 Qianqian Huang
Qianqian Huang Xiao Huang
Xiao Huang Juan Deng
Juan Deng Hegang Liu
Hegang Liu Yanwen Liu
Yanwen Liu Kun Yu
Kun Yu Bisheng Huang
Bisheng Huang- College of Pharmacy, Hubei University of Chinese Medicine, Wuhan, China
The rhizome of Atractylodes lancea is extensively used in the practice of Traditional Chinese Medicine because of its broad pharmacological activities. This study was designed to characterize the transcriptome profiling of the rhizome and leaf of Atractylodes lancea in an attempt to uncover the molecular mechanisms regulating rhizome formation and growth. Over 270 million clean reads were assembled into 92,366 unigenes, 58% of which are homologous with sequences in public protein databases (NR, Swiss-Prot, GO, and KEGG). Analysis of expression levels showed that genes involved in photosynthesis, stress response, and translation were the most abundant transcripts in the leaf, while transcripts involved in stress response, transcription regulation, translation, and metabolism were dominant in the rhizome. Tissue-specific gene analysis identified distinct gene families active in the leaf and rhizome. Differential gene expression analysis revealed a clear difference in gene expression pattern, identifying 1518 up-regulated genes and 3464 down-regulated genes in the rhizome compared with the leaf, including a series of genes related to signal transduction, primary and secondary metabolism. Transcription factor (TF) analysis identified 42 TF families, with 67 and 60 TFs up-regulated in the rhizome and leaf, respectively. A total of 104 unigenes were identified as candidates for regulating rhizome formation and development. These data offer an overview of the gene expression pattern of the rhizome and leaf and provide essential information for future studies on the molecular mechanisms of controlling rhizome formation and growth. The extensive transcriptome data generated in this study will be a valuable resource for further functional genomics studies of A. lancea.
Introduction
Rhizomatous plants comprise a large group, and many of them contribute ecosystem services (e.g., prevention of soil erosion) or have high economic value (e.g., ginger) or significant medicinal uses, such as Paris polyphylla and other rhizomatous medicinal plants (Glover et al., 2010; Yu et al., 2013). Leaf and rhizome are the two most important vegetative organs in rhizomatous plants. It is well known that the main role of leaves is to capture light energy, perform photosynthesis, and accumulate assimilates, while rhizomes primarily store energy reserves (e.g., starch) and allocate nutrients for overwintering and regrowth. The botanical, physiological, and genetic processes in the leaf and rhizome have always drawn intense attention (Bell and Tomlinson, 1980; Yu et al., 2013; Kong et al., 2015). However, the relationship of growth and development between leaf and rhizome and the molecular mechanisms underlying rhizome formation are largely not understood, due to the complexity of the developmental connections and the physiological coordination between the two organs.
Recently developed functional genomics approaches offer an efficient way to dissect complex physiological processes (Baginsky et al., 2010). The large scale of genomic and transcriptomic data have greatly enhanced our understanding of plant growth and development, especially in model plants, such as Arabidopsis and rice. The ability to sequence cDNA libraries has been exploited in functional genomics research in recent years (Fang et al., 2015). The advent of next-generation sequencing technologies has revolutionized functional genomics due to its high-throughput, sensitivity, and accuracy. In particular, RNA sequencing (RNA-Seq), has been widely used to obtain transcriptome data, profile global gene expression, and identify novel genes in both model and non-model plant species, including Arabidopsis (Begara-Morales et al., 2014), rice (Wakasa et al., 2014), Salvia miltiorrhiza (Gao et al., 2014), and Medicago truncatula (Cabeza et al., 2014). With advances in sequencing technology, RNA sequencing has become an effective and powerful tool for transcriptome analysis, especially in non-model species where limited genetic and genomic resources are available (Dillies et al., 2013).
Atractylodes lancea (Thunb.) DC. (Compositae), also called Cangzhu in Chinese, is a well-known and widely prescribed traditional Chinese herb. The rhizome of A. lancea has been used for the treatment of digestive disorders, rheumatic diseases, night blindness, and other conditions which is explained by eliminating dampness, invigorating spleen, and expelling wind according to the theory of traditional Chinese medicine (Committee, 2010). Modern pharmacological studies show that A. lancea has broad pharmacological effects on the nervous, gastrointestinal, and cardiovascular systems (Koonrungsesomboon et al., 2014). Anticancer, antimicrobial, and anti-inflammatory activities have also been demonstrated for the crude extracts of the A. lancea rhizome and its major constituents, such as atractylodin, β-eudesmol, hinesol, and atractylone (Resch et al., 2001; Wang et al., 2009; Zhao et al., 2014). A. lancea is widely distributed in East Asia, especially in China (Shi, 1987). However, natural populations of A. lancea have been rapidly depleted due to intense predatory exploitation. Artificial cultivation is thus imperative to protect the natural resources and achieve a sustainable supply. A crucial question is how to ensure or even improve rhizome quality. Though the phytochemistry, pharmacology, botany, and cultivation of A. lancea have been extensively studied, the molecular mechanisms of the plant's growth and development are poorly understood, largely due to the lack of genomic information (Deng et al., 2014; Koonrungsesomboon et al., 2014).
In this study, we performed high-throughput Illumina sequencing to comprehensively characterize the transcriptome of A. lancea, and reveal differential gene expression profiles between rhizome and leaf, which would facilitate uncovering the molecular mechanisms of regulating rhizome formation and growth of the most important medicinal plants in genus Atractylodes.
Materials and Methods
Plant Materials
Leaves and rhizomes of A. lancea were collected from Mao Mountain, Jiangsu Province, China (31°39′N, 119°19′E). All samples were harvested, washed, surface dried, and then immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction. Two biological replicates were used for RNA extraction and transcriptome sequencing of the leaf and three replicates for the rhizome.
RNA Sequencing and De novo Assembly
Total RNA from each tissue was isolated using the RNAprep Pure Plant Kit (Tiangen, Beijing, China) following the manufacturer's instructions. cDNA library construction and normalization were performed according to published protocols (Zhang et al., 2015). Five cDNA libraries (2 for leaves and 3 for rhizomes) were finally sequenced using an Illumina HiSeq2000 platform, and paired-end reads were generated. Clean reads were obtained by removing the adapter sequences, low quality sequences, and sequences shorter than 35 bases. The remaining high-quality reads were de novo assembled into candidate unigenes using the Trinity program (Grabherr et al., 2011).
Functional Annotation of Unigenes
Assembled unigenes were annotated using BLAST alignment against public databases, including the NCBI non-redundant protein database (NR, http://www.ncbi.nlm.nih.gov), Swiss-Prot (http://www.expasy.ch/sprot), TrEMBL (http://www.ebi.ac.uk/trembl), Gene Ontology (GO, http://www.geneontology.org), and the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg) with an E value cutoff of 10−5. Blast2GO and WEGO program was carried out to perform GO annotation and to obtain GO classifications according to molecular function, biological process, and cellular component (Conesa et al., 2005; Ye et al., 2006). The transcription factor (TF) families were identified by Using known plant transcription factors identified in PlnTFDB (http://plntfdb.bio.uni-potsdam.de/v3.0) based on the annotation.
Sequence Analysis
Analysis of codon usage bias was performed by CodonW (http://codonw.sourceforge.net/). Putative SSR markers were detected using the MISA software package (http://pgrc.ipk-gatersleben.de/misa/). The minimum repeat number was 10 for mono-nucleotides, 6 for di-nucleotide, and 5 for tri-, tetra-, penta-, and hexa-nucleotide.
