 Daniel Vizoso
Daniel Vizoso Chaitanya Deo
Chaitanya Deo- Nuclear and Radiological Engineering Programs, GWW School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA, United States
The use of predictive models to examine defect production and migration in metallic systems requires a thorough understanding of the energetics of defect formation and migration. In fully miscible alloys, atomistic properties will all have a range of values that are heavily dependent on local atomic configurations. In this work we have used the atomistic simulation tool Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) to investigate the impact of first nearest neighbor configuration on vacancy formation energies at 0 K in γ-U-Zr alloys of varying Zr concentrations. The properties of randomly generated alloy microstructures were also compared with those produced as special quasi-random structures (SQS) using the “mcsqs” code within the Alloy Theoretic Automated Toolkit. Results have confirmed that local configuration can have a significant impact on measured properties and must be considered when characterizing miscible alloy systems. Results also indicated that the generation method of the random structure (i.e., via random species assignment or a method of enforced randomness) does not result in a measurable difference in average vacancy formation energies in miscible U-Zr systems.
1 Introduction
The use of predictive models to examine defect production and migration in metallic systems requires a thorough understanding of the energetics of defect formation and migration. In fully miscible alloys, atomistic properties are heavily dependent on local atomic configurations, and can vary significantly across the volume of even a homogeneous alloy mixture. One such atomistic property is the vacancy formation energy (
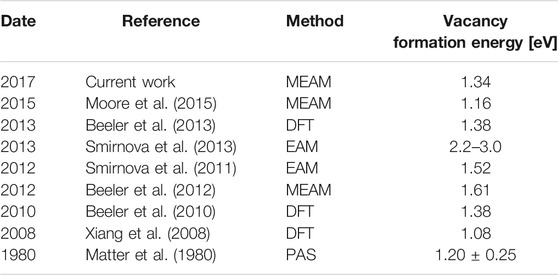
TABLE 1. Vacancy formation energies in bcc uranium.
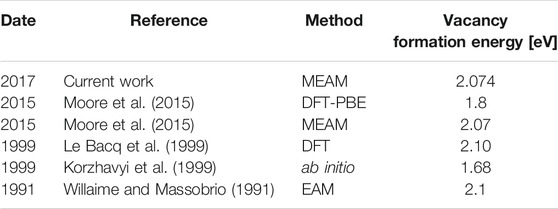
TABLE 2. Vacancy formation energies in bcc zirconium.
For this work, the Modified Embedded Atom Method (MEAM) based interatomic potential for U-Zr developed by Moore et al. (2015) was used. This potential was developed to simulate γ-U-Zr alloys at operational temperatures. The uranium portion of this potential was designed to recreate the properties of γ-phase uranium at high temperatures, but was also fit to the elastic properties and lattice constant of γ-phase uranium at 0 K. However, the γ-phase of pure uranium is unstable at 0 K, and must be stabilized through the application of an external pressure. Due to this mechanical instability, calculations of properties by other common simulation methods such as density functional theory (DFT) or ab initio calculations cannot be performed, due to their reliance on the presence of an equilibrium state at 0 K (Moore et al., 2015). However, through the use of a volume or pressure constraint in molecular dynamics simulations, simulations of pure γ-phase uranium can be performed. It is also important to note that the presence of a small atom percentage of an alloying element can stabilize the γ-phase of uranium, meaning that all of the studied U-Zr alloy compositions in this work were stable without requiring additional constraints.
It is common practice in molecular dynamics (MD) modeling of metallic alloys to populate the lattice structure via random atom placement. Bulk material properties of crystals generated in this way can closely mimic the properties of real crystals; however, due to the finite nature of any modeled supercell, the random population of lattice sites may deviate from “perfect” randomness on a local scale. To address this issue, Wei et al. (1990) proposed a method of generating “special quasirandom structures” (SQS) that would more closely mimic the configuration average of an infinite perfectly random
For any alloy, there exists a distribution of possible vacancy formation energies due to the species arrangement and thermal oscillations of the atoms in the neighboring sites (Zhao et al., 2016). For this work, to examine the impact of different configurations of nearest neighbor sites on vacancy formation energy, all 256 possible nearest neighbor configurations for a binary bcc alloy were examined for each atom percentage and vacancy type tested. However, not all configurations are equally likely to occur in all alloys with differing atom percentages (e.g., all eight nearest neighbor lattice positions of a vacancy in a U-10%Zr alloys being filled by Zr atoms, etc.)
