 Jianhua Huang
Jianhua Huang Yushuai Wang
Yushuai Wang Wenwen Liu1
Wenwen Liu1 Tian Tang
Tian Tang- 1State Key Laboratory of Biocontrol, Guangdong Key Laboratory of Plant Resources School of Life Sciences, Key Laboratory of Biodiversity Dynamics and Conservation of Guangdong Higher Education Institutes, Sun Yat-sen University, Guangzhou, China
- 2South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, China
Long terminal repeat (LTR) retrotransposons constitute the majority of the content of angiosperm genomes, but their evolutionary dynamics remain poorly understood. Here, we report the isolation and characterization of a putative full-length (~9550 bp) Ty1/copia-like retrotransposon in Excoecaria agallocha and its evolution in Euphorbiaceae. The so-called EARE-1 is phylogenetically closely related to RIRE-1 from Oryza australiensis, and has proliferated recently (~7.19 Mya) in the E. agallocha genome. An RT-PCR analysis revealed substantial transcription of EARE-1 in all examined organs (leaves, staminate flowers, pistillate flowers, seeds, and roots) in unstressed E. agallocha plants and indications of elevated expression under stress. We conducted sequence analyses of 256 RT-RH fragments (~860 bp) of EARE-1 from 34 species representing four subfamilies of Euphorbiaceae that exist in China. EARE-1 copies from two Excoecaria species and Phyllanthus urinaria showed incongruent phylogeny with the host species and exhibited high sequence similarity to the host genes, suggesting a horizontal transfer from P. urinaria to the common ancestor of Excoecaria. However, SSAP analysis detected no new insertions of EARE-1 among full-sibling progeny plants of E. agallocha, despite considerable SSAP polymorphisms among half-siblings. EARE-1 is the first transcriptionally active Ty1/copia-like retrotransposon isolated from E. agallocha. Our results provide empirical evidence of the horizontal transfer of LTR retrotransposons in plants, and may suggest a significant role of post-transcriptional host control in the life cycles of transposable elements.
Introduction
Long terminal repeat (LTR) retrotransposons are ubiquitous transposable elements (TEs) that constitute the majority of the content of higher plant genomes (Lee and Kim, 2014). LTR retrotransposons are delimited by LTRs, which contain signals needed for transcription. Autonomous elements contain one open reading frame (ORF) encoding gag and pol proteins, which are necessary for retrotransposition in a “copy-and-paste” mechanism (Wicker et al., 2007). Following the organization of the pol gene, LTR retrotransposons are usually classified into two distinct groups: Ty1/copia and Ty3/gypsy (Kumar and Bennetzen, 1999). Although the dynamics of LTR retrotransposons are critical for the evolution of plant genome size, structure, and function (Liu et al., 2008; Zedek et al., 2010), their own evolution remains poorly understood, especially in non-model organisms with few genomic resources.
The retrotransposon life cycle has been shown to involve multiple steps that are inherently error-prone and mutagenic (Sabot and Schulman, 2006). This leads to numerous remnant elements that are incapable of transposition due to accumulated mutations. Studies have shown that the transposition of retrotransposons is also under strict host control by small RNA-mediated gene silencing at the transcriptional and post-transcriptional levels, which further limits their replication and transmission (Slotkin and Martienssen, 2007; Rigal and Mathieu, 2011). Despite the predominance of inactive elements, it has been reported that bursts of transposition can occur when the host is under genomic shocks such as hybridization, polyploidy, and environmental stresses (Fontdevila, 2005; Casacuberta and González, 2013) or when TEs invade a new “naïve” host genome through horizontal transfer (HT). HT allows TEs to escape host silencing and is thus considered to be an essential step of the TE life cycle, ensuring their long-term survival (Schaack et al., 2010). HT of LTR retrotransposons has been shown to be widespread and frequent in flowering plants (El Baidouri et al., 2014). High sequence similarity between TEs from distantly related taxa, tree incongruence between TEs and the host species and/or patchy distributions of TEs in phylogenies are three lines of evidence commonly used to infer HT (Daniels et al., 1990; Syvanen, 1994; Roulin et al., 2009).
Euphorbiaceae, or the spurge family, is a large, economically important family, with approximately 7500 species organized into 300 genera from five subfamilies: Euphorbioideae, Acalyphoideae, Crotonoideae, Phyllanthoideae, and Oldfieldioideae (Webster, 1994). HT of LTR retrotransposons has been reported in three prominent spurge plants, cassava (Manihot esculenta), caster bean (Ricinus communis), and Barbados nut (Jatropha curcas), for which whole-genome sequences are available (El Baidouri et al., 2014). However, the composition and evolution of TEs remain largely unknown for the vast majority of spurge species, especially those from the subfamily Euphorbioideae, which are characterized by the production of a poisonous milky latex with many medicinal uses (Ernst et al., 2015). Studies on TE dynamics are critical for the understanding of genome evolution and genetic diversity of Euphorbiaceae as a whole.
In this study, we isolated and characterized EARE-1, a Ty1/copia-like retrotransposon in milky mangrove, or blind-your-eye mangrove (Excoecaria agallocha), and surveyed the evolution of EARE-1 in four subfamilies of Euphorbiaceae that exist in China. Our results suggest that both HT and post-transcriptional control mechanisms of the host may play significant roles in the life cycle of EARE-1.
Materials and Methods
Plant Materials and DNA Extraction
Leaves, flowers, seeds and roots of E. agallocha were collected from Qi Ao Island of Zhuhai City, Guangdong Province, China. Seeds from the same fruit but different individuals of E. agallocha were collected and cultivated in a greenhouse until use. Leaves of the Euphorbiaceae species were collected from the Sun Yat-sen University campus and South China Botanical Garden. Voucher specimens were deposited in the Herbarium of Sun Yat-sen University (SYS). DNA was extracted from silica gel-dried leaves using the CTAB method (Doyle and Doyle, 1990).
Isolation and Characterization of EARE-1
Partial reverse transcriptase (RT) fragments of Ty1/copia-like retrotransposons in E. agallocha were amplified using degenerate primer pairs corresponding to the “KTAFLH/NG” and “LLYVDDM/V” conserved motifs (Voytas et al., 1992). PCR amplicons with the expected size (~270 bp) were cloned and sequenced. A total of 32 sequences were acquired. Among them, 25 sequences with a sequence identity of 90–100% were used to design primers for the subsequent inverse PCR. The inverse PCR was conducted as previously described (Syed and Flavell, 2006) except that BamHI, EcoRI, Hind III, or Kpn I (TAKARA) was used for the digestion of the E. agallocha genomic DNA. The sequence assembly was conducted using Lasergene (DNASTAR, Inc., Madison, WI, USA), requiring a minimum overlap of 150 bp and a nucleotide identity over 90%. To confirm the sequence assembly, long-range PCR (LA PCR) was conducted using LA Taq (TAKARA) following the manufacturer's instructions. The primers used for LA PCR are listed in Supplementary Table 1. The putative full sequence of EARE-1 was deposited in GenBank under the accession number KU198316. The structural features of EARE-1 were identified by manual inspection using BLAST (NCBI).
