 Ridhi Goel
Ridhi Goel Ashutosh Pandey
Ashutosh Pandey Prabodh K. Trivedi
Prabodh K. Trivedi Mehar H. Asif
Mehar H. Asif- 1Council of Scientific and Industrial Research-National Botanical Research Institute, Lucknow, India
- 2Academy of Scientific and Innovative Research, New Delhi, India
The WRKY gene family plays an important role in the development and stress responses in plants. As information is not available on the WRKY gene family in Musa species, genome-wide analysis has been carried out in this study using available genomic information from two species, Musa acuminata and Musa balbisiana. Analysis identified 147 and 132 members of the WRKY gene family in M. acuminata and M. balbisiana, respectively. Evolutionary analysis suggests that the WRKY gene family expanded much before the speciation in both the species. Most of the orthologs retained in two species were from the γ duplication event which occurred prior to α and β genome-wide duplication (GWD) events. Analysis also suggests that subtle changes in nucleotide sequences during the course of evolution have led to the development of new motifs which might be involved in neo-functionalization of different WRKY members in two species. Expression and cis-regulatory motif analysis suggest possible involvement of Group II and Group III WRKY members during various stresses and growth/development including fruit ripening process respectively.
Introduction
Transcription factors bind to the specific DNA motifs, regulate gene expression and control various signaling and regulatory networks involved in the proper growth and development as well as stress response in an organism. Out of numerous transcription factor gene families, WRKY gene family is known to be involved in various diverse processes in the plants from germination to senescence (Eulgem et al., 2000; Miao et al., 2007; Niu et al., 2012). Since the discovery of the first member of this gene family about 20 years ago, a number of members have been cloned and characterized from many plant species. In addition, with the advent of whole genome sequencing, the complete WRKY gene family has been analyzed in cotton (Dou et al., 2014), grape (Guo et al., 2014), populus (He et al., 2012), and many other plants.
This transcription factor gene family was named WRKY due to presence of highly conserved sequence WRKYGQK at N-terminus of the proteins (Rushton et al., 1995). In addition, members of this family also contain a conserved metal chelating zinc finger motif (C-X4–5-C-X22–23-H-X-H or C-X5–8-C-X25–28-H-X1–2-C) at the C-terminus of the WRKY motif. Members of this gene family regulate expression of genes through binding to the specific DNA sequence, W-box [(C/T)TGAC(T/C)], present in the promoter region of the target genes. In addition to the W box, WRKY domain also binds to a sugar responsive element, SURE, to regulate transcriptional expression (Sun et al., 2003).
In spite of the presence of highly conserved DNA binding domain, the binding of WRKY proteins to target genes varies due to the presence of variable number of WRKY domains and different pattern of zinc finger motifs (Rushton et al., 1995). Based on these differences, the WRKY genes are divided into three main groups (Eulgem et al., 2000). Group I and Group II WRKY members contain two and one WRKY domains respectively however; members from both the groups contain one zinc finger motif pattern of C2-H2 (C–X4–5–C–X22–23–H–X1–H). Group III WRKY genes contain one WRKY domain but vary in the presence of a different zinc finger motif pattern of C2-HC (C–X7–C–X23–H–X1–C). Based on the presence of different primary amino acid sequence, Group II WRKY genes are further divided into five subgroups (IIa, IIb, IIc, IId, and IIe; Eulgem et al., 2000).
Differential expression of the members of WRKY gene family and their involvement in various regulatory processes associated with biotic and abiotic stresses has been studied in various plants (Eulgem et al., 2000). In rice, OsWRKY71 (Liu et al., 2007), OsWRKY3 (Zhang et al., 2008), OsWRKY 45-1, and OsWRKY45-2 (Tao et al., 2011) have been shown to be induced during bacterial pathogen attack. Similarly, WRKY genes like AtWRKY8 (Chen et al., 2010), AtWRKY3 (Zheng et al., 2006), AtWRKY25 (Zheng et al., 2007), AtWRKY11, and AtWRKY17 (Journot-Catalino et al., 2006) are down-regulated under bacterial pathogen attack. Studies also suggest that OsWRKY11 (Wu et al., 2009), HvWRKY38 (Xiong, 2010) TaWRKY2, and TaWRKY19 (Niu et al., 2012) provide resistance to plants under drought stress. Members of the WRKY gene families are also known to regulate synthesis of the specific metabolites in response to abiotic stresses. Studies elucidated involvement of OsWRKY11 in enhancing raffinose accumulation required for tolerance to plant against desiccation (Wu et al., 2009). In addition to the involvement to stress response and adaptation, involvement of WRKY genes has been demonstrated in the regulation of genes responsible for the proper growth and development of the plant. In Arabidopsis, studies suggest involvement of AtWRKY6, AtWRKY22 and AtWRKY53 in regulating senescence process (Robatzek and Somssich, 2002; Zhou et al., 2011; Miao et al., 2007). In rice, over-expression of OsWRKY89 leads to reduction in the growth and internode length (Wang et al., 2007). OsWRKY72 delays seed germination under normal conditions (Yu et al., 2010) and AtWRKY7 was found to have a calmodulin (CaM)-binding domain (CaMBD) thus interacts with CaM (Park et al., 2005). Various studies suggest that alkaloid biosynthesis is regulated by members of WRKY gene family (Kato et al., 2007; Mishra et al., 2013; Agarwal et al., 2015). In addition to alkaloid, biosynthesis of the sesquiterpenes in cotton has also been demonstrated to be regulated by WRKY genes (Xu et al., 2004).
Therefore, it seems that WRKY genes play important role during plant growth, development and stress response. Studies suggest that during fruit ripening, expression of a number of genes related to stress response is enhanced (Kesari et al., 2007). However, involvement of WRKY gene family in fruit ripening has not been studied. The identification and functional characterization of the WRKY gene family from fruit crops will provide an insight into the regulatory aspects of biochemical and physiological processes operating during fruit ripening as well as response to various environmental stresses. Recently, the genome of two banana species; Musa acuminata (A genome) and Musa balbisiana (B genome) have been completely established (D'Hont et al., 2012; Davey et al., 2013). In this study, we have used these genomic resources to identify members of the WRKY gene family and correlated their expression with plant growth/development and stress response. Gene structure organizations and genome duplication events have been studied in detail using WRKY gene family from M. acuminata and M. balbisiana genomes. Their ortholog and homolog pairs have also been identified and their expression analysis was performed using available transcriptome datasets to identify involvement of specific WRKY gene family members in different processes.
Materials and Methods
Identification and Multiple Sequence Alignment
Protein coding (CDS) and whole genome sequences of two banana species, M. acuminata and M. balbisiana, were downloaded from banana genome database (http://banana-genome.cirad.fr). Hmmer-2.3.2 was used against the protein sequences of both the banana genomes to search the WRKY transcription factor encoding genes using plant Transcription Factor database (http://planttfdb.cbi.pku.edu.cn/; Jin et al., 2014) with E-value ≤ 10−5. All the identified WRKY genes were aligned separately against each other using multiple sequence alignment tool ClustalX2. The 60 amino acid conserved WRKY domain region was extracted from the sequences of identified proteins from M. acuminata and M. balbisiana. The WRKY genes which contain two WRKY domains, the domains were named as “C” or “N” depending on presence of domain at the C- or N-termini of the proteins respectively.
Phylogenetic Analysis
Due to the variable lengths of the complete protein sequences of the WRKY members, WRKY domains extracted from the identified proteins were used to draw the phylogenetic tree. Arabidopsis thaliana (85), Oryza sativa (121), Vitis vinifera (71), Zea mays (185) (Wei et al., 2012; Guo et al., 2014) and those identified from M. acuminata as well as M. balbisiana were used to construct the phylogenetic tree, making it a total of 778 non-redundant WRKY domains. The unrooted phylogenetic tree was generated using Maximum Likelihood algorithm with 1000 bootstrap value using JTT algorithm in MEGA 5.2 (Tamura et al., 2011). The phylogenetic tree generated was divided into the different groups on the basis of Arabidopsis and rice annotations and classification.
Gene Structure Analysis
The gene structure information for each identified WRKY gene was extracted from the general feature format file of their respective genome and the gene structure images were generated using GSDS (Gene Structure Display Server; http://gsds.cbi.pku.edu.cn/; Hu et al., 2015) online server. The chromosomal images showing location of WRKY genes were generated using the MapChart program (Voorrips, 2002).