In a perfectly random binary alloy, the probability of an atom of species A inhabiting any given lattice position can be defined by the concentration of that species in the bulk lattice,
This probability refers to the probability of i species A atoms and
Finally, the expression that defines the probability of having a specific species distribution in z lattice positions in a binary alloy can be expressed as the product of the probability of having the species distribution in a particular configuration and the total number of possible configurations for that species distribution, resulting in a binomial probability distribution (Kim, 1984),
In a BCC structure, there are eight first nearest neighbor sites and 6 s nearest neighbor sites. For a binary BCC alloy, there then exist 256 unique first nearest neighbor configurations and 104 unique second nearest neighbor configurations, combining for a total of 26,624 unique local configurations around a single atomic position, which in a binary system can be one of two different species. To account for this the results of all possible configurations were compared with results for the configurations with atom percentages that were reasonable when compared to the atom percentage of the alloy as a whole.
All MD simulations were performed with a supercell consisting of 10 × 10 × 10 unit cells held in an NVE ensemble.
2 Methods
2.1 Interatomic Potential of Uranium-Zirconium
Molecular dynamics simulations model interactions between atoms via interatomic potential functions that are used to calculate the force on any atom in the system caused by the other N−1 atoms in the system. A primary advantage of interatomic potential functions over first principle calculations is that potentials can be used for the calculation of larger scale atomic properties at temperature, as well as the ability to perform calculations on systems with many atoms. This is particularly advantageous for materials such as γ-uranium, which is mechanically unstable at 0 K (Moore et al., 2015). For this work, a modified embedded atom method, or MEAM, potential was used. MEAM potentials are modified versions of embedded atom method potentials (EAM), which are semi-empirical potentials that express cohesive energy in terms of embedding energies, with the atoms in the system being embedded in an electron gas. The difference between MEAM and EAM potentials is that MEAM potentials account for angular forces, which are present in certain materials that form directional bonds (Beeler et al., 2012). Full descriptions and comparisons of EAM and MEAM potentials have been completed in other works (Beeler et al., 2012; Foiles and Baskes, 2012), and only a short description of the theory will be included here. A more complete description of MEAM potentials is also provided in the Supplementary Material, paraphrased from Beeler et al. (2012). The general equation for the interatomic potential for both EAM and MEAM is shown by Eq. 4.
In this equation, E represents the energy of the atoms i,
Vacancy formation energies can be calculated for a bulk lattice using the following equation,
where
2.2 Special Quasirandom Structures Generation Procedures
For the generation of SQS, three input files were used with the “mcsqs” code from the Alloy Theoretic Automated Toolkit (ATAT) (van de Walle and Asta, 2002; van de Walle et al., 2002; Hart and Forcade, 2008; van de Walle et al., 2013; Liu et al., 2017). The first input file (by default rndstr.in, but this can be changed with the −1 = filename option in the command line) defines the structure of the alloy and the ratio of the components. This input file must be made by the user. The second input file (by default clusters.out, but this can be changed with the −cf = filename option in the command line) tells the code what clusters must be considered while generating the SQS. This input file can be generated by code by using the command
where −2 and −3 define the ranges of pairs and triplets, while in the same directory as rndstr.in. For the purpose of this work, the range of pairs was set to 6 Angstroms, and the range of triplets was set to 8 Angstroms. The third input file used was sqscell. out, which defines the supercell that “mcsqs” will use while generating the SQS. This input file is required only if the user requires a supercell of a specific shape. For the purpose of this work, a cubic supercell was used, to allow for ease of comparison between the SQS structure and the cubic structures that LAMMPS generates. To generate the SQS, the command
must be used, where −n defines the number of atoms that will be placed within the supercell, and −rc tells the code to use the supercell defined in sqscell.out. For the code to work, the value for −n must be an integer, and the number of atoms must match with the size of the supercell provided in sqscell.out.
To run multiple ATAT sequences in parallel, the command −ip = ... can be added to the previous command, with an integer following the equals sign. This integer will be appended to the names of the output files produced by the code. The produced SQS will be found in the output file bestsqs(i). out, which will have the dimensions of the unit cell and the supercell, and then the xyz location and type of each atom in the supercell in units of the specified lattice parameter. The file bestcorr.out tracks the correlations of the best SQS that has been found. For the purpose of this work, the code was stopped after the first SQS was generated.
