Abstract
Collagens form the fibrous component of the extracellular matrix in all multi-cellular animals. Collagen type I is the most abundant collagen present in skin, tendons, vasculature, as well as the organic portion of the calcified tissue of bone and teeth. This review focuses on numerous receptors for which collagen acts as a ligand, including integrins, discoidin domain receptors DDR1 and 2, OSCAR, GPVI, G6b-B, and LAIR-1 of the leukocyte receptor complex (LRC) and mannose family receptor uPARAP/Endo180. We explore the process of collagen production and self-assembly, as well as its degradation by collagenases and gelatinases in order to predict potential temporal and spatial sites of action of different collagen receptors. While the interactions of the mature collagen matrix with integrins and DDR are well-appreciated, potential signals from immature matrix as well as collagen degradation products are possible but not yet described. The role of multiple collagen receptors in physiological processes and their contribution to pathophysiology of diseases affecting collagen homeostasis require further studies.
Introduction
Collagen is the most abundant protein present in mammals, and forms the fibrous component of the extracellular matrix in all multi-cellular animals. In humans, different collagen types are present in connective tissues, including tendons, bones, and dentin, and play critical roles in defining the form and mechanical properties of diverse organs, such as bones, blood vessels, skin, and eyes [1]. In addition to classical fibrillar collagens forming uninterrupted triple helical fibrils, new families of collagen have been characterized as fibril-associated collagens with interrupted triple helices (FACITs), membrane-associated collagens with interrupted triple helices (MACITs), and multiple triple-helix domains and interruptions (MULTIPLEXINs) consisting of triple helical regions interspersed with non-helical domains (for recent in depth reviews of different types of collagen see Shoulders and Raines [2] and Ricard-Blum [3]). The most common fibrillar collagens are collagen types I, II, and III. Collagen type I is present in skin, tendons, vasculature, as well as organs such as lungs, heart and others, and forms the main component in the organic portion of the calcified tissue of bone and teeth [1, 3]. Collagen type II is the primary constituent of cartilage and collagen type III forms reticular fibers, commonly found alongside collagen type I [4]. While many consider fibrillar collagen biology as a textbook topic, it remains an active and exciting field of research. In particular, the recent discovery of numerous receptors for which collagen acts as a ligand indicates a much wider potential role for collagen than just a structural molecule. In this review we focus on collagen type I, and provide an overview of collagen receptors, highlight recent advances in collagen type I synthesis and degradation, discuss potential cellular signaling that can be induced by collagen, and its role in bone matrix physiology and pathology.
Collagen receptors
Extracellular matrix proteins are well-known to interact with cells by directly binding to cell surface receptors [5]. Diverse families of receptors, including integrins, receptor tyrosine kinases, and immunoglobulin-like receptors have now been shown to use collagens as their cognate ligands (Figure 1).
Figure 1
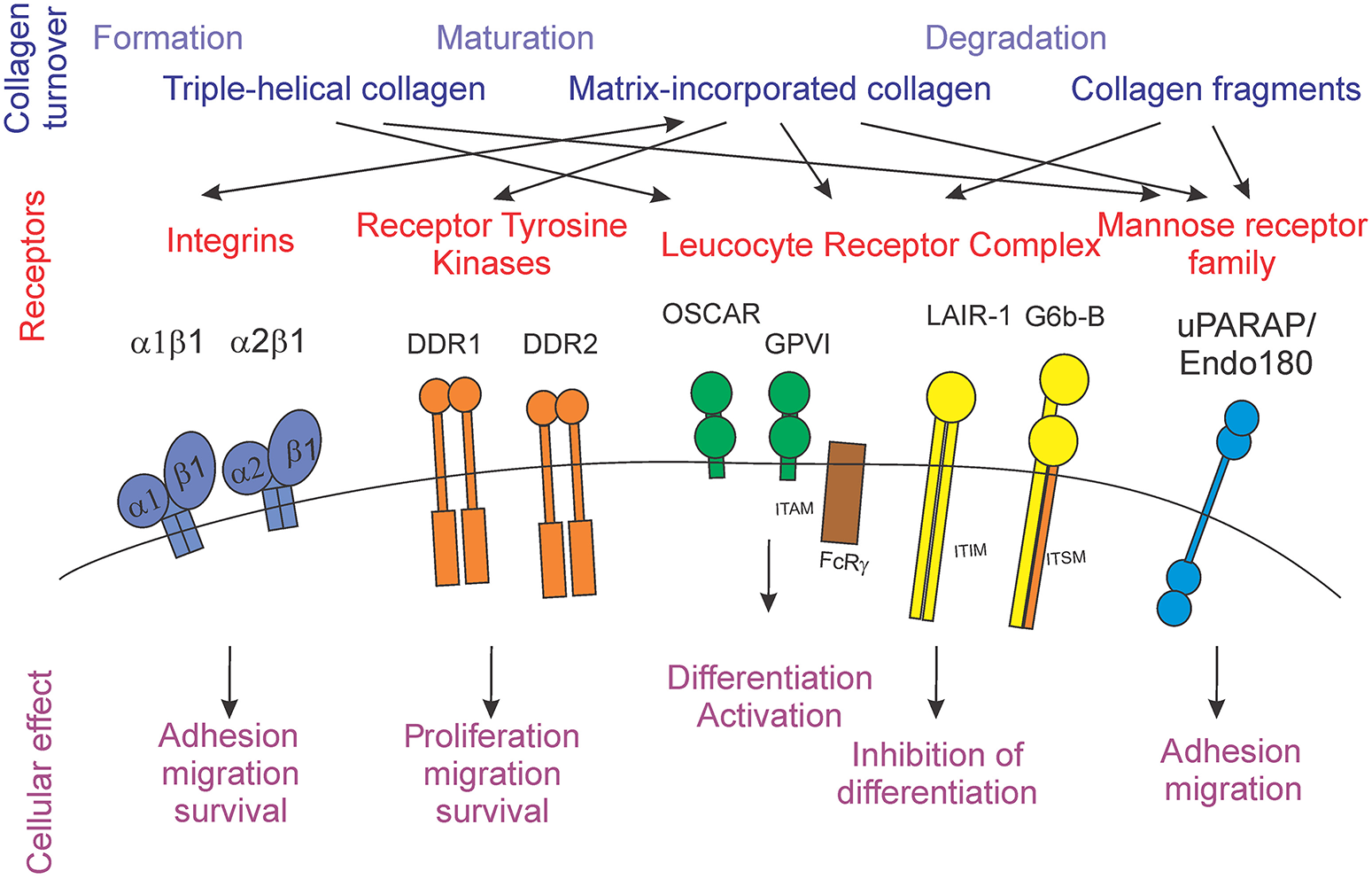
Schematic representation of the stages of collagen turnover creating different forms of collagen (blue), different collagen receptors families (red) and individual receptors, and the cellular outcomes for collagen receptor-mediated signals (purple).
