 Yang Li1
Yang Li1 Peng Peng
Peng Peng Feng Huo
Feng Huo Xiaohong Shao
Xiaohong Shao Zhonghua Xiang
Zhonghua Xiang- 1State Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology, Beijing, China
- 2Beijing Key Laboratory of Ionic Liquids Clean Process, State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, China
- 3College of Science, Beijing University of Chemical Technology, Beijing, China
Two-dimensional Covalent Organic Polymers (COPs) which are used to catalyze oxygen reduction reaction (ORR) in regenerative fuel cells have been researched by efforts. However, due to the lack of closed conjugated molecular structures, most COPs demonstrate limited catalytic performance. In this work, four alternative COPs containing different conjugated π-bonds are designed (termed as COP-Bn, n = 3, 4, 5, 6) and studied based on Charge Density Analysis, Density of States (DOS), Band Structure Analysis, First-principle, and Density Functional Theory (DFT) calculations toward catalyzing ORR. Our outcomes reveal that COPs with larger conjugated structures (e.g., COP-B5 and COP-B6) can achieve higher activity during ORR catalysis.
Introduction
Clean energy technologies, such as fuel cells and metal-air batteries, are promising alternative energy sources for auto industry and electronic products (Sealy, 2008; Dunn et al., 2011; Tachibana et al., 2012; Katsounaros et al., 2014; Kong et al., 2018; Li et al., 2018, 2019). As a vital part for the widespread use of fuel cells, low-cost electrocatalysts with outstanding catalytic activity and high stability for the oxygen reduction reaction (ORR) are indispensable (Bak et al., 2002; Kanan and Nocera, 2008; Gray, 2009; Girishkumar et al., 2010). However, traditional technologies require noble metal (e.g., Pt) catalysts to promote ORR. Thus, the scarce resources and high price of the noble metal catalysts have long limited the clean energy technologies for commercial applications (Winter and Brodd, 2004; Gasteiger et al., 2005).
Recently, nitrogen doped grapheme (Novoselov et al., 2004; Balandin et al., 2008; Lee et al., 2008; Park and Ruoff, 2009; Rao et al., 2009; Liang et al., 2011; Zhu et al., 2011; Yoo et al., 2012; Lung-Hao Hu et al., 2013) and two-dimensional covalent organic polymers (COPs) (Xiang and Cao, 2012, 2013; Xiang et al., 2012a,b; Zhou et al., 2013; Peng et al., 2017) have emerged with high activities for catalyzing ORR. These carbon-based catalysts not only contain abundant sources and flexibility in molecular design, but also demonstrate higher efficiency, stability, and tolerance to crossover/CO-poisoning effects than commercial platinum group metal (PGM) catalysts (Peng et al., 2019a,b). Specially, two-dimensional COPs have been widely developed because of their high thermodynamic stability, planar molecular structures, and versatile elemental environment. Two-dimensional COPs hold the promising potentials of precisely controllable capacities for ORR catalysis. Nevertheless, owing to the poor conductive properties, most reported COPs contain limited catalytic activities (Guo et al., 2018). Thus, pyrolysis is essential to enhance their p-conjugated structures and the electrocatalytic activity, which unfortunately induce undesirable structure changes and even destruct the fine structures, leaving the mechanism of electrocatalysis uncertain. It is urgent to design COPs electrocatalysts with intrinsic conductivity and activity.
In the structure of traditional COPs, the phenyl rings are mostly connected via sigma bonds. Even these irreversible connections offer high stability, the single bonds always lead to the lack of closed conjugated molecular structure. Since the electron fluidity play a decisive role for catalyzing ORR, it is necessary to design two-dimensional COPs with closed conjugated and enhanced conjugative effect.
