 Enrico Bernardo1*
Enrico Bernardo1* Hamada Elsayed1,2
Hamada Elsayed1,2 Acacio Rincon Romero1
Acacio Rincon Romero1 Murilo C. Crovace3
Murilo C. Crovace3 Edgar D. Zanotto3
Edgar D. Zanotto3 Tobias Fey4,5
Tobias Fey4,5- 1Department of Industrial Engineering, Università degli Studi di Padova, Padova, Italy
- 2Ceramics Department, National Research Centre, Cairo, Egypt
- 3Vitreous Materials Laboratory, Department of Materials Engineering, Center for Research, Education, and Technology in Vitreous Materials (CeRTEV), Federal University of São Carlos (UFSCar), São Carlos-SP, Brazil
- 4Department of Materials Science and Engineering, Institute of Glass and Ceramics, University of Erlangen-Nürnberg, Erlangen, Germany
- 5Frontier Research Institute for Materials Science, Nagoya Institute of Technology, Nagoya, Japan
Biosilicate® glass-ceramics are among the most valid alternatives to 45S5 Bioglass. They combine a similar bioactivity and bioresorbability as the 45S5 with superior mechanical strength, owing to the crystallization of a Na–Ca silicate phase. This crystallization may be experienced upon viscous flow sintering of fine glass powders, thus configuring a sinter-crystallization process. As crystallization is seldom complete, sintering can also be applied to semicrystalline powders. The sintering/crystallization combination may be exploited for shaping highly porous bodies, to be used as scaffolds for bone tissue engineering, in the form of foams. The present study aims at exploring a gel-casting process, based on the room temperature foaming of powders suspended in a “weakly alkaline” (1 M NaOH) aqueous solution, followed by sintering at 1,000°C. The gelation of suspensions is attributed to the formation of hydrated compounds, later decomposed upon firing. Amorphous powders provided more intense gelation than semicrystalline ones, promoted a more homogeneous foaming, and stimulated a substantial crystallization upon firing. The homogeneity of foamed samples was assessed using micro-tomography and was further improved by casting foamed suspensions (“foam casting”) before setting.
Introduction
Bioactive glass-ceramics have been established as excellent materials for bone tissue applications since the mid-70s, that is, soon after Hench’s 45S5 Bioglass® (the current “golden standard” for bioceramics) had been proposed (Piotrowski et al. 1975; Hench, 1977; Hench and Polak, 2002; Hench, 2006). Compared to glasses, semicrystalline materials generally exhibit superior mechanical properties (i.e., strength, toughness, and hardness), implying enhanced load-bearing capability, easier machinability, and workability, which are fundamental in dental applications (Albakry et al., 2004; Höland et al., 2007; Höland and Rheinberger, 2008).
Passing from bioactive glasses, that is, 45S5 and alternative formulations, all having applications in the amorphous state (Kolan et al., 2011; Liu et al., 2013; Souza et al., 2017; Bellucci and Cannillo, 2018) to “bioglass-ceramics” actually implied a revision of formulations, to match the desired biocompatibility and bioactivity and enable easy overall processing. Biosilicate® glass-ceramics represent one of the most successful results of this research effort (Crovace et al., 2016; Montazerian and Zanotto, 2016; Pinto et al., 2018). A slight modification of the original composition of 45S5 (23.75 wt% Na2O, 23.75% CaO, 48.5% SiO2, and 4.0% P2O5 instead of 24.5 wt% Na2O, 24.5% CaO, 45% SiO2, and 6.0% P2O5 for 45S5) made the substance, after conversion into a semicrystalline material, absolutely comparable to 45S5 in terms of osteoconductivity, osteoinductivity, biocompatibility, and antibacterial properties (Hench, 2006; Granito et al., 2009).
The control of glass crystallization is delicate, in the manufacturing of complex components by viscous flow sintering of fine powders. Due to the low content of network formers, many bioactive glass powders experience—upon firing—a substantial crystallization, with a negative impact on densification. In addition, when working with glasses that are not specifically designed for controlled crystallization during sintering (“sinter-crystallization”), (Clark and Reed, 1986) the resulting sintered articles may be even less bioactive than the starting powders (Li et al., 1992).
The successful coupling of sintering and crystallization is a necessary but not the decisive point in the overall processing of scaffolds for bone tissue applications. These applications require highly porous open-celled foams, typically achieved by two well-known methods, namely, the replica of sacrificial PU sponges (Baino and Vitale-Brovarone, 2012; Roohani-Esfahani and Zreiqat, 2012; Desimone et al., 2013; Baino and Vitale-Brovarone, 2014) and direct foaming (Sepulveda and Binner, 1999; Gonzenbach et al., 2007).
The replica method is useful for the manufacturing of trabecular structures, with wide openings between cells. Compared to the sintering of ceramic powders, the sinter-crystallization of glass powders is attractive for the filling of voids, left by the burnout of the polymer template at the early stages of sintering, with an enhancement of mechanical properties (Vitale-Brovarone et al., 2009). However, an excessive viscous flow may determine the collapse of the overall structure. Crystallization, yielding rigid crystal inclusions and thus causing a remarkable viscosity increase, can be beneficial. Still, it must occur in an optimum “temperature window,” that is, neither too early (the viscosity increase may hinder the filling of voids) nor too late (the viscosity increase would not prevent the collapse of the struts).
Highly porous foamed scaffolds, with dense struts, are easily achieved by gel-casting, applied to sol–gel formulations at the early stage of gelation (sols) (Li et al., 1992; Desimone et al., 2013), as well as to glass suspensions (Sepulveda and Binner, 1999; Vitale-Brovarone et al., 2009). A cellular structure is formed by air incorporation, using intensive mechanical stirring (“frothing”), with the help of a surfactant. It is subsequently stabilized by the concurrent setting of the liquid, by completion of gelation (transition to the gel state) or polymerization of additives. Recent investigations have highlighted the possibility to avoid organic additives (monomers, cross-linkers, and catalysts) and explored the marked sensitivity of viscosity to the shear rate (“shear-thinning” behavior) exhibited by aqueous suspensions of fine glass powders (“inorganic gel casting”) activated simply with alkali hydroxides (Rincón Romero et al., 2017).