Copy Number Estimation of EARE-1
The copy number of EARE-1 in the E. agallocha genome was determined by real-time PCR (qPCR) using the standard curve method described previously (Liu et al., 2016). Primer pairs specific to the LTR, INT, and RT regions of EARE-1 (Supplementary Table 1) were used for qPCRs using a SYBR® Premix Ex Taq™ kit (TAKARA) on an ABI Prism 7900 HT Real-Time PCR System according to the manufacturer's instructions. DNA samples from three plants were used as three biological replicates. Two technical replicates were run for each sample. Standard curves were established based on a 10-fold dilution series of plasmid DNA, which contained the 5′ LTR of the RT-RH region of EARE-1. After adjusting the baseline cycles and calculating threshold values, the number of cycles halfway through the exponential phase (Ct value) was obtained and plotted against the logarithmic value of the standard sample copy numbers to estimate a linear regression line. The relative copy number of EARE-1 per pg of genomic DNA was then determined based on the linear regression function from the standard curve. The copy number of EARE-1 was calculated as copies per pg DNA.
Expression Analysis of EARE-1
The 5′ and 3′ cDNA ends of EARE-1 were obtained using a SMARTer® RACE cDNA Amplification Kit (Clontech) according to the manufacturer's instructions. The expression of EARE-1 in different organs (leaves, staminate flowers, pistillate flowers, seeds, and roots) of unstressed E. agallocha plants was determined by reverse transcription PCR (RT-PCR) with three biological replicates. RNA was extracted using a CTAB method as previously described (Yang et al., 2008). The purified total RNA was reverse transcribed into cDNA using the SuperScript® III First-Strand Synthesis System (Invitrogen™) following the manufacturer's instructions. A no-reverse-transcriptase RT reaction was used as the negative control. The PCR was performed using PrimeSTAR® HS DNA Polymerase (TAKARA) with primer pairs specific to the LTR, gag, and RT regions of EARE-1. The primers used are shown in Supplementary Table 1.
As for the analysis of stress-responsive expression of EARE-1, small pieces (~1 cm2) of fresh leaves were immersed in 3 mM MES buffer containing 1 mM salicylic acid (SA), 50 μM 1-naphthylacetic acid (NAA), 200 mM NaCl, or 20% PEG 6000 (drought) in petri dishes at room temperature for 8 h. For the wounding and cold treatment, fresh leaves were narrowly cut vertically, or immersed in MES buffer at 4°C. Total RNA was extracted from treated and untreated (control) leaves, purified, and reverse transcribed as described above. Quantitative real-time RT-PCR (qRT-PCR) of EARE-1 transcripts was performed using a SYBR® Premix Ex Taq™ kit (TAKARA) with primers specific to the RT region of EARE-1 (Supplementary Table 1). The relative levels of EARE-1 were calculated using the 2∧−ΔΔCT method (Livak and Schmittgen, 2001). β-actin was used as an internal control. A two-tailed t-test was used to determine the significance of the differences in EARE-1 expression between treated and untreated samples.
Sequence and Phylogenetic Analyses of EARE-1
Multiple alignments of the RT amino acid sequences of EARE-1 and known retrotransposons were generated by MUSCLE (Edgar, 2004). The optimal amino acid substitution model was calculated by the ModelGenerator v0.85 (Keane et al., 2006), and the “LG+G” model was selected to construct a maximum-likelihood (ML) phylogenetic tree using PhyML 3.0 software (Guindon et al., 2010) with 1000 bootstrap replicates. TY3B from the gypsy superfamily was used as an outgroup. The retrotransposon sequences used in the phylogenetic analysis included: Tork4 (EU105455.1), Rider (ABO36622.1), Ta1-3 (X13291), Tnt1-94 (X13777), Tto1 (D83003), Sto-4 (AF082133), SORE-1 (AB370254), RIRE1 (D85597), BARE-1 (Z17327), Angela (AY485644.1), maximus (TREP1654), SIRE1-4 (AY205608.1), HORPIA (AY6615581), leojyg (AY268139.1), Tgmr (U96748), Retrofit (AH005614), TY1B (Z35766.1), Bianca (AF521177.1), and TY3B (CAA97115.1).
Distribution and Sequence Analyses of EARE-1 in Euphorbiaceae
The PCR amplification of the RT-RH fragments of EARE-1 and the host gene rbcL in the Euphorbiaceae species was conducted using PrimeSTAR® HS DNA Polymerase (TAKARA) with primers listed in Supplementary Table 1. The PCR products were visualized on a 1.8% agarose gel, cloned, and sequenced. The sequences obtained in this study were deposited in GenBank under the accession numbers KU242753–KU243006 for the RT-RH fragments of EARE-1 and KU243007–KU243040 for rbcL. Additional matK sequences were retrieved from GenBank: E. agallocha (KM255088.1), E. cochinchinensis (AB233781.1), S. discolor (HQ415366.1), P. urinaria (JX661958.1), M. esculenta (AB233776.1), R. communis (AB233767.1), and Pedilanthus tithymaloides (AB268063.1). The sequences of each gene were aligned by codon using MUSCLE (Edgar, 2004) with a manual check. The ML tree was constructed using Fastree (Price et al., 2009) with default parameters and displayed with Figtree v1.4.2. The number of synonymous substitutions per synonymous site (Ks) and the number of nonsynonymous substitutions per nonsynonymous site (Ka) were calculated using the Nei and Gojobori model (Nei and Gojobori, 1986) as implemented in MEGA 6 (Tamura et al., 2013). Alignment gaps were deleted and stop codons were treated as missing data. The codon bias index (CBI) was calculated by DNAsp v5 (Librado and Rozas, 2009).
SSAP Analysis
The SSAP analysis was performed as previously described (Syed and Flavell, 2006; Liu et al., 2016) with some modifications. Briefly, genomic DNA was digested with MseI and EcoRI (NEB) and ligated with adaptors using T4 DNA ligase (NEB). The adaptor-ligated DNA was then used for pre-amplification with primers with no selective nucleotides (E0 and M0) using Ex Taq polymerase (TAKARA). The pre-amplification product was diluted 50-fold for the selective amplification. The fluorescent PCR for selective amplification was performed with primers containing three selective nucleotides and a specific primer (Supplementary Table 2) labeled with FAM. The PCR products of the selective amplification were resolved by capillary electrophoresis and analyzed using Genemarker (Softgenetics, State College, PA).
Results
Isolation and Characterization of EARE-1 in E. agallocha
The isolation of a putative full-length Ty1/copia-like retrotransposon in E. agallocha was initiated by the amplification of partial RT fragments (Voytas et al., 1992), followed by genome walking and sequence assembly as previously described (Lin et al., 2013). The obtained putative complete retroelement as confirmed by LA PCR was 9555 bp in length and was named EARE-1 (E. agallocha retrotransposable element). EARE-1 included a single ORF of 3920 bp with one stop codon and displayed the gal-pol domain order of Ty1/copia-like retroelements: gag, protease (PR), integrase (INT), reverse transcriptase (RT), and RNase H (Figure 1A). The 5′ and 3′ LTRs of EARE-1 were 1900 bp and 1916 bp, respectively, with 90% sequence identity, and both had a 5′-TG…CA-3′ structure (Figure 1A). 5′ RACE determined the transcription initiation site (TSS) to be position 918 bp of the 5′ LTR while 3′ RACE located the 3′ end of the EARE-1 transcripts to position 690 bp of the 3′ LTR. A putative primer-binding site (PBS) of EARE-1 was located 1 bp downstream of its 5′ LTR and contained a stretch of 5′-TGGTATCAGAGCCT-3′ sequence complementary to the 3′ of tRNAMet (Figure 1A). A putative polypurine tract (PPT), which is required for second-strand DNA synthesis, contained a conserved 5′-TAGTGGGAGAT-3′ sequence just upstream of the 3′ LTR of EARE-1 (Figure 1A). Using the PlantCARE database (Lescot et al., 2002), about 40 TATA-boxes or CAAT-boxes that are common in promoter and enhancer regions and various cis-regulatory motifs involved in light, stress, and phytohormone responsiveness and developmental processes were detected in the 5′ LTR of EARE-1 (Table 1).