Integrins
Integrins are defined as cell adhesion structures, which are important in development and pathological processes. Integrins play critical roles in signaling, migration and survival of different cells (recently reviewed in depth by Barczyk et al. [5] and Iwamoto and Calderwood [6]). The signaling by these receptors is considered bi-directional, involving outside-in and inside-out signaling [5]. Integrins function as heterodimers, which in humans include one of 18 distinct α subunits and one of eight distinct β subunits, and are type I transmembrane glycoproteins with large extracellular and short cytoplasmic domains [6]. Four different integrin heterodimers (α1β1, α2β1, α10β1, and α11β1) have been demonstrated to bind collagen. In particular, α1β1 and α2β1 integrins have been most extensively studied. These integrins bind to both collagen types I and IV, however their affinities differ: α1β1 has a higher affinity for collagen type IV, while α2β1 preferentially binds to collagen type I [5, 7]. Integrin α2β1 has been reported to be one of the main collagen binding integrins present in bone and is critical for bone resorbing cells, osteoclasts. In these cells, α2β1 integrins affect the attachment of the cell to the bone surface and help form a sealing zone around the area to be resorbed, allowing formation of the localized highly acidic environment necessary for bone degradation [7–9].
Receptor tyrosine kinases
The two discoidin domain receptors, DDR1 and DDR2 are receptor tyrosine kinases activated specifically by fibrillar collagens I–III and V, but not by individual α-chains, denatured collagen, de-glycosylated, or degraded collagens [10]. A distinct characteristic of these receptors is their slow and sustained activation upon stimulation. Binding of the discoidin receptors to triple helical collagen leads to tyrosine autophosphorylation with unique activation kinetics, which is followed by receptor internalization [11, 12]. Imbalance or dysregulation of DDR1 has been implicated in the development of diseases such as fibrosis, atherosclerosis, arthritis, and cancer [10]. DDR1-null mice are predisposed to osteoarthritis and temporomandibular joint disorder [13], but are protected from atherosclerosis and smooth muscle mineralization [14]. DDR1 function can be controlled by ADAM10-mediated ectodomain shedding [15]. DDR2 is involved in pathological scarring processes such as wound healing, arthritis, and cancer [16, 17]. DDR2-deficient mice exhibit dwarfism due to reduced proliferation of chondrocytes [18].
Leukocyte receptor complex (LRC)
The leukocyte receptor complex (LRC) consists of a large group of cell surface receptors, essential for a diverse number of immune functions, including antiviral immunity, autoimmunity, and response to grafts [19]. A typical characteristic of these receptors is the occurrence of pairs of antipathetic receptors, which bind to the same ligands but generate opposing signaling responses [20]. The majority of LRC receptors are primarily expressed by immune cells and play diverse roles in modulating their activity [19, 20]. The stimulatory receptors have short cytoplasmic tails and generate positive signals through immunoreceptor tyrosine-based activation motifs (ITAM) present on the required adapter proteins, FcRγ, DAP10, and DAP12. The inhibitory receptors are characterized by long cytoplasmic tails, which contain immunoreceptor tyrosine-based inhibitory motifs (ITIM) [19, 20]. Of interest, collagen has been recently demonstrated to act as a ligand for a number of stimulatory and inhibitory receptors in this family, including osteoclast associated receptor (OSCAR), GPVI, and LAIR-1 [21–23]. The structural basis for collagen recognition by the immune receptors has been investigated in a number of studies [24–27], however some controversy regarding the alignment of collagen-recognition sites among different receptors exists.
OSCAR and GPVI are stimulatory receptors that are activated by collagen. OSCAR is particularly important for osteoclast differentiation, as it acts as a critical co-stimulatory receptor for osteoclast formation and function [23, 26–28]. GPVI is mainly found in platelets and binds to collagen during the process of blood coagulation [21]. The binding of collagen to OSCAR or GPVI results in the recruitment of ITAM-containing FcRγ chains. For OSCAR, the main downstream effect of activation is the initiation of calcium signaling, which is critically important for activation of a key osteoclastogenic transcription factor, nuclear factor of activated T-cells (NFAT) c1. Activation of GPVI leads to the binding of Syk to the FcR-γ chain, which causes an activation of Syk proteins and tyrosine phosphorylation. At the same time, phospholipase C γ2 (PLCγ2) also becomes activated [29, 30].