The setting of alkali-activated glass suspensions is due to the formation of gel-forming compounds at the surface of particles, from variable glass/solution/atmosphere/solution interactions, depending on the chemistry of both activators (e.g., NaOH or KOH) and glass adopted. In fact, the gels comprise hydrated calcium silicate compounds (C-S-H) (Elsayed et al., 2017; Rincón Romero et al., 2017), as well as sodium carbonate–based hydrated compounds (Rincón Romero et al., 2018). The intensity of the alkaline attack may be tuned by controlling the dissolution and “pre-foaming” times. As an example, for soda–lime glass (Rincón Romero et al., 2017), after the same dissolution step (glass powder cast in alkaline solution and left under low-speed magnetic stirring for 3 h), suspensions exhibited a wider thixotropic cycle and higher viscosity, with increasing pre-foaming step (0–4 h at 75°C)—before addition of the surfactant and frothing—due to enhanced formation of gel-forming compounds, resulting in smaller pores.
An interesting finding from previous investigations on Ca-rich glasses, prone to be transformed into bioactive glass-ceramics by sinter-crystallization (Elsayed et al., 2017), concerns the possibility of activation in relatively “weak” alkaline solutions (1 M NaOH). This minimizes modifications in the overall chemical formulation of the base glass, when gel-forming compounds decompose and the related (alkali-rich) inorganic residue is incorporated by the softened glass powders, at the early stages of sintering. The present study is dedicated to the extension of this “weak alkaline activation” to Biosilicate® glass powders, evidencing the changes that occur passing from the processing of already crystallized powders to the processing of amorphous powders. We will show that amorphous powders led to more intense gelation. Foamed suspensions were easily exploited for the achievement of highly homogeneous glass-ceramic foams, which were subjected to extensive microstructural studies, involving advanced micro-tomography, to validate their suitability for bone tissue engineering applications.
Experimental Procedure
Biosilicate®, with a composition of 23.75 Na2O, 23.75 CaO, 48.5 SiO2, and 4 P2O5 (wt%), was prepared according to previous experiences (Granito et al., 2009; Crovace et al., 2016). A mixture of highly pure oxides was melted at 1,450°C, for 3 h, in a Pt crucible. During this period, the melt was poured in a steel plate twice and melted again for a better homogenization. After a total of 3 h of melting, the glass was poured into water. The glass frit obtained was then subjected to a double-stage heat treatment: first at 565°C/100 h for nucleation and then at 665°C/1 h for crystal growth. After cooling, the glass-ceramic frit was milled in a disc mill (MA 700—Marconi) and subsequently in a jet mill (CGS 10 Condux—Netzsch). The resulting Biosilicate® powder (average particle size ∼5 μm) was used to prepare the foams. As an alternative, the powders were remelted in a Pt crucible at 1,450°C for 1 h; fine glass powders were achieved by rapid cooling of the melt by pouring on a Pt foil, followed by dry ball milling and careful sieving. The maximum particle size was 20 μm. The thermal evolution of both types of starting powders was studied by means of differential scanning calorimetry (DSC/TG, 3+ STARe System, Mettler Toledo, Columbus, OH, United States), operating with a heating rate of 10°C/min.
Fine powders were suspended in aqueous solutions of 1.0 M NaOH (reagent grade, Sigma-Aldrich, Gillingham, United Kingdom), for a solid loading of 55–60 wt%. The slurries were first subjected to low-speed mechanical stirring (300 rpm), for 3 h, in polystyrene containers (50 mm diameter), to induce partial dissolution and gelation. In selected cases, a “pre-foaming” conditioning step was applied at 75°C, for 30–60 min. After the addition of 4 wt% Triton X-100 (polyoxyethylene octyl phenyl ether, Sigma-Aldrich, Gillingham, United Kingdom), the slurries were foamed by vigorous mechanical stirring (2,000 rpm), for 5 min, directly in the polystyrene containers, and left to dry at 40°C for 24 h. As an alternative, foamed suspensions were poured in smaller polyethylene containers (25 mm diameter), before drying, as illustrated by Figure 1. “Green” foams were unmolded and subjected to heat treatment in air, comprising a heat treatment at 350°C (5°C/min heating rate) for 1 h, for the burnout of organics, and a sintering stage at 1,000°C (10°C/min heating rate), for 1 h.
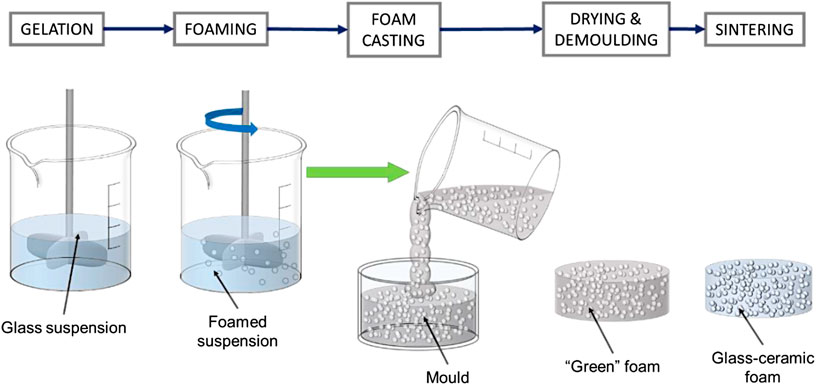
FIGURE 1. Schematic view of “foam casting” process.
Fourier transform infrared spectroscopy (FTIR, FTIR model 2000, Perkin Elmer Waltham, MA, United States) was performed, operating in absorbance mode, in the 4,000–400 cm−1 region, to monitor the low-temperature hardening of suspensions. X-ray diffraction (XRD) (Bruker D8 Advance, Karlsruhe, Germany—CuKα radiation, 0.15418 nm, 40 kV–40 mA, 2θ = 10–70°, step size 0.05°, and 2 s counting time) was applied to evaluate the phase evolution. Phase identification was performed by semi-automatic Match!® program package (Crystal Impact GbR, Bonn, Germany), supported by data from the PDF-2 database (ICDD-International Center for Diffraction Data, Newtown Square, PA, United States).