2.3 Molecular Dynamics Procedures
2.3.1 Random Structures
All of the random structures used for this work were produced by Plimpton (1995) LAMMPS. To produce a random structure, the dimensions of the supercell must be given in terms of unit cells, and the lattice parameter of the unit cell must be provided. Also, the structure of the unit cell must be specified (bcc, fcc, etc…). LAMMPS will produce a homogeneous structure with the specified dimensions and structure. To create a random alloy, the set command is used, which allows the user to specify what percentage of atoms should be replaced by a specified atom type. Each alloy was minimized to energy (unitless) and force (eV/Angstrom) stopping tolerances of 1.0e−15. After running the initial minimization on the as-generated crystal, the atom at the center of the supercell was removed, and the alloy was again run for one time-step, then minimized to the same tolerance limits. For each random supercell generated by LAMMPS, the calculation of the vacancy formation energy was performed for both zirconium and uranium vacancies by setting the atom type of the atom that was to be removed to the desired element. To prevent the mechanical instability of pure uranium at 0 K from influencing the results of simulations performed with pure uranium, the energy minimization step was only performed on the first nearest neighbors and second nearest neighbors of the vacancy.
2.3.2 Special Quasirandom Structures
LAMMPS has the ability to import supercell structures through the use of the read_data command. After the supercell structure is read into the LAMMPS, the same process for the determination of the vacancy formation energy was used. For the 2,000 atom SQS structures, a MATLAB script was used that determined the atom type of the atom at the center of the supercell, and found the xyz coordinates of the closest atom to the center of the supercell that was of the other type. For the 250 SQS structures, the atom at the center of the supercell was removed, and random atoms within the structure were chosen until an atom of the different type was found. Then, two runs were performed for each SQS, one where the atom at the center of the supercell was removed, and then a run where the atom of the opposite type was removed. In this way, the structure of the SQS was preserved.
3 Results and Discussion