The inhibitory receptor LAIR-1 was also shown to be activated by collagen [31]. Another inhibitory receptor G6b-B may also act as a collagen-binding receptor, although the existing evidence is weaker [32]. Both LAIR-1 and G6b-B were first identified to be expressed on megakaryocytes and platelets and to negatively regulate their function [22, 33]. LAIR-1 was also found to be present during osteoclastogenesis and to inhibit this process [34]. LAIR-1 is activated by triple helical collagen, specifically when it encounters the triplet (GPO)10 (glycine-proline-hydroxyproline)10, also known as “collagen related peptide” [31]. LAIR-1 contains two ITIMs, which upon phosphorylation recruit SHP-1 and SHP-2 phosphatases. These phosphatases directly dephosphorylate Syk, Zap70, and PLCγ, preventing ITAM-mediated stimulation of protein kinases and calcium signaling [22, 31]. G6b-B was suggested to be activated by collagen fragments, such as the collagen-related peptide, likely relevant to the microenvironment of damaged epithelium [32, 33]. G6b-B contains one ITIM as well as a newly described immunoreceptor tyrosine-based switch motif (ITSM) [33, 35]. In contrast to ITIM, which generally signals through activation of phosphatases, ITSM interferes with ITAM-mediated signaling by using adaptor molecules. An important characteristic of this motif is the ability to switch between stimulatory and inhibitory signals and to bind SHP1, SHP2, SHIP, and p85 [36]. In platelets, G6b-B interferes with positive signaling induced by collagen binding to GPVI [37].
uPARAP/Endo180
The urokinase plasminogen activator receptor-associated protein (uPARAP/Endo180), a member of the mannose receptor family of type I transmembrane glycoproteins, is a multi-domain transmembrane glycoprotein. Characteristically this family of proteins includes an N-terminal, cysteine-rich/ricin B like domain, a fibronectin type II domain, and a series of 8–10 C-type lectin-like domains. This mesenchymal cell surface receptor has an important function in collagen internalization [38–40]. In addition, uPARAP/Endo180 was shown to aid in the initial adhesion of fibroblasts to collagen and to accelerate the migration of these cells on a fibrillar collagen matrix [38, 39, 41, 42]. In bone, this receptor is highly expressed on osteoblasts and osteocytes at sites of endochondral and intramembranous ossification during development [38].
Thus, multiple receptor families can bind collagen and induce a variety of cellular effects. Although the repertoire of cellular responses affected by collagen receptors appears to be similar, with adhesion, migration and survival being prominent on the list, it is interesting that the receptors can bind to different forms of collagen, including large triple helical fragments, matrix-incorporated collagen fibrils, and small collagen fragments. Next, we will consider the process of collagen turnover and identify the physiological stages during which different forms of collagen can act as effectors of receptor-mediated signaling.
Potential signaling induced during collagen synthesis by osteoblasts
In hard tissues, collagen is produced by highly specialized cells of mesenchymal origin, termed osteoblasts in bone tissue and odontoblasts for dentin. All fibrillar collagen molecules are formed from three polypeptide chains, termed α chains, which are wound into a right-handed triple helix to form a cord-like structure. The triple helical regions of collagen are characterized by the presence of a glycine residue at every third position (Gly-X-Y)n, the other two positions being rich in proline or hydroxyproline. Since glycine residues only have a hydrogen atom as their “side chain,” the polypeptide can pack itself into a super helical structure [1, 43]. The triple helices of collagen type I are formed from two α1 chains and one α2 chain, which are the products of the different genes, COL1A1 and COL1A2. Osteogenesis imperfecta (OI) is a heritable disease characterized by high bone fragility. The large majority of patients with OI have disease-causing dominant mutations in one of the two genes that code for collagen type I alpha chains, COL1A1 and COL1A2 [44]. Interestingly, a number of patients with the clinical presentation of OI were found to have normal collagen type I, but mutations in other proteins with previously unknown function. Investigation of these proteins, which include cartilage-associated protein (CRTAP), prolyl 3-hydroxylase 1 (P3H1), cyclophilin B (CyPB), pigment epithelium-derived factor (PEDF), heat shock protein 47 (HSP47), and FK506 binding protein 65 (FKBP65), resulted in a greater understanding of the regulation of collagen type I production and assembly [45, 46].
