 Cassandra T. Buru
Cassandra T. Buru Jiafei Lyu
Jiafei Lyu Jian Liu
Jian Liu Omar K. Farha
Omar K. Farha- 1Department of Chemistry, Northwestern University, Evanston, IL, United States
- 2Department of Pharmaceutical Engineering, School of Chemical Engineering and Technology, Tianjin University, Tianjin, China
- 3Key Laboratory of Systems Bioengineering, Ministry of Education, Tianjin University, Tianjin, China
Comprised of high valent metal centers connected via oxide bonds, polyoxometalates (POMs) have a rich history in redox reactions. These discrete metal oxide clusters often are immobilized on heterogeneous supports to increase stability and allow for recyclability for catalytic reactions. One such support is the metal-organic framework (MOF), NU-1000. When POMs are installed into NU-1000, they are first sited in the mesoporous channel. Upon application of heat and removal of some physisorbed water, the POMs migrate to the microporous channel, where some active sites are blocked by the MOF linkers, resulting in lowered catalytic activity. To restrict this movement from the mesopore, we report here the use of two MOFs, naphthalene dicarboxylate-modified NU-1000 (NU-1000-NDC) and NU-1008, as supports for the Keggin-type polyoxometalate (POM), H3PW12O40. Upon the application of heat, these frameworks were found to hinder or prevent the movement of the POM between channels by blocking the aperture(s) connecting the mesoporous and microporous channels. The composite POM@MOF materials were used for the oxidation of a mustard gas simulant, 2-cholorethyl ethyl sulfide, using H2O2 as the oxidant. The POM@MOF catalysts exhibit enhanced reactivity when compared to the POM or MOF alone, more than doubling the initial rates. The activity trend in PW12@NU-1000-NDC matched that in PW12@NU-1000, where the scCO2-activated sample poised the POM in the mesopores to give greater substrate accessibility and enhance the reaction rate compared to the heated sample where the POMs are poised in the micropores. Contrary to these observed trends, PW12@NU-1008 prohibits POM migration and performs superior when water has been removed at elevated temperatures as the active sites are more accessible in the mesoporous channel.
Introduction
The active sites in catalysts are often sensitive to their surrounding environments. Consequently, active site engineering has been developed to allow the modulation of a catalyst environment to exploit or inhibit activity (Pikus et al., 1997; Garce et al., 2009; Nasalevich et al., 2014; Liu et al., 2018; Reiner et al., 2019); however, tuning of the solid support, as in single-site heterogeneous catalyst systems, is often difficult to achieve. For this reason, metal-organic frameworks (MOFs) with a high degree of tunability are attractive support materials (Vermoortele et al., 2012; Vandichel et al., 2015; Liu et al., 2018; Palmer et al., 2018). Constructed from metal or metal cluster nodes linked via multitopic organic ligands, MOFs are an increasing popular class of designer materials due to the ability to obtain highly crystalline, porous, and stable structures (Yaghi et al., 2003; Kitagawa et al., 2004; Furukawa et al., 2013; Jiao et al., 2018; Chen et al., 2019; Yang and Gates, 2019).
One ubiquitous class of materials which has benefitted from heterogenization and immobilization on solid supports is polyoxometalates (POMs) (Long et al., 2007; Frenzel et al., 2016; Hou et al., 2018). POMs are isolated metal oxide clusters, whose tunable size, charge density, and acid strength allow for a wide range of applications, primarily in catalysis (Pope, 1983; Wang and Yang, 2015; Anyushin et al., 2018; Gumerova and Rompel, 2018; Sullivan et al., 2018). However, POMs suffer from low surface areas, high solubility in polar solvents, and limited stability in aqueous environments, making POMs difficult to exploit as reusable catalysts (Kholdeeva et al., 2010). To increase surface area and impart some stability, POMs can be supported on porous heterogeneous materials, among which are metal-organic frameworks (MOFs) (Salomon et al., 2018; Wang et al., 2018; Zhong et al., 2018).
As support materials (Sosa et al., 2018), MOFs can interact with POMs and other guest molecules via chemical bonds, steric encapsulation within pores, or electrostatic interactions. With chemical bonding as found in polyoxometalate open frameworks, POMs serve as the nodes of the framework (Nohra et al., 2011; Miras et al., 2014), and consequently the active sites are blocked by the structural linkers. Steric trapping relies on a MOF whose pores can accommodate a POM, but whose apertures are much smaller than the POM thus preventing POM diffusion out of the MOF (Song et al., 2011; Salomon et al., 2015; Cai et al., 2019). This aperture dependence was investigated in a recent report (Lin et al., 2018). Lastly, functionalities in MOFs can impart favorable electrostatic interactions (Férey et al., 2005). For example, in Zr-based MOFs, the nodes have an affinity for oxyanions (Howarth et al., 2015). Without these weak interactions, the POM guests are able to freely diffuse in and out of the MOF, making a composite structure difficult to isolate and recycle.
NU-1000 is a Zr-based csq-net MOF comprised of mesoporous 31 Å hexagonal channels and microporous 12 Å triangular channels (Figure 1). We have recently reported the impregnation of NU-1000 with phosphotungstic acid (H3PW12O40) resulting in the composite material, PW12@NU-1000, which was stable to leaching (Buru et al., 2017, 2018; Ahn et al., 2018) due to favorable electrostatic interactions. When the material activated (removing the solvent guest molecules) using supercritical CO2 (scCO2) drying, the [PW12O40]3− clusters were found in the easily accessible mesopore. Evidence of this location was observed in the pore-size distribution calculated using the N2 isotherm and confirmed via difference envelope density analysis, where the high intensity, low angle peaks of a powder X-ray diffraction (PXRD) pattern generated from a synchrotron X-ray source were used to create an electron envelope from with the known parent MOF was subtracted and the residual electron density mapped onto the parent MOF indicated the location of the guest molecules (Yakovenko et al., 2014; Gallington et al., 2016). Upon heating the scCO2-dried material, the [PW12O40]3− clusters migrate from the mesoporous channel to the microporous channel, likely owing to the enhanced electrostatic and van der Waals interactions when cited in the micropore. The movement was also accompanied by the loss of water weight from thermogravimetric analysis (TGA). We proposed that this movement occurs because the POM can fit in the c-pores, the 8 × 10 Å windows between the channels, since this transformation readily occurs in the absence of solvent. Different activations of these materials yielded different rates and selectivities in the oxidation of 2-chloroethyl ethyl sulfide (CEES), a mustard gas (bis(2-chloroethyl)sulfide) simulant (Figure 2). Interestingly, due to mechanistic differences (Al-Ajlouni et al., 2007; Lousada et al., 2012), the homogeneous H3PW12O40 promoted the selective oxidation of CEES, while the MOF alone catalyzed the over-oxidation. This allowed for insight into which clusters were more accessible in the composite material through analysis of reaction selectivity.