Using LAMMPS, vacancy formation energies were calculated for the random structures and the SQS by minimizing the energy of the lattice as it was produced, then removing an atom from the center of the structure, minimizing the energy, and then taking the difference between the energies before and after the removal of the atom, as described by Eq. 5 in the Section Methods. All of these simulations were performed at a temperature of 0 K.
Figures 1, 2 compare the vacancy formation energies for the removal of zirconium and uranium atoms, respectively, for structures that were generated using the SQS method and by random atom placement. SQS can not be generated for pure metals, so only a single LAMMPS simulation was performed to determine the
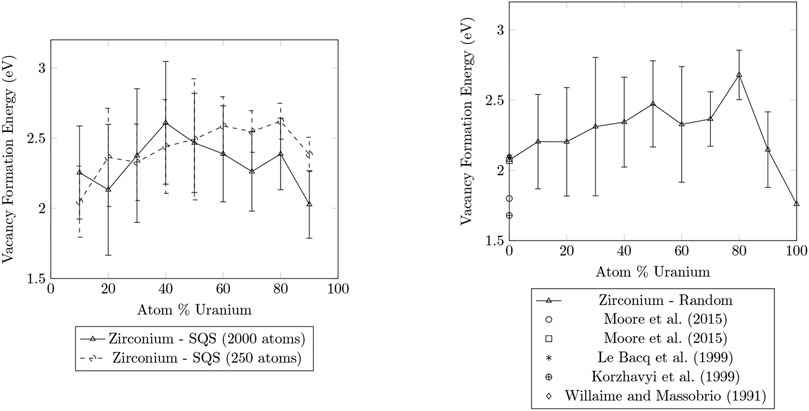
FIGURE 1. Plots of the vacancy formation energies for the removal of zirconium calculated for SQS and random structures of U-Zr alloy ranging from 0
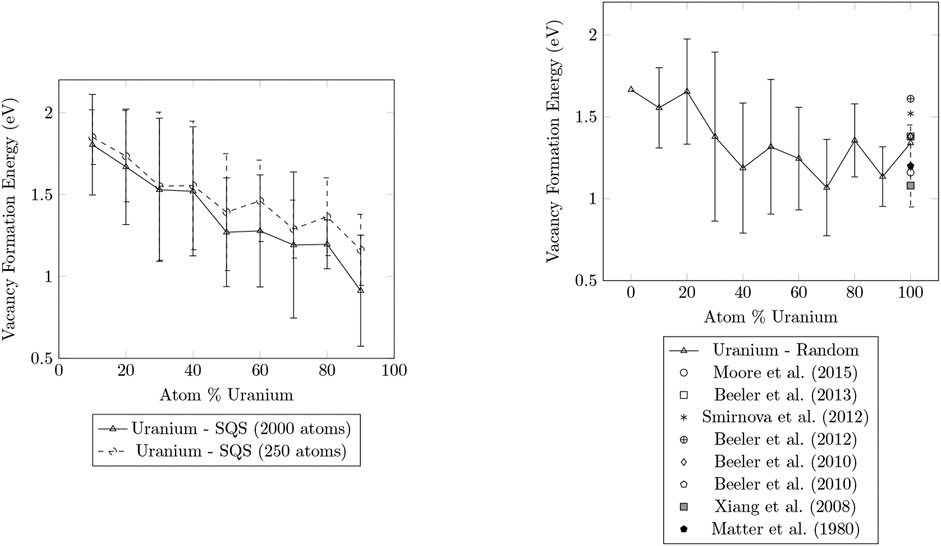
FIGURE 2. Plots of the vacancy formation energies for the removal of uranium calculated for SQS and random structures of U-Zr alloy ranging from 0
For the
The results for the vacancy formation energies of the 2000 atom SQS structures and the random structures were in good agreement. The
To examine the impact of local configuration on the vacancy formation energy, all possible nearest neighbor configurations were tested for the U-Zr alloy system for both uranium and zirconium vacancies. It has been shown in previous studies that the vacancy formation energy in binary alloys is highly dependent on the species distribution in both the first and second nearest neighbor sites (del Rio et al., 2011). For the simulations performed for this work only the first nearest neighbor configurations were varied, while keeping the second nearest neighbor configurations as they were generated. Therefore, the impact of second nearest neighbor configurations on the vacancy formation energy cannot be determined from the collected data.
It is important to note that not all local atomic configurations are equally likely in an alloy with a set concentration. For example based on the probabilities provided in Table 3, a U-10%Zr alloy will have a 96.2% chance that there will be three or fewer zirconium atoms in nearest neighbor sites, and a 99.55% chance that there will be four or fewer zirconium atoms in nearest neighbor positions. Therefore, when considering the impact that different configurations have on the average vacancy formation energy for a given material composition, it is valid to sample configurations based on their probability of occuring.
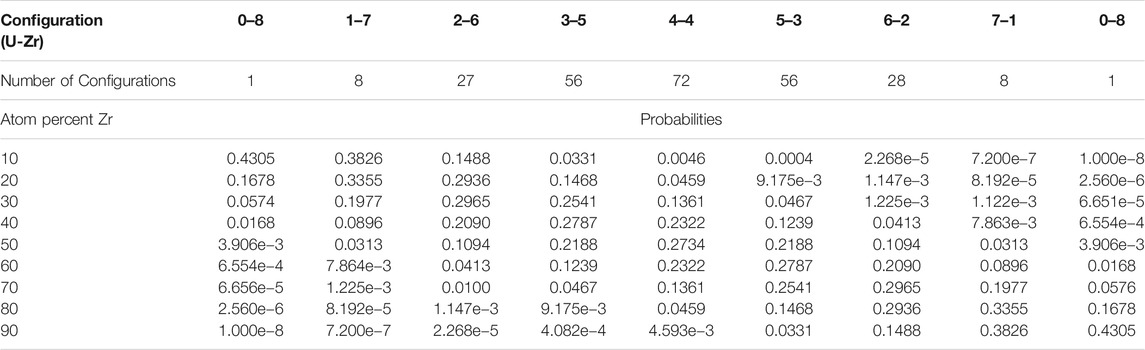
TABLE 3. Probability of zirconium inhabiting a number of nearest neighbor positions in U-Zr alloy.