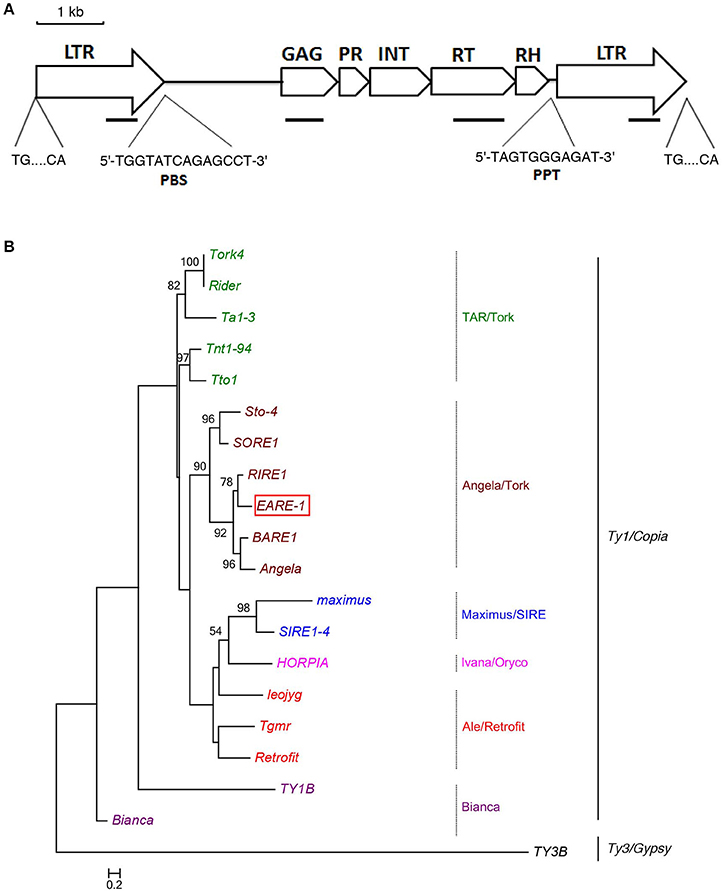
Figure 1. Structural characteristics of EARE-1 from E. agallocha. (A) Schematic presentation of EARE-1. LTRs and coding regions are indicated with arrows and boxes, respectively. The sequences of PBS and PPT are underlined. Lines indicate the locations of PCR products that were used to estimate the copy number of EARE-1 in E. agallocha. (B) ML tree of EARE-1 and known LTR retrotransposons based on the RT amino acid sequences. Numbers above branches indicate bootstrap values >50% based on 1000 replicates. The retrotransposon sequences used were: EARE-1 (GenBank accession number: KU198316), Tork4 (EU105455.1), Rider (ABO36622.1), Ta1-3 (X13291), Tnt1-94 (X13777), Tto1 (D83003), Sto-4 (AF082133), SORE-1 (AB370254), RIRE1 (D85597), BARE-1 (Z17327), Angela (AY485644.1), maximus (TREP1654), SIRE1-4 (AY205608.1), HORPIA (AY6615581), leojyg (AY268139.1), Tgmr (U96748), Retrofit (AH005614), TY1B (Z35766.1), Bianca (AF521177.1), and TY3B (CAA97115.1).
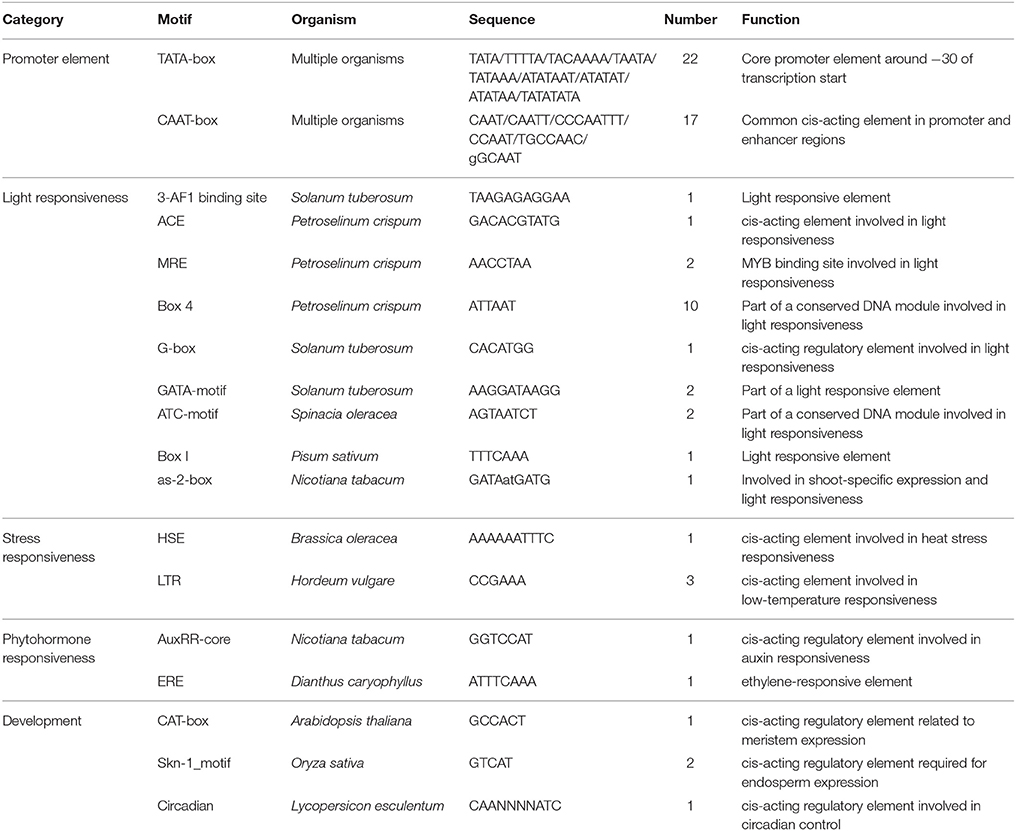
Table 1. Types and numbers of putative cis-regulatory elements in the 5′ LTR of EARE-1.