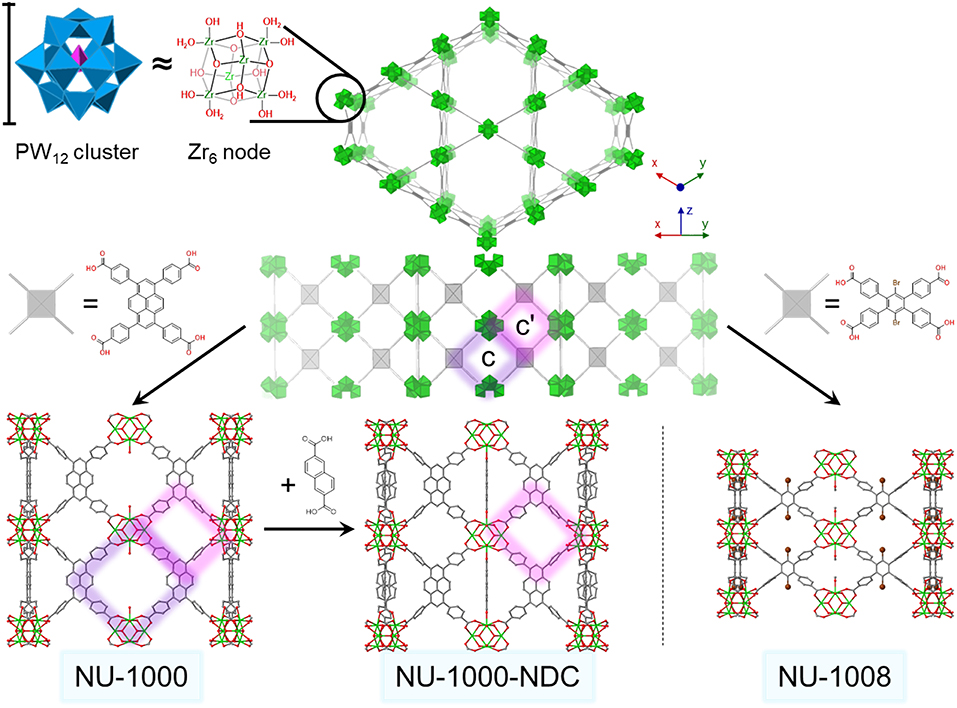
Figure 1. Generic csq structure (top) viewed down the c-axis and an orthogonal view. Structures for zirconium node and size comparison to PW12 cluster (top left). Crystal structures and structural linkers of NU-1000 (lower left), NU-1000-NDC (lower center), and NU-1008 (lower right) viewed from the ab direction. The c- and c′- pores are highlighted in purple and pink, respectively. For clarity, hydrogens are omitted, and one possible conformation of NDC has been extracted. Green polyhedron = Zr6 node, blue polyhedron = WO5, pink polyhedron = PO4, green sphere = Zr, red = O, gray = C, brown = Br.
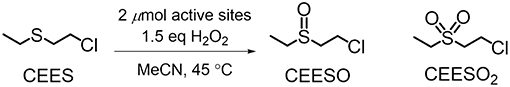
Figure 2. Oxidation of CEES to CEESO and CEESO2 under the conditions reported in Buru et al. (2018).
To determine a more exact nature of this migration and the effect the POM location has on catalysis, we employ here two MOFs, NU-1000-NDC and NU-1008 (Figure 1) (Lyu et al., 2018; Peters et al., 2018) to immobilize [PW12O40]3− clusters. These MOFs have the hierarchical channel-type structure that NU-1000 possesses, except the apertures connecting the channels are blocked in NU-1000-NDC (c-pores in Figure 1) or are smaller in NU-1008 (c- and c′-pores in Figure 1). This would inhibit [PW12O40]3− movement between channels if the mechanism of POM migration was allowed by movement through these windows, so that the composites activated via supercritical CO2 drying and via heating under vacuum should only differ in the amount of physisorbed water. The influence of this residual water is investigated in the catalytic oxidation of CEES, using hydrogen peroxide.
Results and Discussion
NU-1000-NDC and NU-1008 were selected for this study to investigate whether the [PW12O40]3− moves through the c- and c′-pores (Figure 1) as previously proposed. In NU-1000-NDC, 2,6-naphthalene dicarboxylate (NDC) bridges two nodes in the c-pore, effectively reducing the size of that aperture (Peters et al., 2018). Both the c- and c′-pore diameters in NU-1008 are reduced in size compared to those in NU-1000-NDC (Lyu et al., 2018; Wasson et al., 2018). If the mechanism involved complete desorption from the mesopore and re-adsorption into the microporous channel of the POM, the size and accessibility to the c/c′-pores would have negligible effect on POM movement.
PW12@NU-1000-NDC and PW12@NU-1008 were prepared in a similar manner to PW12@NU-1000 (Buru et al., 2017). Briefly, the MOFs were soaked in an aqueous H3PW12O40 solution for 3 days, followed by rigorous washing with water and ethanol. After soaking in ethanol overnight, the composites were subjected to supercritical CO2 drying. Half the material was then heated to 120°C for at least 2 h and designated PW12@NU-1008-120°C and PW12@NU-1000-NDC-120°C, while the other half was used “as-is” for subsequent characterization and designated PW12@NU-1008-scCO2 and PW12@NU-1000-NDC-scCO2.