The procollagen type I α chain genes are transcribed and processed from COL1A1 and COL1A2 to mRNAs. Direct translation to the pro-α1 and α2 chains occurs in the rough endoplasmic reticulum [2]. The procollagen has extensions on each end, termed amino and carboxyl procollagen propeptides. These extensions increase the solubility of the peptide and assist its movement within the cell during the process of post-translational modification. One of these modifications is the hydroxylation of specific proline and lysine residues [47, 48]. The prolyl 3-hydroxylation complex is a post-translational collagen modification system present in the endoplasmic reticulum, and consists of CRTAP, P3H1, and CyPB. The complex modifies a single proline residue (Pro 986) to 3-hydroxyproline on each α1 chain of type I and II collagen [49, 50]. Mutations in CRTAP, P3H1/LEPRE1, or PPIB (the gene that encodes cyclophilin B) strongly affect post-translational modifications of collagen resulting in a complete absence of proline 3-hydroxylation in the case of mutations in CRTAP, P3H1, and site-specific alterations in the hydroxylation and glycosylation of collagen, in the case of mutations in PPIB [49, 51, 52]. Consequently, collagen folding is delayed [49, 52–54] and a change in fibril assembly, cross-linking, and bone mineralization occurs [51, 52, 54]. The newly formed hydroxylysine residues are glycosylated by the addition of monosaccharides, such as galactose and glucose. This step is carried out by glycosyl transferases and gives the new collagen molecule unique chemical and structural characteristics [55]. The C-terminal region of the procollagen molecules contains cysteine residues that form intermolecular disulfide bonds, facilitating the registration of the three procollagen chains. When the appropriate alignment is reached, the three chains wrap around each other to form a string-like structure [3]. After all the modifications are complete and the triple helix is formed, the molecules of procollagen are transported along microtubules, organized in the Golgi apparatus, and eventually secreted into the extracellular space [56]. In a homozygous patient with HSP47 missense mutation, it was demonstrated that HSP47, potentially acting in cooperation with immunophilin FKBP65, encoded by FKBP10, is important for proper trafficking of type I procollagen to the Golgi [57]. Mutations in FKBP10 also cause moderately severe osteogenesis imperfecta [58, 59] with decreased collagen cross-linking, resulting in sparsity and disorder of collagen fibril deposition [60].
Extracellularly, procollagen is processed by procollagen proteases, which are responsible for removing the extension peptides from both ends of the molecule. The N-terminal is processed by enzymes like a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)-2, -3, and -14, while bone matrix protein-1 (BMP-1) is responsible for the C-terminal processing, resulting in the formation of N-telopeptide (NTP) and C-telopeptide (CTP), respectively [61–63]. Mutations in the collagen type I C-propeptide cleavage site disrupt extracellular collagen processing, resulting in osteogenesis imperfecta [64]. Mutations in BMP1 similarly result in decreased collagen maturation, hyperosteoidosis, and hypermineralization [65]. Interestingly, mutations in SERPINF1, the gene that encodes PEDF, lead to osteogenesis imperfecta type VI, which has a phenotype of a disorganized bone matrix, large amount of unmineralized osteoid, and abnormal mineralization pattern, similar to the phenotype observed in OI due to mutations at the collagen type I C-propeptide cleavage site or in BMP1, suggesting that PEDF may also play a role in procollagen processing [66–69]. It has been noted that the rate of bone formation positively correlates with CTP levels, which lead to its use as a marker of osteoblastic bone formation [70, 71]. Once the triple helical collagen molecules lacking their extension peptides are formed in the extracellular space, the process of the fiber formation commences [3].
Receptors potentially activated by triple-helical collagen
Two classes of receptors, including those of LRC and of mannose-receptor family, have been shown respond to triple-helical collagen which is not necessarily incorporated in the matrix (Figure 1). In particular, the uPARAP/Endo180 receptor was found on early osteoblast precursors as well as actively matrix-producing osteoblasts [72, 73]. While this receptor has been implicated in osteoblast recruitment to the remodeling sites [73], this function can likely be attributed only to early precursors, but not to mature cells. It is thus possible that additional regulation may be exerted by this receptor present on mature osteoblasts. From the LRC family, OSCAR and LAIR-1 have been shown to be expressed by osteoclasts [28, 34]. It is possible that LAIR-1-mediated inhibition of osteoclastogenesis contributes to the prevention of premature activation of resorption at the sites of freshly laid down osteoid. It is also interesting to speculate that formation of the two fragments, NTP and CTP, during procollagen processing may generate soluble and thus longer-reaching signals for the receptors activated by smaller collagen fragments. Abnormal signaling though collagen receptors can also potentially contribute to the pathophysiology of OI, as it commonly results in abnormal collagen modification and thus would significantly alter receptor-ligand interactions.
Formation of collagenous bone matrix
Individual collagen molecules are first assembled into collagen fibrils, which in turn combine to form fibers, which give the tissue its structural properties. Collagen fibril formation begins with the post-translational modification by the copper-containing enzyme lysyl oxidase, which oxidizes the peptidyl lysine residues to facilitate the formation of covalent intra- and inter-molecular bonds, also known as crosslinks [74]. Lysyl oxidase has been found extracellularly, intracellularly as well as in the nucleus, and was reported to have diverse and important roles in the human body such as developmental regulation, tumor suppression, cell motility, and cellular senescence [74]. After the covalent bonds are formed between the tropocollagen chains, triple helical molecules line up and collagen fibrils are formed. The triple helical tropocollagen molecule is 300 nm long, 1.5 nm in diameter, and consists of two α1(I) chains and one α2(I) chain [75, 76]. Another enzyme important for the crosslinking of the collagen fibers is tissue transglutaminase 2, which specifically mediates the reaction between the side chains of glutamine and lysine residues of adjacent fibers resulting in formation of ε(γ-glutamyl) lysine crosslinks, covalent amide bonds that reinforce the three dimensional structure [77, 78]. The collagen fibers are cable-like bundles, 50–200 μm in diameter that are visible under the light microscope [75, 76]. Collagen type I in skin and bone is formed of the same two α1 and one α2 chains. However, post-translational modifications and crosslinking differ between bone and skin fibrils, giving tissue-specific properties to the final extracellular matrix [75].
In bone, after collagen maturation is completed, matrix mineralization during which calcium and phosphate precipitate to form crystals of hydroxyapatite [Ca10(PO4)6(OH)2] within the organic matrix proceeds [79]. The localization and orientation of individual crystals is guided by specific organic moieties on collagen type I and non-collagenous proteins present in bone matrix [80]. Finally, bone tissue is formed as a composite material containing a precise mixture of macromolecules and hydroxyapatite crystals.