Determination of Unigene Expression Level
Since no reference genome was available for A. lancea, the clean reads from each sequencing library were mapped back to the assembled unigenes using Bowtie with a maximum mismatch of 2 nucleotides (Langmead et al., 2009). The expression level of each unigene were normalized and calculated as the value of fragments per transcript kilobase per million fragments mapped (FPKM), which eliminates the influence of different gene lengths and sequencing discrepancies (Trapnell et al., 2010).
Analysis of Unigene Differential Expression
The differential gene expression analysis of two assigned libraries was performed using the edgeR package (Robinson et al., 2010). The differentially expressed genes (DEGs) were screened with the threshold false discovery rate (FDR) < 0.05 and the absolute value of log2FoldChange > 1. Subsequently, GO functional enrichment analysis and KEGG pathway analysis of the DEGs were performed using GOseq and KOBAS, respectively (Young et al., 2010; Xie et al., 2011).
Quantitative Real-Time PCR
Twenty unigenes (c40786_g1, c53153_g2, c45414_g1, c40381_g1, c33812_g1, c37348_g1, c29120_g1, c36168_g1, c41101_g1, c44073_g2, c41696_g3, c45627_g1, c37834_g1, c39104_g1,c47165_g4, c49171_g1,c43275_g1, c38241_g1, c51805_g3, and c45003_g1) were selected for verification of the sequencing and computational results by quantitative real-time PCR (qPCR). All reactions were carried out in 96-well plates in the StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using the SYBR Premix Ex Taq II (TaKaRa, Dalian, China) kit with four replicates. Cycling conditions were 95°C for 10 min followed by 45 cycles of 94°C for 30 s and 60°C for 45 s. The relative expression levels of the selected unigenes were normalized to the internal control gene Tubulin (c50304_g2), and determined by the ΔΔCt-method. All primers used are shown in Supplementary Table 1.
Results
Sequence Analysis and Assembly
To obtain a comprehensive overview of the A. lancea transcriptome, RNAseq libraries were constructed from leaves and rhizomes and sequenced using Illumina paired-end sequencing technology. After the removal of adaptor sequences and low-quality reads, approximately 118.4 and 152.7 million clean reads were acquired for the leaf and rhizome transcriptomes, respectively. Thus, a total of 33,885 Mb valid data were acquired with an average length of 125 bp. An overview of the sequencing statistics is shown in Table 1. All clean reads were subsequently subjected to de novo assembly with the Trinity program resulting in 185,544 transcripts. A total of 92,366 unigenes with an average length of 721 bp, a maximum size of 15.9 kb, and an N50 of 1.1 kb (i.e., 50% of the assembled bases were incorporated into unigenes of 1.1 kb or longer) were obtained (Table 2). The GC content of the reads and unigenes distributed within 41–45% (Tables 1, 2). The size distribution of the A. lancea unigenes is given in Figure 1A, with 27% of all unigenes showing lengths longer than 1 kb. A Venn diagram of the expressed unigenes with FPKM ≥1 is shown in Figure 1B. A total of 42,517 unigenes were found to be both expressed in leaf and rhizome samples of A. lancea. All reads generated in this study have been deposited in the National Center for Biotechnology Information (NCBI) and can be accessed in the Short Read Archive (SRA) Sequence Database under accession number SRP068251.
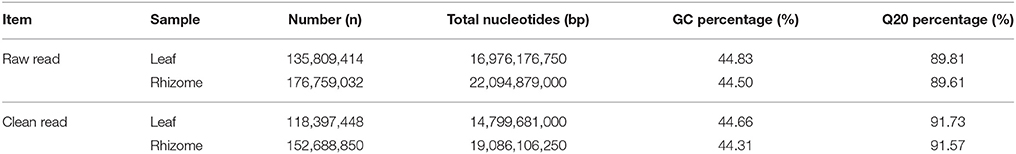
Table 1. Summary of transcriptomes from leaf and rhizome in A. lancea.
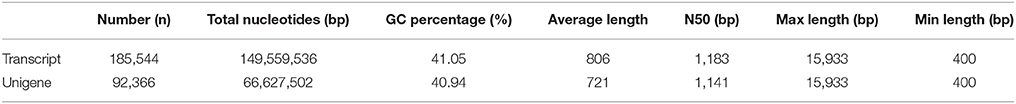
Table 2. Assembly results of clean reads.
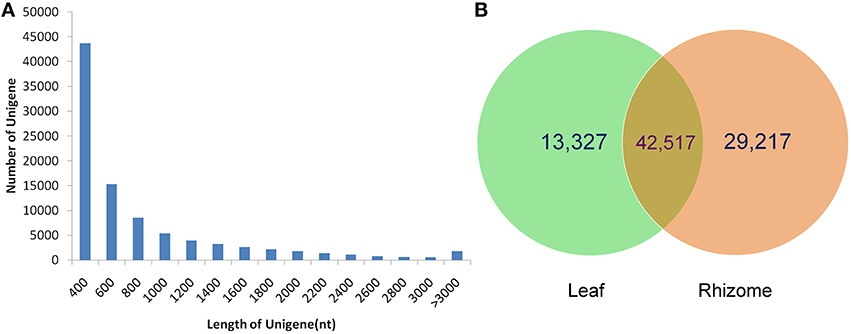
Figure 1. Length distribution of unigenes from samples of leaf and rhizome (A). The Venn diagram shows the number of expressed genes (FPKM >1) in samples of leaf and rhizome (B).
Functional Annotation and Classification
The unigenes were aligned against public protein databases (NR, Swiss-Prot, GO, and KEGG) using BLAST with a cut-off E-value of 1.0e−5. A total of 39,664 unigenes (42.90% of the total assembled unigenes) had a match in the NR database, and 38,699 (41.19%), 26,159 (28.32%), and 10,508 (11.38%) unigenes showed significant similarity to sequences in the Swiss-Prot, GO, and KEGG databases, respectively (Table 3).
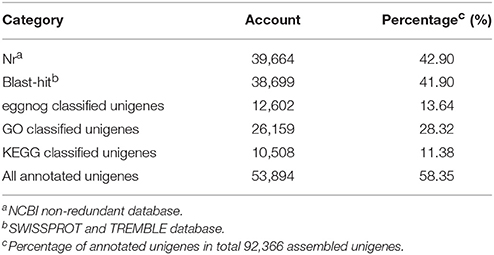
Table 3. Statistics of annotations for assembled unigenes.
GO classification was used to classify unigene functions based on the Nr annotation, and 26,159 (28.32%) unigenes were assigned to one or more GO terms (Figure 2). Within the “biological process” domain, the assignments were mostly enriched in the terms “cellular process” (21,094, 22.84%), “metabolic process” (16,157, 17.49%), “response to stimulus” (7133, 7.72%), and “biological regulation” (6762, 7.32%). In the “molecular function” domain, the terms “catalytic activity” (14,221, 15.40%) and “binding” (17,944, 19.43%) were mostly assigned. For the “cellular component” domain, the most evident matches were to the terms “cell” (19,140, 20.72%), “cell part” (19,098, 20.68%), “organelle” (14,672, 15.88%), and “organelle part” (7317, 7.92%).