Identification of Motifs and Tandem Genome Duplication
The conserve motif patterns were generated for the identified WRKY genes using MEME server (Bailey et al., 2009) with zoops models of minimum and maximum weight of 6 and 60 amino acid residues, respectively. These motifs were annotated using The Eukaryotic Linear Motif (ELM; http://elm.eu.org/) resource (Dinkel et al., 2014). Whole genome duplications were analyzed within the M. acuminata and M. balbisiana genomes using Musa ancestral blocks available at Plant Genome Duplication Database (PGDD; http://chibba.agtec.uga.edu/duplication/; Lee et al., 2013) and the visualization was carried out with CIRCOS (Krzywinski et al., 2009). The possible duplications between M. acuminata and A. thaliana as well as M. acuminata and O. sativa were also identified and visualized.
Identification of cis-Regulatory Elements
Custom Perl script was used to extract the 1.5 kb upstream regions from the translation start codon of the genes and was considered as proximal promoter sequences. To identify cis-regulatory elements, PLACE cis-regulatory element online server (http://www.dna.affrc.go.jp/PLACE/; Higo et al., 1999) was used.
In silico Expression Analysis
To identify the ripening related WRKY genes, expression levels in ethylene treated and untreated fruit transcriptome datasets of M. acuminata; dwarf Cavendish, genome AAA, var. Robusta, Harichhal (Asif et al., 2014b) was analyzed. Reads from both the datasets were mapped on the MaWRKY proteins using GSMapper (version 2.5.) and the TPM (transcript per million) and fold change for the annotated contigs were calculated. Ripening related expression of the identified WRKY genes was also analyzed using transcriptome datasets of M. acuminata fruit under acetylene response for 40, 60, and 90 days (D'Hont et al., 2012). In addition, to study expression of identified WRKY genes in response to biotic stress; root transcriptome datasets of M. acuminata under Fusarium oxysporum fsp cubense stress (Li et al., 2012) were used. The heat map for the annotated transcripts with their expression values were generated using MeV version 4.2 (Saeed et al., 2003).
To study the expression of the ortholog pairs of M. acuminata WRKY genes with the rice WRKY genes, the microarray cel files were downloaded for the rice development (GSE6893) abiotic stress (GSE6901) and heavy metal stress (GSE25206) condition. The microarray cel files were normalized using RMA in AffylmGUI package (Wettenhall et al., 2006) in R-bioC (ver 2.15) and normalized data was used for differential gene expression.
Plant Material and Treatments
Dessert (M. acuminata) and cooking (M. paradisiaca) varieties of banana were used to study gene expression during development and fruit ripening. M. paradisiaca is the hybrid of M. acuminata and M. balbisiana belonging to AAB genome. Fruits from three independent plants of each cultivar at different developmental stages were harvested to collect the samples. Banana fingers from the same whorl of the hand representing similar developmental stage were treated with 100 μL/L ethylene for 24 h at 22°C in dark and then allowed to ripen in air as described in Lohani et al. (2004). Other vegetative tissues were harvested from 6 month old banana plants growing in the field. For RNA isolation, samples were quickly frozen in liquid nitrogen before extraction or stored at −80°C for further use.
RNA Isolation and Expression Analysis
Total RNA was isolated from banana tissues according to previously described protocol (Asif et al., 2006). Each RNA sample was treated with DNase I Digest kit (Sigma-Aldrich, USA) to eliminate DNA contamination. The integrity and size distribution of total RNA was analyzed by agarose gel electrophoresis. A NanoQuant (Infinite® 200 PRO NanoQuant, Austria) was used for RNA quantification. DNA-free RNA (5 μg) was used for synthesis of first strand of cDNA by using Revert Aid First Strand cDNA synthesis Kit (Fermentas, USA) as per manufacturer's recommendations. The quantitative RT-PCR was carried out with an ABI 7700 Sequence Detector (Applied Biosystems, USA) using SYBR Green chemistry. The amount of cDNA was normalized by using amplification of housekeeping banana actin as an internal control. Differential expression of five M. acuminata WRKY genes (MaWRKY121, MaWRKY38, MaWRKY61, MaWRKY83, and MaWRKY119) during fruit ripening was studied. Details of the oligonucleotide primers used in the study are provided in Supplementary Table 1. These MaWRKY genes were subjected to RT-PCR to validate their response in various stages of M. acuminata (AAA genome; dessert variety) and M. paradisiaca (ABB genome; cooking variety) during their course of ripening. The data from real-time PCR amplification was analyzed in terms of comparative fold expression following 2−△△ct method. All the experiments were repeated using three biological replicates and the data were analyzed statistically (± Standard Deviation).
Results and Discussion
Identification of the WRKY Family Members
The WRKY domain sequences were downloaded from Plant Transcription Factor database (http://planttfdb.cbi.pku.edu.cn/; Jin et al., 2014) and used to construct the HMM profile using HMMER (version 2.3.2). This profile was used for the identification of WRKY gene family members from the protein sequence of M. acuminata and M. balbisiana. After removal of partial and redundant sequences, a total of 147 and 132 WRKY genes were identified from M. acuminata and M. balbisiana, respectively (Supplementary Table 2). These MaWRKY and MbWRKY genes were named on the basis of their location on their respective chromosomes (Supplementary Table 2). Interestingly, though the WRKY genes are present on all the chromosomes, higher abundance was observed on chromosome 7 and 4 in M. acuminata while only in chromosome 7 in M. balbisiana.
There have been three rounds of genome duplication in Musa sp after the divergence of Musa from Zingiberales (D'Hont et al., 2012). Though, these duplication events resulted in the expansion of many gene families in Musa, there have also been simultaneous loss of many genes resulting in the control of gene family members (D'Hont et al., 2012). Some of the footprints of the genome duplication events were also observed in the WRKY gene family members. Our analysis suggests presence of duplicons in WRKY genes family members throughout the genome. The two Musa genome duplication blocks span chromosome 1, 2, 4 and 5, 8, 11, respectively. A total of 118 genes were present as duplication pairs throughout the genome, some of the genes had more than one duplicons.
Classification of the WRKY Family Members
Based on the presence of the WRKY and Zinc finger domains, WRKY gene family members were classified into three groups. Group I WRKY gene family consists of two WRKY domains while Group II contains only one WRKY domain. Both these groups have same zinc finger motif pattern of C2-H2. Group III WRKY genes also contain one WRKY domain but with a different zinc finger motif pattern of C2-HC (Rushton et al., 1995; Eulgem et al., 2000). The M. acuminata and M. balbisiana WRKY genes were classified into these three groups on the basis on their similarity to A. thaliana (72) (http://www.arabidopsis.org/) and O. sativa (101) (http://systemsbiology.usm.edu/BrachyWRKY/WRKY/Rice.html) WRKY domains.
WRKY domains from 147 to 132 WRKY members from M. acuminata and M. balbisiana respectively were aligned with each other using multiple sequence alignment tool ClustalX2 (Supplementary Figures 1, 2). The identified WRKY members which contain two WRKY domains were named as “CTD” or “NTD” on the basis of presence of domain at the C- or N-termini of the protein respectively. This led to identification of 167 and 150 separate MaWRKY and MbWRKY domains, respectively.
Further, the conserved 60 amino acid region of the WRKY domains was extracted from A. thaliana, O. sativa, V. vinifera, and Z. mays WRKY genes. This led to development of dataset comprising 778 non-redundant WRKY domains including M. acuminata and M. balbisiana WRKY gene domains (Table 1). An unrooted ML phylogenetic tree was constructed using these WRKY domains (Figure 1). Based on sequence similarity, the WRKY domains could be classified into Group I, II, and III genes. The Group II was further classified in Group II a-e. In M. acuminata, 24 genes were categorized in Group I whereas 106 and 16 identified as members of Group II and Group III, respectively. In M. balbisiana, 20, 87, and 21 WRKY genes were categorized in Group I, Group II, and Group III, respectively (Table 1). There were few WRKY genes (3 in M. balbisiana and 2 in grape) which were not assigned to any group and these were named as NG (not grouped).