Inductively coupled plasma optical emission spectroscopy (ICP-OES) analyses of the digested MOFs show that [PW12O40]3− does not leach during heating. The loadings were found to be 0.7 PW12/Zr6 node for NU-1000-NDC and 0.8 PW12/Zr6 node in NU-1008. Scanning electron microscopy (SEM) images reveal that the size and shape of the MOF crystallites remains the same before and after POM loading (Figures S1, S2). Energy dispersive X-ray spectroscopy (EDS) line scans display that the POM distribution in the MOF is homogeneous throughout the crystallites.
From the TGA traces (Figure S3) of the materials under nitrogen, both scCO2-activated MOFs lost ~5 wt% more than the 120°C-activated counterparts at 250°C. This initial loss has been attributed to residual water physisorbed to the POM species, which remain after removal of solvent. Once heated to 120°C, the elevated temperature activation step removes the physisorbed water which was difficult to remove from the POM after scCO2-drying alone. These residual waters were factored into catalyst loading calculations.
In the N2 sorption isotherms (Figures S4, S5), the maximum uptakes of the composites are significantly lower gravimetrically than the parent MOFs. Since the POMs constitute >50 wt% according to ICP analyses, volumetric equivalents were obtained after estimating densities to determine whether the decreased uptake was a result of mass gain or the degradation of the MOF (Figure 3). For NU-1000-NDC, the MOF has a Brunauer-Emmett-Teller (BET) area of 1,150 m2/cm3 which slightly decreases to 990 m2/cm3 in PW12@NU-1000-NDC-scCO2 and 1,100 m2/cm3 for PW12@NU-1000-NDC-120°C. Similarly, in NU-1008 the MOF has a BET area of 910 m2/cm3, and the scCO2 and 120°C activated composites have areas of 820 and 880 m2/cm3, respectively. With these marginal decreases in BET areas, the decreased gravimetric uptake can be attributed primarily to the additional mass of the POMs and not the degradation of the MOF. In the density functional theory (DFT)-calculated pore size distribution (PSD) for NU-1000-NDC, the mesopore around 30 Å decreases after POM loading and scCO2-drying (Figure 3C). Once heated to 120°C, some of the mesopore volume is regained. This hints that the POM occupies the mesopore after scCO2 drying and some POM leaves after heating. The PSD for NU-1008 shows a similar behavior from the MOF to the scCO2-dried sample (Figure 3D); however, after heating the mesopore volume is retained, suggesting that the POMs are unable to move from the mesopore.
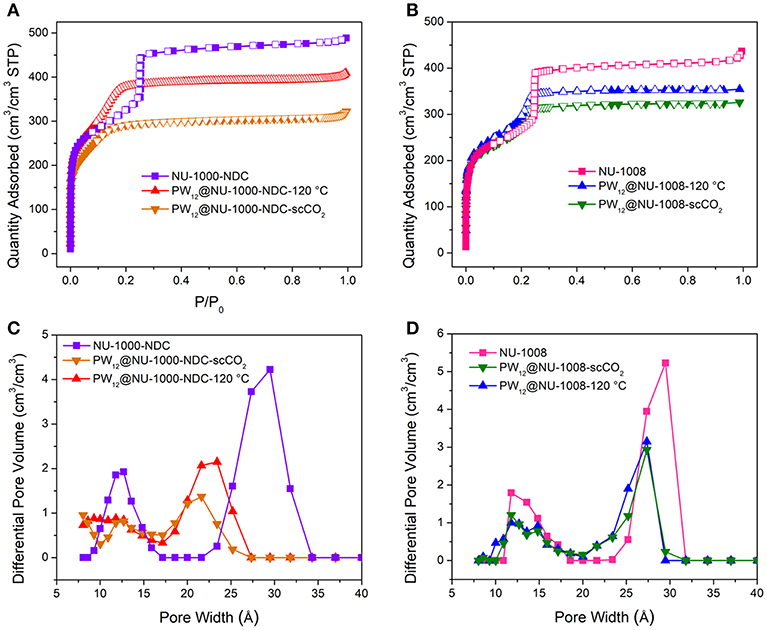
Figure 3. N2 sorption isotherms of (A) NU-1000-NDC with and without POM and (B) NU-1008 with and without POM. Corresponding DFT-calculated PSD of (C) NU-1000-NDC and its composites and (D) NU-1008 and its composites.
The PXRD patterns for the parent NU-1000-NDC and NU-1008 are similar (Figure S6); the most prominent peaks arise from the (100) plane around 2θ = 2.5° and (200) peak around 5°. Once the POM is installed, the highest intensity is the (2–10) Bragg peak appearing around 2θ = 4°, indicating the extra electron density from the POMs resides in the mesopore (Buru et al., 2018). ScCO2 drying has minimal effect on the composites PXRD pattern (Figure 4), suggesting little to no POM movement during activation. After heating to 120°C for 2 h, neither composite appears to have changed. However, after 12 h of heating at 120°C, the PW12@NU-1000-NDC closely resembles the parent MOF, indicating that the POMs now occupy the micropores (Figure 5). This change is indicative of the c′-pores being flexible enough to allow POM movement. In PW12@NU-1000, complete movement of the POM from the meso- to micropores occurs, suggesting that PW12@NU-1000-NDC requires more energy to undergo the same transformation, so while blocking the c-pore does not prevent the POM from moving, it does force the POM through a higher energy barrier intermediate. Interestingly, PW12@NU-1008 appears to have retained its PXRD pattern under the same heating conditions, indicating the POMs remain in the mesopore (Figure 5) and supporting the previously proposed mechanism where the POMs move through the c- or c′-pores. In situ variable temperature PXRD patterns (Figure S7) show this transformation occurs gradually over time.