Receptors potentially activated by matrix-incorporated collagen
The majority of the collagen receptors are assumed to be activated by collagen present in mature matrix. The evidence for a physiological significance of such interactions is strongest for members of the integrin receptor family, which are well-known for their substrate-recognition roles [81]. Positive responses, such as support of cell adhesion, survival, migration, and proliferation result from collagen interacting with integrin receptors α1β1 and α2β1 [5], receptor tyrosine kinases DDR1 and DDR2 [12], LRC member OSCAR [23], and mannose family receptor uPARAP [38]. Inhibitory cellular effects have been observed upon stimulation of LAIR-1 with collagen [22, 32]. It is particularly interesting, that osteoclasts express both the stimulatory collagen receptor OSCAR [28] and an inhibitory receptor LAIR-1 [34]. However, if we assume that OSCAR and LAIR-1 are expressed at different stages of osteoclastogenesis, we can attempt to reconcile how contradictory collagen signals can be perceived by these cells. We can speculate that LAIR-1 is present on early precursors, and that its role is to prohibit osteoclast differentiation on immature matrix, while OSCAR is expressed later during osteoclastogenesis and is engaged by the matrix-incorporated collagen to support osteoclast formation on the correct substrate. While extensive studies on the role of OSCAR during osteoclast formation have been performed [23, 26, 28], much less is known about the temporal and spatial aspects of regulation of osteoclastogenesis by LAIR-1. In diseases associated with abnormal collagen synthesis, such as OI, as well as abnormal mineralization, for example osteomalacia due to calcium and phosphate deficiency, the structure of the mature tissue matrix is altered, which may potentially result in changes in receptor-ligand binding for collagen receptors, contributing to the pathophysiology of these disorders.
Potential signaling induced by collagen degradation
Bone mineral together with the organic matrix is physiologically removed by osteoclasts. Similarly, during tooth eruption, odontoclasts resorb the deciduous tooth roots in order to provide space for the eruption of permanent teeth [82]. By tightly attaching to the bone matrix, osteoclasts form a sealed phagolysosomal compartment underneath the cell, where vacuolar type H+-ATPase in the plasma membrane of ruffled borders releases protons to lower the extracellular pH and dissolve hydroxyapatite mineral. Concomitantly, proteolytic enzymes are released to digest the organic matrix [83, 84]. Proteases (also termed peptidases or proteinases) hydrolyze the peptide bonds that link amino acids together in the polypeptide chains forming proteins. Due to the closely packed nature of the mature triple helix, collagens are resistant to attack by most proteases, however, specialized proteases termed collagenases are able to hydrolyze these molecules [76]. The most important collagenases are the papain-like cysteine protease cathepsin K and matrix metalloproteinases (MMP) [85, 86]. Within the MMP family, MMP-1, MMP-8, MMP-13, and MMP-14 are specifically capable of degrading triple-helical fibrillar collagens, while MMP-2 and MMP-9 act after the triple helix is unwound and are termed gelatinases. MMP-13 and MMP-9 are particularly important for bone resorption (Table 1).
Table 1
| Enzyme | Other designations | Properties |
|---|---|---|
| CYSTEINE PROTEASES | ||
| Cathepsin K | Cathepsin O, O2, X (obsolete designations) | As a complex with glycosaminoglycans cleaves triple helix, very effective gelatinase |
| Zn2+ METALLOPROTEASES | ||
| MMP-1 | Interstitial collagenase Collagenase-1 | Cleaves triple helix, preference for collagen types I and III |
| MMP-2 | Gelatinase A Type IV collagenase | Degrades gelatin |
| MMP-8 | Neutrophil collagenase Collagenase-2 | Cleaves triple helix, preference for collagen type I |
| MMP-9 | Gelatinase B | Degrades gelatin |
| MMP-13 | Collagenase-3 | Cleaves triple helix, preference for collagen type II |
| MMP-14 | MT1-MMP | Cleaves triple helix, membrane-anchored |
Collagen-degrading enzymes.
Cathepsin K
Cathepsin K, a member of the C1 peptidase family, is expressed by osteoclasts at the cell surface adjacent to the bone and was shown to be critical for matrix breakdown in the resorption compartment during bone remodeling [86, 87]. The protease works optimally at low pH and has been shown to degrade type I collagen [85]. Both the α1 and α2 collagen type I chains are cleaved by cathepsin K [88]. The protein cleavage site of papain-like cysteine proteases, including cathepsin K, is determined by the amino acids occupying the two positions before (N-terminal to) the cleavage site and one or two positions after the cleavage site. Generally, a hydrophobic side chain such as valine, leucine, or proline is found in the second residue before the cleavage site, whereas the amino acid directly before the cleavage site is usually glutamic acid, alanine, or glycine. Finally, in the position after the cleavage site, the most common amino acids are glycine, glutamic acid, and/or isoleucine [87, 89]. Overall, it has been found that cathepsin K performs a cleavage after helical cross-linking residues, which in collagen type I is most likely to occur after glycine due to high prevalence of GXY repeats [90]. Cathepsin K was shown to cleave substrates with proline in the second amino acid position after the cleavage site [91], which is critical for its ability to target collagen [87, 92].