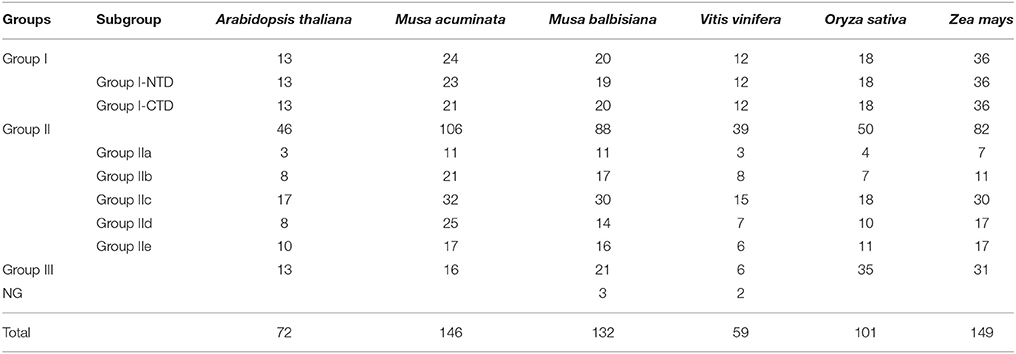
Table 1. Number of WRKY TF gene family members present in various monocot and dicot plants.
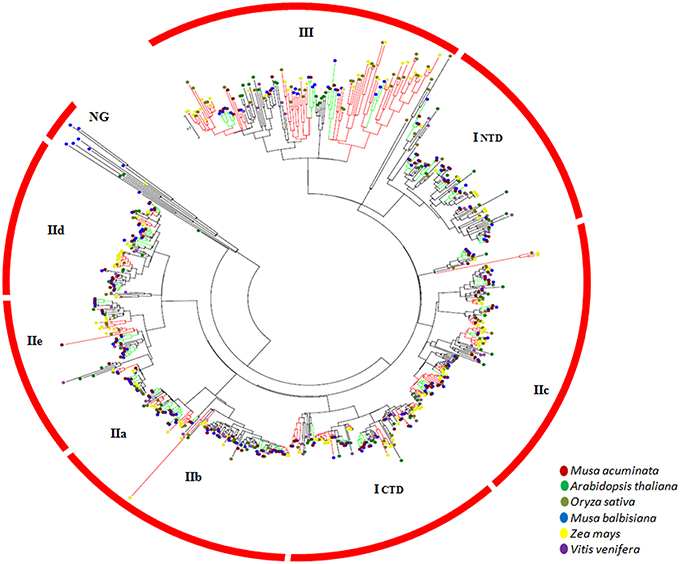
Figure 1. Unrooted phylogenetic tree of WRKY domains. The tree was constructed using WRKY domains from the complete WRKY gene families of Musa acuminate, Musa balbisiana, Arabidopsis thaliana, Oryza sativa, Zea mays, and Vitis vinifera. A total of 778 WRKY domains were used for the analysis. The unrooted phylogenetic tree was generated using Maximum Likelihood algorithm with 1000 bootstrap value using JTT algorithm in MEGA 5.2.
The Group II WRKY genes were further divided in five (a–e) subgroups. Analysis suggested that five MaWRKY genes (MaWRKY118, MaWRKY85, MaWRKY144, MaWRKY11, and MaWRKY99) were mis-annotated as Group IId in banana Genome hub on the basis of their annotation with A. thaliana. The phylogenetic analysis suggested that these WRKY genes are the members of the Group IIc. Therefore, the MaWRKY genes present in Group IIa, Group IIb, Group IIc, Group IId, and Group IIe contains 11, 21, 32, 25, and 17 members respectively. At the same time, 11, 16, 30, 14, and 16 genes were present in Group IIa, Group IIb, Group IIc, Group IId, and Group IIe, respectively in M. balbisiana. In Group I, all the C-terminal and the N-terminal WRKY domain were grouped independently (Figure 1).
WRKY genes formed monocot- and dicot-specific clades depicting that the WRKY genes have evolved simultaneously in both monocot and dicot plants from the common lineage (Figure 1). Interestingly, except for Group I NTD, phylogenetic analysis led to the formation of monocot-specific clades for all the groups. Group III had the largest monocot-specific clade comprising Musa-specific clades (Figure 1). There were few WRKY members which did not cluster in any of the three groups and were named Not Grouped (NG). Similar observation was also made in Setaria where such genes were included in Group IV (Muthamilarasan et al., 2015).
Within three groups of WRKY genes, there are four major lineages comprising Group IIa + Group IIb, Group IId + Group IIe, Group IIc + Group I (NTD and CTD), and Group III (Zhang and Wang, 2005; Rinerson et al., 2015). Till now, there were two proposed hypothesis regarding evolution of WRKY gene family in plants. According to Group I hypothesis, all the WRKY genes are evolved from the Group I-CTD (Zhang and Wang, 2005). In addition, alternative Group IIa+b hypothesis state that Group IIa and Group IIb evolved directly from the single domain ancestral algal WRKY genes separately from Group I WRKY genes (Rinerson et al., 2015). The phylogenetic analysis of the monocot and dicot plants favors the Group I hypothesis, stating that in the five plants selected, the evolution of the WRKY genes had occurred from the Group I ancestral WRKY genes (Figure 1).
Duplication and Ks Analysis
The gene duplication pairs for M. acuminata were downloaded from the PGDD (http://chibba.agtec.uga.edu/duplication/) database and the genes containing WRKY domains were filtered. There were 136 WRKY gene duplication pairs which corresponded to 118 genes. This suggests that some of the WRKY genes have more than one duplicated genes (Supplementary Table 3). This could be due to multiple rounds of whole genome duplication events which have occurred in the Musa genome. The Ks (substitution rate) of these gene pairs was plotted against the number of gene pairs. Analysis revealed presence of two peaks corresponding to α/β and γ duplication events (Figure 2). It is interesting to note that more duplication pairs were present from the γ duplication event which could have occurred earlier. The analysis also suggests that recent duplicates of WRKY genes might have been lost due to the genome rearrangements (Figure 2). Interestingly, mapping of duplication pairs on the chromosomes suggested that many duplication pairs correspond to block 4 and block 7 of the Musa duplication blocks as suggested by D'Hont et al. (2012).
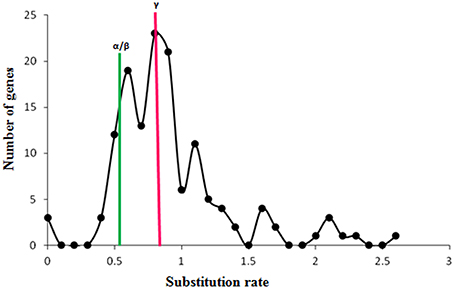
Figure 2. Substitution rate of WRKY genes in Musa acuminate. The graph represents the ratio of synonymous (Ka) vs. non-synonymous (Ks) variations within the Musa acuminate WRKY gene family. X- and Y-axis represent the substitution rate and number of genes respectively. Green bars represent the substitution rate at the time α/β WGD and the red bar represents the substitution rate at the time of γ WGD.
The orthologs of WRKY genes from O. sativa were also identified from the PGDD database. A total of 101 ortholog pairs corresponding to 62 MaWRKY genes were identified. The presence of additional 42 WRKY genes in O. sativa suggests that some genes have more than one orthologs (Supplementary Table 3). Analysis also suggests presence of 112 homolog pairs of WRKY genes between M. acuminata and M. balbisiana. Out of these, 111 and 112 were from M. acuminata and M. balbisiana WRKY genes, respectively. This suggests that there are specific WRKY genes which might have transferred and were conserved during the course of evolution within the Musaceae family (Supplementary Table 3). It was also identified that the ortholog genes within a chromosome of M. acuminata follow similar pattern as the M. balbisiana genome, depicting the conserved nature in the WRKY gene pattern.
Gene Structure and Chromosome Localization
The gene structures of the identified WRKY genes were retrieved from the genomic sequence of M. acuminata and M. balbisiana. The coding sequences of all the identified WRKY genes were mapped on to their respective complete genomes to identify intron-exon boundaries. The gene structures of the WRKY genes showed group-specific intron-exon patterns similar to A. thaliana and O. sativa (Wu et al., 2005). The number of exons varied from 1 to 9 in most of the WRKY genes of both the Musa species. Maximum number of exons, 23 numbers, was identified in MaWRKY14 (Supplementary Table 4). In M. balbisiana, twelve genes contain more than nine introns and the numbers ranged from 11 to 25. It was also identified that a set of the WRKY genes in both the species contain an intron in between the WRKY domain as reported in A. thaliana and O. sativa (Wu et al., 2005).