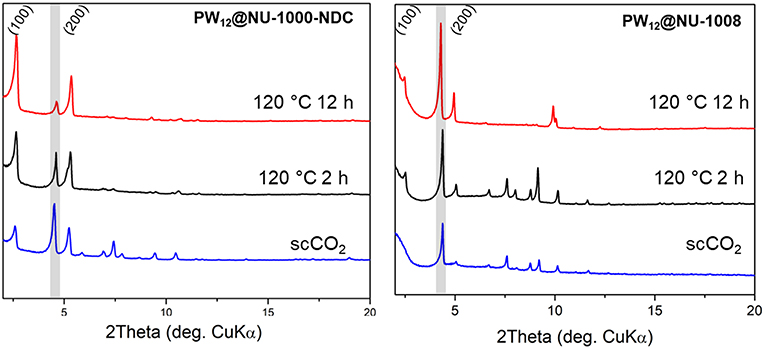
Figure 4. Ex situ PXRD patterns of PW12@NU-1000-NDC (Left) and NU-1008 (Right) under specified activation conditions. Gray boxes the (2–10) reflection.
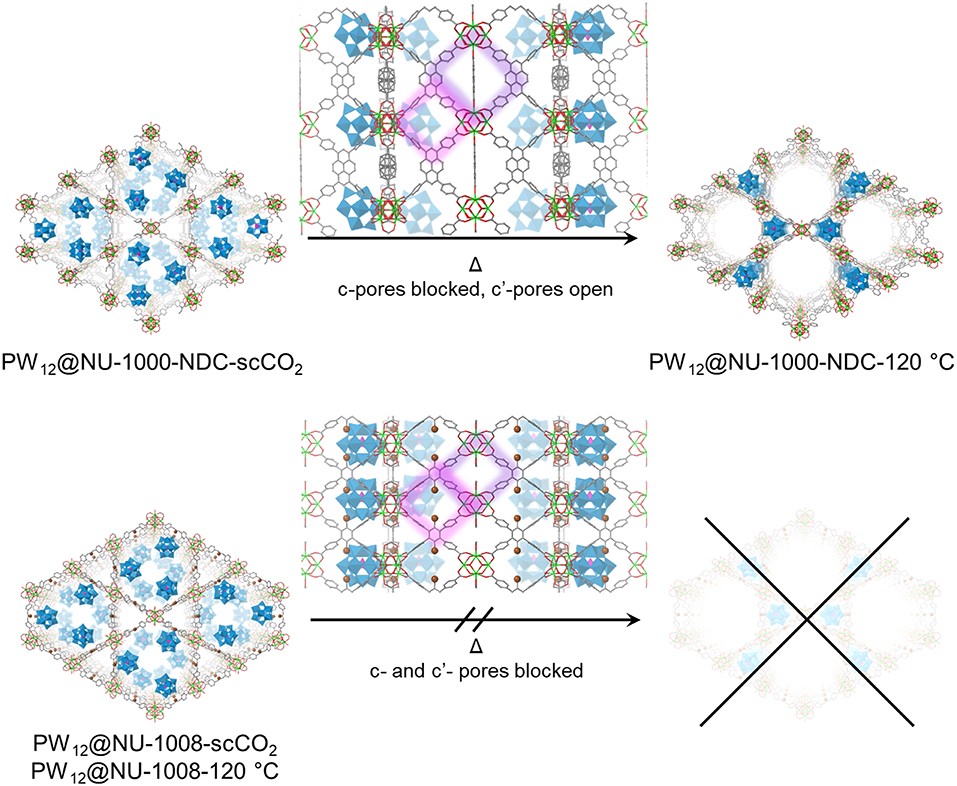
Figure 5. Proposed structures and transitions for PW12@NU-1000-NDC (top) and PW12@NU-1008 (bottom) under heating conditions. The c- and c′- pores are highlighted in purple and pink, respectively. For clarity, hydrogens are omitted. Green polyhedron = Zr6 node, blue polyhedron = WO5, pink polyhedron = PO4, green sphere = Zr, red = O, gray = C, brown = Br.
The oxidation of CEES was selected as a model reaction (Figure 2). The catalytic setup consists of 1 mol% catalyst (0.86 μmol active clusters, i.e., a node or POM) in a vial suspended in acetonitrile and heated to 45°C. CEES and internal standard were then added once the vial reached the set temperature. Then to start the reaction, 1.5 eq of 30% aqueous H2O2 was added. The disappearance of CEES was monitored by GC-FID. From the kinetic traces (Figure 6), the H3PW12O40, NU-1008, and NU-1000-NDC alone exhibit similar rates, achieving ~50% conversion after 30 min. With the same number of active clusters, PW12@NU-1000-NDC and PW12@NU-1008 convert CEES at faster rates than the individual components alone. For PW12@NU-1000-NDC (Figure 6 left), the reactivity trend is consistent with PW12@NU-1000, where the scCO2-activated sample goes to full conversion at the highest rate, and the 120°C-activated sample has slightly lower activity. The marginal difference in initial turnover frequencies (TOF; 25 and 29 min−1 for scCO2- and 120°C-activated, respectively) suggests that the POMs exist in both meso- and microporous channels; this can be compared to the 3-fold difference in initial rates in NU-1000 when the POM is located exclusively in one or the other channel. Interestingly, PW12@NU-1008-scCO2 is slower for CEES oxidation than PW12@NU-1008-120°C, exhibiting TOFs of 14 and 21 min−1, respectively. In this case, the POMs are located only in the mesopore, unable to migrate to the micropores. This finding suggests that the removal of water when not accompanied by POM movement leads to rate enhancement, and that limiting POM migration to a more thermodynamically favorable site allows for better accessibility of the active sites. Note, the apparent enhanced rate of the NU-1000-NDC over NU-1008 can be attributed to particle size differences and the poor catalyst accessibility of the substrate from the micropore, both hindering substrate diffusion.