The bulk density was calculated from the weight-to-volume ratios on regular blocks (approximately 12 mm × 12 mm × 12 mm, cut from bigger foamed samples), later used for compressive tests, after careful determination of weight and dimensions using an analytical balance and a digital caliper. The apparent and real densities were measured by He gas pycnometry (Micromeritics AccuPyc 1330, Norcross, GA, United States), applied on samples in bulk form (before crushing tests) and powder form (after manual milling of fragments from crushing tests), respectively. Each data point represents the average value of at least five individual tests.
Morphological and microstructural characterizations were performed by optical stereomicroscopy (AxioCam ERc 5s Microscope Camera, Carl Zeiss Microscopy, Thornwood, New York, NY, United States) and scanning electron microscopy (FEI Quanta 200 ESEM, Eindhoven, Netherlands).
The 3D microstructure of the bioglass samples was additionally characterized by micro-computed tomography (µCT, Skyscan 1172, Bruker Micro CT, Kontich, Belgium) with a resolution of 6.68 µm/voxel. The tungsten X-ray tube was operated with 80 kV, and 100 mA; a wavelength of λ = 0.024 nm, an 11 MP detector, and an Al filter were used. The samples were rotated for 180° with a rotation step size of 0.25°. The recorded 2D sinograms were reconstructed with NRecon (version 1.6, Bruker Micro CT, Kontich, Belgium) and 3D visualized using the software Amira (version 5.6, Thermo Fisher Scientific, Inc., Waltham, United States). The evaluation of the porous morphology was carried out using the CT Analyzer (version 1.18.4, Bruker Micro CT, Kontich, Belgium).
The crushing strength of the foamed scaffolds obtained was measured at room temperature using an Instron 1121 UTM (Instron, Danvers, MA, United States) operating with a crosshead speed of 0.5 mm/min. Each data point represents the average value of at least 10 individual tests.
Results and Discussion
Processing of Biosilicate® Powders
The XRD analysis in Figure 2A (bottom pattern) testifies that the starting material was already highly crystallized, with all diffraction peaks attributed to sodium–calcium silicate (Na2CaSi2O6, i.e., Na2O·CaO·2SiO2, PDF#77-2189), previously known as the characteristic phase in Biosilicate® glass-ceramics. More precisely, Biosilicate® glass-ceramics, in the “1P” variant of Biosilicate® glass-ceramics, feature just Na2CaSi2O6; in the “2P” variant; according to different heat treatment conditions, the sodium–calcium silicate crystal phase is accompanied by a second phase, such as sodium calcium phosphate (NaCaPO4, i.e., Na2O·2CaO·P2O5) (Montazerian and Zanotto, 2016).
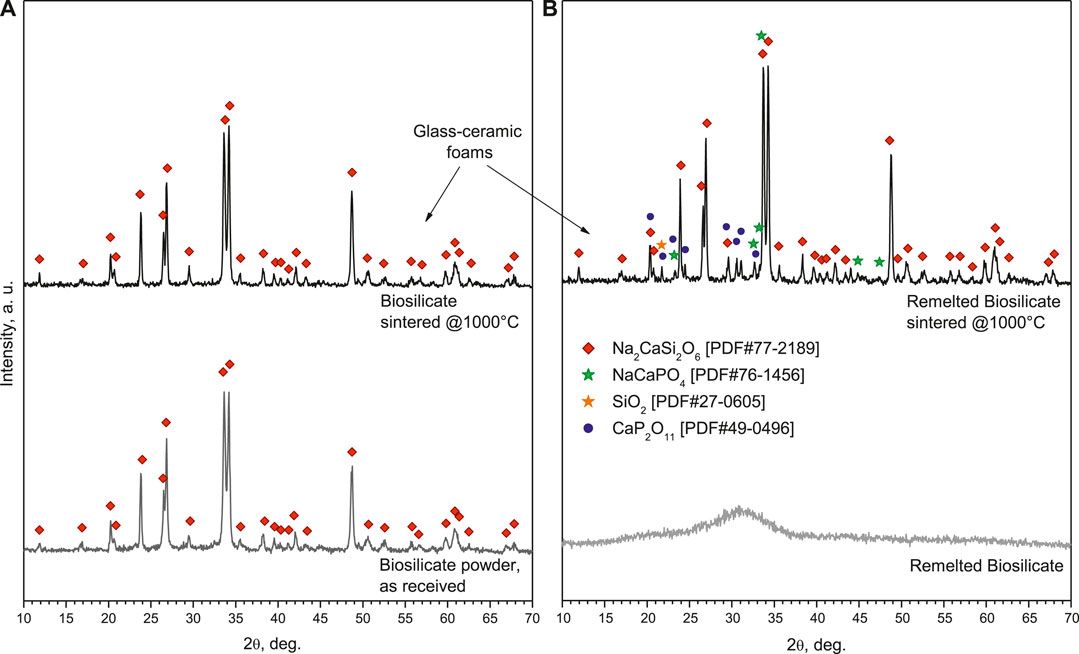
FIGURE 2. Phase evolution from raw materials to glass-ceramic foams: (A) Biosilicate® powders; (B) amorphous powders.
The phosphate-enriched residual glass phase, in agreement with previous observations (Desimone et al., 2013), was significant in enabling liquid phase–assisted sintering of Biosilicate® powders. As shown by Figure 3, a pellet deriving from uniaxial pressing (at 40 MPa), fired at 1,000°C, for 1 h (10°C/min heating rate), achieved an optimum coupling between densification—inferable from the porosity (<2%, determined by pycnometry analysis) and from the remarkable translucency (Figure 3A)—and shape retention—evident from the neat contours (polished cross section in Figure 3B). The sintering treatment did not cause any change in the crystallization (the diffraction pattern—not shown—was identical to that of starting powders).

FIGURE 3. Morphology of pellet from sintering of as received Biosilicate® powders.
Concerning the alkali activation, the Biosilicate® powders did not behave like glasses, leading to bioactive wollastonite-diopside glass-ceramics (Elsayed et al., 2017). These glasses could be activated by a weak alkaline solution, avoiding any pre-foaming step after dissolution at low-speed mechanical stirring (i.e., stage at 75°C to enhance dissolution and gelation), originally applied in the first “inorganic gel casting” experiments, on soda–lime glass, by Rincón Romero et al. (2017). Suspensions of Biosilicate® powders, without any step at 75°C, did not undergo an appreciable setting so that the bubbles generated by the frothing step collapsed easily upon drying. On the contrary, a pre-foaming step at 75°C, lasting 30 min, sufficed in stabilizing the “wet” foams, which remained unaltered after drying (i.e., in “green” state), as shown in the optical micrograph in Figure 4A.