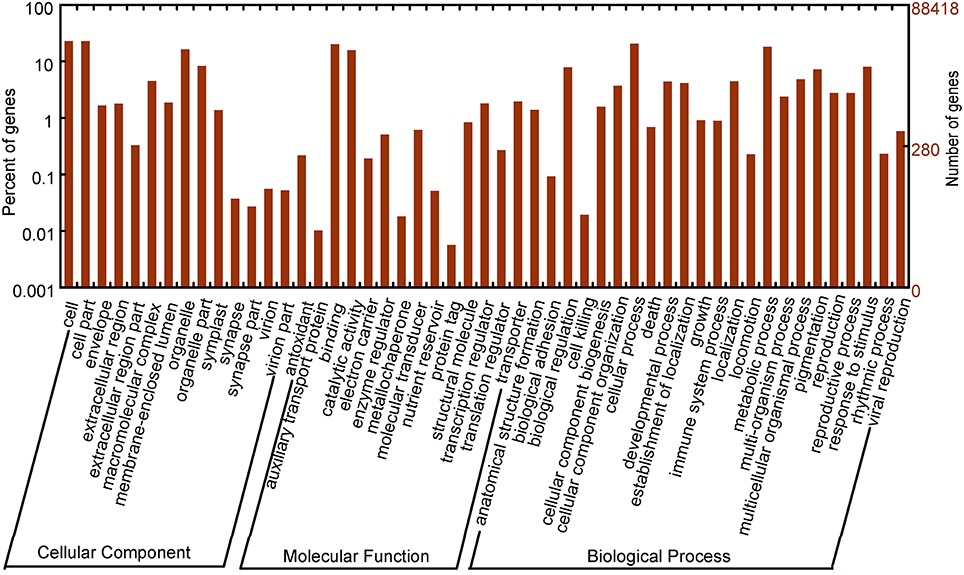
Figure 2. Gene ontology classification of assembled unigenes.
KEGG pathway analysis was performed to identify the biochemical pathways active in the leaf and rhizome of A. lancea. A total of 10,504 unigenes were annotated and assigned to 289 KEGG pathways. Unigenes classified to the five main KEGG biochemical pathways, metabolism, genetic information processing, environmental information processing, cellular processes, and organismal systems pathways are presented in Figure 3, with unigenes associated with Human Diseases filtered out. The three most highly represented pathways are “carbon metabolism” (ko01200), “Ribosome” (ko03010), and “Biosynthesis of amino acids” (ko01230).
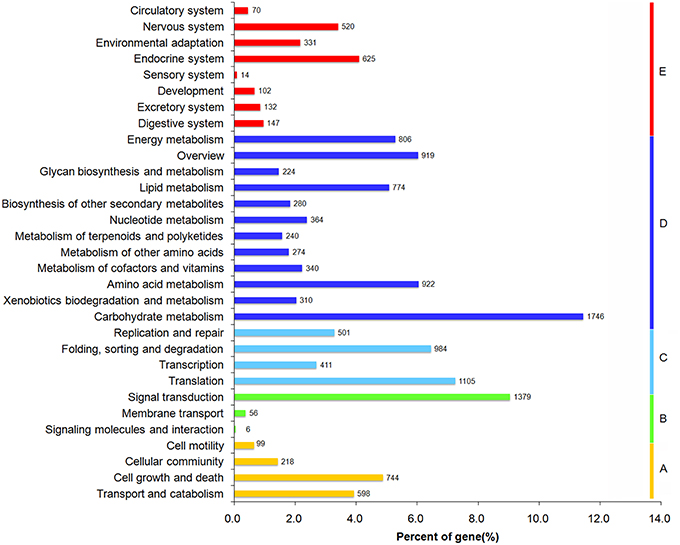
Figure 3. Functional classification and pathway assignment of assembled unigenes by KEGG.
Cytochromes P450 (CYP450s) form by far the largest superfamily of plant enzymes and take part in numerous primary and secondary metabolic processes (Weitzel and Simonsen, 2015). In the A. lancea transcriptome data, 161 unigenes were functionally annotated as CYP450s; the genes belong to 71 CYP family categories, with the majority in the CYP72A219 family (Supplementary Table 2).
Characterization of Codon Usage and SSR Markers
Codon usage analysis was based on 1556 full-length sequences with ORF ≥600 bp. The codon usage Table was created from 9.2 million codons (Supplementary Table 3). GAT was the most frequently used codon with the occurrence frequency 3.81% followed by GAA (3.54%) and AAG (3.05%).
A total of 10,103 SSRs were identified in 92,366 unigenes, 1074 of which contained more than one SSR, and 406 SSRs were present in compound form (Table 4). The most abundant repeat motifs were mono-nucleotides (4440, 43.94%), followed by di-nucleotides (3604, 35.67%) and tri-nucleotides (1892, 19.62%).

Table 4. Summary of SSR searching results.
Highly Expressed and Tissue-Specific Genes
We identified 227 transcripts in the leaf and 105 in the rhizome with an FPKM value greater than 1000, of these, 49 were in both tissues. Most of the highly expressed genes in the leaf were predominantly involved in photosynthesis, stress response, and translation; in the rhizome, transcripts involved in stress response were dominant, followed by those related to transcription regulation, translation, and metabolism (Supplementary Table 4). The 10 most abundant transcripts in leaf and rhizome are listed in Table 5.
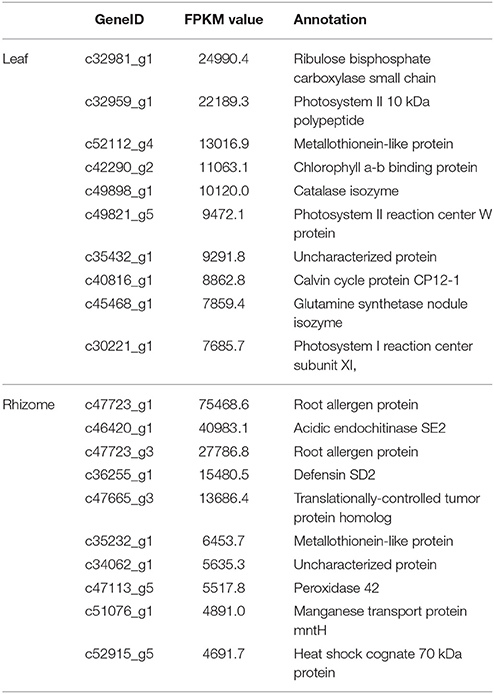
Table 5. Top 10 abundant transcripts in leaf and rhizome of A. lancea.
Genes that are represented by more than 10 reads in one tissue and no more than one read in another are considered tissue specific (Zhang et al., 2014). According to these criteria, we identified 697 leaf-specific genes from several gene families, including UDP-glycosyl transferases, CYP450s, and ethylene responsive transcription factors (Supplementary Table 5). Chlorophyll binding proteins and photosystem proteins were shown to be highly expressed in the leaf, as expected. Several transcription factors were also specifically expressed in leaf, such as bHLH87-like, WRKY, MYB, and TCP5-like TFs. Of the 469 rhizome-specific genes we identified, we recorded high FPKM values for zinc finger genes, non-specific lipid transfer genes, and many secondary metabolism-related genes (e.g., isocomene synthase, vinorine synthase-like protein, N-benzoyltransferase, etc.). Several gene families were also enriched in the rhizome-specific set, such as leucine-rich repeat receptor-like kinases, heat shock proteins, and transcription factors (Supplementary Table 6).