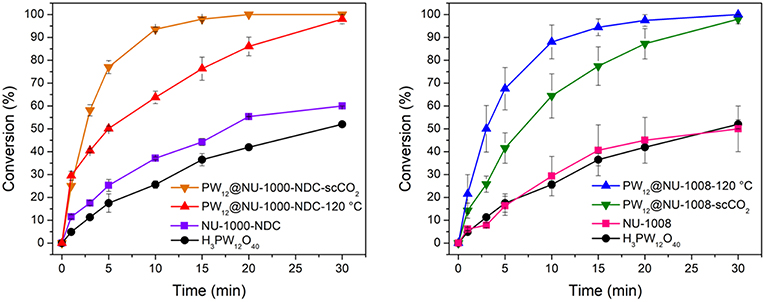
Figure 6. CEES conversion over time with (Left) NU-1000-NDC and its POM composites and (Right) NU-1008 and its POM composites. Error bars represent standard deviation of at least 2 trials.
Conclusions
Two MOFs, NU-1000-NDC and NU-1008 were used to immobilize H3PW12O40. The composite materials were activated via supercritical drying and/or heating to 120°C, resulting in PW12@NU-1008-scCO2, PW12@NU-1008-120°C, PW12@NU-1000-NDC-scCO2 and PW12@NU-1000-NDC-120°C. POM location in each was confirmed via PXRD pattern analysis. For NU-1000-NDC, the POM required longer heating for POM movement to occur than in previously reported NU-1000. In NU-1008, POM movement is completely inhibited and the POM remains in the mesopore after heating. The composite materials were more active than the individual components alone. Among the composites, the location of POM and water content in composite was found to affect the oxidation rate of a mustard gas simulant, CEES. In NU-1000-NDC, the [PW12O40]3− cluster in the mesopore is more active than when the material is heated and the cluster resides in the micropore. In contrast, when comparing the NU-1008 composites where both materials have the [PW12O40]3− cluster in the mesopore, heating the sample to remove water yields the more reactive catalyst. These findings highlight the importance of active site location on a heterogeneous support, and future studies aim to exploit the effect of POM location on catalysis.
Experimental
Materials
NU-1000, NU-1000-NDC, and NU-1008 were synthesized via literature procedure (Wang et al., 2015; Lyu et al., 2018; Peters et al., 2018). NDC incorporation was confirmed by NMR. All other reagents were purchased from Fisher Scientific or Sigma Aldrich and used as received. All gases for activation and quantification were Ultra High Purity Grade 5.
Composite Syntheses
PW12@NU-1000-NDC
H3PW12O40 (130 mg, 0.04 mmol) was dissolved in 10 mL deionized water in a centrifuge tube. NU-1000-NDC (50 mg, 0.02 mmol) was added to the solution and sonicated for 1 min. The mixture was allowed to sit for 3 days, with occasional agitation. Then, the tube was centrifuged, and the solid material was washed with water (3 × 10 mL) and allowed to sit in dry ethanol overnight. The powder was rinsed with dry ethanol (3 × 10 mL) and suspended in a minimum amount of ethanol and kept in a minimal amount of ethanol.
PW12@NU-1008
H3PW12O40 (130 mg, 0.04 mmol) was dissolved in 10 mL deionized water in a centrifuge tube. NU-1008 (50 mg, 0.02 mmol) was added to the solution and sonicated for 1 min. The mixture was allowed to sit for 3 days, with occasional agitation. Then, the tube was centrifuged, and the solid material was washed with water (3 × 10 mL) and allowed to sit in dry ethanol overnight. The powder was rinsed with dry ethanol (3 × 10 mL) and kept in a minimal amount of ethanol.
Composite Activation
ScCO2 Drying
PW12@MOF suspended in a minimal amount of absolute ethanol was placed in a glass dish and loaded into the critical point drier. Liquid CO2 was used to exchange ethanol 4 times over 8 h. The material was then heated above 31°C (P = 73 atm), the critical point of CO2 before the instrument was evacuated at a rate of 0.1 sccm (Nelson et al., 2009; Farha et al., 2011; Shultz et al., 2011). The powder was transferred to a sorption tube, where the material was kept at room temperature under dynamic vacuum for at least 1 h prior to subsequent characterization.
120°C Heating
PW12@MOF-scCO2 (~½ of the batch) was heated in a 120°C oven for 2–12 h. The supercritically dried materials can also be heated under dynamic vacuum to afford the same result.
Methods
Supercritical CO2 drying was done on a TousimisTM Samdri PVT-3D critical point drier.
Inductively coupled plasma optical emission spectroscopy (ICP-OES) samples of solids were prepared in a 2–5 mL Biotage microwave vial by dissolving 1–2 mg of composite in 1 mL HNO3. The vial was cringe-capped and heated to 150°C for 15 min in a microwave reactor. H2O2 (0.1 mL, 30 wt%) was added and the solution was heated in a sand bath at 110°C for 1 h. If the solution was still colored, 0.1 mL increments of H2O2 were added until the solution became colorless. The resulting solutions were diluted to 10 mL with deionized water and analyzed with Thermo iCap7600 ICP-OES spectrometer, equipped with a CCD detector and Ar plasma covering 175–785 nm range.
Powder X-ray diffraction (PXRD) patterns were collected on a STOE STADI P instrument using a CuKα1 source. Variable temperature PXRD patterns were taken on a STOE STADI MP instrument using a MoKα1 source. The VT samples were loaded into a capillary and flame sealed.
Solution NMR spectra were collected on a 500 MHz Bruker Avance III system equipped with DCH CryoProbe or on a 400 MHz Bruker Avance III HD Nanobay system at IMSERC (Integrated Molecular Structure Education and Research Center) at Northwestern University.
Scanning electron microscopy (SEM) images and energy dispersive spectroscopy (EDS) line scans were collected using a Hitachi SU8030 FE-SEM microscope at Northwestern University's EPIC/NUANCE facility. All samples were coated with ~15 nm of OsO4 or Au/Pd immediately prior to imaging.