Since cathepsin K is the only papain-like cysteine protease capable of cleaving triple helical collagen, it is of significant interest as a pharmaceutical target [90, 93]. Structural analysis revealed that for cathepsin K to demonstrate its collagenase activity, a dimer has to form an oligomeric complex with a glycosaminoglycan and dock onto a collagen molecule with its central grove [92, 94, 95]. The presence of glycosaminoglycans allows access to the triple helix, leading to the cutting of the fibril into smaller sub-fibrils and a simultaneous release of glycosaminoglycans. This process eventually results in a progressive unfolding of the fibrils, making them less stable and therefore accessible for further degradation [96]. Cathepsins that do not exhibit collagenase activity, such as cathepsins L, V, S, and B have only a limited effect on the fibril structures [96]. In addition to bone, cathepsin K is also expressed in hematopoietic, epithelial, and fibroblast cells, and was shown to play a role in arthritis, obesity, schizophrenia, bone metastases, and various other pathological conditions [89].
In humans, mutations in the cathepsin K gene were shown to underlie the skeletal disorder pycnodysostosis, which is characterized by osteopetrosis, bone fragility, short stature, acrosteolysis of the distal phalanges, delayed cranial suture closure, clavicular dysplasia, and dental abnormalities [97, 98]. In animal models, cathepsin K deficiency results in an osteopetrotic bone phenotype [99–101] and has been shown to predispose mice to lung fibrosis [102], abnormal airway morphologies [103] and deficiencies in learning and memory aptitudes [104]. Even though the osteoclast numbers were not changed in cathepsin K deficient mice, large areas of demineralized bone matrix underlying the ruffled borders of osteoclasts were frequently found, suggesting that only the degradation of organic bone matrix is impaired in these animals [99, 100]. Once cathepsin K cleaves the triple helix, it unwinds and becomes available for degradation by any protease with gelatinolytic activity. Interestingly in cathepsin K knockout mice, osteoclastic resorption, though severely impaired, was still present [99] due to involvement of other proteases with collagenase activity, such as the matrix metalloproteinases [101].
Matrix metalloproteinases
The matrix metalloproteinases (MMP), peptidase family M10, is composed of zinc-dependent endopeptidases, also known as the metzincin superfamily. The main task of MMPs is to degrade extracellular matrix proteins, as well as a number of bioactive molecules [87, 105, 106]. Within the MMP family of endopeptidases, MMP-1, MMP-8, MMP-13, and MMP-14 exhibit collagenase activity and are specifically capable of degrading triple-helical fibrillar collagens [106, 107]. MMP-1 and MMP-13 are produced by osteoclasts and play important roles in bone matrix degradation. After the triple helix is separated, MMP-2 and MMP-9 can further cleave collagen chains working as gelatinases. The MMP family members with collagenase activity require at least three structural components to successfully achieve their function: (i) a hemopexin-like C-terminal domain, (ii) a linker or hinge region between the catalytic and hemopexin domain, and (iii) a specific peptide loop in the catalytic domain [108, 109]. Collagen type I is cleaved by MMP-1, -8, and -13 at a characteristic site located between Gly775/Ile776, three quarters of the distance from the N-terminus, leading to the formation of two fragments—a larger fragment of ¾ and a smaller ¼ fragment [107, 110, 111].
MMP-1, also known as interstitial collagenase or fibroblast collagenase, is involved in the breakdown of extracellular matrix in normal physiological processes, such as embryonic development, reproduction, and tissue remodeling, as well as in disease processes, such as arthritis and cancer metastasis [106, 112, 113]. Osteoblasts have been demonstrated to produce MMP-1, which may affect their differentiation [114]. In addition, MMP-1 was shown to be upregulated in response to mechanical loading [115]. However, it is not clear if MMP-1 activity toward collagen type I is important for osteoblasts. While MMP-1 is not generally found to be expressed by osteoclasts, there is evidence that it may be involved in bone resorption in pathological conditions [113, 116, 117].
MMP-13 is involved in the degradation of extracellular matrix for tumor invasion and metastasis [84] and is a critical collagenase MMP for osteoclastic bone resorption [86]. Mutations in MMP-13 cause metaphyseal anadysplasia 1, which includes the Missouri type of spondyloepimetaphyseal dysplasia, a spectrum of diseases characterized by defective growth and severe skeletal changes that resolve spontaneously with age [118, 119]. This protease is more aggressive to collagen type II than to type I, therefore the effects are more prominent in the joints [120, 121]. In animal models, overexpression of MMP-13 produces arthritis with cartilage erosion [122]. Knockout of MMP-13 shows temporary anomalies in cartilage resorption in long bone growth and fracture healing. While these animals have a normal lifespan and sufficient fertility, microscopic analyses of the skeletal system verified profound defects in the growth plate cartilage with a clear intensification in the hypertrophic chondrocyte zone and a delay in primary ossification [121, 123].
Gelatinases
The main gelatinase expressed and released by osteoclasts is MMP-9 [124]. MMP-9 participates in dissolution of bone collagens working inside the sealing zone located underneath the osteoclast, in concert with collagenases MMP-13 and cathepsin K [125]. MMP-9-deficient animals exhibit a skeletal development phenotype, and chondrocyte apoptosis [126]. In addition, vascularization and ossification of cartilage is significantly delayed [126]. The role of MMP-9 in bone resorption is not clear, however it was demonstrated that in the absence of MMP-9 osteoclastic recruitment is delayed and that MMP-9 is required for osteoclast invasion into the discontinuously mineralized hypertrophic cartilage [127]. Mutations in MMP-9 result in metaphyseal anadysplasia 2, which is phenotypically indistinguishable from metaphyseal anadysplasia 1 due to mutations in MMP-13 [119]. A dominant, more severe phenotype of metaphyseal anadysplasia was found to be associated with deactivation of both MMP-13 and MMP-9 [119].