It has already been reported that there are two types (R- and V-type) of introns which are present in between WRKY domain. R-type introns, the phase-2 introns, have a splice site directly on the R residue of the WRKY domain. On the other hand, V-type introns, phase-0 introns, have a splice site before the V residue, at the sixth amino acid after the second Cys residue, in the C2H2 zinc finger motif (Wu et al., 2005). In M. acuminata, 105 WRKY genes contain R-type intron while only 31 WRKY genes have V-type intron and 11 WRKY genes do not contain introns in between the WRKY domains. Similarly, in M. balbisiana, 88 and 25 WRKY genes contain R- and V-type introns respectively while 17 WRKY genes do not have any intron in between WRKY domain. Analysis suggested that V-type introns are present mostly in Group IIa and Group IIb while R-type introns are present mostly in all the other groups (Group I, IIc, IId, IIe, and III). WRKY genes which do not contain any intron in the WRKY domains, in both the genomes, are those WRKY genes in which domain are mostly present at the N-terminal of the group I. This loss of introns was considered as the result of intron turnover or due to reverse transcription of the mature mRNA followed by homologous recombination with intron-containing alleles (Wu et al., 2005; He et al., 2012). The positioning of the introns in between the WRKY domains was found in the ancestral WRKY genes. Thus, it can be hypothesized that the WRKY domains in the WRKY family member might have duplicated from the ancestral WRKY genes which contain introns, followed by the divergence rather than the formation of the similar genes through convergence events (Wu et al., 2005).
The genomic position of each identified WRKY gene in both the genomes was also used to identify the chromosome localization. Analysis suggests that WRKY genes are distributed throughout the genomes of the M. acuminata and M. balbisiana (Figure 3 and Supplementary Figure 3). It was found that the maximum number of the identified MaWRKY (22) are localized on to the chromosome 7 followed by chromosome 4 and 10 with 18 and 16 MaWRKY genes, respectively. Six MaWRKY genes are present in the uncharacterized region of the sequenced M. acuminata genome (Figure 3). In M. balbisiana, maximum number (18) of the WRKY genes is present on the chromosome 7 while minimum (6) on chromosome number 1 and 2 (Supplementary Figure 3). Ten MbWRKY genes were present in the uncharacterized region of the sequenced M. balbisiana genome. It was observed that MaWRKY75 and MaWRKY76 on chromosome 7 as well as MaWRKY120 and MaWRKY121 on chromosome 10 are clustered together as the difference between them is 30 and 3 kb, respectively (Figure 3). Analysis also suggests that the identified MaWRKY and MbWRKY genes are evenly distributed on the different chromosomes and the uncharacterized region of both the genomes in accordance with the size of each chromosome (Figure 3 and Supplementary Figure 3).
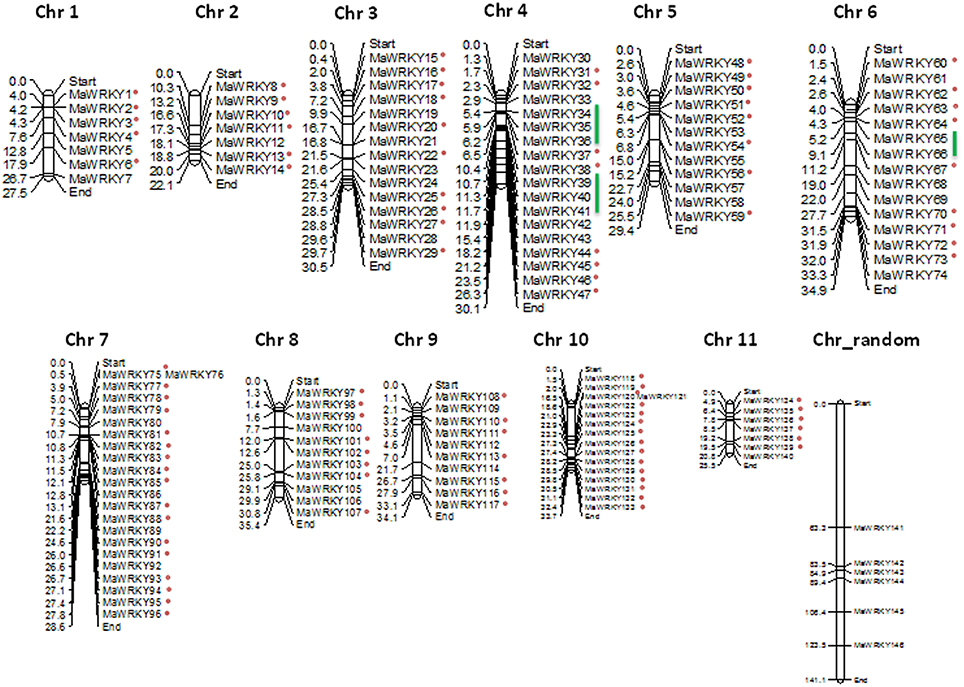
Figure 3. Physical map of MaWRKY genes showing their chromosomal locations. The genomic position of each identified WRKY gene was used to identify the chromosome localization on different chromosomes and the uncharacterized region. Vertical bars represent the chromosomes and numbers at the left indicate the position of genes (in Mb). The segmental duplicated genes are shown with red dots, and tandem duplicated with green lines.
Unique Motif Analysis
Ten conserved motif patterns were identified for both M. acuminata and M. balbisiana (Figure 4) WRKY genes. Of these 10 motifs in WRKY members from both the species, motifs 1, 2, 3, 4, 7, and 10 were identical in M. acuminata and M. balbisiana. Motif 6 of M. acuminata was identical to motif 8 of M. balbisiana (Figure 4). Motifs 5, 8, and 9 of M. acuminata and motifs 5, 6, and 9 of M. balbisiana were species specific.
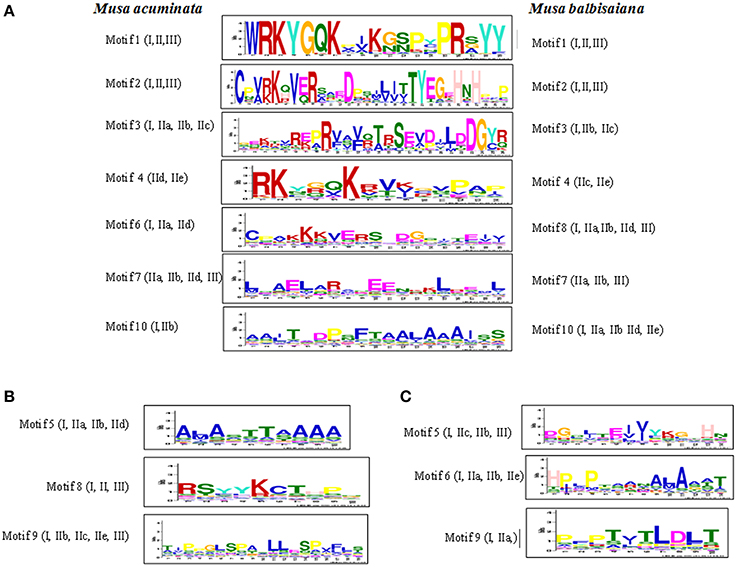
Figure 4. Conserved motifs in WRKY proteins of Musa sp. The conserved motifs were identified in 147 MaWRKY and 132 MbWRKY proteins using MEME server with zoops models of minimum and maximum weight of 6 and 60 amino acid residues, respectively. The motifs that were present in both the Musa sp are represented in (A). Specific motifs that were present in only Musa acuminate are represented in (B) and those only in Musa balbisiana are represented in (C).
In M. acuminata and M. balbisiana, motif 1 and 2 are found to be conserved in all the identified WRKY genes while motifs 3 and 10 were conserved in Group I and few sub-groups of Group II WRKY members. Motif 4 was found to be kexin isozyme cleavage site while motif 10 is a pro-apoptotic protein. Motif sequence 1 is the basic WRKY domain of 20 amino acids which is found in all the WRKY genes. However, motif sequence pattern 2 is the variable sequence found in the zinc finger motif C2H2/C2CH (Figure 4). Motif 3 is the C2H2 conserve zinc finger motif which was found to be conserved at the N-terminal of the Group I WRKY genes (Figure 4). The Motif 6 of M. acuminata was similar to Motif 8 of M. balbisiana which is a fungi specific variant and was present in Groups IIa, IIb, IId, and III.