GC-FID measurements were carried out on an Agilent Technologies 7820A GC system equipped with an Agilent J&W GC HP-5 capillary column (30 m × 320 μm × 0.25 μm film thickness). All samples were filtered and diluted with dichloromethane prior to injection. Starting temperature: 70°C, Hold: 0.5 min, Ramp: 30°C/min, Time: 1 min, Ramp: 75°C/min, End temperature: 250°C. The disappearance of the reactant was calculated relative to a 0-min time point.
Thermogravimetric analyses (TGA) were performed on a Mettler Toldeo STARe TGA/DSC 1 under a N2 or air flow at a 10°C/min ramp rate from 25 to 700 to 25°C.
All MOF samples were evacuated under high vacuum on a Micromeritics Smart VacPrep instrument. N2 adsorption and desorption isotherm measurements were performed on a Micromeritics Tristar II at 77 K. To calculate volumetric plots, the density of NU-1000-NDC was reported to be 0.533 g/cm3 and PW12@NU-1000-NDC was calculated to be 1.01 g/cm3; the density of NU-1008 was reported to be 0.675 g/cm3 and PW12@NU-1008 was calculated to be 1.36 g/cm3.
Catalysis Procedure
Since both the MOF node and the POM are active for catalysis, the catalyst loadings were 1 mol% by active clusters (0.86 μmol). This normalized the loadings so that the number of clusters (node or POM) remained constant for comparison.
The catalyst was weighed out in a 2 dram vial, and 1 mL acetonitrile was added. The mixture was sonicated for 1 m in. Then, CEES (10 μL, 85 μmol) and an internal standard (1-bromo-3,5-difluorobenzene, 5 μL) was added to the reaction vial. For GC quantification, the zero time point was taken. Then, hydrogen peroxide (30 wt% in water, 13 μL, 1.5 eq) was added. The vial was placed in a sand bath, which was pre-heated to 45°C. Aliquots, ~10 μL, were taken, filtered, and diluted with dichloromethane for GC-FID.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
All experimental work was performed by CB, JLy, and JLi under guidance of OF. Data analysis was performed by CB with valuable contributions from OF. The manuscript was written by CB with contributions and corrections from JLy, JLi, and OF.
Funding
Defense Threat Reduction Agency (HDTRA1-18-1-0003).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
OF gratefully acknowledges support from the Defense Threat Reduction Agency (HDTRA1-18-1-0003). Metal analysis was performed at the Northwestern University Quantitative Bio-element Imaging Center. This work made use of the IMSERC at Northwestern University, which has received support from the NSF (CHE-1048773); Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the State of Illinois and International Institute for Nanotechnology (IIN). This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1720139) at the Materials Research Center; IIN; the Keck Foundation; and the State of Illinois, through the IIN.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmats.2019.00152/full#supplementary-material
References
Ahn, S., Nauert, S. L., Buru, C. T., Rimoldi, M., Choi, H., Schweitzer, N. M., et al. (2018). Pushing the limits on metal–organic frameworks as a catalyst support: NU-1000 supported tungsten catalysts for o-Xylene isomerization and disproportionation. J. Am. Chem. Soc. 140, 8535–8543. doi: 10.1021/jacs.8b04059
Al-Ajlouni, A. M., Daiafla, T. M., and El-Khateeb, M. (2007). New nitrophenyl-substituted polyperoxotungstate catalyst : a more active and selective for the oxidation of sulfides by hydrogen peroxide. J. Mol. Catal. A 275, 139–147. doi: 10.1016/j.molcata.2007.05.041
Anyushin, A. V., Sap, A., Quanten, T., Proost, P., and Parac-Vogt, T. N. (2018). Selective hydrolysis of ovalbumin promoted by Hf(IV)-substituted Wells-Dawson-type polyoxometalate. Front. Chem. 6:614. doi: 10.3389/fchem.2018.00614
Buru, C. T., Li, P., Mehdi, B. L., Dohnalkova, A., Platero-Prats, A. E., Browning, N. D., et al. (2017). Adsorption of a catalytically accessible polyoxometalate in a channel-type metal-organic framework. Chem. Mater. 29, 5174–5181. doi: 10.1021/acs.chemmater.7b00750
Buru, C. T., Platero-Prats, A. E., Chica, D. G., Kanatzidis, M. G., Chapman, W., and Farha, O. K. (2018). Thermally induced migration of a polyoxometalate within a metal– organic framework and its catalytic. J. Mater. Chem. 6, 7389–7394. doi: 10.1039/C8TA02562B
Cai, X., Xu, Q., Tu, G., Fu, Y., Zhang, F., and Zhu, W. (2019). Synergistic catalysis of ruthenium nanoparticles and polyoxometalate integrated within single UiO−66 microcrystals for boosting the efficiency of methyl levulinate to γ -valerolactone. Front. Chem. 7:42. doi: 10.3389/fchem.2019.00042
Chen, Z., Hanna, S. L., Redfern, L. R., Alezi, D., Islamoglu, T., and Farha, O. K. (2019). Reticular chemistry in the rational synthesis of functional zirconium cluster-based MOFs. Coord. Chem. Rev. 386, 32–49. doi: 10.1016/j.ccr.2019.01.017
Farha, O. K., Shultz, A. M., Sarjeant, A. A., Nguyen, S. T., and Hupp, J. T. (2011). Active-site-accessible, porphyrinic metal À organic framework materials. J. Am. Chem. Soc. 133, 5652–5655. doi: 10.1021/ja111042f