In this study, we have designed a series of two-dimensional COPs (termed as COP-Bn) with closed conjugated molecular structures and calculated their possible activities in catalyzing ORR. Different to traditional COPs, the phenyl rings in COP-Bn are connected via π-bonds instead of sigma bonds. Four alternative COP-Bn (n = 3, 4, 5, 6; Figure 1, Figure S1) are studied by different characterization methods, such as Charge Density Analysis, Density of States (DOS) and Band Structure Analysis, First-principle, and Density Functional Theory (DFT) calculations. Our findings point the directions to design proper COPs possessing enough electron fluidity for efficient catalyzing ORR.
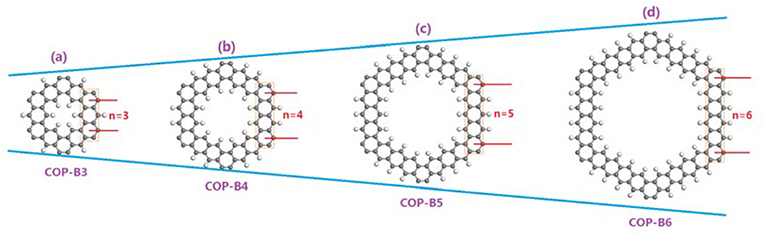
Figure 1. The molecular structures of two-dimensional closed conjugated COPs (a, COP-B3; b, COP-B4; c, COP-B5; d, COP-B6); The gray atoms stand for carbon atoms, and the white atoms stand for hydrogen atoms.
Calculation and Simulation Methods
First-principle calculations were performed within the framework of DFT as implemented in the plane wave set VASP code. The detailed implementation was described in reference (Li et al., 2014). All structures were modeled as three-dimensional periodic structures, where a rectangle periodic box was represented with a dashed line. To avoid interaction between slabs, the vacuum layers were set as 18 Å in the y-and z-directions, respectively. The k-point sampling of the Brillioun zone was obtained using a 4 × 1 × 1 grid generating meshes with their origin centered at the gamma (Γ) point. The plane wave basis set contained a high cut off energy of 450 eV throughout the computations. The k point meshes and energy cut off were chosen to ensure that the energies were converged within 1 meV/per atom. The Fermi level was slightly broadened using a Fermi-Dirac smearing of 50 meV. All calculations were spin polarized and completed when the force of the system converged to ~0.02 eV/Å.
The oxygen reduction reaction proceeds either through a two-step two-electron pathway that O2 is reduced to hydrogen peroxide, or completely via a four-electron process in which O2 is reduced directly to water without involvement of hydrogen peroxide intermediate. Herein, since previous results showed that the ORR proceeds on graphene and related COPs through the four-electron mechanism (Zhang and Xia, 2011), we took the complete reduction cycle into account. In an acid environment the oxygen reduction reaction (ORR) could be written as:
We considered two possible ORR mechanisms: associative mechanism that involved a HOO* species and a direct O2 dissociation mechanism. The associative mechanism was conducted through the following elementary steps:
Where * was the active site on the graphene surface (l), and (g) referred to liquid and gas phases, respectively. O*, OH*, and OOH* were adsorbed intermediates.
In an alkaline electrolyte, it was the H2O acted as the proton donor instead of H3O+. Thus, the oxygen reduction could be written as:
According to the associative mechanism, equation (7) could then be split into the following elementary steps in ORR:
Furthermore, the mechanism via direct O2 dissociation of oxygen atoms started with the following elementary step:
followed by steps (5) and (6) in acidic environment or (11) and (12) in alkaline solution.
According to previous reports, the rate determining step of a typical ORR could either be the adsorption of O2 as OOH* or the desorption of OH* as water (Calle-Vallejo and Koper, 2012). Here, we took reactions (8)–(12) to derive the thermochemistry for ORR.
The thermochemistry of these electrochemical reactions was obtained using DFT calculations in conjunction with Standard Hydrogen Electrode model developed by Norskov et al. (2004) and Man et al. (2011). This thermodynamic approach established a minimum set of requirements for the facile reactions based on the binding of the intermediates with the assumption that there were no extra barriers from, e.g., adsorption/dissociation of O2 or proton/electron transfer reactions. In our calculations, the ORR was analyzed using intermediate species associated with one electron transfer at a time, which was energetically more favorable than the simultaneous transfer of more than one electron.