The Evolutionary Relationship between EARE-1 and Major Ty1/Copia Lineages
To identify the evolutionary relationship between EARE-1 and the known copia retrotransposons, we constructed an ML phylogenetic tree based on their deduced RT amino acid sequences, using TY3B from the Ty3/gypsy superfamily as an outgroup. As shown in Figure 1B, EARE-1 was most similar to RIRE-1 from the wild rice Oryza australiensis (Noma et al., 1997) and also similar (slightly less so) to the clade of BARE-1 (Manninen and Schulman, 1993) and Angela (Yan et al., 2004). This clustering is supported with high bootstrap values, placing EARE-1 in the Angela/Tork superfamily (Figure 1B). A homology matrix comparison detected high similarity between the deduced amino acid sequences of EARE-1 and RIRE-1: 45, 49, 70, 68, and 68% for gag, PR, IN, RT, and RNase H, respectively, with an overall identity of 61% across 1317 amino acids. Therefore, EARE-1 is closely related to RIRE-1, which is present in O. australiensis in an extraordinary number of copies (Noma et al., 1997).
Low Ratio of LTR to Internal Region of EARE-1 in E. agallocha
To estimate the copy number of EARE-1 in the E. agallocha genome, we conducted qPCR analyses using primers specific to the partial fragments of LTR, IN, and RT. The standard curves, which were generated using a 10-fold dilution series of plasmid DNA containing EARE-1, indicated high amplification efficiency for each primer pair (Figures 2A–C). Based on the standard curves, the estimated EARE-1 copy number per pg DNA was 216 ± 19, 100 ± 14, and 87 ± 12 based on the LTR region and the ORF domains INT and RT, respectively (Figure 2D). The average terminal-to-internal ratio of EARE-1 was 2.16 (LTR: INT) or 2.48 (LTR: RT), indicating few copies of solo LTRs relative to the full length. These numbers are consistent with, and most simply explained by, the assumption that the majority of copies of EARE-1 are intact.
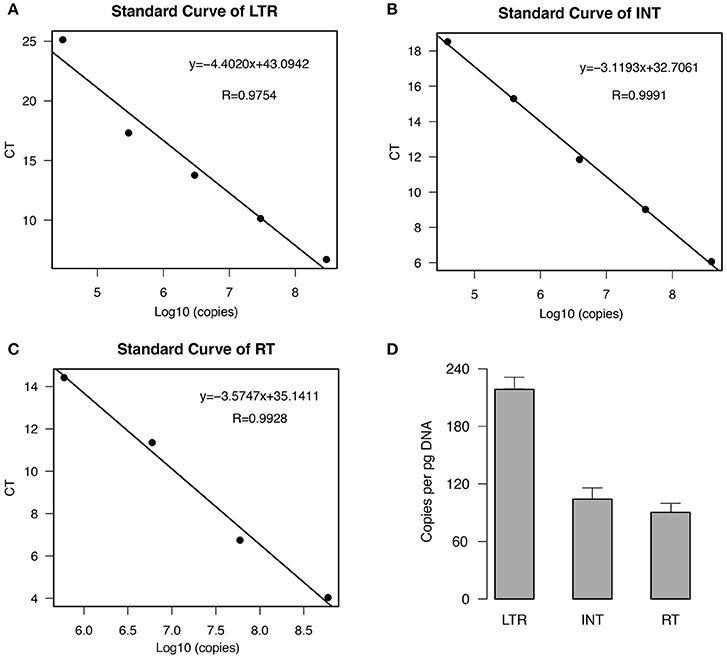
Figure 2. Estimation of the copy number of EARE-1 in E. agallocha by qPCR. (A–C) qPCR standard curves from a serial dilution of plasmid DNAs harboring the cloned EARE-1 and amplification with LTR/INT/RT primers. The CT values were plotted against the logarithmic value of the standard sample copy numbers. The linear regression function and the correlation coefficient (R) are given for each standard curve. (D) Estimation of the EARE-1 copy number based on the LTR, INT, and RT regions. Error bars indicate the standard deviation (n = 3).
Expression of EARE-1 in Different Organs and under Stress Treatments in E. agallocha
The abundant cis-regulatory elements found in the LTRs (Table 1) suggested that EARE-1 may be transcriptionally active and responsive to different internal and external stimuli. Indeed, we found that EARE-1 was constitutively and ubiquitously expressed at a considerable level in roots, staminate flowers, pistillate flowers, leaves, and seeds of E. agallocha using RT-PCR. All three primers specific for the LTR, gag and RT regions amplified amplicons with the expected size (Figure 3A), and these were further confirmed by cloning sequencing. The sequence similarity ranged from 94 to 98% for particular primer pairs, indicating a diverse pool of EARE-1 transcripts in E. agallocha. Using qRT-PCR, we further examined the expression of EARE-1 in leaves of E. agallocha under various stress treatments, including 50 μM 1-naphthaleneacetic acid (NAA), 1 mM salicylic acid (SA), 200 mM NaCl, 20% PEG6000, wounding and cold (see Materials and Methods). EARE-1 was slightly upregulated in all stress treatments except for wounding, whereas the upregulation was significant only for drought (two-tailed t-test, P < 0.01) and NAA due to considerable expression variation (two-tailed t-test, P < 0.05, Figure 3B). These data may indicate constitution expression of EARE-1 in E. agallocha and its elevated expression under stress. However, one needs to be cautious about the stress responsiveness of EARE-1 as only one internal gene β-actin was used in the qRT-PCR analysis.
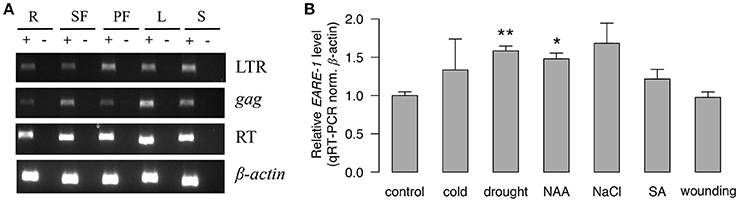
Figure 3. The transcriptional activity of EARE-1 in E. agallocha. (A) Expression of EARE-1 in different organs of E. agallocha. R, root; SF, staminate flower; PF, pistillate flower; L, leaf and S, seed. β-actin was used as an internal control. (+): reactions with reverse transcriptase; (−): reactions without reverse transcriptase. (B) Stress-responsive expression of EARE-1 determined by qRT-PCR. The data shown are the relative expression levels of EARE-1 in the stress-treated vs. untreated (control) leaves of E. agallocha normalized using the expression levels of β-actin. Cold, 4°C treatment; drought, 20% PEG 6000; NAA, 50 μM 1-naphthylacetic acid; NaCl, 200 mM NaCl; SA, 1 mM salicylic acid and wounding. Three biological replicates were conducted for each treatment. The significance determined by a two-tailed t-test is shown as *, P < 0.05; **, P < 0.01.
The Distribution of EARE-1 in Euphorbiaceae and Phylogenetic Incongruence between EARE-1 and Host Species
To understand the evolutionary history of EARE-1, we sequenced a partial RT-RNaseH fragment (~860 bp) of the EARE-1 homologs in 34 species from 31 genera of Euphorbiaceae that represent the four subfamilies that exist in China (Table 2). Amplicons with the expected size were successfully obtained in 27 out of 34 species. Eight to ten clones were randomly chosen and sequenced for each amplicon, resulting in a total of 256 sequences. Two sequences with nucleotide identity <50% with EARE-1 from E. agallocha were removed from further analyses. Species that failed to produce EARE-1 homologs mainly came from Phyllanthoideae, with an additional two from Crotonoideae or Acalyphoideae (Table 2). The absence of EARE-1 in J. curcas was confirmed by BLAST screening against the spurge species with whole-genome sequences.