Férey, G., Mellot-Draznieks, C., Serre, C., Millange, F., Dutour, J., Surblé, S., et al. (2005). A chromium terephthalate-based solid with unusually large pore volumes and surface area. Science 309, 2040–2042. doi: 10.1126/science.1116275
Frenzel, R., Morales, D., Romanelli, G., Sathicq, G., Blanco, M., and Pizzio, L. (2016). Synthesis, characterization and catalytic evaluation of H3PW12O40 included in acrylic acid/acrylamide polymer for the selective oxidation of sulfides. J. Mol. Catal. Chem. 420, 124–133. doi: 10.1016/j.molcata.2016.01.026
Furukawa, H., Cordova, K. E., O'Keeffe, M., and Yaghi, O. M. (2013). The chemistry and applications of metal-organic frameworks. Science 341:1230444. doi: 10.1126/science.1230444
Gallington, L. C., Kim, I. S., Liu, W. G., Yakovenko, A. A., Platero-Prats, A. E., Li, Z., et al. (2016). Regioselective atomic layer deposition in metal-organic frameworks directed by dispersion interactions. J. Am. Chem. Soc. 138, 13513–13516. doi: 10.1021/jacs.6b08711
Garce, J. M., Meima, G. R., Jacobs, P. A, Martens, J. A., and Lee, G. J. (2009). Shape selective chemistries with modified mordenite zeolites. Top Catal. 52, 1175–1181. doi: 10.1007/s11244-009-9269-2
Gumerova, N. I., and Rompel, A. (2018). Synthesis, structures and applications of electron-rich polyoxometalates. Nat. Rev. Chem. 2:0112. doi: 10.1038/s41570-018-0112
Hou, Y., An, H., Zhang, Y., Hu, T., Yang, W., and Chang, S. (2018). Rapid destruction of two types of chemical warfare agent simulants by hybrid polyoxomolybdates modified by carboxylic acid ligands. ACS Catal. 8, 6062–6069. doi: 10.1021/acscatal.8b00972
Howarth, A. J., Katz, M. J., Wang, T. C., Platero-Prats, A. E., Chapman, K. W., Hupp, J. T., et al. (2015). High efficiency adsorption and removal of selenate and selenite from water using metal-organic frameworks. J. Am. Chem. Soc. 137, 7488–7494. doi: 10.1021/jacs.5b03904
Jiao, L., Yen, J., Seow, R., Skinner, W. S., Wang, Z. U., and Jiang, H. (2018). Metal–organic frameworks : structures and functional applications. Mater. Today. doi: 10.1016/j.mattod.2018.10.038
Kholdeeva, O. A., Maksimchuk, N. V., and Maksimov, G. M. (2010). Polyoxometalate-based heterogeneous catalysts for liquid phase selective oxidations: comparison of different strategies. Catal. Today 157, 107–113. doi: 10.1016/j.cattod.2009.12.016
Kitagawa, S., Kitaura, R., and Noro, S. (2004). Functional porous coordination polymers. Angew. Chem. Int. Ed. 43, 2334–2375. doi: 10.1002/anie.200300610
Lin, Z., Zheng, H., Chen, J., Zhuang, W., Lin, Y., Su, J., et al. (2018). Encapsulation of phosphotungstic acid into metal–organic frameworks with tunable window sizes: screening of PTA@MOF catalysts for efficient oxidative desulfurization. Inorg. Chem. 57, 13009–13019. doi: 10.1021/acs.inorgchem.8b02272
Liu, J., Ye, J., Li, Z., Otake, K., Liao, Y., Peters, A. W., et al. (2018). Beyond the active site: tuning the activity and selectivity of a metal–organic framework-supported ni catalyst for ethylene dimerization. J. Am. Chem. Soc. 140, 11174–11178. doi: 10.1021/jacs.8b06006
Long, D.-L., Burkholder, E., and Cronin, L. (2007). Polyoxometalate clusters, nanostructures and materials: from self assembly to designer materials and devices. Chem. Soc. Rev. 36, 105–121. doi: 10.1039/B502666K
Lousada, C. M., Johansson, A. J., Brinck, T., and Jonsson, M. (2012). Mechanism of H 2 O 2 decomposition on transition metal oxide surfaces. J. Phys. Chem. C 116, 9533–9543. doi: 10.1021/jp300255h
Lyu, J., Zhang, X., Otake, K., Wang, X., Li, P., Li, Z., et al. (2018). Topology and porosity control of metal–organic frameworks through linker functionalization. Chem. Sci. 10, 1186–1192. doi: 10.1039/C8SC04220a
Miras, H. N., Vilà-Nadal, L., and Cronin, L. (2014). Polyoxometalate based open-frameworks (POM-OFs). Chem. Soc. Rev. 43, 5679–5699. doi: 10.1039/C4CS00097H
Nasalevich, M. A., Veen, M. Van Der, Kapteijn, F., and Gascon, J. (2014). Metal–organic frameworks as heterogeneous photocatalysts: advantages and challenges. CrystEngComm. 16, 4919–4926. doi: 10.1039/C4CE00032C
Nelson, A. P., Farha, O. K., Mulfort, K. L., and Hupp, J. T. (2009). Supercritical processing as a route to high internal surface areas and permanent microporosity in metal - organic framework materials. J. Am. Chem. Soc. 131, 458–460. doi: 10.1021/ja808853q
Nohra, B., Moll, H., El Albelo, L. M. R., Mialane, P., Mellot-draznieks, C., Kee, M. O., et al. (2011). Polyoxometalate-Based Metal Organic Frameworks (POMOFs): structural trends, energetics, and high electrocatalytic efficiency for hydrogen evolution reaction. J. Am. Chem. Soc. 133, 13363–13374. doi: 10.1021/ja201165c
Palmer, R. H., Kung, C., Liu, J., Farha, O. K., and Hupp, J. T. (2018). Nickel – Carbon – zirconium material derived from nickel-oxide clusters installed in a metal – organic framework sca ff old by atomic layer deposition. Langmuir 34, 14143–14150. doi: 10.1021/acs.langmuir.8b02166