DEGs between Rhizomes and Leaves
To identify different expression levels of genes between rhizomes and leaves of A. lancea, we calculated the FPKM values of assembled unigenes. We found 4982 differentially expressed unigenes between the two tissues, including 1518 genes and 3464 genes down-regulated in the rhizome compared with the leaf. A volcano plot was constructed to illustrate the distribution of significantly regulated genes (Figure 4).
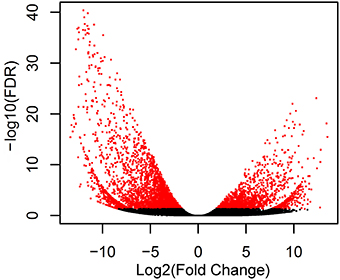
Figure 4. Volcano plot of the transcriptome in leaf and rhizome. The horizontal line and vertical lines indicate the significance threshold (FDR < 0.05) and two-fold change threshold (|log2FoldChange|>1), respectively. The DEGs are shown with blue dots while non-DEGs are in black.
In order to further understand the biological functions of the DEGs, enrichment analyses based on GO and KEGG pathways were performed. When the 4982 DEGs were checked against the GO database, 262 GO terms were significantly enriched (Supplementary Figure S1). In the KEGG analysis, the 1518 up-regulated unigenes were linked to 159 KEGG pathways. The pathway assigned the largest number of unigenes (29) was “plant hormone signal transduction” (ko04075), followed by “starch and sucrose metabolism” (ko00500), “protein processing in endoplasmic reticulum” (ko04141), and “biosynthesis of amino acids” (ko01230; Table 6). 39 unigenes were mapped to secondary metabolism pathways, including 18 unigenes which might be involved in “metabolism of terpenoids and polyketides” (Supplementary Table 7).
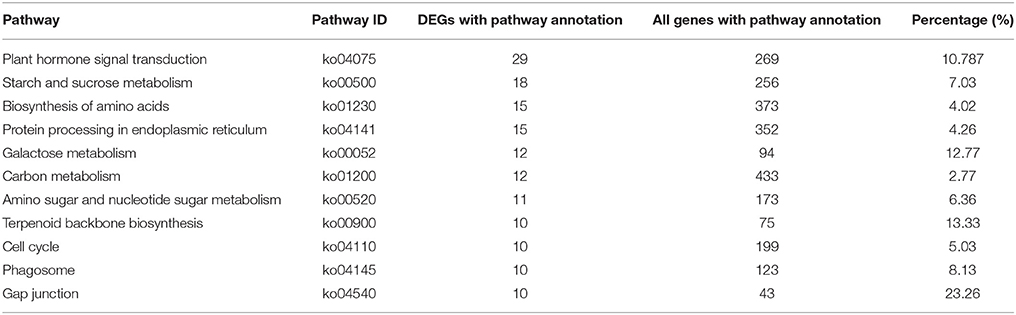
Table 6. List of KEGG Pathway with more than 10 DEGs annotated.
The top 30 up-regulated unigenes in the leaf and rhizome are shown in Figure 5. Unigenes involved in photosynthesis (e.g., ribulose bisphosphate carboxylase, chlorophyll a-b binding protein) the main up-regulated genes in the leaf, while unigenes associated with plant hormones (e.g., cytokinin hydroxylase and auxin-responsive proteins) and secondary metabolism (such as farnesene synthase and cinnamoyl-CoA reductase) were up-regulated in the rhizome.

Figure 5. List of top 30 up-regulated transcripts in leaf (A) and rhizome (B). L, leaf; R, rhizome.
Validation of DEGs by qPCR
The RNA-Seq and computational results were verified by qPCR using 20 selected DEGs. The expression patterns of all the selected genes show the same trend in the transcriptome analysis and the qRT-PCR (Supplementary Figure S2). We also tested the correlations of these genes and found a significant positive correlation between them, with the correlation coefficient reaching 0.81.
Identification of TF Families
A total of 42 TF families were identified when aligning the annotated A. lancea transcripts to the AGRIS database (Figure 6A). Members of the MYB, MYB-related, AP2-EREBP, bHLH, NAC, WRKY, C3H, GRAS, ABI3VP1, and mTERF families were the top 10 classes, each with more than 48 unigenes. There were 60 TFs up-regulated in the leaf, mainly from the NAC, WRKY, AP2-EREBP, mTERF, and TCP families. A total of 67 TFs were up-regulated in the rhizome, mainly from the AP2-EREBP, bHLH, C2H2, and SBP families (Figure 6B).
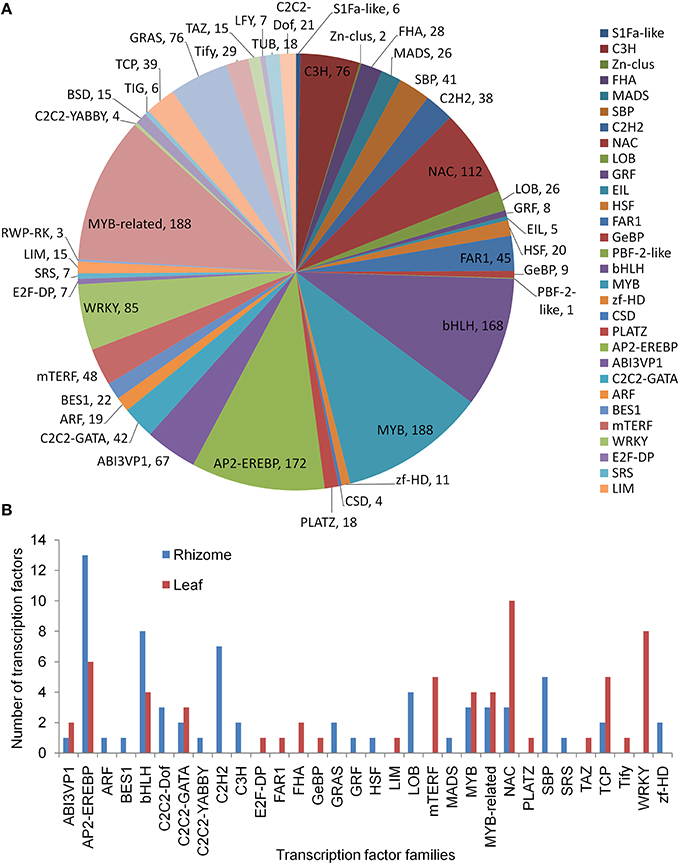
Figure 6. Transcription factor analysis. (A) Distribution of transcription factor families. (B) Up-regulated transcription factors in leaf and rhizome.
Genes Involved in Rhizome Formation and Growth
DEGs were further analyzed to screen candidate genes involved in rhizome formation and development. A total of 104 genes involved in organ development, hormone biosynthesis, and hormone signal transduction were identified, including some transcription factors (e.g., 2 MADS-box proteins, 12 AP2-like transcription factors). Unigenes encoding sucrose synthase, lipoxygenase, and gibberellin (GA) 20-oxidase were also identified as candidates (Table 7), as well as a number of genes involving in hormone response, biosynthesis, and signal transduction. Candidate genes in the auxin (IAA), GA, abscisic acid (ABA), ethylene (ETH), cytokinin (CTK), jasmonic acid (JA), and brassinosteroid (BR) pathways are shown in Figure 7.