Figures 3, 4 show the vacancy formation energy as a function of local atomic composition and configuration calculated for a zirconium and uranium vacancy in U-10%Zr alloy, respectively. Each data point represents the
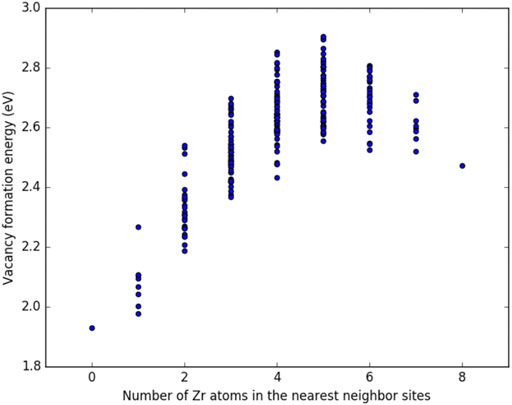
FIGURE 3. Plot of the vacancy formation energy for removing a zirconium atom from a U-10
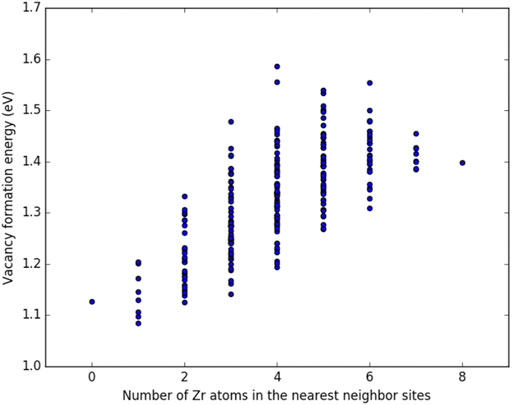
FIGURE 4. Plot of the vacancy formation energy for removing a uranium atom from a U-10
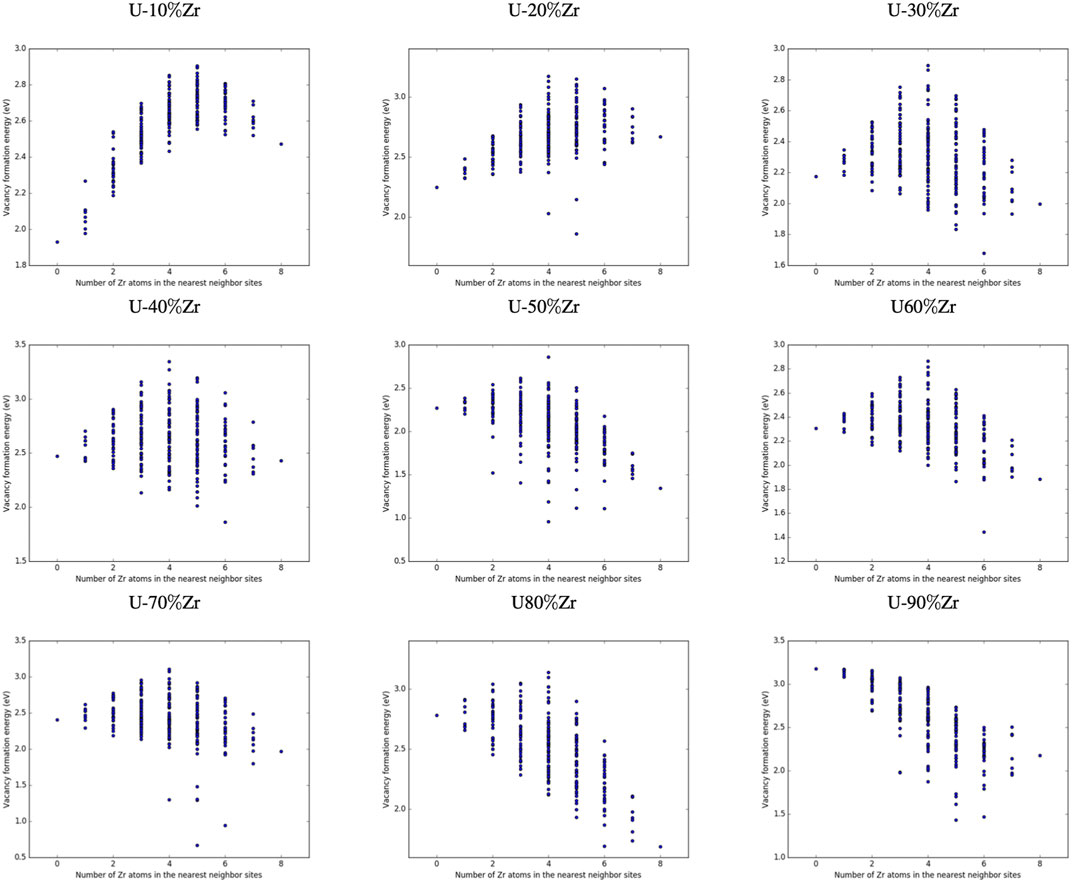
FIGURE 5. Configuration
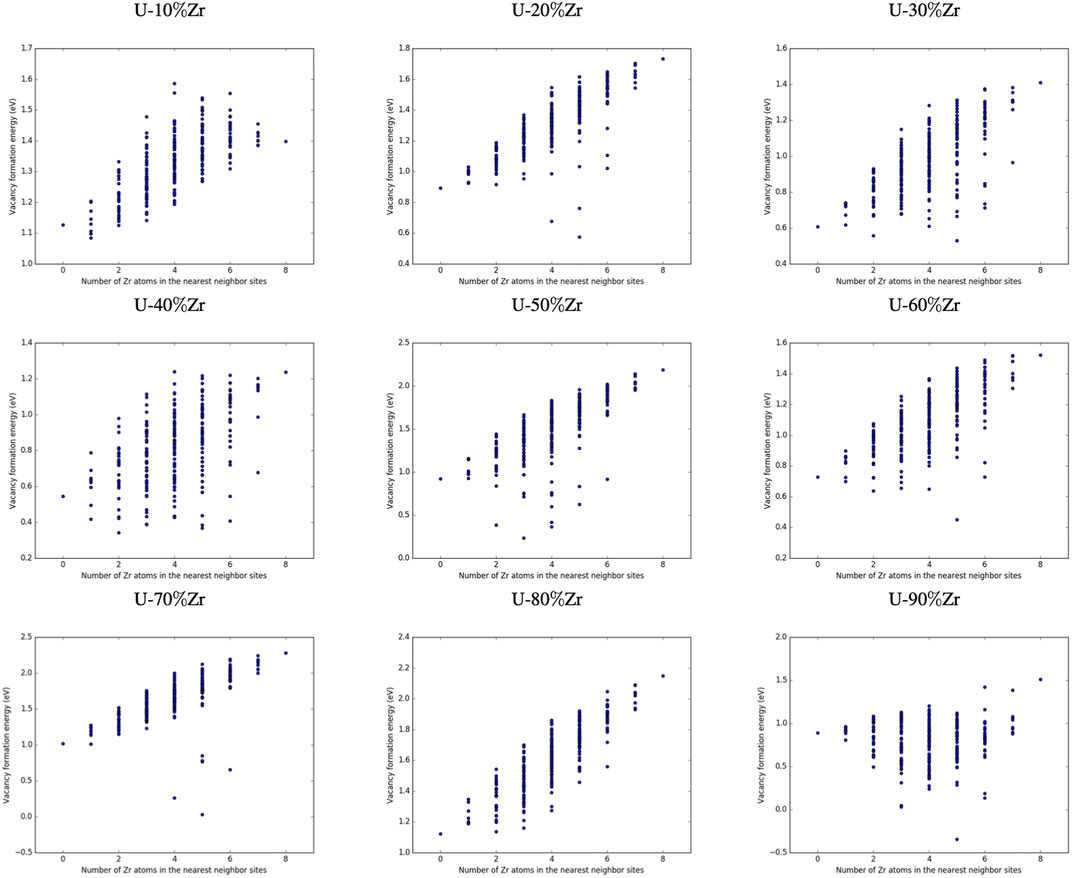
FIGURE 6. Configuration
By examining Figures 3, 4, it can be seen that the energy required to remove either a uranium or zirconium atom from a U-10%Zr alloy generally increases as the total number of Zr atoms in nearest neighbor positions increases. In the case of removal of Zr, there is a slight drop in
Determining specific trends in how the local arrangement of the species (i.e., species clumped together or distributed evenly across the nearest neighbor sites) impacts the formation energy of vacancies is difficult, particularly since this will also depend on the composition and arrangement of the second nearest neighbor atoms, which were not manipulated in this study, as well as the alloy composition. Examination of specific configurations by hand was performed for a series of initial alloy compositions and configurations, and it was determined that
4 Conclusions
The formation energies of uranium and zirconium vacancies in U-Zr binary bcc alloys were determined using MD simulations with a MEAM potential. Non-linear dependence on atom percent was observed for both uranium and zirconium vacancies. Good agreement was found for the
The impact of crystal generation method on
The effect of local atomic configuration on
With a better understanding of the
Data Availability Statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author Contributions
CD conceptualized and initiated the ideas in this work, DV performed calculations and made modifications to initial ideas based on results and consultation with CD. DV wrote the initial draft and CD edited and revised the paper draft.