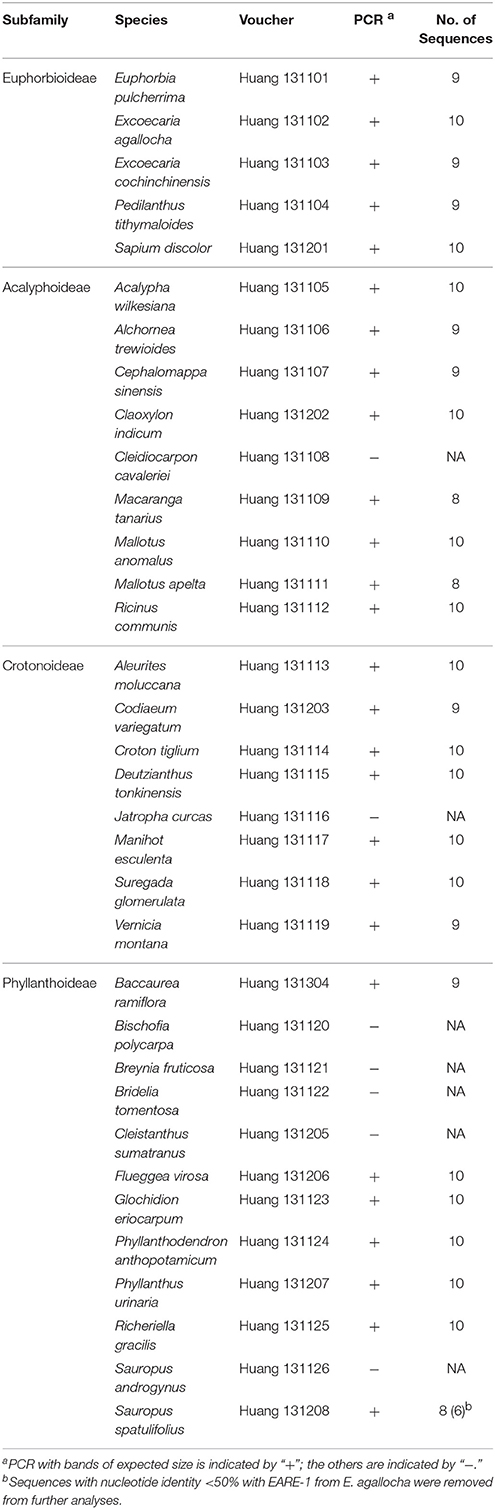
Table 2. Summary of the distribution of EARE-1 in Euphorbiaceae.
The distribution of EARE-1 in the taxa studied is shown in an ML phylogenetic tree based on host ribulose-1,5-bisphosphate carboxylase/oxygenase (rbcL), using Erodium stephanianum as an outgroup (Figure 4A). Taxa from Euphorbioideae and Acalyphoideae constituted sister groups and then clustered with those from Crotonoideae, while taxa from Phyllanthoideae diverged distantly from the others. The widespread distribution of EARE-1 in Euphorbioideae suggests that EARE-1 entered the common ancestor of Euphorbioideae, was transmitted vertically and underwent independent losses in specific lineages.
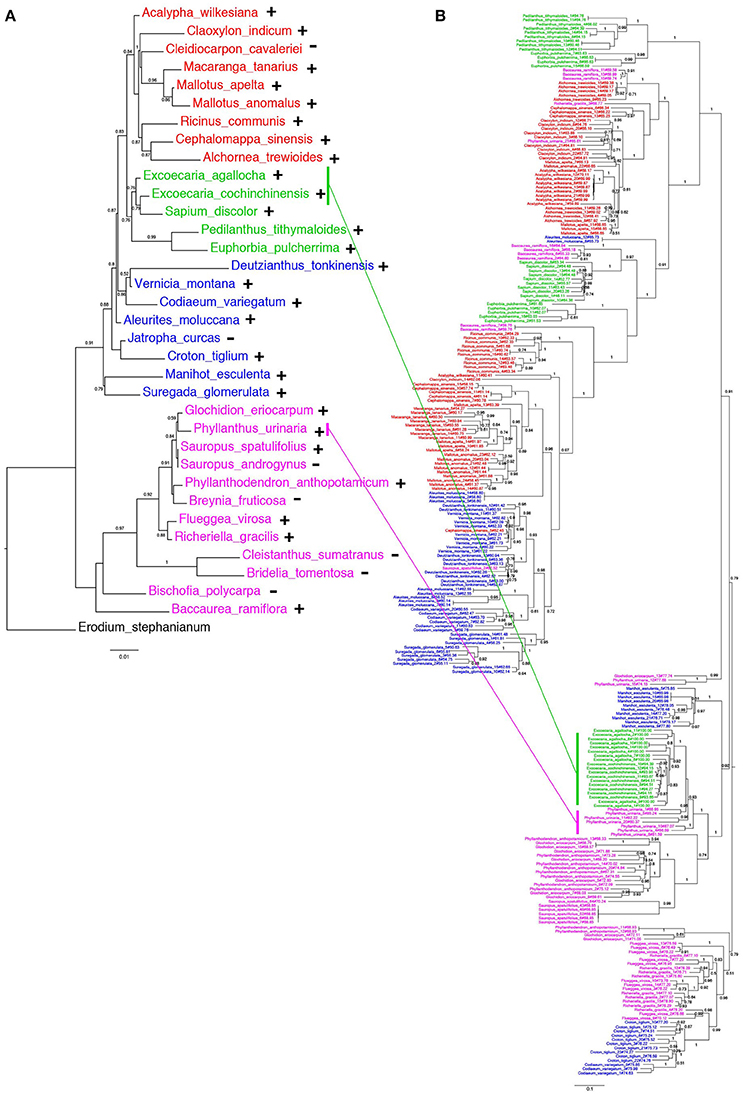
Figure 4. Comparison of species and EARE-1 phylogenetic histories. (A) ML tree of the host species Euphorbiaceae based on the partial rbcL sequences. The presence or absence of EARE-1 in each species is labeled with “+” or “−.” (B) ML tree of EARE-1 based on the sequences of the partial RT-RH fragments. The Shimodaira-Hasegawa support values of 0.5 and greater are indicated above the branches. Names of species from the Acalyphoideae, Euphorbioideae, Crotonoideae, Phyllanthoideae subfamilies of Euphorbiaceae are indicated in red, green, blue, and pink, respectively. The sequences of EARE-1 likely to present a HT event in the gene tree and their corresponding host species in the species tree are connected with straight lines.
In contrast, while individual copies isolated from each of the species mainly formed monophyletic clades in the ML tree of EARE-1, the relationships among these clades were not fully congruent with those that connect their host species (Figure 4A vs. Figure 4B). This is most evident when the EARE-1 sequences from species of distantly related subfamilies of Euphorbiaceae were clustered together (Figure 4B). The phylogenetic incongruence can be explained by either we compared paralogous sequences of EARE-1, which were missed in certain lineages due to the limited number of sequenced clones, or HT of EARE-1 might have occurred between distantly related Euphorbiaceae species.