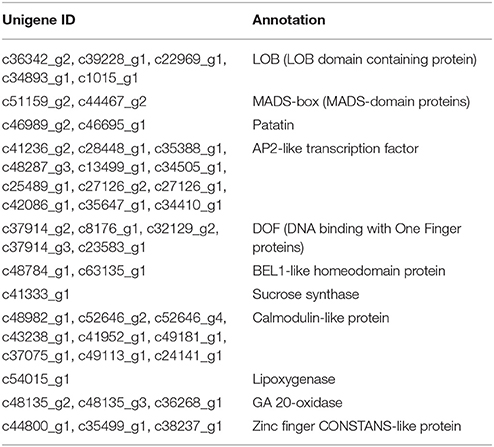
Table 7. Putative unigenes involved in the rhizome formation and growth.
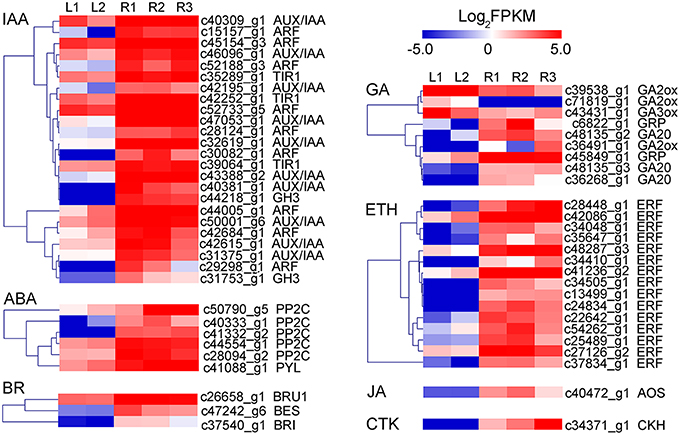
Figure 7. Heatmap of the expressed genes assigned to hormone responsive protein, hormone biosynthesis pathway, and signal transduction pathway in leaf and rhizome transcriptomes. AOS, allene oxide synthase; ARF, auxin response factor; BES, brassinazole-resistant protein; BRI, BR Insensitive 1; BRU1, Brassinosteroid-regulated protein; CKH, cytokinin hydroxylase; ERF, Ethylene-responsive transcription factor; GA20, Gibberellin 20-oxidase; GA2ox, Gibberellin 2-beta-dioxygenase; GA3ox, Gibberellin 3-beta-dioxygenase; GH3, IAA-amido synthetase; GRP, Gibberellin-regulated protein; PP2C, 2C type protein phosphatase; PYL, abscisic acid receptor; TIR1, transport inhibitor response 1.
Discussion
Global Gene Transcription in the Leaf and Rhizome of A. lancea
Deep RNA sequencing is currently an effective choice for studying the transcriptome of non-model plant species, including A. lancea. Transcript profiling and comparative transcriptome analysis have frequently been used to identify differentially expressed genetic networks and the expression patterns of genes in different organs or tissues of plants. As genome resources for the genus Atractylodes are not yet available, Illumina-based RNA sequencing was used to profile the transcriptome of A. lancea, one of the most important medicinal plants in the genus Atractylodes. We obtained 271 million clean sequencing reads which were assembled de novo into 92,366 unigenes. Of these, 53,894 unigenes (about 58.4% of the assembled unigenes) could be functionally annotated against public protein databases (NR, Swiss-Prot, GO, and KEGG), while no functional annotation was found for 41.6% of the assembled unigenes, either due to a match with a protein of unknown function or because no homologous nucleotide sequence matched (Table 2). These unigenes may be of great importance for further research, since they may be considered novel transcripts or alternative splice variants.
Analysis of gene expression levels was employed to profile global gene expression in the leaf and rhizome, the two main vegetative tissues of A. lancea, analysis of gene expression levels was employed. Genes related to photosynthesis and stress response, such as ribulose bisphosphate carboxylase, photosystem II polypeptide, and metallothionein-like protein, had the highest expression levels in the leaf. This observation is in agreement with previous reports (Mizrachi et al., 2010; Brown et al., 2012; Zhang et al., 2014). Several gene families were identified to be leaf-specific, including UDP-glycosyl transferases, cytochrome P450 proteins, and ethylene responsive transcription factors. UDP-glycosyl transferases play important roles in the biosynthesis of natural plant products, such as terpenoids and flavonoids, and in the regulation of plant hormones (Yonekura-Sakakibara and Hanada, 2011). Plant cytochrome P450s participate in a wide range of biochemical pathways that produce primary and secondary metabolites, such as lipids, terpenoids, and plant hormones (Mizutani and Ohta, 2010). Ethylene responsive transcription factors, members of the AP2/ERF superfamily, are implicated in diverse biological events, such as hormonal signal transduction, response to biotic and abiotic stress, and metabolism regulation (Nakano et al., 2006; Mizoi et al., 2012). These leaf-specific genes may all be functionally related to leaf growth, development, and metabolic processes.
In the rhizome, highly expressed genes were predominantly those involved in stress response, such as defensin, allergen, antioxidative enzymes, acidic endochitinase, and metallothionein-like protein. Similar results were found in the rhizomes of Ligusticum chuanxiong and Sorghum propinquum (Zhang et al., 2014; Song et al., 2015). The tissue-specific genes were largely distinct between leaf and rhizome. The plant zinc finger proteins, which belong to a large family of transcription factors, play a variety of important roles in growth and development, hormone response, and response to abiotic and biotic stresses (Li et al., 2013). Non-specific lipid transfer proteins are known to play key roles in plant defense, growth and development (Liu et al., 2015). A variety of enzymes involved in secondary metabolism, such as the biosynthesis of terpenoids, alkaloids, and isoflavone, were also identified as rhizome-specific, which is similar to the results in S. propinquum (Zhang et al., 2014).
DEGs in Leaf and Rhizome Transcriptome of A. lancea
Differential gene expression patterns were investigated to further profile global gene expression differences between leaf and rhizome. Most of the DEGs identified in the rhizome were assigned to hormone signal transduction, primary metabolic pathways (carbohydrate metabolism and protein biosynthesis), or some secondary metabolic pathways, such as terpenoid biosynthesis and phenylpropanoid biosynthesis, which is in accord with the rhizome's physiological function as a storage organ for carbohydrates and essential oils. The list of the top 30 up-regulated rhizome transcripts is consistent with this (Figure 5). In addition, other genes involved in primary metabolism (non-specific lipid transfer protein, lysine histidine transporter, etc.) and secondary metabolism (vinorine synthase, farnesene synthase, cinnamoyl-CoA reductase, etc.) were also up-regulated in the rhizome. Notably, some stress response-related genes were remarkably up-regulated. Heavy metal-associated isoprenylated plant proteins have been demonstrated to be involved in heavy metal homeostasis and detoxification, response to cold and drought, and plant–pathogen interactions (de Abreu-Neto et al., 2013). Polygalacturonase and bidirectional sugar transporters were both found to be responsible for plant-microbe interactions and important physiological processes in plants, including cell separation, and phloem transport (Yu et al., 2014; Chen et al., 2015).
Gene expression patterns in the leaf were quite different. Genes associated with photosynthesis (ribulose bisphosphate carboxylase, chlorophyll binding protein, etc.) and fundamental metabolism (carboxyl-terminal proteinase, lysosomal pro-x carboxypeptidase, etc.) were greatly up-regulated, in addition to genes associated with plant developmental events and stress tolerance. The zinc finger protein CONSTANS is known to play a role in flowering time and stress tolerance (Yang et al., 2014). MLO proteins have been found to be associated with various developmental pathways and biotic and abiotic stresses (Deshmukh et al., 2014). Alkenal reductase has been proposed to have a key role in the detoxification of reactive carbonyls (Mano et al., 2005). The expression pattern of these genes suggests that they play specific roles in physiological processes in the rhizome and leaf.