Funding
CD would like to acknowledge funding from the G.W. Woodruff Fellowship. CD and DV acknowledge support from Department of Energy Nuclear Energy UNiveristy Progranms Office.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the reviewers of this work for their help in improving the quality of this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmats.2021.692660/full#supplementary-material
References
Badillo, A., Bellon, P., and Averback, R. S. (2015). A Phase Field Model for Segregation and Precipitation Induced by Irradiation in Alloys. Model. Simul. Mater. Sci. Eng. 23, 035008. doi:10.1088/0965-0393/23/3/035008
Beeler, B., Deo, C., Baskes, M., and Okuniewski, M. (2012). Atomistic Properties of γ Uranium. J. Phys. Condens. Matter 24, 075401. doi:10.1088/0953-8984/24/7/075401
Beeler, B., Deo, C., Baskes, M., and Okuniewski, M. (2013). First Principles Calculations of the Structure and Elastic Constants of α, β and γ Uranium. J. Nucl. Mater. 433, 143–151. doi:10.1016/j.jnucmat.2012.09.019
Beeler, B., Good, B., Rashkeev, S., Deo, C., Baskes, M., and Okuniewski, M. (2010). First Principles Calculations for Defects in U. J. Phys. Condens. Matter 22, 505703. doi:10.1088/0953-8984/22/50/505703
del Rio, E., Sampedro, J. M., Caturla, M. J., Caro, M., et al. (2011). Formation Energy of Vacancies in FeCr Alloys: Dependence on Cr Concentration. J. Nucl. Mater. 408, 18–24. doi:10.1016/j.jnucmat.2010.10.021
Foiles, S. M., and Baskes, M. I. (2012). Contributions of the Embedded-Atom Method to Materials Science and Engineering. MRS Bull. 37, 485–491. doi:10.1557/mrs.2012.93
Hart, G. L. W., and Forcade, R. W. (2008). Algorithm for Generating Derivative Structures. Phys. Rev. B 77, 224115. doi:10.1103/PhysRevB.77.224115
Kim, S. M. (1984). Vacancy Formation Energies in Disordered Alloys. Phys. Rev. B 30, 4829–4832. doi:10.1103/PhysRevB.30.4829
Korzhavyi, P. A., Abrikosov, I. A., Johansson, B., Ruban, A. V., and Skriver, H. L. (1999). First-principles Calculations of the Vacancy Formation Energy in Transition and noble Metals. Phys. Rev. B 59, 11693–11703. doi:10.1103/PhysRevB.59.11693
Le Bacq, O., Willaime, F., and Pasturel, A. (1999). Unrelaxed Vacancy Formation Energies in Group-IV Elements Calculated by the Full-Potential Linear Muffin-Tin Orbital Method: Invariance with crystal Structure. Phys. Rev. B 59, 8508–8515. doi:10.1103/PhysRevB.59.8508
Liu, Z. T. Y., Burton, B. P., Khare, S. V., and Gall, D. (2017). First-principles Phase Diagram Calculations for the Rocksalt-Structure Quasibinary Systems TiN-ZrN, TiN-HfN and ZrN-HfN. J. Phys. Condens. Matter 29, 035401. doi:10.1088/0953-8984/29/3/035401
Matter, H., Winter, J., and Triftshäuser, W. (1980). Investigation of Vacancy Formation and Phase Transformations in Uranium by Positron Annihilation. J. Nucl. Mater. 88, 273–278. doi:10.1016/0022-3115(80)90283-4
Moore, A. P., Beeler, B., Deo, C., Baskes, M. I., and Okuniewski, M. A. (2015). Atomistic Modeling of High Temperature Uranium-Zirconium alloy Structure and Thermodynamics. J. Nucl. Mater. 467, 802–819. doi:10.1016/j.jnucmat.2015.10.016