High Sequence Similarity of EARE-1 between Excoecaria Species and the Divergent P. urinaria
As shown in Figure 4, the EARE-1 sequences from two Exocaria species (E. agallocha and E. cochinchinensis) of Euphorbioideae are surprisingly similar to those from Phyllanthus urinaria of Phyllanthoideae. This promoted us to investigate the possibility of HT. HT can be inferred whenever the divergence among TE sequences is significantly lower than that observed for the host genes, assuming similar or higher levels of selective constraints on the latter. To this end, two chloroplast genes, rbcL and matK, were used for the comparisons of EARE-1 and host gene divergences between pairs of taxa. The number of synonymous substitutions per synonymous site (Ks) between Excoecaria and P. urinaria was 0.296 ± 0.032 for EARE-1, 0.207 ± 0.042 for rbcl and 0.272 ± 0.045 for matK, indicating comparable divergence between EARE-1 and the two host genes (Figure 5). This is quite striking since chloroplast genes are under strong selective constraint, while TEs usually degenerate quickly and thus lack constraint. In contrast, EARE-1 exhibited a much greater Ks than the host genes when comparing Excoecaria with species from other subfamilies of Euphorbiaceae: 6.9–7.9 times greater between Excoecaria and M. esculenta, Crotonoideae, and 27.2–36.4 times greater between Excoecaria and R. communis, Acalyphoideae (Figure 5). In addition, the divergence of EARE-1 between Excoecaria and P. urinaria was smaller than that between Excoecaria and other species within the Euphorbioideae subfamily, while the divergence of host genes showed the opposite trend (Figure 5).
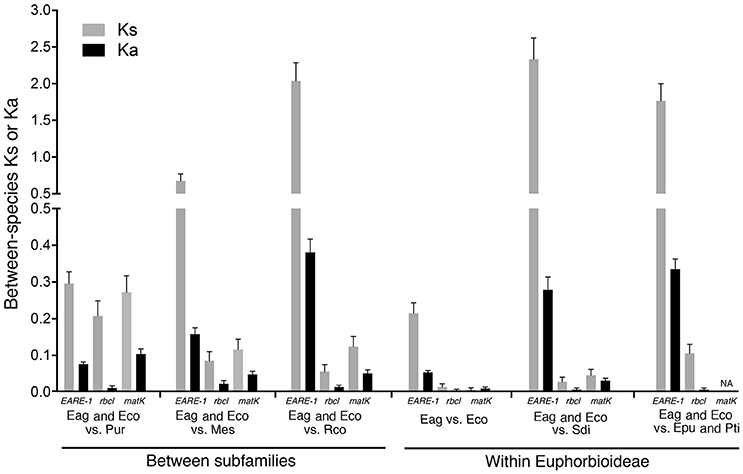
Figure 5. Mean numbers of substitutions per synonymous site (Ks) and substitutions per nonsynonymous site (Ka) for EARE-1 copies, rbcl, and matk between Excoecaria and species from different subfamilies of Euphorbioideae. Eag, Excoecaria agallocha; Eco, E. cochinchinensis; Epu, Euphorbia pulcherrima; Mes, Manihot esculenta; Pti, Pedilanthus tithymaloides; Pur, Phyllanthus urinaria; Sdi, Sapium discolor; Rco, Ricinus communis.
The observed low synonymous divergence of EARE-1 could be caused by purifying selection for codon usage bias. To rule out this possibility, we calculated the codon bias index (CBI) for both EARE-1 and host genes in each species of E. agallocha, E. cochinchinensis, and P. urinaria. A CBI of 0 indicates no codon bias, and a CBI of 1 indicates the most severe codon bias. The CBI values of EARE-1 (0.33 ± 0.02) were less than that of rbcL (CBI = 0.45) or matK (CBI = 0.43), indicating that purifying selection cannot explain the low divergence of EARE-1 between Excoecaria species and P. urinaria. Based on these results, we concluded that a horizontal transfer occurred between P. urinaria and Excoecaria species. The date at which the HT might have occurred can be estimated by calculating the pairwise divergence between all individual EARE-1 copies from E. agallocha and E. cochinchinensis and the ancestral founder copy of P. urinaria (Ks = 0.296 ± 0.032). The result corresponds to an HT occurring at 11.38 Mya based on a molecular clock of 1.3 × 10−8 per synonymous substitutions (Ma and Bennetzen, 2004). The small pairwise Ks of EARE-1 sequences within Excoecaria species (Ks = 0.187 ± 0.022), corresponding to a burst time of 7.19 Mya, suggest a scenario of recent amplification after the HT.
The Transpositional Activity of EARE-1 in E. agallocha
We next looked at whether EARE-1 is still capable of retrotransposition after its invasion into the E. agallocha genome using sequence-specific amplification polymorphism (SSAP) analysis. E. agallocha are dioecious trees with three-lobed fruit capsules, where each portion contains a seed. Every three seeds from a fruit produce full-sibling progeny plants, while seeds from different fruits of an E. agallocha tree produce half-sibling progeny plants. A total of 153 offspring from 51 fruits of four maternal plants of E. agallocha were used for the SSAP analysis. Six primer combinations yielded 268–323 PCR bands for each plant (Supplementary Table 3). Full-sibling plants displayed identical SSAP banding patterns, though half-sibling plants exhibited considerable SSAP polymorphisms, with the percentage of polymorphic bands (P%) ranging from 12.31 to 19.50% (Supplementary Table 3). These results indicate that the transpositional activity of EARE-1, if any, is too low to be reliably detected using the current sampling of E. agallocha.
Discussion
We have isolated the first full-length Ty1/copia retrotransposon from the milky mangrove E. agallocha. Despite its ancient origin, EARE-1 is transcriptionally active and has only recently reached hundreds of copies in the genome of E. agallocha. Most importantly, phylogenetic incongruence and high sequence similarity provide strong empirical evidence that HT has played a critical role in the evolution of EARE-1 in E. agallocha. The observed low ratio of LTR to the internal region (2.16–2.48) of EARE-1 indicates either that the efficiency of removal of EARE-1 by unequal intra-strand homologous recombination (UHR) is low or that a recent burst of transposition has occurred that outpaces DNA removal by recombination. We considered the latter scenario more plausible, since EARE-1 has long LTRs approximately 1900 bp in length that should be readily subject to UHR.
HT has been shown to preferentially occur in several TE types (Schaack et al., 2010). Consistent with this notion, we found that EARE-1 is phylogenetically close to RIRE1, which is extremely successful (approximately 30,000 complete copies and 10,000 solo-LTRs) in the wild rice O. australiensis (Noma et al., 1997; Piegu et al., 2006). After the RIRE1 burst in O. australiensis, multiple HTs of RIRE1 occurred from O. australiensis into other reproductively isolated Oryza species (Roulin et al., 2008). Unlike RIRE1, the bursts of EARE-1 in E. agallocha and E. cochinchinensis probably occurred after the HT from P. urinaria into the common ancestor of Excoecaria. The estimated HT time of 11.38 Mya, predating the estimated divergence time of E. agallocha and E. cochinchinensis (5.38 Mya) using a molecular clock of 1.3 × 10−9 for rbcL (Zurawski and Clegg, 1987), supports this speculation. The fact that the cluster of all horizontally transferred EARE-1 copies from E. agallocha and E. cochinchinensis is included in the larger cluster of copies from the Phyllanthoideae subspecies suggests that E. agallocha and E. cochinchinensis are the recipient species of the HT event. Moreover, the extensive incongruence between the phylogeny of EARE-1 and that of various host species, such as Baccaurea ramiflora, Alchornea trewioides, Cephalomappa sinensis, and Vernicia montana (Figure 4), suggests that HTs of EARE-1 may be frequent within Euphorbiaceae. A comprehensive sampling of more taxa and further sequence analyses of longer or full-length segments of EARE-1 copies may help to validate this hypothesis.
