019.
Edited by:
Christian Johannes Cyron, Hamburg University of Technology, GermanyReviewed by:
David Cereceda, Villanova University, United StatesYouyong Li, Soochow University, China
Copyright © 2019 Menon, Thompson-Colón and Washburn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Newell R. Washburn, d2FzaGJ1cm5AYW5kcmV3LmNtdS5lZHU=