Peters, A. W., Otake, K., Platero-Prats, A. E., Li, Z., Destefano, M. R., Chapman, K. W., et al. (2018). Site-directed synthesis of cobalt oxide clusters in a metal–organic framework. ACS Appl. Mater. Interfaces 10, 15073–15078. doi: 10.1021/acsami.8b02825
Pikus, J. D., Studts, J. M., Mcclay, K., Steffan, R. J., and Fox, B. G. (1997). Changes in the Regiospecificity of aromatic hydroxylation produced by active site engineering in the diiron enzyme toluene 4-monooxygenase. Biochemistry 36, 9283–9289. doi: 10.1021/bi971049t
Reiner, B. R., Kassie, A. A., and Wade, C. R. (2019). Unveiling reactive metal sites in a Pd Pincer MOF: insights into lewis acid catalysis. Dalt. Trans. doi: 10.1039/C8DT03801E
Salomon, W., Dolbecq, A., Roch-marchal, C., Paille, G., Dessapt, R., Mialane, P., et al. (2018). A multifunctional dual-luminescent polyoxometalate @ Metal-Organic Framework EuW10@UiO-67 composite as chemical probe and temperature sensor. Front. Chem. 6:425. doi: 10.3389/fchem.2018.00425
Salomon, W., Roch-Marchal, C., Mialane, P., Rouschmeyer, P., Serre, C., Haouas, M., et al. (2015). Immobilization of polyoxometalates in the Zr-based metal organic framework UiO-67. Chem. Commun. 51, 2972–2975. doi: 10.1039/C4CC09986A
Shultz, A. M., Farha, O. K., Adhikari, D., Sarjeant, A. A., Hupp, J. T., and Nguyen, S. T. (2011). Selective surface and near-surface modification of a noncatenated, catalytically active metal-organic framework material based on Mn(salen) struts. Inorg. Chem. 50, 3174–3176. doi: 10.1021/ic101952y
Song, J., Luo, Z., Britt, D. K., Furukawa, H., Yaghi, O. M., Hardcastle, K. I., et al. (2011). A multiunit catalyst with synergistic stability and reactivity: a polyoxometalate-metal organic framework for aerobic decontamination. J. Am. Chem. Soc. 133, 16839–16846. doi: 10.1021/ja203695h
Sosa, J. D., Bennett, T. F., Nelms, K. J., Tovar, R. C., and Liu, Y. (2018). Metal–organic framework hybrid materials and their applications. Crystals 8:325. doi: 10.3390/cryst8080325
Sullivan, K. P., Yin, Q., Collins-Wildman, D. L., Tao, M., Geletii, Y. V., Musaev, D. G., et al. (2018). Multi-tasking POM systems. Front. Chem. 6:365. doi: 10.3389/fchem.2018.00365
Vandichel, M., Hajek, J., Vermoortele, F., Waroquier, M., De Vos, D. E., and Speybroeck, V. (2015). Active site engineering in UiO-66 type metal–organic frameworks by intentional creation of defects: a theoretical rationalization. CrystEngComm 17, 395–406. doi: 10.1039/C4CE01672F
Vermoortele, F., Ameloot, R., Alaerts, L., Matthessen, R., Carlier, B., Fernandez, E. V. R., et al. (2012). Tuning the catalytic performance of metal–organic frameworks in fine chemistry by active site engineering. J. Mater. Chem. 22, 10313–10321. doi: 10.1039/c2jm16030g
Wang, C., Zhou, M., Ma, Y., Tan, H., Wang, Y., and Li, Y. (2018). Hybridized Polyoxometalate-based metal–organic framework with ketjenblackfor the nonenzymatic detection of H2O2. Chem. Asian J. 13, 2054–2059. doi: 10.1002/asia.201800758
Wang, S., and Yang, G. (2015). Recent advances in polyoxometalate-catalyzed reactions. Chem. Rev. 115, 4893–4962. doi: 10.1021/cr500390v
Wang, T. C., Vermeulen, N. A., Kim, I. S., Martinson, A. B. F., Stoddart, J. F., Hupp, J. T., et al. (2015). Scalable synthesis and post-modification of a mesoporous metal-organic framework called NU-1000. Nat. Protoc. 11, 149–162. doi: 10.1038/nprot.2016.001
Wasson, M. C., Lyu, J., Islamoglu, T., and Farha, O. K. (2018). Linker competition within a metal–organic framework for topological insights. Inorg. Chem. 58, 1513–1517. doi: 10.1021/acs.inorgchem.8b03025
Yaghi, O. M., O'Keeffe, M., Ockwig, N. W., Chae, H. K., Eddaoudi, M., and Kim, J. (2003). Reticular synthesis and the design of new materials. Nature 423, 705–714. doi: 10.1038/nature01650
Yakovenko, A. A., Wei, Z., Wriedt, M., Li, J.-R., Halder, G. J., and Zhou, H. (2014). Study of guest molecules in metal – organic frameworks by powder x-ray diffraction: analysis of difference envelope density. Cryst. Growth Des. 14, 5397–5407. doi: 10.1021/cg500525g
Yang, D., and Gates, B. C. (2019). Catalysis by metal organic frameworks: perspective and suggestions for future research. ACS Catal. 9, 1779–1798. doi: 10.1021/acscatal.8b04515
Keywords: metal-organic framework, polyoxometalate, sulfide oxidation, chemical warfare agent, catalysis, host-guest
Citation: Buru CT, Lyu J, Liu J and Farha OK (2019) Restricting Polyoxometalate Movement Within Metal-Organic Frameworks to Assess the Role of Residual Water in Catalytic Thioether Oxidation Using These Dynamic Composites. Front. Mater. 6:152. doi: 10.3389/fmats.2019.00152
Received: 22 February 2019; Accepted: 13 June 2019;
Published: 27 June 2019.
Edited by:
Seong Huh, Hankuk University of Foreign Studies, South KoreaReviewed by:
Steve Suib, University of Connecticut, United StatesLaia Vilà Nadal, University of Glasgow, United Kingdom
Yangyang Liu, California State University, Los Angeles, United States
Copyright © 2019 Buru, Lyu, Liu and Farha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Omar K. Farha, by1mYXJoYUBub3J0aHdlc3Rlcm4uZWR1