Some taxa-specific motifs were also identified between M. acuminata and M. balbisiana. In M. acuminata motif sequence 5 was found mostly in Group I and IIc while motif sequence 8 was present mostly in Group IIa and IIe. This motif is known to be responsible for the initiation of protein degradation. Motif 9 is phosphorylation recognition site present mostly in Group III, IId, and IIe (Figure 4). Likewise in M. balbisiana, motifs 5 and 6 are present on Group I, IIa, and IId while motif 5 is present in the Group IIb which is a phosphothreonine motif binding site. Motif 9 present in Group I, IIb, IIc, IIe, and III is a phosphorylation recognition site (Figure 4).
Apart from the basic 60 amino acid residue domain present in all the WRKY genes of different groups, there are differences in the WRKY domains present at the N and C terminal regions (Figure 4). These motifs are conserved in a particular group and thus might be involved in performing specific physiological and biological processes in plants. Therefore, it can be hypothesized that a specific group of WRKY genes might be responsible for the specific biological processes in plants (Kalde et al., 2003).
Analysis also revealed that the common motifs within M. acuminata and M. balbisiana WRKY members are the basic domain and the zinc finger motif. Also, the function of some of the motifs is similar in spite of the differences in their motif sequence pattern. Interestingly, Motif 9 present in both the Musa species have different sequence pattern but their function seems to be similar. This suggests that the function of the WRKY genes in a particular group is conserved and evolved simultaneously in specific taxa.
Apart from the basic WRKYGQK conserve motif in all the WRKY family members in M. acuminata and M. Balbisiana, WRKYGKK (in MaWRKY23, MaWRKY80, MaWRKY91, MaWRKY106, MbWRKY42, and MbWRKY92) and WRKYGRK (in MaWRKY37 and MbWRKY53) motifs were also identified. These variations in the WRKY domains are also reported in A. thaliana (Dong et al., 2003). Apart from these motifs, M. balbisiana WRKY members also contain WRKYGNK (MbWRKY59), WRKYGHK (MbWRKY54 and MbWRKY104), and WRKYGEK (MbWRKY122, MbWRKY123, and MbWRKY124) motifs (Supplementary Figure 1).
cis-Regulatory Elements
The 1.5 kb upstream regions of the WRKY genes were extracted and used for the identification of cis-regulatory elements using PLACE server and the cis-regulatory elements database. Many of the regulatory elements related to SA, cold, drought, ABA, and fungal response were identified in different genes. Interestingly, promoters from more than 50% of the WRKY genes harbor such elements indicating their important role during stress response (Figure 5). A set of WRKY genes also contain W-box and related motifs therefore regulation of these genes through other WRKY genes cannot be ruled out (Figure 5). A set of WRKY genes contain hormone-responsive elements of which majority of them are auxin and gibberellin response elements. A large number of genes also contain cytokinin, ethylene, and copper responsive elements (Figure 5). Previous reports have demonstrated that copper plays an important role in the signal transduction cascade of different hormones. A large number of genes also contain sulfur responsive element binding site which is known for essential for auxin dependent regulatory processes (Maruyama-Nakashita et al., 2005). Our analysis and previous studies (Knoth et al., 2007; Jiang and Deyholos, 2009; Levee et al., 2009) clearly suggest that WRKY genes play an important role during growth and development and stress responses. Interestingly, number of cis-regulatory elements identified in M. balbisiana is lesser as compared to those present in M. acuminate in the proximal promoter region used for the analysis (Figure 5).
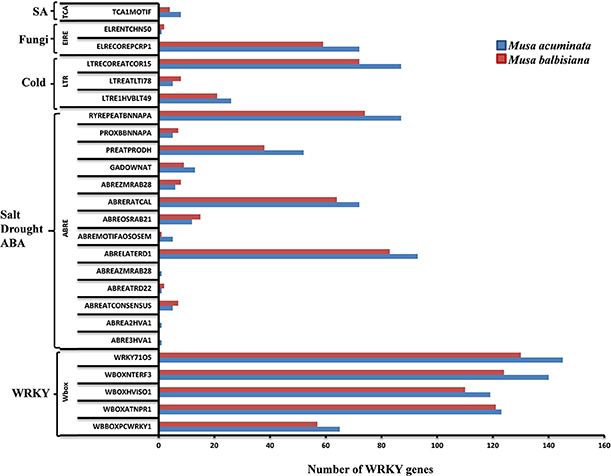
Figure 5. cis-regulatory elements in WRKY genes of Musa sp. The cis-regulatory elements were identified in the 147 MaWRKY genes and 132 MbWRKY genes, respectively, using the PLACE server. Custom Perl script was used to extract the 1.5 kbp upstream regions from the translation start codon of the genes and was considered as proximal promoter sequences. Graph was plotted on the basis of presence of cis-regulatory element responsive to specific elicitors/conditions (x-axis) in WRKY gene family members (y-axis). The blue bars represent Musa acuminate WRKY genes and the red bar represents the Musa balbisiana WRKY genes.
The cis-regulatory elements identified, in this study, are evenly distributed throughout the promoter regions of the WRKY genes. The presence of SA, JA, ABA and ethylene related cis-regulatory elements suggest that these WRKY genes may be involved in various signaling pathways (Figure 5). The modulated expression of AtWRKY18 (Chen and Chen, 2002), AtWRKY70 (Knoth et al., 2007), and PtWRKY23 (Levee et al., 2009) has already been reported during fungi infection as well as in response to SA and JA. This suggests that a set of MaWRKY and MbWRKY genes might be involved in various defense related pathways. The other WRKY genes like GsWRKY20 (Luo et al., 2013), AtWRKY25, and AtWRKY33 (Jiang and Deyholos, 2009) GmWRKY13, GmWRKY21, and GmWRKY54 (Zhou et al., 2008) were also found to be induced during abiotic stresses. In M. acuminata, Group I and Group III WRKY genes contains regulatory elements responsive to salt, drought, and ABA stresses while Group II WRKY genes contain regulatory elements responsive to cold stress and pathogen infection. In M. balbisiana the same pattern was also observed apart from Group II genes which contained elements responsive to salt, drought, and ABA stresses (Figure 5).
In silico Expression Analysis
WRKY genes are known to be involved in growth and development as well as in stress responses (Maruyama-Nakashita et al., 2005; Jiang and Deyholos, 2009; Levee et al., 2009). In this study, involvement of WRKY genes in fruit ripening has been studied using the transcriptome data set of M. acuminata (available at the banana genome hub; (D'Hont et al., 2012)) as well as our in house transcriptome dataset of ripe and unripe fruit (Asif et al., 2014b). Thirty-one WRKY genes showed differential expression in the transcriptome data of D'Hont et al. (2012) of which 20 genes showed increase in expression during ripening with a fold change of ≥ 1.5 (Figure 6 and Supplementary Table 5). The WRKY genes were also mapped to the transcriptome dataset of ripe and unripe fruit (Asif et al., 2014b). A total of 64 genes mapped successfully in ripe and unripe fruit transcriptome of M. acuminata of which 14 genes showed enhanced expression (fold change ≥ 1.5) during fruit ripening (Figure 6 and Supplementary Table 5). Analysis using both the datasets suggests that a set of WRKY genes are differentially regulated during ripening process in M. acuminata. Analysis revealed that during ethylene induced fruit ripening, Group I MaWRKY genes are down-regulated while a set of Group II MaWRKY genes and most of the Group III MaWRKY genes are up-regulated (Figures 6A,B).

Figure 6. Expression of MaWRKY genes in response the ethylene, acetylene, and fusarium infection. To identify the ripening related WRKY genes, expression levels in ethylene treated and untreated fruit transcriptome datasets of banana (Asif et al., 2014a) was analyzed. Ripening related expression in response to acetylene exposure was analyzed using transcriptome datasets of M. acuminata fruit under acetylene response for 40, 60, and 90 days (D'Hont et al., 2012). Root transcriptome datasets under F. oxysporum stress (Li et al., 2012) were used to analyse biotic stress response of WRKY gene family. Log2 transformed count value are used for heat map construction. Panel (A) represents fold change between control (untreated) and ethylene treated (after 4 days of ethylene treatment. Panels (B–D) represent fold change between control (untreated) and acetylene treated after 40, 60, and 90 days, respectively. Panels (E), (G), (I) represent differential expression between control (untreated) and Foc1 treated after 3, 27, and 51 h, respectively. Panel (F), (H), and (J) represent differential expression between control (untreated) and Foc4 treated samples for 3, 27, and 51 h, respectively.)