The absorption energies were calculated as follows (Li et al., 2014),
in which, E(*), E(OH*), E(O*), and E(OOH*) were the ground state energies of a clean surface and the surfaces adsorbed with OH*, O*, and OOH*, respectively. EH2O and EH2 were the calculated DFT energies of H2O and H2 molecules in the gas phase. We also considered the zero-point energy (ZPE) and entropy correction. These calculations transformed DFT binding energies (ΔEads−DFT) into free energies of adsorption (ΔGads) through the following equation (Li et al., 2014),
Where T was the temperature and ΔS was the entropy change. For the ZPE, the vibrational frequencies of adsorbed species (O*, OH*, and OOH*) were calculated with the graphene or COPs that remain fixed to obtain ZPE contribution in the free energy expression. Moreover, only vibration entropy contributions were considered for adsorbates and total entropies for solvent molecules were taken from standard thermodynamic tables.
For each step the reaction free energy ΔG was defined as the difference between free energies of the initial and final states, which was given by the expression (Li et al., 2014):
Where ΔE was the reaction energy of reactant and product molecules adsorbed on catalyst surface. ΔGU was obtained from DFT calculations (ΔGU = −eU, where U was the potential at the electrode, and e was the number of transferred). ΔGpH was the correction of the H+ free energy by the concentration dependence of the entropy:
Notice that neglecting the electric field, there was no difference in the free energy of the ORR intermediates calculated in acidic and alkaline environment at fixed potential on the Reversible Hydrogen Electrode scale (Rossmeisl et al., 2007). We emphasized that our model neglects the effect of the electric field in the double layer and didn't treat barriers that may depend on whether the proton donor was H2O or H3O+.
Results and Discussion
Charge Density Analysis
Firstly, we analyzed the related charge density of COP-Bn. In these two-dimensional and colorful charge density distribution pictures (Figure 2), the red area suggested the area which possessed the lowest charge density in the corresponding COP molecular structure. Obviously, the red area of COP-B5 (Figure 2c) and COP-B6 (Figure 2d) was darkest among these three COP models. Moreover, a kind of COP model with large low charge density area was likely to benefit the oxygen adsorption for ORR. Meanwhile, this sort of COP model was also likely to possess preferable electron fluidity which could facilitate the ORR process. Through Charge Density Analysis, we found an important law firstly. Specially, the charge density became lower with the addition of phenyl rings in two-dimensional closed conjugated molecular structure, and the electron fluidity became better at the same time.
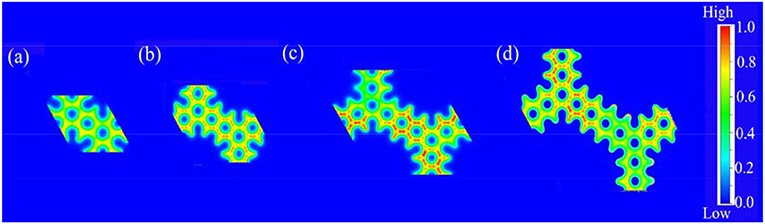
Figure 2. Charge density distribution pictures of two-dimensional closed conjugated COPs (a, COP-B3; b, COP-B4; c, COP-B5; d, COP-B6).
DOS and Band Structure Analysis
Whereas, the electron fluidity for a kind of molecular model was closely associated with its electrical conductivity, an ideal characterization parameter was the energy gap value obtained by DOS figure which could be calculated by VASP. In detail, for some kind of molecular model, lower energy gap value indicated a better electrical conductivity. Therefore, the energy gap value can characterize the electron fluidity of these COP models well, which was essential for the ORR process. In this case, Figures 3, 4 indicated that COP-B5 and COP-B6 possessed the lower energy gap value (0.876, 0.526 eV) among these four COP models, suggesting that COP-B5 and COP-B6 have the better electron fluidity. These Band Structure Figures also revealed the same law. For high symmetry points (lines, G, F, Q, Z), the density of curves became higher with the addition of phenyl rings in two-dimensional closed conjugated molecular structure.