FIGURE 4. Microstructural details of foam from as received Biosilicate® powders (55% solid loading, 30 min activation): (A) “green” foam (after drying); (B, C) fired foam (1,000°C/1 h).
The activation was evaluated by means of infrared spectroscopy. As shown by Figure 5A, there was a limited change in the spectrum passing from as received to activated conditions (30 min conditioning), expressed by the wide absorption band centered at about 3,400 cm−1 and absorption peaks at 1,400–1,450 and 500 cm−1. In our opinion, these bands could not be attributed to any hydrated calcium silicate compound; the signals are consistent with hydrated carbonate and phosphate phases. In particular, the quite intense peak at 1,400–1,450 cm−1 is attributed to C-O stretching vibrations in carbonates (Garcia Lodeiro et al., 2010), whereas the peak at about 500 cm−1 is probably due to hydrated phosphates (Joshi and Joshi, 2003); the wide band at higher wavenumbers corresponds to water of crystallization (Joshi and Joshi, 2003; Garcia Lodeiro et al., 2010).
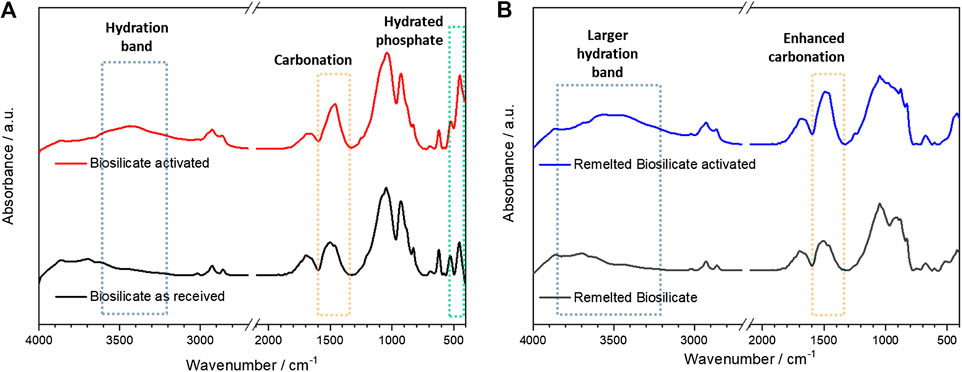
FIGURE 5. Assessment of activation by comparing infrared spectra, before and after activation: (A) as received Biosilicate® powders; (B) amorphous powders.
Owing to the high crystallization degree (again, no change in the phase assemblage occurred upon firing, see Figure 2B, top pattern), the sintering at 1,000°C confirmed the open porosity achieved with low-temperature foaming, as shown by the micrographs in Figures 4B,C. However, the crushing strength of the foams was not remarkable for any variant, based on the solid loading (55–60 wt%) and duration of pre-foaming step (30–60 min) applied to the starting suspension, as reported in Table 1.
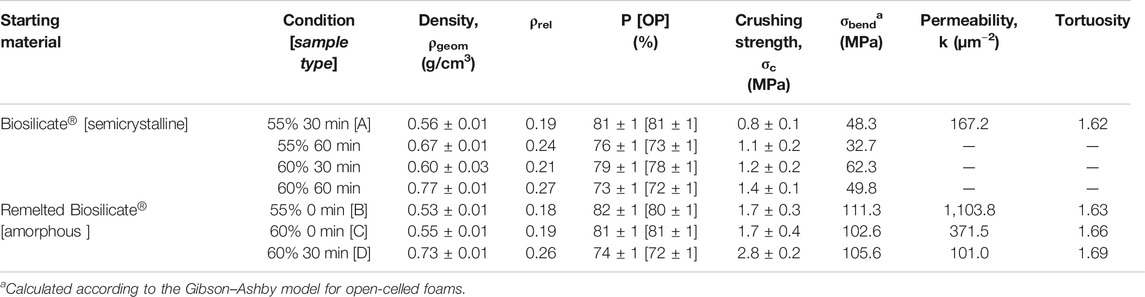
TABLE 1. Physical and mechanical properties of Biosilicate® glass-ceramic foams.
The direct comparison of crushing strength data, for porous bodies, was not appropriate, considering the variability of porosity, but the relative weakness of the foams could be easily understood by analyzing the experimental data according to the well-recognized Gibson–Ashby model (Gibson and Ashby, 1999), for open-celled foams. The model predicts the crushing strength of foams (σc) as ruled by the bending strength of the solid phase (σbend), “downscaled” by the relative density (ρrel = 1 − P/100, where P is the total porosity, in vol%), as follows:
Reversing Eq. 1 and introducing the experimental data (crushing strength and relative density), we could calculate a bending strength of the solid phase well below 70 MPa, that is, a typical value for soda–lime glass (Bernardo et al. 2004).
The calculated values could not be interpreted as the real bending strength of the solid phase. In fact, the strength/density correlation, according to the Gibson–Ashby model, assumes foams as ideal lattices, corresponding to the assemblage of mono-dimensional elements (struts), with uniform cell size. The effects of cell size and structural inhomogeneity, leading to local stress concentrations, are neglected. Keeping the simplicity of the Gibson–Ashby model, we could think at an enhanced downscaling of strength with porosity, and update Eq. 1 by including a structural factor (SF), below 1, as follows:
The inhomogeneity of the cellular structure was confirmed by the microstructural details shown in Figure 3C. The cell walls comprised a multitude of poorly sintered particles, with irregularly shaped openings between adjacent cells.
Processing of Remelted Biosilicate® Powders
To adjust the interaction with the alkaline solutions and develop glass-ceramics by a real sinter-crystallization process, the Biosilicate® material was considered also after remelting, rapid cooling, and milling. As shown by the bottom pattern in Figure 2B, featuring the typical diffraction “halo” of glass, the newly obtained powders were completely amorphous. According to the DSC plots, in Figure 6, amorphous Biosilicate® powders (exhibiting a transition temperature Tg ∼550°C) were prone to “recover” the crystallization during the sintering, testified by the strong exothermic peak centered at Tc∼700°C, consistent with the formation of the already discussed Na2CaSi2O6 phase (Huang et al., 2007). A similar exothermic effect was not exhibited by the original Biosilicate® powders, already featuring the sodium–calcium silicate phase. Weak endothermic peaks at ∼1,200°C (marked with Tm) were attributed to the melting of crystal phases, for both materials.