TFs play a paramount role in governing plant growth and development by specifically binding to the cis-acting elements in the promoters of downstream genes (Yang et al., 2012). RNA-seq emerged as a powerful tool for the identification of various TF families not only in model plants but also in medicinal plants, such as Salvia miltiorrhiza (Gao et al., 2014) and Bupleurum chinense (Sui et al., 2015). Of the 42 TF families identified in total in this study, TFs belonging to 18 and 22 families were found to be up-regulated in the leaf and rhizome, respectively. The MYB and bHLH TFs are members of two of the largest plant TF families, and function in diverse biological processes, such as the regulation of primary and secondary metabolism, hormone signal transduction, defense, and stress response (Feller et al., 2011). The NAC, WRKY, AP2-EREBP, and C2H2 zinc-finger TFs have all been shown to function in plant developmental processes and stress responses (Chen et al., 2012; Mizoi et al., 2012; Nakashima et al., 2012; Razin et al., 2012), while the mTERF transcription factors are mainly targeted to mitochondria or chloroplasts (Kleine and Leister, 2015). Among the TF families identified in our data, the MYB, bHLH, WRKY, NAC, and AP2-EREBP TFs have been reported to be involved in regulating secondary metabolic pathways (Yang et al., 2012). However, further studies are needed to ascertain and unravel their underlying mechanism of action in A. lancea.
Complex Regulation of Rhizome Formation and Development in A. lancea
As storage organ derived from modified stems, the rhizome serves as a deposit for photosynthates and other compounds, such as essential oils in A. lancea. However, the mechanisms governing rhizome formation and growth are poorly understood. The development of other storage organs, such as potato tubers and lotus rhizomes, has been extensively studied (Fernie and Willmitzer, 2001; Yang et al., 2015). Rhizome formation and development are complex developmental processes in rhizomatous plants; they have been reported to be regulated by a combination of environmental stimuli (e.g., photoperiod) and endogenous factors (Cheng et al., 2013; Yang et al., 2015).
Short day conditions promote the formation of storage organs through the regulation of proteins related to photoperiod signal transduction, such as CONSTANS, cycling Dof factors, and the AP2-like TFs (Martinez-Garcia et al., 2002; Imaizumi and Kay, 2006; Cheng et al., 2013). Patatin, MADS-box transcription factors, BEL1-like homeodomain proteins, and calmodulin-like proteins are also believed to be involved in storage organ development (Hannapel et al., 1985; Bamfalvi et al., 1994; Banerjee et al., 2006; Kim et al., 2009). Plant lateral organ development could be regulated by LOB domain-containing proteins (Majer and Hochholdinger, 2011). Sucrose synthase, a starch biosynthesis enzyme, and lipoxygenase also play important roles in the growth of storage organs (Fernie and Willmitzer, 2001; Kolomiets et al., 2001). Candidate genes encoding such proteins were identified in this study and may participate in rhizome formation and growth of A. lancea.
Plant hormones have been reported to play important roles in the formation of storage organs (Fernie and Willmitzer, 2001). GA and ABA act antagonistically during plant development, including the process of storage organ formation (Yu et al., 2009). GA prevents the formation of potato tubers and lotus rhizomes, while ABA promotes these processes (Xu et al., 1998; Yang et al., 2015). The effects of JA on the induction of storage organs have been extensively studied in potato, yam, garlic, and lotus (Koda, 1992; Zel et al., 1997; Cheng et al., 2013). Moreover, ethylene, auxin, cytokinins, and brassinosteroids also have positive effects on tuber initiation and development (Vreugdenhil and Struik, 1989; Peres et al., 2005). In our study, a range of GA, ABA, ethylene, cytokinin, JA, and brassinosteroid related genes were detected, indicating the complexity of the gene regulatory networks and developmental processes involved in rhizome formation in A. lancea. Further studies on these candidate genes might be useful to uncover the mechanisms of rhizome formation and development in A. lancea.
Conclusions
In this study, 92,366 unigenes, including 38,472 novel genes, were assembled, and 4982 DEGs were identified. Highly expressed and tissue-specific genes were also identified, as well as TF families in leaves and rhizomes. The comparative transcriptome analysis revealed clear differences in global gene expression profile between the leaf and rhizome, suggesting specific and complex molecular mechanisms regulating the growth and development of these two organs. In addition, 104 DEGs were identified to be relevant to rhizome formation and development. These results reveal the coordination of the vegetative organs of rhizomatous plants at the transcriptional level. The sequence datasets and analysis reported here will facilitate functional genomics, gene discovery, transcriptional regulation, and applied studies in A. lancea and other Atractylodes species. DEGs and potential candidate genes involving in rhizome formation and development will help for illustrating the molecular mechanisms underlying rhizome formation and growth.
Author Contributions
QH, XH, and JD prepared the material for sequencing and analyzed the data. HL participated in data analysis. YL is the main coordinator of the project and participated in the conception of the study together with KY and BH. KY and BH were responsible for drafting and revising the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was financially supported by the National Science Foundation of China (Grant No. 31300277), Specialized Research Fund for the Doctoral Program of Higher Education (Grant No. 20124230120002), Construction Project of Heritage Studio of Famous TCM Expert, and Educational Commission of Hubei Province, China (Grant No. Q20121161).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00348
References
Baginsky, S., Hennig, L., Zimmermann, P., and Gruissem, W. (2010). Gene expression analysis, proteomics, and network discovery. Plant Physiol. 152, 402–410. doi: 10.1104/pp.109.150433
Bamfalvi, Z., Kostyal, Z., and Barta, E. (1994). Solanum brevidens possesses a non-sucrose inducible patatin gene. Mol. Gen. Genet. 245, 517–522. doi: 10.1007/BF00302265
Banerjee, A. K., Chatterjee, M., Yu, Y., Suh, S. G., Miller, W. A., and Hannapek, D. J. (2006). Dynamics of a mobile RNA of potato involved in a long distance signaling pathway. Plant Cell 18, 3443–3457. doi: 10.1105/tpc.106.042473
Begara-Morales, J. C., Sánchez-Calvo, B., Luque, F., Leyva-Pérez, M. O., Leterrier, M., Corpas, F. J., et al. (2014). Differential transcriptomic analysis by RNA-seq of GSNO-responsive genes between Arabidopsis roots and leaves. Plant Cell Physiol. 55, 1080–1095. doi: 10.1093/pcp/pcu044
Bell, A. D., and Tomlinson, P. B. (1980). Adaptive architecture in rhizomatous plants. Bot. J. Linn. Soc. 80, 125–160. doi: 10.1111/j.1095-8339.1980.tb01662.x
Brown, A. P., Kroon, J. T., Swarbreck, D., Febrer, M., Larson, T. R., Graham, I. A., et al. (2012). Tissue-specific whole transcriptome sequencing in castor, directed at understanding triacylglycerol lipid biosynthetic pathways. PLoS ONE 7:e30100. doi: 10.1371/journal.pone.0030100
Cabeza, R., Koester, B., Liese, R., Lingner, A., Baumgarten, V., Dirks, J., et al. (2014). An RNA sequencing transcriptome analysis reveals novel insights into molecular aspects of the nitrate impact on the nodule activity of Medicago truncatula. Plant Physiol. 164, 400–411. doi: 10.1104/pp.113.228312
Chen, L., Song, Y., Li, S., Zhang, L., Zou, C., Yu, D., et al. (2012). The role of WRKY transcription factors in plant abiotic stresses. BBA Gene Regul. Mech. 1819, 120–128. doi: 10.1016/j.bbagrm.2011.09.002
Chen, L. Q., Cheung, L. S., Feng, L., Tanner, W., and Frommer, W. B. (2015). Transport of sugars. Annu. Rev. Biochem. 84, 865–894. doi: 10.1146/annurev-biochem-060614-033904
Cheng, L., Li, S., Yin, J., Li, L., and Chen, X. (2013). Genome-wide analysis of differentially expressed genes relevant to rhizome formation in lotus root (Nelumbo nucifera Gaertn). PLoS ONE 8:e67116. doi: 10.1371/journal.pone.0067116
Committee, S. P. (2010). Pharmacopoeia of the People's Republic of China. Beijing: People's Medical Publishing House.