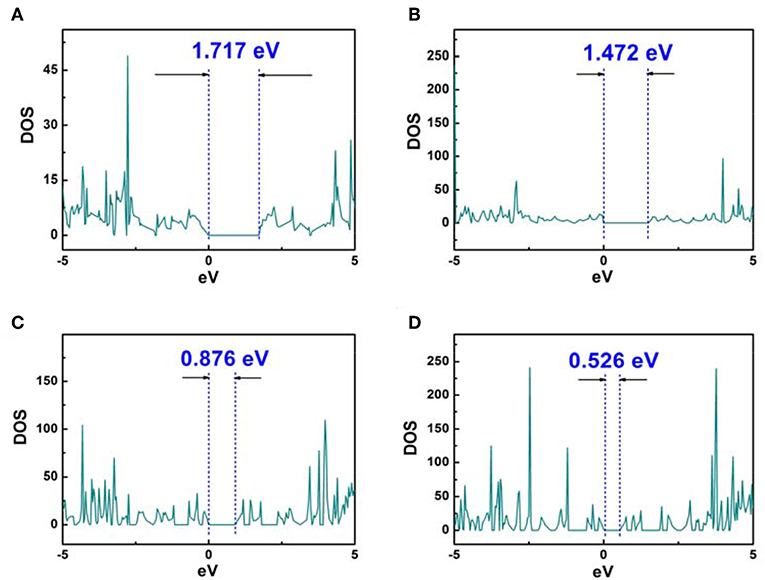
Figure 3. DOS Analysis Figures of two-dimensional closed conjugated COPs (A, COP-B3; B, COP-B4; C, COP-B5; D, COP-B6).
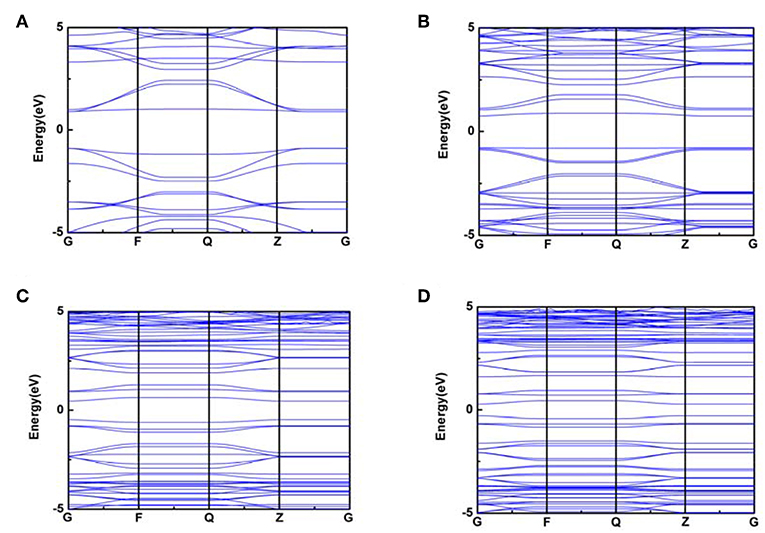
Figure 4. Band Structure Figures of two-dimensional closed conjugated COPs (A, COP-B3; B, COP-B4; C, COP-B5; D, COP-B6).