Piochaud, J. B., Nastar, M., Soisson, F., Thuinet, L., and Legris, A. (2016). Atomic-based Phase-Field Method for the Modeling of Radiation Induced Segregation in Fe-Cr. Comput. Mater. Sci. 122, 249–262. doi:10.1016/j.comatsci.2016.05.02110.1016/j.commatsci.2016.05.021
Plimpton, S. (1995). Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 117, 1–19. doi:10.1006/jcph.1995.1039
Ruban, A. V. (2016). Thermal Vacancies in Random Alloys in the Single-Site Mean-Field Approximation. Phys. Rev. B 93, 134115. doi:10.1103/PhysRevB.93.134115
Smirnova, D. E., Starikov, S. V., and Stegailov, V. V. (2011). Interatomic Potential for Uranium in a Wide Range of Pressures and Temperatures. J. Phys. Condens. Matter 24, 015702. doi:10.1088/0953-8984/24/1/015702
Smirnova, D. E., Kuksin, Y. A., Starikov, S. V., Stegailov, V. V., Rest, J., Insepov, Z., et al. (2013). A Ternary EAM Interatomic Potential for U-Mo Alloys with Xenon. Model. Simul. Mater. Sci. Eng. 21, 035011. doi:10.1088/0965-0393/21/3/035011
Soisson, F. (2006). Kinetic Monte Carlo Simulations of Radiation Induced Segregation and Precipitation. J. Nucl. Mater. 349, 235–250. doi:10.1016/j.jnucmat.2005.11.003
van de Walle, A., Asta, M., and Ceder, G. (2002). The alloy Theoretic Automated Toolkit: A User Guide. Calphad 26, 539–553. doi:10.1016/S0364-5916(02)80006-2
van de Walle, A., Tiwary, P., de Jong, M., Olmsted, D. L., Asta, M., Dick, A., et al. (2013). Efficient Stochastic Generation of Special Quasirandom Structures. Calphad 42, 13–18. doi:10.1016/j.calphad.2013.06.006
Walle, A. v. d., and Asta, M. (2002). Self-driven Lattice-Model Monte Carlo Simulations of alloy Thermodynamic Properties and Phase Diagrams. Model. Simul. Mater. Sci. Eng. 10, 521–538. doi:10.1088/0965-0393/10/5/304
Wei, S.-H., Ferreira, L. G., Bernard, J. E., and Zunger, A. (1990). Electronic Properties of Random Alloys: Special Quasirandom Structures. Phys. Rev. B 42, 9622–9649. doi:10.1103/PhysRevB.42.9622
Willaime, F., and Massobrio, C. (1991). Development of anN-Body Interatomic Potential for Hcp and Bcc Zirconium. Phys. Rev. B 43, 11653–11665. doi:10.1103/PhysRevB.43.11653
Xiang, S., Huang, H., and Hsiung, L. M. (2008). Quantum Mechanical Calculations of Uranium Phases and Niobium Defects in γ-uranium. J. Nucl. Mater. 375, 113–119. doi:10.1016/j.jnucmat.2007.11.003
Zhang, X., and Sluiter, M. H. F. (2015). Ab Initioprediction of Vacancy Properties in Concentrated Alloys: The Case of Fcc Cu-Ni. Phys. Rev. B 91, 174107. doi:10.1103/PhysRevB.91.174107
Keywords: molecular dynamics, point defects, uranium-zirconium, SQS, vacancy
Citation: Vizoso D and Deo C (2021) Determination of Vacancy Formation Energies in Binary UZr Alloys Using Special Quasirandom Structure Methods. Front. Mater. 8:692660. doi: 10.3389/fmats.2021.692660
Received: 08 April 2021; Accepted: 17 June 2021;
Published: 06 July 2021.
Edited by:
Shijun Zhao, City University of Hong Kong, Hong Kong, SAR ChinaReviewed by:
Anupam Neogi, Interdisciplinary Center for Advanced Materials Simulation (ICAMS), GermanyChao Jiang, Idaho National Laboratory (DOE), United States
Jingming SHI, Hokkaido University, Japan
Copyright © 2021 Vizoso and Deo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chaitanya Deo, Y2hhaXRhbnlhLmRlb0BtZS5nYXRlY2guZWR1