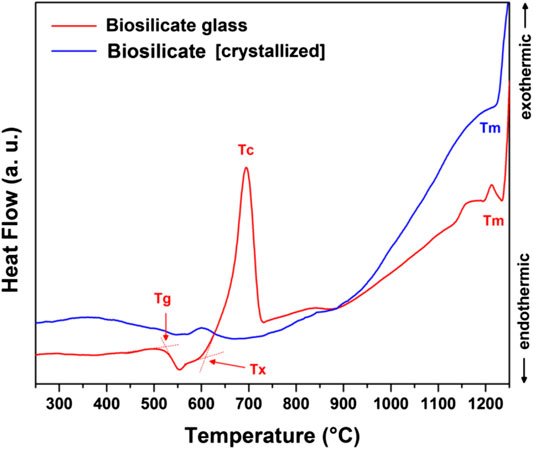
FIGURE 6. DSC plots of as received Biosilicate® powders and amorphous powders.
Starting from amorphous Biosilicate® powders, “green” foams could be achieved by frothing even without the pre-foaming step, due to the formation of gels with a different formulation, testified by changes in the infrared spectra. As shown by Figure 4B, there was an enhanced absorption above 3,500 cm−1, consistent with the formation of hydrated calcium silicate compounds (Garcia Lodeiro et al., 2010). Carbonation, according to the band at 1,400–1,450 cm−1, also increased.
The enhanced gelation motivated a further upgrade. Instead of drying foamed suspensions in the same plastic containers used for preliminary dissolution, a “foam casting” process was applied according to the scheme in Figure 1. Foamed suspensions were cast in plastic molds, later used just for the drying step. This was intended to favor the homogeneity, since the pouring could determine some remixing, thus preventing gradients arising from different distances from the stirrer (removed after foaming), in the original container.
Examples of the new cellular structures are shown in Figure 7. A high homogeneity was achieved, in both “green” (optical image in Figure 6A) and sintered state (SEM micrographs in Figures 6B–D). The stabilization of the open-celled structures, upon firing (Figure 6B), was caused again by the viscosity increase associated with crystallization. As expected from the DSC in Figure 5 and illustrated by Figure 2B, remelted Biosilicate® powders underwent a substantial devitrification upon viscous flow sintering. Considering the more intense diffraction peaks related to sodium–calcium silicate, crystallization was even enhanced. This is justified by the appearance of some extra peaks corresponding to the previously mentioned characteristic crystal phase of the “2P” variant of Biosilicate® glass-ceramics, that is, sodium–calcium phosphate (NaCaPO4, that is, Na2O·2CaO·P2O5, PDF#76-1456). Traces of cristobalite (SiO2, PDF#27-0605) and calcium phosphate (CaP2O11, i.e., CaO·2P2O5, PDF#49-0496) were detected as well. The additional calcium phosphate phase, known for its high resorbability (Safronova and Putlyaev, 2017), has been already detected in glass-ceramic derivatives of 45S5 (Ouis et al., 2012).

FIGURE 7. Microstructural details of a porous glass-ceramic from remelted Biosilicate® (sample [C], 60 wt% solid loading, 0 h): (A) green foam; (B–D) foam after firing at 1,000°C for 1 h.
Remelted Biosilicate® powders, after alkali activation and firing, confirmed what previously observed with porous glass-ceramics based on wollastonite and diopside (CaSiO3-CaMgSi2O6) (Elsayed et al., 2017). In the case of semicrystalline Biosilicate® powder activation, gelation and sintering relied on phosphate-enriched residual glass phase so that the overall process could be considered as “selective.” With amorphous powders, on the contrary, the process involved all glass components. The new gel formulation promoted both densification, evident from more compact cell walls (Figure 7C), and crystallization. The latter is testified, besides by diffraction patterns, by a multitude of tiny crystals immersed in former glass powders, well bound together (Figure 7D). The alkali-enriched surface gels, as previously observed (Elsayed et al., 2017), reasonably transformed in an alkali-rich low viscosity liquid surrounding glass particles, promoting the ionic interdiffusion, and reducing the activation energy for crystal growth (Watanabe et al., 2008).
The enhanced densification of walls and crystallization determined a substantial improvement of the mechanical properties, as reported by Table 1. Reversing Eq. 1, according to the crushing strength of foams from the processing of remelted Biosilicate®, combined with relative density, yielded a reference bending strength exceeding 100 MPa, more appropriate for a glass-ceramic solid phase. In other words, the updated processing led to more “mechanically efficient” cellular structures (SF closer to 1).
Advanced Microstructural Characterization
The cell and strut size distribution in selected samples was carefully studied using advanced micro-tomography. The technique led to reconstructed images, shown in Figure 8, later subjected to analysis of the pore connectivity, using a skeletonization algorithm—within the Amira Software—on the binarized µCT images as shown by Figures 9A,B. All determinations were performed to assess the suitability of the proposed approach, according to well-known requirements, in bone tissue engineering, concerning pore size distribution, permeability and tortuosity (i.e., ratio of the actual path length of channels within the scaffold to the thickness of the scaffold in the macroscopic flow direction) (Innocentini et al., 2010). In particular, effective cell ingrowth and vascularization are typically achieved with foamed scaffolds exhibiting a distinctive similarity with the trabecular bone. This is expressed by relatively big pores (with a diameter of about 500 µm and an interconnect diameter exceeding 100 μm; Jones et al., 2007), high permeability (k exceeding 3·10–11 m2, i.e., 30 µm−2), and specified tortuosity (in the 1.26–2.64 range; Innocentini et al., 2010).