First-Principle and DFT Calculations
To studied the four COP models toward catalyzing ORR, the corresponding figures named Free Energy Curve were essential, which could be obtained by First-principle and DFT calculations. These figures were calculated based on zero electrode potential (U = 0 V) and equilibrium electrode potential (U = 1.23 V). Two essential free energy curves were shown as inside. Besides, acid (pH = 0) and alkaline (pH = 14) conditions was also considered, respectively. For every free energy curve, the final state (H2O or OH−) free energy value was set as zero according to the related references (Norskov et al., 2004; Zhang and Xia, 2011; Li et al., 2014). It should be noted that when the electrode potential was zero, the original state (O2+*) was set as 4.92 eV. If the former was equilibrium electrode potential, the latter was still set as zero. Moreover, the free energy values of three transition states (OOH*, O*, OH*) was also calculated by VASP. In particular, if some material model possessed ORR performance theoretically, no matter the condition was acid or alkaline, there would be the third free energy curve based on a vital potential value, which was named onset potential value (0 < U < 1.23 V), as shown in the related figures. The onset potential value was the maximum which ORR can occur. Hence, if this maximum was negative, the third free energy curve would not exist, meaning that the corresponding material model didn't possess ORR performance. The fact was that all of the others do not possess ORR performance except COP-B5 (Figures 5A,B) and COP-B6 (Figures 5C,D), because there was no one chosen oxygen adsorbed site which was satisfied with related conditions. Although COP-B5 possess certain ORR performance theoretically among the four chosen oxygen adsorbed sites (Figure S2), there were only two sites (No. 3 and No. 4, Figure 6) which could act as the activated sites for ORR. In detail, for COP-B5, when the condition was acid, no matter the activated site was No.3 or No.4, the obtained onset potential value was 0.19 V (Figure S3). If the condition was alkaline, COP-B5 also didn't possess ORR performance. For COP-B6, there were four sites (Figure S2) which could be used for catalyzing ORR, the maximum of onset potential can reach 0.3 V (Figure 6, Figure S4). Therefore, in these three COP models, COP-B5 and COP-B6 were the COPs model which can catalyze ORR process theoretically. Objectively, although in acid condition, the ORR performance of COP-B5 and COP-B6 were not enough high, and there was no ORR performance for them in alkaline condition, they were still a sort of COP model which can be used for modification in catalyzing ORR process. For the conjugated COP-Bn (n = 3, 4, 5, 6) models, only when the value of n was five and six, the proper conjugated COP associated with ORR catalysis can be obtained. In other words, for two-dimensional closed conjugated COPs, the more phenyl rings would lead to more active sites for ORR.
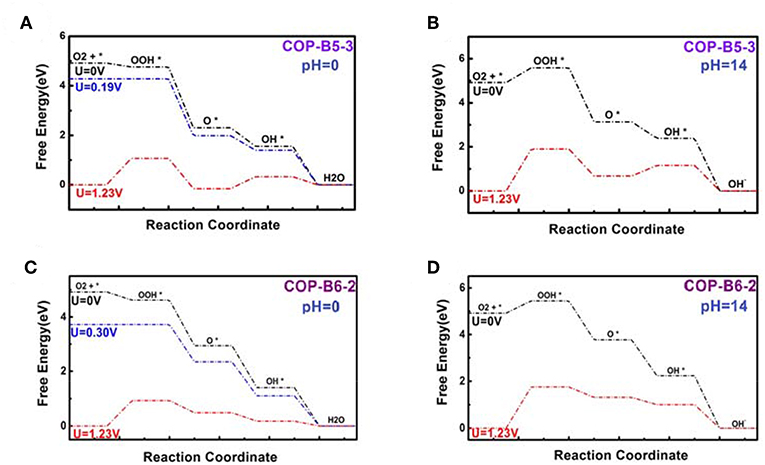
Figure 5. Free energy curves of COP-B5 (Sites No. 3, A, pH = 0; B, pH = 14) and COP-B6 (Site No. 2, C, pH = 0; D, pH = 14).