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., and Talon, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
de Abreu-Neto, J. B., Turchetto-Zolet, A. C., de Oliveira, L. F. V., Zanettini, M. H. B., and Margis-Pinheiro, M. M. (2013). Heavy metal-associated isoprenylated plant protein (HIPP): characterization of a family of proteins exclusive to plants. FEBS J. 280, 1604–1616. doi: 10.1111/febs.12159
Deng, Z., Huang, H. J., Wang, W. J., Liu, H. G., and Yu, K. (2014). Morphological Study of Five Types of Atractylodes lancea. North. Horticul. 19, 147–150.
Deshmukh, R., Singh, V. K., and Singh, B. D. (2014). Comparative phylogenetic analysis of genome-wide Mlo gene family members from Glycine max and Arabidopsis thaliana. Mol. Genet. Genomics 289, 345–359. doi: 10.1007/s00438-014-0811-y
Dillies, M. A., Rau, A., Aubert, J., Hennequet-Antier, C., Jeanmougin, M., Servant, N., et al. (2013). A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 14, 671–683. doi: 10.1093/bib/bbs046
Fang, Y., Huang, J., Huang, X., Chen, S. H., Zou, P. C., Li, W. S., et al. (2015). Generation of expressed sequence tags from a cDNA Library of Coleus forskohlii for identification of genes involved in terpene biosynthesis. Biol. Plantarum 59, 463–468. doi: 10.1007/s10535-015-0526-x
Feller, A., Machemer, K., Braun, E. L., and Grotewold, E. (2011). Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 66, 94–116. doi: 10.1111/j.1365-313X.2010.04459.x
Fernie, A. R., and Willmitzer, L. (2001). Molecular and biochemical triggers of potato tuber development. Plant Physiol. 127, 1459–1465. doi: 10.1104/pp.010764
Gao, W., Sun, H. X., Xiao, H., Cui, G., Hillwig, M. L., Jackson, A., et al. (2014). Combining metabolomics and transcriptomics to characterize tanshinone biosynthesis in Salvia miltiorrhiza. BMC Genomics 15:73. doi: 10.1186/1471-2164-15-73
Glover, J. D., Reganold, J. P., Bell, L. W., Borevitz, J., Brummer, E. C., Buckler, E. S., et al. (2010). Increased food and ecosystem security via perennial grains. Science 328, 1638–1639. doi: 10.1126/science.1188761
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., and Thompson, D. A. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Hannapel, D. J., Miller, J. C., and Park, W. D. (1985). Regulation of potato tuber protein accumulation by gibberellic acid. Plant Physiol. 78, 700–703. doi: 10.1104/pp.78.4.700
Imaizumi, T., and Kay, S. A. (2006). Photoperiodic control of flowering: not only by coincidence. Trands Plant Sci. 11, 550–558. doi: 10.1016/j.tplants.2006.09.004
Kim, M. C., Chung, W. S., Yun, D. J., and Cho, M. J. (2009). Calcium and calmodulin mediated regulation of gene expression in plants. Mol. Plant. 2, 13–21. doi: 10.1093/mp/ssn091
Kleine, T., and Leister, D. (2015). Emerging functions of mammalian and plant mTERFs. BBA Bioenergetics 1847, 786–797 doi: 10.1016/j.bbabio.2014.12.009
Koda, Y. (1992). The role of jasmonic acid and related compounds in the regulation of plant development. Int. Rev. Cytol. 135, 155–199. doi: 10.1016/S0074-7696(08)62040-9
Kolomiets, M. V., Hannapel, D. J., Chen, H., Tymeson, M., and Gladon, R. J. (2001). Lipoxygenase is involved in the control of potato tuber development. Plant Cell 13, 613–626. doi: 10.1105/tpc.13.3.613
Kong, W., Kim, C., Goff, V. H., Zhang, D., and Paterson, A. H. (2015). Genetic analysis of rhizomatousness and its relationship with vegetative branching of recombinant inbred linesof Sorghum bicolor × S. propinquum. Am. J. Bot. 102, 718–724. doi: 10.3732/ajb.1500035
Koonrungsesomboon, N., Na-Bangchang, K., and Karbwang, J. (2014). Therapeutic potential and pharmacological activities of Atractylodes lancea (Thunb.) DC. Asian Pac. J. Trop. Med. 7, 421–428. doi: 10.1016/S1995-7645(14)60069-9
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Li, W. T., He, M., Wang, J., and Wang, Y. P. (2013). Zinc finger protein (ZFP) in plants-A review. Plant Omics 6, 474.