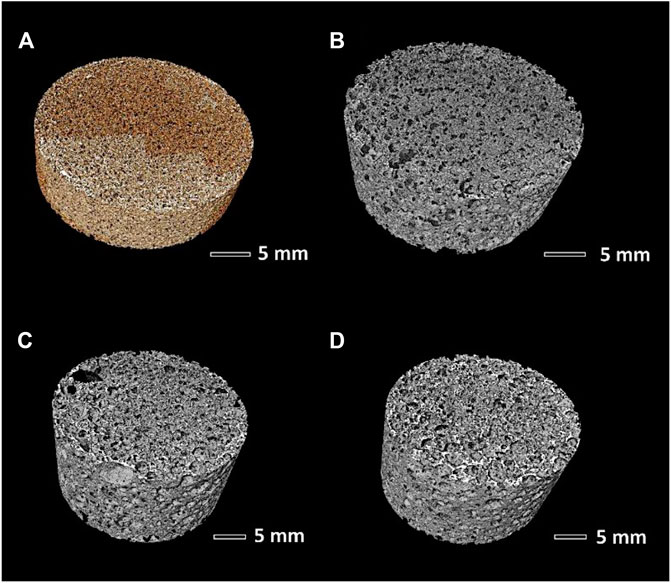
FIGURE 8. Microtomography reconstructed images: (A) sample A (Biosilicate® glass-ceramics); (B–D) samples B, C, and D (remelted Biosilicate®).
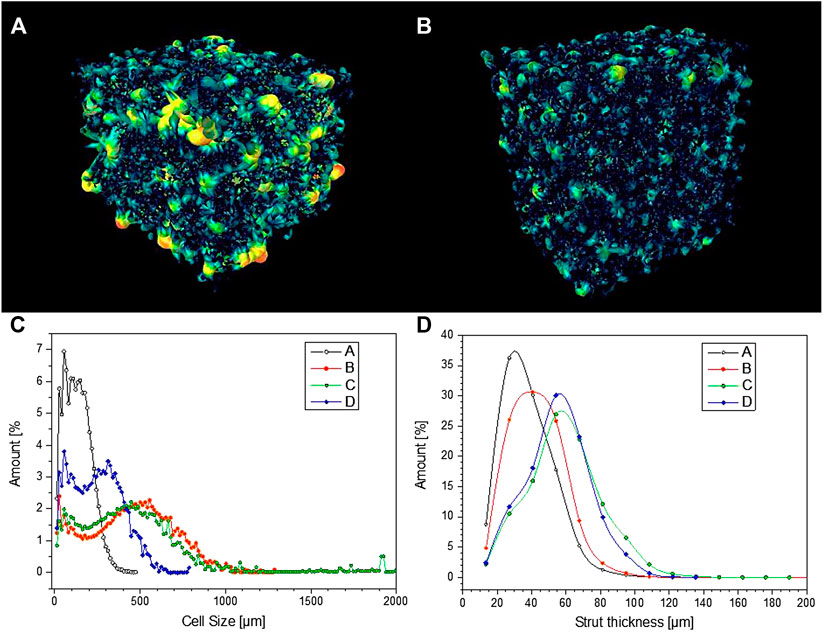
FIGURE 9. (A, B) pore connectivity of samples C and D; (C, D) pore size distribution and strut thickness of selected porous Biosilicate® glass-ceramics.
From Figure 9C, it can be observed that glass-ceramic samples B and C, from amorphous powders, well fulfilled the requirement of cell size, as previously mentioned. More specifically, they exhibited a similar cell size distribution, with an average cell size of 450–470 µm. The distribution was bimodal, that is, derived from the overlapping contributions from two populations, with size below 100 µm and slightly above 500 µm.
According to Table 1, the permeability of sample B (k > 1,100 μm−2) was much more substantial than that of sample C, already quite remarkable, owing to a wide distribution of interconnect (cell window) size, with a significant fraction above 100 μm, as illustrated by Figure 10A. The measurement of cell windows derived from a careful selection of cross-sectional images (Figure 10B), inversion (Figure 10C), application of watershed transformation (Figure 10D), and image subtraction (Figure 10E). Although highly permeable, the sample from glass-ceramic powders (sample A) could not be accepted, owing to the limited mean cell size (136 µm) and limited cell windows.
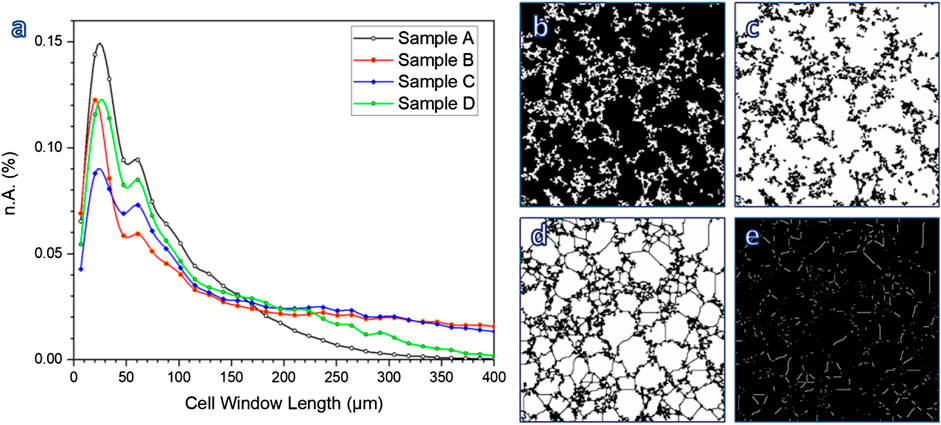
FIGURE 10. (A) Determination of cell window length; (B–E) examples of image treatment for calculations.
The “inorganic gel casting” is confirmed in offering a high “tunability” (Rincón Romero et al., 2017), expressed by the interplay of many parameters. In particular, the shear-thinning behavior of activated suspensions, essential for the “freezing” of the cellular structure, established utilizing intensive mechanical stirring, before sintering, is known to increase with both solid loading (Deng et al., 2015) and reaction degree (Rincon Romero et al., 2017). At a fixed reaction degree (no pre-foaming step), passing from sample B (lower solid loading) to sample C (higher solid loading) led to an increase of strut size (from 43 to 59 µm maybe these values could be round up), as illustrated by Figure 9D. An increase of reaction degree (30 min pre-foaming step added, from sample C to sample D) confirmed the strut size but reduced the cell size (249 µm for sample D).
Conclusion
Our results confirm “weak alkali activation” (attack of glass powders in weakly alkaline solutions, followed by gelation), as a method for the easy manufacturing of highly porous, open-celled glass-ceramic bodies. Applied to semicrystalline Biosilicate® powders, however, it is hardly controllable.