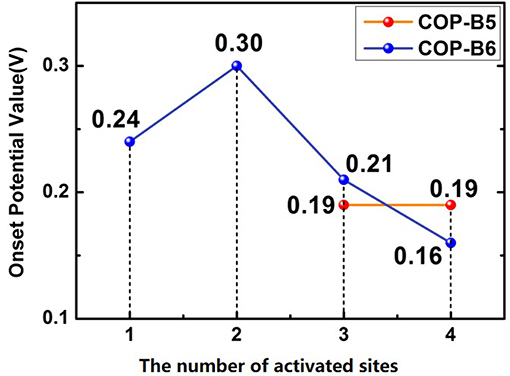
Figure 6. The number of activated sites and the corresponding onset potential values for ORR. The possible ORR sites were marked in Figure S2.
Conclusions
In this study, we designed four COP models (COP-Bn, n = 3, 4, 5, 6) to study their ORR performance. Through charge density analysis, DOS and Band Structure analysis, we proved that COP-B5 and COP-B6 were likely to be a proper COP model associated with ORR catalysis. Consistently, the related free energy curve figures also indicated that COP-B5 and COP-B6 were the COP models which can catalyze ORR. For the conjugated COP-Bn (n = 3, 4, 5, 6) models, only when the value of n achieved five, a proper conjugated COP which is really associated with ORR could be obtained. Although the conjugated COPs such as COP-B5 and COP-B6 may not possess enough high performance for ORR, they can still be used for modification in catalyzing ORR process. Two-dimensional closed conjugated COPs such COP-B5 and COP-B6 were capable to realize the positive transition of electrical conductivity and catalysis for ORR in theory.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
YL, FH, and XS performed the DFT calculations. YL and PP wrote this papers. ZX supervised and led this project. All authors provided critical feedback, helped shape the research and manuscript, and commented on the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFA0206500), the Natural Science Foundation of China (21676020), the Beijing Natural Science Foundation (17L20060), the Fundamental Research Funds for the Central Universities (buctrc201420 and buctrc201714), the BUCT Fund for Disciplines Construction and Development (XK1502), the Young Elite Scientists Sponsorship Program by CAST (2017QNRC001), the Talent cultivation and open project (OIC-201801007) of State Key Laboratory of Organic-Inorganic Composites, the Distinguished Scientist Program at BUCT (buctylkxj02); Double-First-Class construction projects (XK180301; XK1804-02) and the 111 Project of China (B14004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmats.2019.00244/full#supplementary-material
References
Bak, T., Nowotny, J., Rekas, M., and Sorrell, C. C. (2002). Photo-electrochemical hydrogen generation from water using solar energy. Materials-related aspects. Int. J. Hydrogen Energy 27, 991–1022. doi: 10.1016/S0360-3199(02)00022-8
Balandin, A. A., Ghosh, S., Bao, W., Calizo, I., Teweldebrhan, D., Miao, F., et al. (2008). Superior thermal conductivity of single-layer graphene. Nano Lett. 8, 902–907. doi: 10.1021/nl0731872
Calle-Vallejo, F., and Koper, M. T. M. (2012). First-principles computational electrochemistry: achievements and challenges. Electrochim. Acta 84, 3–11. doi: 10.1016/j.electacta.2012.04.062
Dunn, B., Kamath, H., and Tarascon, J.-M. (2011). Electrical energy storage for the grid: a battery of choices. Science 334, 928–935. doi: 10.1126/science.1212741
Gasteiger, H. A., Kocha, S. S., Sompalli, B., and Wagner, F. T. (2005). Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 56, 9–35. doi: 10.1016/j.apcatb.2004.06.021
Girishkumar, G., McCloskey, B., Luntz, A. C. Swanson, S., and Wilcke, W. (2010). Lithium– air battery: promise and challenges. J. Phys. Chem. Lett. 1, 2193–2203. doi: 10.1021/jz1005384