Receptors potentially activated by degraded collagen
Physiological degradation of collagen can result in production of shorter triple-helical proteins, along with single strand fragments of different molecular weight [86, 128]. Such fragments can potentially activate the receptors of LRC, GPVI, LAIR-1, and G6b-B, as well as uPARAP/Endo180. Since LAIR-1 is expressed by osteoclasts [34] and uPARAP/Endo180 by osteoblasts [129], it is possible that such signals mediate temporal and spatial coordination of collagen matrix formation and degradation. In addition, formation of circulating fragments, such as those used as biomarkers of bone resorption [130], raises a possibility of long-distance signals generated during collagen degradation. In pycnodysostosis and metaphyseal anadysplasia, mutations in cathepsin K and MMP-9 and MMP-13 result in altered collagen degradation, thus producing different collagen fragments, potentially interfering with collagen receptor signaling.
Conclusions
Collagen is the main component in many tissues in the human body, particularly in bone tissue where this protein forms 90–95% of the organic matrix. Even though collagen is one of the best studied molecules, many questions remain regarding its physiology as highlighted by a high number of poorly understood disorders associated with collagen production and degradation, including genetic diseases such as osteogenesis imperfecta and metaphyseal anadysplasia, and inflammatory disorders such as arthritis and periodontitis. From the perspective of its temporally and spatially controlled self-assembly, collagen is a fascinating molecule that undergoes many precise, yet not fully understood transitions from its initial translation product to the final mature fiber. Moreover, bone tissue is remodeled continuously, which brings into focus the importance of a regulated degradation of collagen, as well as other components of the extracellular matrix. The collagen substrate structural signals mediated by integrins have been well-appreciated since the 1980s. The discovery of numerous novel receptors for collagen highlights the possibility of previously unknown aspects of collagen biology. The role of these receptors in physiological processes and their contribution to pathophysiology of diseases affecting collagen homeostasis require further studies.
Funding
This work was supported by the Canadian Institutes of Health Research (grant MOP-137091 to SK). IB is supported by the Faculty of Dentistry, McGill University, and by Réseau de Recherche en Santé Buccodentaire et Osseuse.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Statements
Author contributions
IB, JM, and SK contributed to the conceptual planning; IB and JW designed and performed literature analysis, IB wrote the first draft of the manuscript. All authors critically revised the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
HulmesDJC. Collagen Diversity, Synthesis and Assembly. In: FratzlP. editor. Collagen: Structure and Mechanics, Springer Science + Business Media, LLC (2008).
2.
ShouldersMDRainesRT. Collagen structure and stability. Annu Rev Biochem. (2009) 78:929. 10.1146/annurev.biochem.77.032207.120833
3.
Ricard-BlumS. The collagen family. Cold Spring Harb Perspect Biol. (2011) 3:a004978. 10.1101/cshperspect.a004978
4.
WessTJCollagen Fibrillar Structure and Hierarchies. In FratzlP. editor. Collagen: Structure and Mechanics, Springer Science + Business Media, LLC (2008).
5.
BarczykMCarracedoSGullbergD. Integrins. Cell Tissue Res. (2010) 339:269–80. 10.1007/s00441-009-0834-6
6.
IwamotoDVCalderwoodDA. Regulation of integrin-mediated adhesions. Curr Opin Cell Biol. (2015) 36:41–7. 10.1016/j.ceb.2015.06.009
7.
XuYGurusiddappaSRichRLOwensRTKeeneDRMayneRet al. Multiple binding sites in collagen type I for the integrins α1β1 and α2β1. J. Biol Chem. (2000) 275:38981–9. 10.1074/jbc.M007668200
8.
HelfrichMHNesbittSALakkakorpiPTBarnesMJBodarySCShankarGet al. β1 integrins and osteoclast function: involvement in collagen recognition and bone resorption. Bone (1996) 19:317–28. 10.1016/S8756-3282(96)00223-2
9.
RodanSBRodanGA. Integrin function in osteoclasts. J Endocrinol. (1997) 154:S47–56.
10.
IwaiLLuczynskiMHuangP. Discoidin domain receptors: a proteomic portrait. Cell Mol Life Sci. (2014) 71:3269–79. 10.1007/s00018-014-1616-1
11.
CarafoliFHohenesterE. Collagen recognition and transmembrane signalling by discoidin domain receptors. Biochim Biophys Acta (2013) 1834:2187–94. 10.1016/j.bbapap.2012.10.014
12.
FuHLValiathanRRArkwrightRSohailAMihaiCKumarasiriMet al. Discoidin domain receptors: unique receptor tyrosine kinases in collagen-mediated signaling. J Biol Chem. (2013) 288:7430–7. 10.1074/jbc.R112.444158
13.
SchminkeBMuhammadHBodeCSadowskiBGerterRGersdorffNet al. A discoidin domain receptor 1 knock-out mouse as a novel model for osteoarthritis of the temporomandibular joint. Cell Mol Life Sci. (2014) 71:1081–96. 10.1007/s00018-013-1436-8
14.
AhmadPJTrckaDXueSFrancoCSpeerMYGiachelliCMet al. Discoidin domain receptor-1 deficiency attenuates atherosclerotic calcification and smooth muscle cell-mediated mineralization. Am J Pathol. (2009) 175:2686–96. 10.2353/ajpath.2009.080734
15.