Liu, F., Zhang, X., Lu, C., Zeng, X., Li, Y., Fu, D., et al. (2015). Non-specific lipid transfer proteins in plants: presenting new advances and an integrated functional analysis. J. Exp. Bot. 66, 5663–5681. doi: 10.1093/jxb/erv313
Majer, C., and Hochholdinger, F. (2011). Defining the boundaries: structure and function of LOB domain proteins. Trends Plant Sci. 16, 47–52. doi: 10.1016/j.tplants.2010.09.009
Mano, J., Belles-Boix, E., Babiychuk, E., Inzé, D., Torii, Y., Hiraoka, E., et al. (2005). Protection against photooxidative injury of tobacco leaves by 2-alkenal reductase. Detoxication of lipid peroxide-derived reactive carbonyls. Plant Physiol. 139, 1773–1783. doi: 10.1104/pp.105.070391
Martinez-Garcia, J. F., Virgos-Soler, A., and Prat, S. (2002). Control of photoperiod regulated tuberization in potato by the Arabidopsis flowering-time gene constans. Proc. Natl. Acad. Sci. U.S.A. 99, 15211–15216. doi: 10.1073/pnas.222390599
Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). AP2/ERF family transcription factors in plant abiotic stress responses. BBA Gene Regul. Mech. 1819, 86–96. doi: 10.1016/j.bbagrm.2011.08.004
Mizrachi, E., Hefer, C. A., Ranik, M., Joubert, F., and Myburg, A. A. (2010). De novo assembled expressed gene catalog of a fast-growing Eucalyptus tree produced by Illumina mRNA-Seq. BMC genomics 11:681. doi: 10.1186/1471-2164-11-681
Mizutani, M., and Ohta, D. (2010). Diversification of P450 genes during land plant evolution. Annu. Rev. Plant Biol. 61, 291–315. doi: 10.1146/annurev-arplant-042809-112305
Nakano, T., Suzuki, K., Fujimura, T., and Shinshi, H. (2006). Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 140, 411–432. doi: 10.1104/pp.105.073783
Nakashima, K., Takasaki, H., Mizoi, J., Shinozaki, K., and Yamaguchi-Shinozaki, K. (2012). NAC transcription factors in plant abiotic stress responses. BBA Gene Regul. Mech. 1819, 97–103. doi: 10.1016/j.bbagrm.2011.10.005
Razin, S. V., Borunova, V. V., Maksimenko, O. G., and Kantidze, O. L. (2012). Cys2His2 zinc finger protein family: classification, functions, and major members. Biochemistry (Moscow) 77, 217–226. doi: 10.1134/S0006297912030017
Peres, L. E., Carvalho, R. F., Zsögön, A., Bermúdez-Zambrano, O. D., Robles, W. G., and Tavares, S. (2005). Grafting of tomato mutants onto potato rootstocks: an approach to study leaf-derived signaling on tuberization. Plant Sci. 169, 680–688. doi: 10.1016/j.plantsci.2005.05.017
Resch, M., Heilmann, J., Steigel, A., and Bauer, R. (2001). Further phenols and polyacetylenes from the rhizomes of Atractylodes lancea and their anti-inflammatory activity. Planta Med. 67, 437–442. doi: 10.1055/s-2001-15817
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Song, T., Liu, Z. B., Li, J. J., Zhu, Q. K., Tan, R., Chen, J. S., et al. (2015). Comparative transcriptome of rhizome and leaf in Ligusticum Chuanxiong. Plant Syst. Evol. 301, 2073–2085. doi: 10.1007/s00606-015-1211-4
Sui, C., Chen, M., Xu, J., Wei, J., Jin, Y., Xu, Y., et al. (2015). Comparison of root transcriptomes and expressions of genes involved in main medicinal secondary metabolites from Bupleurum chinense and Bupleurum scorzonerifolium, the two Chinese official Radix bupleuri source species. Physiol. Plant. 153, 230–242. doi: 10.1111/ppl.12254
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., Mortazavi, A., et al. (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Vreugdenhil, D., and Struik, P. C. (1989). An integrated view of the hormonal regulation of tuber formation in potato (Solanum tuberosum). Physiol. Plant. 75, 525–531. doi: 10.1111/j.1399-3054.1989.tb05619.x
Wakasa, Y., Oono, Y., Yazawa, T., Hayashi, S., Ozawa, K., Hayashi, S., et al. (2014). RNA sequencing-mediated transcriptome analysis of rice plants in endoplasmic reticulum stress conditions. BMC Plant Biol. 14:101. doi: 10.1186/1471-2229-14-101
Wang, Y., Dai, C. C., and Chen, Y. (2009). Antimicrobial activity of volatile oil from Atractylodes lancea against three species of endophytic fungi and seven species of exogenous fungi. Ying Yong Sheng Tai Xue Bao 20, 2778–2784.
Weitzel, C., and Simonsen, H. T. (2015). Cytochrome P450-enzymes involved in the biosynthesis of mono- and sesquiterpenes. Phytochem. Rev. 14, 7–24. doi: 10.1007/s11101-013-9280-x
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Xu, X., van Lammeren, A. A. M., Vermeer, E., and Vreugdenhil, D. (1998). The role of gibberellin, abscisic acid, and sucrose in the regulation of potato tuber formation in vitro. Plant Physiol. 117, 575–584. doi: 10.1104/pp.117.2.575
Yang, C. Q., Fang, X., Wu, X. M., Mao, Y. B., Wang, L. J., and Chen, X. Y. (2012). Transcriptional regulation of plant secondary metabolism. J. Integr. Plant. Biol. 54, 703–712. doi: 10.1111/j.1744-7909.2012.01161.x
Yang, M., Zhu, L., Pan, C., Xu, L., Liu, Y., Ke, W., et al. (2015). Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical lotus (Nelumbo nucifera). Sci. Rep. 5:13059. doi: 10.1038/srep13059
Yang, Y., Ma, C., Xu, Y., Wei, Q., Imtiaz, M., Lan, H., et al. (2014). A zinc finger protein regulates flowering time and abiotic stress tolerance in chrysanthemum by modulating gibberellin biosynthesis. Plant Cell 23, 2038–2054. doi: 10.1105/tpc.114.124867
Ye, J., Fang, L., Zheng, H., Zhang, Y., and Chen, J. (2006). WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 34, 293–297. doi: 10.1093/nar/gkl031
Yonekura-Sakakibara, K., and Hanada, K. (2011). An evolutionary view of functional diversity in family 1 glycosyltransferases. Plant J. 66, 182–193. doi: 10.1111/j.1365-313X.2011.04493.x
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R4. doi: 10.1186/gb-2010-11-2-r14
Yu, K., Fan, Q., Wang, Y., Wei, J., Ma, Q., Yu, D., et al. (2013). Function of leafy sepals in Paris polyphylla: photosynthate allocation and partitioning to the fruit and rhizome. Funct.Plant Biol. 40, 393–399. doi: 10.1071/FP12257
Yu, K., Wei, J., Ma, Q., Yu, D., and Li, J. (2009). Senescence of aerial parts is impeded by exogenous gibberellic acid in herbaceous perennial Paris polyphylla. J. Plant Physiol. 166, 819–830. doi: 10.1016/j.jplph.2008.11.002
Yu, Y., Liang, Y., Lv, M., Wu, J., Lu, G., and Cao, J. (2014). Genome-wide identification and characterization of polygalacturonase genes in Cucumis sativus and Citrullus lanatus. Plant Physiol. Biochem. 74, 263–275. doi: 10.1016/j.plaphy.2013.11.022
Zhang, G. H., Ma, C. H., Zhang, J. J., Chen, J. W., Tang, Q. Y., He, M. H., et al. (2015). Transcriptome analysis of Panax vietnamensis var. fuscidicus discovers putative ocotillol-type ginsenosides biosynthesis genes and genetic markers. BMC Genomics 16:159. doi: 10.1186/s12864-015-1332-8
Zhang, T., Zhao, X., Wang, W., Huang, L., Liu, X., Huang, L., et al. (2014). Deep transcriptome sequencing of rhizome and aerial-shoot in Sorghum propinquum. Plant Mol. Biol. 84, 315–327. doi: 10.1007/s11103-013-0135-z
Zhao, M., Wang, Q., Ouyang, Z., Han, B., Wang, W., Wei, Y., et al. (2014). Selective fraction of Atractylodes lancea (Thunb.) DC. and its growth inhibitory effect on human gastric cancer cells. Cytotechnology 66, 201–208. doi: 10.1007/s10616-013-9559-1
Keywords: differentially expressed gene, Illumina sequencing, rhizome formation, rhizomatous plants, tissue-specific genes, transcription factor
Citation: Huang Q, Huang X, Deng J, Liu H, Liu Y, Yu K and Huang B (2016) Differential Gene Expression between Leaf and Rhizome in Atractylodes lancea: A Comparative Transcriptome Analysis. Front. Plant Sci. 7:348. doi: 10.3389/fpls.2016.00348
Received: 08 November 2015; Accepted: 07 March 2016;
Published: 30 March 2016.
Edited by:
Daniel Pinero, Universidad Nacional Autónoma de México, MexicoReviewed by:
Rupesh Kailasrao Deshmukh, Laval University, CanadaRicardo A. Cabeza, Universidad de Chile, Chile
Copyright © 2016 Huang, Huang, Deng, Liu, Liu, Yu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kun Yu, yukun_hbtcm@163.com;
Bisheng Huang, hbsh1963@163.com
†These authors have contributed equally to this work.