Guo, J., Lin, C., Xia, Z., and Xiang, Z. (2018). A pyrolysis-free covalent organic polymer for oxygen reduction. Angew. Chem. Int. Edn. 57, 12567–12572. doi: 10.1002/anie.201808226
Kanan, M. W., and Nocera, D. G. (2008). In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 321, 1072–1075. doi: 10.1126/science.1162018
Katsounaros, I., Cherevko, S., Zeradjanin, A. R., and Mayrhofer, K. J. J. (2014). Oxygen electrochemistry as a cornerstone for sustainable energy conversion. Angew. Chem. Int. Edn. 53, 102–121. doi: 10.1002/anie.201306588
Kong, L., Li, B. Q., Peng, H. J., Zhang, R., Xie, J., Huang, J. Q., et al. (2018). Porphyrin-derived graphene-based nanosheets enabling strong polysulfide chemisorption and rapid kinetics in lithium–sulfur batteries. Adv. Energy Mater. 8:1800849. doi: 10.1002/aenm.201800849
Lee, C., Wei, X., Kysar, J. W., and Hone, J. (2008). Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science 321, 385–388. doi: 10.1126/science.1157996
Li, B. Q., Peng, H. J., Chen, X., Zhang, S. Y., Xie, J., Zhao, C. X., et al. (2019). Polysulfide electrocatalysis on framework porphyrin in high-capacity and high-stable lithium–sulfur batteries. CCS Chem. 1, 128–137. doi: 10.31635/ccschem.019.20180016
Li, B. Q., Zhang, S. Y., Kong, L., Peng, H. J., and Zhang, Q. (2018). Porphyrin organic framework hollow spheres and their applications in lithium–sulfur batteries. Adv. Mater. 30:1707483. doi: 10.1002/adma.201707483
Li, M., Zhang, L., Xu, Q., Niu, J., and Xia, Z. (2014). N-doped graphene as catalysts for oxygen reduction and oxygen evolution reactions: theoretical considerations. J. Catal. 314, 66–72. doi: 10.1016/j.jcat.2014.03.011
Liang, Y., Li, Y., Wang, H., Zhou, J., Wang, J., Regier, T., et al. (2011). Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 10, 780–786. doi: 10.1038/nmat3087
Lung-Hao Hu, B., Wu, F.-,Y., Lin, C.-T., Khlobystov, A. N., and Li, L.-J. (2013). Graphene-modified LiFePO4 cathode beyond theoretical capacity. Nat. Commun. 4, 1687–1687. doi: 10.1038/ncomms2705
Man, I. C., Su, H.-Y., Calle-Vallejo, F., Hansen, H. A., Martinez, J. I., Inoglu, N. G., et al. (2011). Universality in oxygen evolution electrocatalysis on oxide surfaces. Chemcatchem 3, 1159–1165. doi: 10.1002/cctc.201000397
Norskov, J. K., Rossmeisl, J., Logadottir, A., Lindqvist, L., Kitchin, J. R., Bligaard, T., et al. (2004). Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B. 108, 17886–17892. doi: 10.1021/jp047349j
Novoselov, K. S., Geim, A. K., Morozov, S. V., Jiang, D., Zhang, Y., Dubonos, S. V., et al. (2004). Electric field effect in atomically thin carbon films. Science 306, 666–669. doi: 10.1126/science.1102896
Park, S., and Ruoff, R. S. (2009). Chemical methods for the production of graphenes. Nat. Nanotechnol. 4, 217–224. doi: 10.1038/nnano.2009.58
Peng, P., Shi, L., Huo, F., Mi, C., Cheng, Y., Wu, X., et al. (2019b). A pyrolysis-free path toward superiorly catalytic nitrogen-coordinated single atom. Sci. Adv. 5:eaaw2322. doi: 10.1126/sciadv.aaw2322
Peng, P., Shi, L., Huo, F., Zhang, S., Mi, C., Cheng, Y., et al. (2019a). In situ charge exfoliated soluble covalent organic framework directly used for Zn–air flow battery. ACS Nano 13, 878–884. doi: 10.1021/acsnano.8b08667