To study the role of WRKY genes during biotic stress, WRKY gene expression was analyzed in transcriptome datasets related to F. oxysporum (Li et al., 2012) infection, available in public domain. It was observed that a large number (56) of WRKY genes expressed during F. oxysporum infected root tissue (Figure 6 and Supplementary Table 5). However, expression of WRKY genes was dependent on time duration post infection. There are 2 races (Foc1 and Foc4) of F. oxysporum which were studied at three time points (3, 27, and 51 h post infection; Figure 6 and Supplementary Table 5). While most of WRKY genes were up-regulated, decreased expression was observed for a set of WRKY genes post infection. Race specific differences in expression pattern were also observed as a set of WRKY genes expressed during early infection in Foc1 but not in Foc4 (Figure 6 and Supplementary Table 5). Expression of most of the WRKY genes continued to increase with time (Figure 6). During the pathogen attack in the M. acuminata plant, all the MaWRKY genes from Group II and Group III as well as some of the group I were up-regulated (Figure 6) suggesting response of specific banana genes during biotic stress response.
Since no public data is available to study the WRKY gene family expression during plant growth and development and other stresses in M. acuminata, the orthologs of the MaWRKY genes from O. sativa were retrieved and their expression was studied in available databases. Out of 147 WRKY genes in M. acuminate, only 62 had orthologs in rice database corresponding to 42 O. sativa WRKY genes (Supplementary Table 2). Analysis suggested that these WRKY genes are involved in the overall growth and development of the plant as well as in stress response (Supplementary Figure 4 and Supplementary Table 6). O. sativa WRKY genes were mostly up-regulated during abiotic stress (cold stress, drought stress, and salinity stress; Supplementary Figure 5A and Supplementary Table 6). In addition, data available in public domain (Chakrabarty et al., 2009; Dubey et al., 2010) was also used to study responsiveness of WRKY genes toward heavy metal stress. Analysis suggested that most of the WRKY genes are down-regulated during heavy metal stress (chromium, cadmium, arsenic, and lead; Supplementary Figure 5B and Supplementary Table 4). During the course of growth and development, Group I and III WRKY gene family members were up-regulated while Group II members were down-regulated (Supplementary Figure 4 and Supplementary Table 6).
In M. acuminata, most of the Group I WRKY genes were down-regulated during ripening and pathogen attack. Interestingly, their orthologos in O. sativa were up-regulated during abiotic stress and developmental processes (Figure 6, Supplementary Figures 4, 5). This suggests that the Group I WRKY genes were mainly involved in the developmental processes as well as in the abiotic stress response. Group IIc and Group IId genes are abundant in the set of WRKY genes regulating ethylene and acetylene related ripening. Our analysis suggests that in addition to involvement of Group II genes in fungal stress response, this group might also be responsible for ripening related gene regulation (Figure 6, Supplementary Figures 4, 5). Group III WRKY genes were found to be differentially regulated during early development of pollen and throughout the shoot development. Analysis also suggests Group III WRKY may not be involved in abiotic stress response (Figure 6, Supplementary Figures 4, 5).
The genes which showed differential expression under ethylene and acetylene exposure and fungal stress response had a high incidence of cis-regulatory elements related to elicitor response, ABREs and WRKY itself. This indicates that WRKY genes might be self-regulated by other WRKY members. Out of 147 WRKY genes, 26 genes contain the ethylene response element (ERELEE4). Out of these, 12 and 11 genes showed ethylene and acetylene responsive expression. Of these, five WRKY family members were commonly regulated by ethylene and acetylene.
Validation of MaWRKY Differential Expression
The differential expression of five M. acuminata WRKY (MaWRKY121, MaWRKY38, MaWRKY61, MaWRKY83, and MaWRKY119) genes was analyzed during fruit ripening using qRT-PCR. Our in silico analysis using different datasets of M. acuminata (dessert variety) revealed that these WRKY genes are differentially regulated during fruit ripening. RT-PCR analysis suggested that expression of MaWRKY121 is enhanced gradually during the banana fruit ripening process. In addition, MaWRKY83 and MaWRKY61 were late responsive as their expression enhanced significantly at the 6 and 8 day of ethylene induced fruit ripening respectively. No significant difference was observed in the expression of MaWRKY38 during course of fruit ripening. This is in contrast to the down regulation of MaWRKY38 gene in transcriptome analysis for ethylene and acetylene stress (Figures 6A,B).
Dessert (M. acuminata) and cooking (M. paradisiaca) varieties were also used to identify the possible role of banana WRKY genes in fruit development and ripening. These varieties have been used to study expression of members AP2/ERF and HDZIV gene families during fruit ripening (Lakhwani et al., 2015; Pandey et al., 2016). Expression analysis of five WRKY genes was carried out in different fruit developmental stages (3, 6, 15, 18, 21, and 24 weeks) and ripening stage of pulp (Figure 7). During fruit development, expression of MaWRKY38 gradually increased in the cooking variety and was highest at 21 week. Distinctly, in dessert variety, expression was significantly lesser and gradually decreased after 15 weeks (Figure 7A). The significantly higher expression of MaWRKY83 was observed at the 6 weeks in dessert variety (Figure 7A). This ripening related expression of MaWRKY83 is similar to as observed during ethylene induced ripening (Figure 7A). The expression of MaWRKY121 and MaWRKY61 did not vary much during the course of ripening in both dessert and cooking variety.
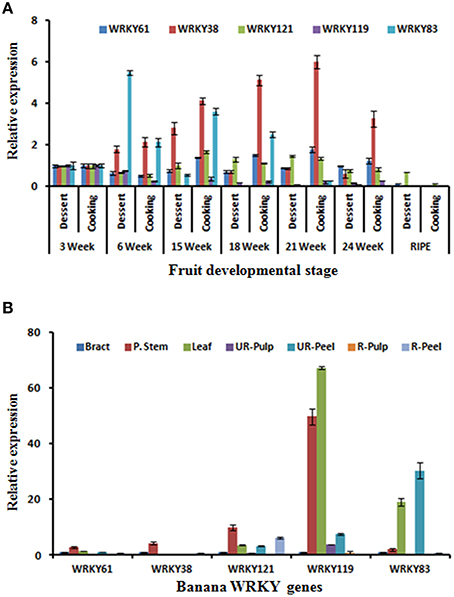
Figure 7. Expression analysis of selected WRKY genes during fruit development stages, ripening, and in different tissues. (A) Dessert (M. acuminata) and cooking (M. paradisiaca) varieties of banana were used to study gene expression during fruit development (in different weeks of development) and ripening (B) Expression analysis of WRKY genes in different tissues. Total RNA isolated from different tissues including Bract, Psudostem, Leaf as well as peel, and pulp tissues of ripe and unripe banana fruit was used for the analysis. The relative transcript abundance was normalized using banana actin gene.
The expression of selected MaWRKY genes was also studied at various tissues of the M. acuminata. It was observed that all the selected MaWRKY genes express mostly in stem as compared to other parts of the plant (Figure 7B). Highest expression of MaWRKY119 gene was observed in stem and leaf while MaWRKY83 gene was expressed mostly in leaf and unripe peel and was absent in unripe pulp (Figure 7B). Similar to WRKY gene family, genome-wide studies have been carried on few other gene families (Asif et al., 2014a; Lakhwani et al., 2015; Pandey et al., 2016) which has provided some inside about regulatory role of their members during fruit ripening and stress response.
In conclusion, this study suggests that WRKY gene family in M. acuminata and M. balbisiana is a large gene family. The gene family seems to have expanded before speciation, with minor changes in gene family after speciation. The expansion of the WRKY gene family was mainly due to the earlier γ duplication event of which most of the duplicons were lost in the recent α β duplication events. Our study suggests involvement of WRKY family members in various processes from development to ripening of fruit as well as biotic stress response in banana.