Remelting of Biosilicate® powders led to a fully amorphous material, with a much different response under alkaline attack; in particular, gelation could be activated in shorter times and lead to gelling compounds with different composition, compared to the case of semicrystalline powders.
The easier control of gelation conditions, for glass powders resulting from remelting of Biosilicate®, enabled the application of the “foam casting” approach, leading to homogeneous open-celled foams. These vitreous powders underwent sinter-crystallization upon firing at 1,000°C; such process did not lead to the simple recovery of the crystal phase already present in Biosilicate® powders; instead, it also led to extra phases. The sinter-crystallization process enabled merging of glass powders at cell walls, with the formation of relatively thick struts, and the viscosity increase deriving from the precipitation of crystals impeded the collapse of the open-celled structure achieved with foam casting.
Owing to the adequate cell size distribution and cell connectivity, optimized porous Biosilicate® glass-ceramics obtained from remelting and sinter-crystallization well match the requirements for foamed scaffolds applied in bone tissue engineering.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
HE and AR: manufacturing of foams and characterization (except micro-tomography). AR: editing of schemes. HE: editing of draft. MC and EZ: preparation of starting materials and manuscript editing. TF: micro-tomography characterization and manuscript editing. EB: concept and manuscript editing.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past collaboration with one of the authors EZ.
Acknowledgments
The authors thank Mr. Matteo Pinton (University of Padova) for experimental assistance. We also thank the Sao Paulo Research Foundation, Fapesp, Process #2013/007796-3 for financial support.
References
Albakry, M., Guazzato, M., and Swain, M. V. (2004). Influence of hot pressing on the microstructure and fracture toughness of two pressable dental glass–ceramics. J. Biomat. Res. Part B. 71, 99–107. doi:10.1002/jbm.b.30066
Baino, F., and Vitale-Brovarone, C. (2012). “New trends in bone tissue engineering scaffolds: hierarchically porous glass and glass-ceramic structures,” in Surface tailoring of inorganic materials for biomedical applications. Editors L. Rimondini, C. L. Bianchi, and E. Verné (Sharjah, UAE: Bentham Science Publishers), 376–391.
Baino, F., and Vitale-Brovarone, C. (2014). “Bioactive glass and glass-ceramic foam scaffolds for bone tissue restoration” in Biomedical foams for tissue engineering applications. 1st Edn. Editor P. Netti (Cambridge, UK: Woodhead Publishing), 213–248.
Bellucci, D., and Cannillo, V. (2018). A novel bioactive glass containing strontium and magnesium with ultra-high crystallization temperature. Mater. Lett. 213, 67–70. doi:10.1016/j.matlet.2017.11.020
Bernardo, E., Scarinci, G., Maddalena, A., and Hreglich, S. (2004). Development and mechanical properties of metal-particulate glass matrix composites from recycled glasses. Compos. Part A Appl. Sci. Manuf. 35, 17–22. doi:10.1016/j.compositesa.2003.09.022
Clark, T. J., and Reed, J. S. (1986). Kinetic processes involved in the sintering and crystallization of glass powders. J. Am. Ceram. Soc. 69, 837–846. doi:10.1111/j.1151-2916.1986.tb07370.x
Crovace, M. C., Souza, M. T., Chinaglia, C. R., Peitl, O., and Zanotto, E. D. (2016). Biosilicate®—a multipurpose, highly bioactive glass-ceramic. In vitro, in vivo and clinical trials. J. Non-Cryst. Sol. 432, 90–110. doi:10.1016/j.jnoncrysol.2015.03.022
Deng, X., Wang, J., Liu, J., Zhang, H., Li, F., Duan, H., et al. (2015). Preparation and characterization of porous mullite ceramics via foam-gelcasting, Ceram. Int. 41, 9009–9017. doi:10.1016/j.ceramint.2015.03.237
Desimone, D., Li, W., Roether, J. A., Schubert, D. W., Crovace, M. C., Rodrigues, A. C. M., et al. (2013). Biosilicate-gelatine bone scaffolds by the foam replica technique: development andcharacterization. Sci. Technol. Adv. Mater. 14, 045008. doi:10.1088/1468-6996/14/4/045008
Elsayed, H., Rincón Romero, A., Ferroni, L., Gardin, C., Zavan, B., and Bernardo, E. (2017). Bioactive glass-ceramic scaffolds from novel ‘inorganic gel casting’ and sinter-crystallization. Materials 10, 171. doi:10.3390/ma11030349
Garcia Lodeiro, I., Fernandez-Jimenez, A., Palomo, A., and Macphee, D. E. (2010). Effect on fresh C-S-H gels of the simultaneous addition of alkali and aluminium. Cem. Concr. Res. 40, 27–32. doi:10.1016/j.cemconres.2009.08.004
Gibson, L. J., and Ashby, M. F. (1999). Cellular solids, structure and properties. 2nd Edn. Cambridge, UK: Cambridge University Press.
Gonzenbach, U. T., Studart, A. R., Steinlin, D., Tervoort, E., and Gauckler, L. J. (2007). Processing of particle‐stabilized wet foams into porous ceramics. J. Am. Ceram. Soc. 90, 3407–3414. doi:10.1111/j.1551-2916.2007.01907.x
Granito, R. N., Ribeiro, D. A., Rennó, A. C. M., Ravagnani, C., Bossini, P. S., Peitl-Filho, O., et al. (2009). Effects of biosilicate and bioglass 45S5 on tibial bone consolidation on rats: a biomechanical and a histological study. J. Mater. Sci. Mater. Med. 20, 2521–2526. doi:10.1007/s10856-009-3824-z |
Höland, W., and Rheinberger, V. (2008). “Dental glass-ceramics,”in Bioceramics and their clinical applications. Sawston, England: Woodhead Publishing Series in Biomaterials, 548–568.