Peng, P., Zhou, Z., Guo, J., and Xiang, Z. (2017). Well-defined 2D covalent organic polymers for energy electrocatalysis. ACS Energy Lett. 2, 1308–1314. doi: 10.1021/acsenergylett.7b00267
Rao, C. N. R., Sood, A. K., Subrahmanyam, K. S., and Govindaraj, A. (2009). Graphene: the new two-dimensional nanomaterial. Angew. Chem. Int. Edn. 48, 7752–7777. doi: 10.1002/anie.200901678
Rossmeisl, J., Qu, Z. W., Zhu, H., Kroes, G. J., and Norskov, J. K. (2007). Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 607, 83–89. doi: 10.1016/j.jelechem.2006.11.008
Sealy, C. (2008). The problem with platinum. Mater. Today 11, 65–68. doi: 10.1016/S1369-7021(08)70254-2
Tachibana, Y., Vayssieres, L., and Durrant, J. R. (2012). Artificial photosynthesis for solar water-splitting. Nat. Photonics 6, 511–518. doi: 10.1038/nphoton.2012.175
Winter, M., and Brodd, R. J. (2004). What are batteries, fuel cells, and supercapacitors? Chem. Rev. 104, 4245–4269. doi: 10.1021/cr040110e
Xiang, Z., and Cao, D. (2012). Synthesis of luminescent covalent–organic polymers for detecting nitroaromatic explosives and small organic molecules. Macromol. Rapid Commun. 33, 1184–1190. doi: 10.1002/marc.201100865
Xiang, Z., and Cao, D. (2013). Porous covalent–organic materials: synthesis, clean energy application and design. J. Mater. Chem. A. 1, 2691–2718. doi: 10.1039/C2TA00063F
Xiang, Z., Cao, D., Wang, W., Yang, W., Han, B., and Lu, J. (2012a). Postsynthetic lithium modification of covalent-organic polymers for enhancing hydrogen and carbon dioxide storage. J. Phys. Chem. C. 116, 5974–5980. doi: 10.1021/jp300137e
Xiang, Z., Zhou, X., Zhou, C., Zhong, S., He, X., Qin, C., et al. (2012b). Covalent-organic polymers for carbon dioxide capture. J. Mater. Chem. 22, 22663–22669. doi: 10.1039/c2jm35446b
Yoo, E., Nakamura, J., and Zhou, H. (2012). N-Doped graphene nanosheets for Li–air fuel cells under acidic conditions. Energy Environ. Sci. 5, 6928–6932. doi: 10.1039/c2ee02830a
Zhang, L., and Xia, Z. (2011). Mechanisms of oxygen reduction reaction on nitrogen-doped graphene for fuel cells. J. Phys. Chem. C. 115, 11170–11176. doi: 10.1021/jp201991j
Zhou, Y., Xiang, Z., Cao, D., and Liu, C.-J. (2013). Covalent organic polymer supported palladium catalysts for CO oxidation. Chem. Commun. 49, 5633–5635. doi: 10.1039/c3cc00287j
Keywords: covalent organic polymers, oxygen reduction reaction, first principle calculation, 2D materials, conjugated system
Citation: Li Y, Peng P, Huo F, Shao X and Xiang Z (2019) Two-Dimensional Closed Conjugated Covalent Organic Polymers for Oxygen Reduction Reaction. Front. Mater. 6:244. doi: 10.3389/fmats.2019.00244
Received: 31 July 2019; Accepted: 18 September 2019;
Published: 04 October 2019.
Edited by:
Dingshan Yu, Sun Yat-sen University, ChinaReviewed by:
Qiang Zhang, Tsinghua University, ChinaJiantie Xu, South China University of Technology, China
Copyright © 2019 Li, Peng, Huo, Shao and Xiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Huo, aHVvZmVuZ0BpcGUuYWMuY24=; Xiaohong Shao, c2hhb3hoQG1haWwuYnVjdC5lZHUuY24=; Zhonghua Xiang, eGlhbmd6aEBtYWlsLmJ1Y3QuZWR1LmNu