Author Contributions
MA and PT conceived and planned study. RG and AP carried out experiments. RG, AP, PT, and MA analyzed data. RG, MA, and PKT wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a research grant from the Council of Scientific and Industrial Research, New Delhi, as Network Project (BSC-107).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00299
References
Asif, M., Trivedi, P. K., Solomos, T., and Tucker, M. (2006). Isolation of high-quality RNA from apple (Malus domestica) fruit. J. Agric. Food Chem. 54, 5227–5229. doi: 10.1021/jf053137n
Asif, M. H., Lakhwani, D., Pathak, S., Gupta, P., Bag, S. K., and Trivedi, P. K. (2014b). Transcriptome analysis of ripe and unripe fruit tissue of banana identifies major metabolic networks involved in fruit ripening process. BMC Plant Biol. 14:316. doi: 10.1186/s12870-014-0316-1
Asif, M. H., Lakhwani, D., Sumya, P. S., Bhambhani, S., Bag, S. K., and Trivedi, P. K. (2014a). Genome-wide identification and expression analysis of the mitogen-activated protein kinase gene family from banana suggest involvement of specific members in different stages of fruit ripening. Funct. Integr. Genomics 14, 161–175. doi: 10.1007/s10142-013-0349-9
Agarwal, P., Pathak, S., Lakhwani, D., Gupta, P., Asif, M. H., and Trivedi, P. K. (2015). Comparative analysis of transcription factor gene families from Papaver somniferum: identification of regulatory factors involved in benzylisoquinoline alkaloid biosynthesis. Protoplasma. doi: 10.1007/s00709-015-0848-8. [Epub ahead of print].
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Chakrabarty, D., Trivedi, P. K., Misra, P., Tiwari, M., Shri, M., Shukla, D., et al. (2009). Comparative transcriptome analysis of arsenate and arsenite stresses in rice seedlings. Chemosphere 74, 688–702. doi: 10.1016/j.chemosphere.2008.09.082
Chen, C., and Chen, Z. (2002). Potentiation of developmentally regulated plant defense response by AtWRKY18, a pathogen-induced Arabidopsis transcription factor. Plant Physiol. 129, 706–716. doi: 10.1104/pp.001057
Chen, L., Zhang, L., and Yu, D. (2010). Wounding-induced WRKY8 is involved in basal defense in Arabidopsis. Mol. Plant Microbe Interact. 23, 558–565. doi: 10.1094/MPMI-23-5-0558
Davey, M. W., Gudimella, R., Harikrishna, J. A., Sin, L. W., Khalid, N., and Keulemans, J. (2013). A draft Musa balbisiana genome sequence for molecular genetics in polyploid, inter- and intra-specific Musa hybrids. BMC Genomics 14:683. doi: 10.1186/1471-2164-14-683
D'Hont, A., Denoeud, F., Aury, J.-M., Baurens, F.-C., Carreel, F., Garsmeur, O., et al. (2012). The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488, 213–217. doi: 10.1038/nature11241
Dinkel, H., Van, K. R., Michael, S., Davey, N. E., Weatheritt, R. J., Born, D., et al. (2014). The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 42, D259–D266. doi: 10.1093/nar/gkt1047
Dong, J., Chen, C., and Chen, Z. (2003). Expression profiles of the ArabidopsisWRKY gene superfamily during plant defense response. Plant Mol. Biol. 51, 21–37. doi: 10.1023/A:1020780022549
Dou, L., Zhang, X., Pang, C., Song, M., Wei, H., Fan, S., et al. (2014). Genome-wide analysis of the WRKY gene family in cotton. Mol. Genet. Genomics 289, 1103–1121. doi: 10.1007/s00438-014-0872-y
Dubey, S., Misra, P., Dwivedi, S., Chatterjee, S., Bag, S. K., Mantri, S., et al. (2010). Transcriptomic and metabolomic shifts in rice roots in response to Cr (VI) stress. BMC Genomics 11:648. doi: 10.1186/1471-2164-11-648
Eulgem, T., Rushton, P. J., Robatzek, S., and Somssich, I. E. (2000). The WRKY superfamily of plant transcription factors. Trends Plant Sci. 5, 199–206. doi: 10.1016/S1360-1385(00)01600-9
Guo, C., Guo, R., Xu, X., Gao, M., Li, X., Song, J., et al. (2014). Evolution and expression analysis of the grape (Vitis vinifera L.) WRKY gene family. J. Exp. Bot. 65, 1513–1528. doi: 10.1093/jxb/eru007
He, H., Dong, Q., Shao, Y., Jiang, H., Zhu, S., Cheng, B., et al. (2012). Genome-wide survey and characterization of the WRKY gene family in Populus trichocarpa. Plant Cell Rep. 31, 1199–1121. doi: 10.1007/s00299-012-1241-0
Higo, K., Ugawa, Y., Iwamoto, M., and Korenaga, T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database. Nucleic Acids Res. 27, 297–300. doi: 10.1093/nar/27.1.297
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., and Gao, G. (2015). GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
Jiang, Y., and Deyholos, M. K. (2009). Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol. Biol. 69, 91–105. doi: 10.1007/s11103-008-9408-3
Jin, J. P., Zhang, H., Kong, L., Gao, G., and Luo, J. C. (2014). PlantTFDB 3.0: A portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi: 10.1093/nar/gkt1016
Journot-Catalino, N., Somssich, I. E., Roby, D., and Kroj, T. (2006). The transcription factors WRKY11 and WRKY17 act as negative regulators of basal resistance in Arabidopsis thaliana. Plant Cell 18, 3289–3302. doi: 10.1105/tpc.106.044149
Kalde, M., Barth, M., Somssich, I. E., and Lippok, B. (2003). Members of the Arabidopsis WRKY group III transcription factors are part of different plant defense signaling pathways. Mol. Plant Microbe Interact. 16, 295–305. doi: 10.1094/MPMI.2003.16.4.295
Kato, N., Dubouzet, E., Kokabu, Y., Yoshida, S., Taniguchi, Y., Dubouzet, J. G., et al. (2007). Identification of a WRKY protein as a transcriptional regulator of benzylisoquinoline alkaloid biosynthesis in Coptis japonica. Plant Cell Physiol. 48, 8–18. doi: 10.1093/pcp/pcl041
Kesari, R., Trivedi, P. K., and Nath, P. (2007). Ethylene-induced ripening in banana evokes expression of defense and stress related genes in fruit tissue. Postharvest Biol. Technol. 46, 136–143. doi: 10.1016/j.postharvbio.2007.04.010
Knoth, C., Ringler, J., Dangl, J. L., and Eulgem, T. (2007). Arabidopsis WRKY70 is required for full RPP4-mediated disease resistance and basal defense against Hyaloperonospora parasitica. Mol. Plant Microbe Interact. 20, 120–128. doi: 10.1094/MPMI-20-2-0120
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lakhwani, D., Pandey, A., Dhar, Y. B., Bag, S. K., Trivedi, P. K., and Asif, M. H. (2015). Genome-wide analysis of the AP2/ERF family in Musa species reveals divergence and neofunctionalisation during evolution. Sci. Rep. 5:18878. doi: 10.1038/srep18878
Lee, T. H., Tang, H., Wang, X., and Paterson, A. H. (2013). PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res 41, D1152–D11588. doi: 10.1093/nar/gks1104
Levee, V., Major, I., Levasseur, C., Tremblay, L., MacKay, J., and Seguin, A. (2009). Expression profiling and functional analysis of Populus WRKY23 reveals a regulatory role in defense. New Phytol. 184, 48–70. doi: 10.1111/j.1469-8137.2009.02955.x
Li, C. Y., Deng, G. M., Yang, J., Viljoen, A., Jin, Y., Kuang, R. B., et al. (2012). Transcriptome profiling of resistant and susceptible Cavendish banana roots following inoculation with Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genomics 13:374. doi: 10.1186/1471-2164-13-374
Liu, X., Bai, X., Wang, X., and Chu, C. (2007). OsWRKY71, a rice transcription factor, is involved in rice defense response. J. Plant Physiol. 164, 969–979. doi: 10.1016/j.jplph.2006.07.006
Lohani, S., Trivedi, P. K., and Nath, P. (2004). Changes in activities of cell wall hydrolases during ethylene-induced ripening in banana: effect of 1-MCP, ABA and IAA. Postharvest Biol. Technol. 31, 119–126. doi: 10.1016/j.postharvbio.2003.08.001