An active TE copy can experience rapid amplification once it invades into the naive genome through HT. A well-known example is Rider, which quickly amplified to approximately 2000 copies after it transferred into the tomato genome from Arabidopsis (Cheng et al., 2009; Jiang et al., 2009). In this study, EARE-1 had reached about 100 copies pre pg DNA in E. agallocha (Figure 2) and most of the copies of EARE-1 are likely to be intact. Considering E. agallocha has a large genome (2n = 130, Das et al., 2011), the copy number of EARE-1 in E. agallocha would greatly exceeded that in cassava (4 copies) or caster bean (59 copies) estimated by LTR_finder. Interestingly, our results indicate that the life cycle of EARE-1 is firmly controlled at the post-transcriptional level. This is evident from the contrast between the substantial constitutive expression of EARE-1 in all analyzed organs of E. agallocha—owing to the numerous cis promoter and enhancer elements in its LTRs—and its transpositional rate, which was too low to be detected among full-sibling progeny plants. High transcriptional but low transpositional activities were also observed in sunflower hybrid species, in which the proliferation of retrotransposons led to genome expansion (Ungerer et al., 2006; Vukich et al., 2009). Considering the stress-responsive expression of EARE-1 and the stressful habitats (mangrove swamps) of E. agallocha, it would be interesting to study how genomic shock may be associated with HTs in shaping the life cycle of EARE-1 in E. agallocha. A recent study of mangrove retrotransposons may suggest such an association (Liu et al., 2016).
It is unclear how the transfer may have occurred. Considering that Excoecaria plants such as E. agallocha are well-protected by chemical defenses (Zou et al., 2006), bacteria, fungi, or sapsucking insects that are often believed to be the vectors of HT (Won and Renner, 2003; Fortune et al., 2008; Sun et al., 2013) are less likely to mediate DNA transfer into these species. A possible vehicle may be viruses. In animals, poxviruses, a family of double-stranded DNA viruses, may have acted as vectors for the HT of a SINE element from reptiles to mammals (Piskurek and Okada, 2007). Another group of dsDNA viruses, polydnaviruses (PDVs), are thought to mediate the HT of mariner-like elements between a parasitoid braconid wasp and its lepidopteran host (Yoshiyama et al., 2001; Dupuy et al., 2011). EARE-1 is an attractive candidate for testing whether similar mechanisms of horizontal transposon transfer exist in plants.
In conclusion, EARE-1 is the first transcriptionally active Ty1/copia-like retrotransposon isolated from E. agallocha. Both horizontal transfer and post-transcriptional host control might have played significant roles in the life cycle of EARE-1. These mechanisms may be important in understanding the evolution of TEs and TE-driven genomic evolution.
Author Contributions
JH, YW, and TT planned and designed the research; JH, YW, WL, and XS conducted the experiments and data analyses; QF and SJ provided the plant materials; JH, YW, and TT wrote the manuscript. All authors have read and approved the manuscript.
Funding
This study was funded by the National Science Foundation of China (31170308, 91231117, and 31301010), the Science Foundation for Outstanding Young Teachers in Higher Education of Guangdong (Yq2013005), the Fundamental Research Funds for the Central Universities (16lgjc75) and the Chang Hungta Science Foundation of Sun Yat-sen University.
Conflict of Interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00045/full#supplementary-material
References
Casacuberta, E., and González, J. (2013). The impact of transposable elements in environmental adaptation. Mol. Ecol. 22, 1503–1517. doi: 10.1111/mec.12170
Cheng, X., Zhang, D., Cheng, Z., Keller, B., and Ling, H. Q. (2009). A new family of Ty1-copia-like retrotransposons originated in the tomato genome by a recent horizontal transfer event. Genetics 181, 1183–1193. doi: 10.1534/genetics.108.099150
Daniels, S. B., Peterson, K. R., Strausbaugh, L. D., Kidwell, M. G., and Chovnick, A. (1990). Evidence for horizontal transmission of the P transposable element between Drosophila species. Genetics 124, 339–355.
Das, A. B., Jena, S., Pradhan, C., and Chand, P. K. (2011). Genetic variability among male populations of a minor mangrove Excoecaria agallocha L. as evident by chromosome morphology and DNA markers. Nucleus 54, 39–47. doi: 10.1007/s13237-011-0027-z
Dupuy, C., Periquet, G., Serbielle, C., Bézier, A., Louis, F., Drezen, J. M., et al. (2011). Transfer of a chromosomal Maverick to endogenous bracovirus in a parasitoid wasp. Genetica 139, 489–496. doi: 10.1007/s10709-011-9569-x
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
El Baidouri, M., Carpentier, M. C., Cooke, R., Gao, D., Lasserre, E., Llauro, C., et al. (2014). Widespread and frequent horizontal transfers of transposable elements in plants. Genome Res. 24, 831–838. doi: 10.1101/gr.164400.113
Ernst, M., Grace, O. M., Saslis-Lagoudakis, C. H., Nilsson, N., Simonsen, H. T., and Rønsted, N. (2015). Global medicinal uses of Euphorbia L. (Euphorbiaceae). J. Ethnopharmacol. 176, 90–101. doi: 10.1016/j.jep.2015.10.025
Fontdevila, A. (2005). Hybrid genome evolution by transposition. Cytogenet. Genome Res. 110, 49–55. doi: 10.1159/000084937
Fortune, S., Sinclair, J., and Hawton, K. (2008). Help-seeking before and after episodes of self-harm: a descriptive study in school pupils in England. BMC Public Health 8:369. doi: 10.1186/1471-2458-8-369
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., and Hordijk, W. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Jiang, N., Gao, D., Xiao, H., and van der Knaap, E. (2009). Genome organization of the tomato sun locus and characterization of the unusual retrotransposon Rider. Plant J. 60, 181–193. doi: 10.1111/j.1365-313X.2009.03946.x
Keane, T. M., Creevey, C. J., Pentony, M. M., Naughton, T. J., and McLnerney, J. O. (2006). Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 6:29. doi: 10.1186/1471-2148-6-29
Kumar, A., and Bennetzen, J. L. (1999). Plant retrotransposons. Annu. Rev. Genet. 33, 479–532. doi: 10.1146/annurev.genet.33.1.479
Lee, S. I., and Kim, N. S. (2014). Transposable elements and genome size variations in plants. Genomics Inform. 12, 87–97. doi: 10.5808/GI.2014.12.3.87
Lescot, M., Déhais, P., Thijs, G., Marchal, K., Moreau, Y., Van de Peer, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lin, X. F. P., Huang, J., Zhang, T., Shia, S., and Tang, T. (2013). Isolation and characterization of RARE-1, a Ty1/copia-like retrotransposon domesticated in the genome of Rhizophora apiculata. Biochem. Syst. Ecol. 50, 248–257. doi: 10.1016/j.bse.2013.03.039
Liu, W., Wang, Y., Shen, X., and Tang, T. (2016). Isolation, characterization, and marker utility of KCRE1, a transcriptionally active Ty1/copia retrotransposon from Kandelia candel. Mol. Genet. Genomics. 291, 2031–2042. doi: 10.1007/s00438-016-1237-5
Liu, Z., Yue, W., Li, D., Wang, R. R., Kong, X., Lu, K., et al. (2008). Structure and dynamics of retrotransposons at wheat centromeres and pericentromeres. Chromosoma 117, 445–456. doi: 10.1007/s00412-008-0161-9
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Ma, J., and Bennetzen, J. L. (2004). Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl. Acad. Sci. U.S.A. 101, 12404–12410. doi: 10.1073/pnas.0403715101
Manninen, I., and Schulman, A. H. (1993). BARE-1, a copia-like retroelement in barley (Hordeum vulgare L.). Plant Mol. Biol. 22, 829–846. doi: 10.1007/BF00027369
Nei, M., and Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426.