Höland, W., Rheinberger, V., Apel, E., and van’t Hoen., C. (2007). Principles and phenomena of bioengineering with glass-ceramics for dental restoration. J. Eur. Ceram. Soc. 27, 1521–1526. doi:10.1016/j.jeurceramsoc.2006.04.101
Hench, L. L. (1977). Analysis of bioglass fixation of hip prostheses. J. Biomed. Mater. Res. 11, 267–282. doi:10.1002/jbm.820110211 |
Hench, L. L. (2006). The story of Bioglass®. J. Mater. Sci. Mater. Med. 17, 967–978. doi:10.1007/s10856-006-0432-z |
Hench, L. L., and Polak, J. M. (2002). Third-generation biomedical materials. Science 295, 1014–1017. doi:10.1126/science.1067404 |
Huang, L.-C., Lin, C.-C., and Shen, P. (2007). Crystallization and stoichiometry of crystals in Na2CaSi2O6–P2O5 based bioactive glasses. Mat. Sci. Eng. A 453–453, 326–333. doi:10.1016/j.msea.2006.10.136 |
Innocentini, M. D. M., Faleiros, R. K., Pisani, R., Thijs, I., Luyten, J., and Mullens, S. (2010). Permeability of porous gelcast scaffolds for bone tissue engineering. J. Por. Mat. 17, 615–627. doi:10.1007/s10934-009-9331-2
Jones, J. R., Poologasundarampillai, G., Atwood, R. C., Bernard, D., and Lee, P. D. (2007). Non-destructive quantitative 3D analysis for the optimisation of tissue scaffolds. Biomaterials 28, 1404–1413. doi:10.1016/j.biomaterials.2006.11.014 |
Joshi, V. S., and Joshi, M. J. (2003). FTIR spectroscopic, thermal and growth morphological studies of calcium hydrogen phosphate dihydrate crystals. Crystal 38 (2003), 817–821. doi:10.1002/crat.200310100
Kolan, K. C. R., Leu, M. C., Hilmas, G. E., Brown, R. F., and Velez, M. (2011). Fabrication of 13-93 bioactive glass scaffold for bone tissue engineering using indirect selective laser sintering. Biofabrication 3, 025004. doi:10.1088/1758-5082/3/2/025004
Li, P., Yang, Q., Zhang, F., and Kokubo, T. (1992). The effect of residual glassy phase in a bioactive glass-ceramic on the formation of its surface apatite layer in vitro. J. Mater. Sci. Mater. Med. 3, 452–456. doi:10.1007/BF00701242
Liu, X., Rahaman, M. N., Hilmas, G. E., and Bal, B. S. (2013). Mechanical properties of bioactive glass (13-93) scaffolds fabricated by robotic deposition for structural bone repair. Acta Biomat. 9, 7025–7034. doi:10.1016/j.actbio.2013.02.026
Montazerian, M., and Zanotto, E. D. (2016). History and trends of bioactive glass‐ceramics. J. Biomed. Mat. Res. A 104, 1231–1249. doi:10.1002/jbm.a.35639
Ouis, M. A., Abdelghany, A. M., and ElBatal, H. A. (2012). Corrosion mechanism and bioactivity of borate glasses analogue to Hench’s bioglass. Process. Appl. Ceram. 6, 141–149. doi:10.2298/PAC1203141O
Pinto, K. N. Z., Tim, C. R., Crovace, M. C., Orsini Rossi, B. R., Kido, H. W., Parizotto, N. A., et al. (2018). Scaffolds of bioactive glass-ceramic (Biosilicate®) and bone healing: a biological evaluation in an experimental model of tibial bone defect in rats. Bio Med. Mater. Eng. 29, 665–683. doi:10.3233/BME-181016
Piotrowski, G., Hench, L. L., Allen, W. C., and Miller, G. J. (1975). Mechanical studies of the bone bioglass interfacial bond. J. Biomed. Mater. Res. 9, 47–61. doi:10.1002/jbm.820090408 |
Rincón Romero, A., Giacomello, G., Pasetto, M., and Bernardo, E. (2017). Novel ‘inorganic gel casting’ process for the manufacturing of glass foams. J. Eur. Ceram. Soc. 37, 2227–2234. doi:10.1016/j.jeurceramsoc.2017.01.012
Rincón Romero, A., Salvo, M., and Bernardo, E. (2018). Up-cycling of vitrified bottom ash from MSWI into glass-ceramic foams by means of ‘inorganic gel casting’ and sinter-crystallization. Construct. Build. Mater. 192, 133–140. doi:10.1016/j.conbuildmat.2018.10.135
Roohani-Esfahani, S. I., and Zreiqat, H. H. (2012). “Ceramic scaffolds, current issues and future trends,” in Integrated biomaterials in tissue engineering. Editors M. Ramalingamet al. (New York, NY: Wiley), 25–46.
Safronova, T. V., and Putlyaev, V. I. (2017). Powder systems for calcium phosphate ceramics. Inorg. Mater. 53, 17–26. doi:10.1134/S0020168516130057
Sepulveda, P., and Binner, J. G. P. (1999). Processing of cellular ceramics by foaming and in situ polymerisation of organic monomers. J. Eur. Ceram. Soc. 19, 2059–2066. doi:10.1016/S0955-2219(99)00024-2
Souza, M. T., Rennó, A. C. M., Peitl, O., and Zanotto, E. D. (2017). Focus on biomaterials new highly bioactive crystallization-resistant glass for tissue engineering applications. Transl. Mater. Res. 4, 014002. doi:10.1088/2053-1613/aa53b5
Vitale-Brovarone, C., Baino, F., and Verné, E. (2009). High strength bioactive glass-ceramic scaffolds for bone regeneration. J. Mater. Sci. Mater. Med. 20, 643–653. doi:10.1007/s10856-008-3605-0 |
Keywords: glass-ceramics, sintering, alkali activation, gel casting, foams (froth)
Citation: Bernardo E, Elsayed H, Romero AR, Crovace MC, Zanotto ED and Fey T (2021) Biosilicate® Glass-Ceramic Foams From Refined Alkali Activation and Gel Casting. 7:588789. doi: 10.3389/fmats.2020.588789
Received: 29 July 2020; Accepted: 28 October 2020;
Published: 12 March 2021.
Edited by:
Francesco Baino, Politecnico di Torino, ItalyReviewed by:
Ashutosh Goel, Rutgers, The State University of New Jersey, United StatesSara Banijamali, Materials and Energy Research Center, Iran
Copyright © 2021 Bernardo, Elsayed, Romero, Crovace, Zanotto and Fey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enrico Bernardo, ZW5yaWNvLmJlcm5hcmRvQHVuaXBkLml0