Luo, X., Bai, X., Sun, X., Zhu, D., Liu, B., Ji, W., et al. (2013). Expression of wild soybean WRKY20 in Arabidopsis enhances drought tolerance and regulates ABA signaling. J. Exp. Bot. 64, 2155–2169. doi: 10.1093/jxb/ert073
Maruyama-Nakashita, A., Nakamura, Y., Watanabe-Takahashi, A., Inoue, E., Yamaya, T., and Takahashi, H. (2005). Identification of a novel cis-acting element conferring sulfur deficiency response in Arabidopsis roots. Plant J. 42, 305–314. doi: 10.1111/j.1365-313X.2005.02363.x
Miao, Y., Laun, T., Smykowski, A., and Zentgraf, U. (2007). Arabidopsis MEKK1 can take a short cut: it can directly interact with senescence-related WRKY53 transcription factor on the protein level and can bind to its promoter. Plant Mol. Biol. 65, 63–76. doi: 10.1007/s11103-007-9198-z
Mishra, S., Triptahi, V., Singh, S., Phukan, U. J., Gupta, M. M., Shanker, K., et al. (2013). Wound induced transcriptional regulation of benzylisoquinoline pathway and characterization of wound inducible PsWRKY transcription factor from Papaver somniferum. PLoS ONE 8:e52784. doi: 10.1371/journal.pone.0052784
Muthamilarasan, M., Bonthala, V. S., Khandelwal, R., Jaishankar, J., Shweta, S., Nawaz, K., et al. (2015). Global analysis of WRKY transcription factor superfamily in Setaria identifies potential candidates involved in abiotic stress signaling. Front. Plant Sci. 6:910. doi: 10.3389/fpls.2015.00910
Niu, C.-F., Wei, W., Zhou, Q.-Y., Tian, A. G., Hao, Y. J., Zhang, W. K., et al. (2012). Wheat WRKY genes TaWRKY2and TaWRKY19 regulate abiotic stress tolerance in transgenic Arabidopsis plants. Plant Cell Environ. 35, 1156–1170. doi: 10.1111/j.1365-3040.2012.02480.x
Pandey, A., Misra, P., Alok, A., Kaur, N., Sharma, S., Lakhwani, D., et al. (2016). Genome wide identification and expression analysis of Homeodomain leucine zipper subfamily IV (HDZIV) gene family from Musa accuminata. Front. Plant Sci. 7:20. doi: 10.3389/fpls.2016.00020
Park, C. Y., Lee, J. H., Yoo, J. H., Moon, B. C., Choi, M. S., Kang, Y. H., et al. (2005). WRKY group IId transcription factors interact with calmodulin. FEBS Lett. 579, 1545–1550. doi: 10.1016/j.febslet.2005.01.057
Rinerson, C. I., Rabara, R. C., Tripathi, P., Shen, Q. J., and Rushton, P. J. (2015). The evolution of WRKY transcription factors. BMC Plant Biol. 15:66. doi: 10.1186/s12870-015-0456-y
Robatzek, S., and Somssich, I. E. (2002). Targets of AtWRKY6 regulation during plant senescence and pathogen defense. Genes Dev. 16, 1139–1149. doi: 10.1101/gad.222702
Rushton, P. J., MacDonald, H., Huttly, A. K., Lazarus, C. M., and Hooley, R. (1995). Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of α-Amy2 genes. Plant Mol. Biol. 29, 691–702. doi: 10.1007/BF00041160
Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., et al. (2003). TM4: A free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378.
Sun, C., Palmqvist, S., Olsson, H., Boren, M., Ahlandsberg, S., and Jansson, C. (2003). A novel WRKY transcription factor, SUSIBA2, participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter. Plant Cell 15, 2076–2092. doi: 10.1105/tpc.014597
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Tao, Z., Kou, Y., Liu, H., Li, X., Xiao, J., and Wang, S. (2011). OsWRKY45 alleles play different roles in abscisic acid signalling and salt stress tolerance but similar roles in drought and cold tolerance in rice. J. Exp. Bot. 62, 4863–4874. doi: 10.1093/jxb/err144
Voorrips, R. E. (2002). MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Heredity 93, 77–78. doi: 10.1093/jhered/93.1.77
Wang, H., Hao, J., Chen, X., Hao, Z., Wang, X., Lou, Y., et al. (2007). Overexpression of rice WRKY89 enhances ultraviolet B tolerance and disease resistance in rice plants. Plant Mol. Biol. 65, 799–815. doi: 10.1007/s11103-007-9244-x
Wei, K. F., Chen, J., Chen, Y. F., Wu, L. J., and Xie, D. X. (2012). Molecular phylogenetic and expression analysis of the complete WRKY transcription factor family in maize. DNA Res. 19, 153–164. doi: 10.1093/dnares/dsr048
Wettenhall, J. M., Simpson, K. M., Satterley, K., and Smyth, G. K. (2006). affylmGUI: a graphical user interface for linear modeling of single channel microarray data. Bioinformatics 22, 897–899. doi: 10.1093/bioinformatics/btl025
Wu, K. L., Guo, Z. J., Wang, H. H., and Li, J. (2005). The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. 12, 9–26. doi: 10.1093/dnares/12.1.9
Wu, X., Shiroto, Y., Kishitani, S., Ito, Y., and Toriyama, K. (2009). Enhanced heat and drought tolerance in transgenic rice seedlings overexpressing OsWRKY11 under the control of HSP101 promoter. Plant Cell Rep. 28, 21–30. doi: 10.1007/s00299-008-0614-x
Xiong, X. (2010). Constitutive expression of the barley HvWRKY38 transcription factor enhances drought tolerance in turf and forage grass (Paspalumnotatum Flugge). Mol. Breed. 25, 419–432. doi: 10.1007/s11032-009-9341-4
Xu, Y. H., Wang, J. W., Wang, S., Wang, J. Y., and Chen, X. Y. (2004). Characterization of GaWRKY1, a cotton transcription factor that regulates the sesquiterpene synthase gene (+)-delta-cadinene synthase-A. Plant Physiol. 135, 507–515. doi: 10.1104/pp.104.038612
Yu, S., Ligang, C., Liping, Z., and Diqiu, Y. (2010). Overexpression of OsWRKY72 gene interferes in the abscisic acid signal and auxin transport pathway of Arabidopsis. J. Biosci. 35, 459–471. doi: 10.1007/s12038-010-0051-1
Zhang, J., Peng, Y., and Guo, Z. (2008). Constitutive expression of pathogen-inducible OsWRKY31 enhances disease resistance and affects root growth and auxin response in transgenic rice plants. Cell Res. 18, 508–521. doi: 10.1038/cr.2007.104
Zhang, Y., and Wang, L. (2005). The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol. Biol. 5:1. doi: 10.1186/1471-2148-5-1
Zheng, Z., Qamar, S. A., Chen, Z., and Mengiste, T. (2006). Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 48, 592–605. doi: 10.1111/j.1365-313X.2006.02901.x
Zheng, Z., Mosher, S. L., Fan, B., Klessig, D. F., and Chen, Z. (2007). Functional analysis of Arabidopsis WRKY25 transcription factor in plant defense against Pseudomonas syringae. BMC Plant Biol. 7:2. doi: 10.1186/1471-2229-7-2
Zhou, Q. Y., Tian, A. G., Zou, H. F., Xie, Z. M., Lei, G., Huang, J., et al. (2008). Soybean WRKY-type transcription factor genes, GmWRKY13, GmWRKY21, and GmWRKY54, confer differential tolerance to abiotic stresses in transgenic Arabidopsis plants. Plant Biotechnol. J. 6, 486–503. doi: 10.1111/j.1467-7652.2008.00336.x
Keywords: abiotic stress, banana, differential gene expression, fruit ripening, WRKY gene family
Citation: Goel R, Pandey A, Trivedi PK and Asif MH (2016) Genome-Wide Analysis of the Musa WRKY Gene Family: Evolution and Differential Expression during Development and Stress. Front. Plant Sci. 7:299. doi: 10.3389/fpls.2016.00299
Received: 31 December 2015; Accepted: 25 February 2016;
Published: 14 March 2016.
Edited by:
Shabir Hussain Wani, Sher-e-Kashmir University of Agricultural Sciences and Technology of Kashmir, IndiaReviewed by:
Abu Hena Mostafa Kamal, University of Texas at Arlington, USAMukesh Jain, Jawaharlal Nehru University, India
Copyright © 2016 Goel, Pandey, Trivedi and Asif. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prabodh K. Trivedi, cHJhYm9kaHRAbmJyaS5yZXMuaW4=; cHJhYm9kaHRAaG90bWFpbC5jb20=;
Mehar H. Asif, bWguYXNpZkBuYnJpLnJlcy5pbg==
†Present Address: Ashutoh Pandey, Department of Biotechnology, National Agri-Food Biotechnology Institute, Government of India, Mohali, India