Noma, K., Nakajima, R., Ohtsubo, H., and Ohtsubo, E. (1997). RIRE1, a retrotransposon from wild rice Oryza australiensis. Genes Genet. Syst. 72, 131–140. doi: 10.1266/ggs.72.131
Piegu, B., Guyot, R., Picault, N., Roulin, A., Sanyal, A., Kim, H., et al. (2006). Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 16, 1262–1269. doi: 10.1101/gr.5290206
Piskurek, O., and Okada, N. (2007). Poxviruses as possible vectors for horizontal transfer of retroposons from reptiles to mammals. Proc. Natl. Acad. Sci. U.S.A. 104, 12046–12051. doi: 10.1073/pnas.0700531104
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Rigal, M., and Mathieu, O. (2011). A “mille-feuille” of silencing: epigenetic control of transposable elements. Biochim. Biophys. Acta 1809, 452–458. doi: 10.1016/j.bbagrm.2011.04.001
Roulin, A., Piegu, B., Fortune, P. M., Sabot, F., D'Hont, A., Manicacci, D., et al. (2009). Whole genome surveys of rice, maize and sorghum reveal multiple horizontal transfers of the LTR-retrotransposon Route66 in Poaceae. BMC Evol. Biol. 9:58. doi: 10.1186/1471-2148-9-58
Roulin, A., Piegu, B., Wing, R. A., and Panaud, O. (2008). Evidence of multiple horizontal transfers of the long terminal repeat retrotransposon RIRE1 within the genus Oryza. Plant J. 53, 950–959. doi: 10.1111/j.1365-313X.2007.03388.x
Sabot, F., and Schulman, A. H. (2006). Parasitism and the retrotransposon life cycle in plants: a hitchhiker's guide to the genome. Heredity (Edinb). 97, 381–388. doi: 10.1038/sj.hdy.6800903
Schaack, S., Gilbert, C., and Feschotte, C. (2010). Promiscuous DNA: horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. (Amst). 25, 537–546. doi: 10.1016/j.tree.2010.06.001
Slotkin, R. K., and Martienssen, R. (2007). Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–285. doi: 10.1038/nrg2072
Sun, B. F., Xiao, J. H., He, S., Liu, L., Murphy, R. W., and Huang, D. W. (2013). Multiple interkingdom horizontal gene transfers in Pyrenophora and closely related species and their contributions to phytopathogenic lifestyles. PLoS ONE 8:e60029. doi: 10.1371/journal.pone.0060029
Syed, N. H., and Flavell, A. J. (2006). Sequence-specific amplification polymorphisms (SSAPs): a multi-locus approach for analyzing transposon insertions. Nat. Protoc. 1, 2746–2752. doi: 10.1038/nprot.2006.407
Syvanen, M. (1994). Horizontal gene transfer: evidence and possible consequences. Annu. Rev. Genet. 28, 237–261. doi: 10.1146/annurev.ge.28.120194.001321
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Ungerer, M. C., Strakosh, S. C., and Zhen, Y. (2006). Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr. Biol. 16, R872–R873. doi: 10.1016/j.cub.2006.09.020
Voytas, D. F., Cummings, M. P., Koniczny, A., Ausubel, F. M., and Rodermel, S. R. (1992). Copia-like retrotransposons are ubiquitous among plants. Proc. Natl. Acad. Sci. U.S.A. 89, 7124–7128. doi: 10.1073/pnas.89.15.7124
Vukich, M., Giordani, T., Natali, L., and Cavallini, A. (2009). Copia and Gypsy retrotransposons activity in sunflower (Helianthus annuus L.). BMC Plant Biol. 9:150. doi: 10.1186/1471-2229-9-150
Webster, G. L. (1994). Classification of the Euphorbiaceae. Ann. Mo. Bot. Gard. 81, 3–32. doi: 10.2307/2399908
Wicker, T., Sabot, F., Hua-Van, A., Bennetzen, J. L., Capy, P., Chalhoub, B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982. doi: 10.1038/nrg2165
Won, H., and Renner, S. S. (2003). Horizontal gene transfer from flowering plants to Gnetum. Proc. Natl. Acad. Sci. U.S.A. 100, 10824–10829. doi: 10.1073/pnas.1833775100
Yan, L., Loukoianov, A., Blechl, A., Tranquilli, G., Ramakrishna, W., SanMiguel, P., et al. (2004). The wheat VRN2 gene is a flowering repressor down-regulated by vernalization. Science 303, 1640–1644. doi: 10.1126/science.1094305
Yang, G., Zhou, R., Tang, T., and Shi, S. (2008). Simple and efficient isolation of high-quality total RNA from Hibiscus tiliaceus, a mangrove associate and its relatives. Prep. Biochem. Biotechnol. 38, 257–264. doi: 10.1080/10826060802164991
Yoshiyama, M., Tu, Z., Kainoh, Y., Honda, H., Shono, T., and Kimura, K. (2001). Possible horizontal transfer of a transposable element from host to parasitoid. Mol. Biol. Evol. 18, 1952–1958. doi: 10.1093/oxfordjournals.molbev.a003735
Zedek, F., Smerda, J., Smarda, P., and Bureš, P. (2010). Correlated evolution of LTR retrotransposons and genome size in the genus Eleocharis. BMC Plant Biol. 10:265. doi: 10.1186/1471-2229-10-265
Zou, J. H., Dai, J., Chen, X., and Yuan, J. Q. (2006). Pentacyclic triterpenoids from leaves of Excoecaria agallocha. Chem. Pharm. Bull. 54, 920–921. doi: 10.1248/cpb.54.920
Keywords: LTR retrotransposon, transcriptional and transpositional activities, horizontal transfer, Excoecaria agallocha, euphorbiaceae
Citation: Huang J, Wang Y, Liu W, Shen X, Fan Q, Jian S and Tang T (2017) EARE-1, a Transcriptionally Active Ty1/Copia-Like Retrotransposon Has Colonized the Genome of Excoecaria agallocha through Horizontal Transfer. Front. Plant Sci. 8:45. doi: 10.3389/fpls.2017.00045
Received: 11 October 2016; Accepted: 09 January 2017;
Published: 24 January 2017.
Edited by:
Alessio Mengoni, University of Florence, ItalyReviewed by:
Jan Schmid, Massey University, New ZealandMilind Ratnaparkhe, Directorate of Soybean Research, India
Copyright © 2017 Huang, Wang, Liu, Shen, Fan, Jian and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tian Tang, lsstt@mail.sysu.edu.cn
†These authors have contributed equally to this work.