ShitomiYThøgersenIBItoNLeitingerBEnghildJJItohY. ADAM10 controls collagen signaling and cell migration on collagen by shedding the ectodomain of discoidin domain receptor 1 (DDR1). Mol Biol Cell (2015) 26:659–73. 10.1091/mbc.E14-10-1463
16.
VogelW. Discoidin domain receptors: structural relations and functional implications. FASEB J (1999) 13:77–82.
17.
OlasoELinHCWangLHFriedmanSL. Impaired dermal wound healing in discoidin domain receptor 2-deficient mice associated with defective extracellular matrix remodeling. Fibrogenesis Tissue Repair (2011) 4:5. 10.1186/1755-1536-4-5
18.
YancopoulosGLabradorJPAngelPAzcoitiaVTuckermannJMartínezACet al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. (2001) 2:446–52. 10.1093/embo-reports/kve094
19.
MartinAMKulskiJKWittCPontarottiPChristiansenFT. Leukocyte Ig-like receptor complex (LRC) in mice and men. Trends Immunol. (2002) 23:81–8. 10.1016/S1471-4906(01)02155-X
20.
BarrowADTrowsdaleJ. The extended human leukocyte receptor complex: diverse ways of modulating immune responses. Immunol. Rev. (2008) 224:98–123. 10.1111/j.1600-065X.2008.00653.x
21.
NieswandtBWatsonSP. Platelet-collagen interaction: is GPVI the central receptor?Blood (2003) 102:449–61. 10.1182/blood-2002-12-3882
22.
MeyaardL. The inhibitory collagen receptor LAIR-1 (CD305). J Leukoc Biol. (2008) 83:799–803. 10.1189/jlb.0907609
23.
BarrowADRaynalNAndersenTLSlatterDABihanDPughNet al. OSCAR is a collagen receptor that costimulates osteoclastogenesis in DAP12-deficient humans and mice. J Clin Invest. (2011) 121:3505–16. 10.1172/JCI45913
24.
HoriiKKahnMLHerrAB. Structural basis for platelet collagen responses by the immune-type receptor glycoprotein VI. Blood (2006) 108:936–42. 10.1182/blood-2006-01-010215
25.
BrondijkTHde RuiterTBalleringJWienkHLebbinkRJvan IngenH.et al. Crystal structure and collagen-binding site of immune inhibitory receptor LAIR-1: unexpected implications for collagen binding by platelet receptor GPVI. Blood (2010) 115:1364–73. 10.1182/blood-2009-10-246322
26.
HaywoodJQiJChenCCLuGLiuYYanJet al. Structural basis of collagen recognition by human osteoclast-associated receptor and design of osteoclastogenesis inhibitors. Proc Natl Acad Sci USA. (2016) 113:1038–43. 10.1073/pnas.1522572113
27.
ZhouLHinermanJMBlaszczykMMillerJLConradyDGBarrowADet al. Structural basis for collagen recognition by the immune receptor OSCAR. Blood (2016) 127:529–37. 10.1182/blood-2015-08-667055
28.
KimNTakamiMRhoJJosienRChoiY. A novel member of the leukocyte receptor complex regulates osteoclast differentiation. J Exp Med. (2002) 195:201–9. 10.1084/jem.20011681
29.
AsselinJGibbinsJMAchisonMLeeYHMortonLFFarndaleRWet al. A collagen-like peptide stimulates tyrosine phosphorylation of syk and phospholipase Cγ2 in platelets independent of the integrin α2β1. Blood (1997) 89:1235–42.
30.
WatsonSPAugerJMMcCartyOJTPearceAC. GPVI and integrin αIIbβ3 signaling in platelets. J Thromb Haemost. (2005) 3:1752–62. 10.1111/j.1538-7836.2005.01429.x
31.
LebbinkRJde RuiterTAdelmeijerJBrenkmanABvan HelvoortJMKochMet al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J Exp Med. (2006) 203:1419–25. 10.1084/jem.20052554
32.
SenisYATomlinsonMGGarcíaÁDumonSHeathVLHerbertJet al. A comprehensive proteomics and genomics analysis reveals novel transmembrane proteins in human platelets and mouse megakaryocytes including G6b-B, a novel immunoreceptor tyrosine-based inhibitory motif protein. Mol Cell Prot. (2007) 6:548–64. 10.1074/mcp.D600007-MCP200
33.
CoxonCHSadlerAJHuoJCampbellRDBudayL. An Investigation of hierachical protein recruitment to the inhibitory platelet receptor, G6B-b. PLoS ONE (2012) 7:e49543. 10.1371/journal.pone.0049543
34.
ZhangYDingYHuangYZhangCBoquanJRanZ. Expression of leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1) on osteoclasts and its potential role in rheumatoid arthritis. Clinics (2013) 68:475–81. 10.6061/clinics/2013(04)07
35.
NewlandSAMacAulayICFlotoARde VetECOuwehandWHWatkinsNAet al. The novel inhibitory receptor G6B is expressed on the surface of platelets and attenuates platelet function in vitro. Blood (2007) 109:4806–9. 10.1182/blood-2006-09-047449
36.
El FirarAVoisinTRouyer-FessardCOstuniMACouvineauALaburtheM. Discovery of a functional immunoreceptor tyrosine-based switch motif in a 7-transmembrane-spanning receptor: role in the orexin receptor OX1R-driven apoptosis. FASEB J. (2009) 23:4069–80. 10.1096/fj.09-131367
37.
MoriJPearceACSpaltonJCGrygielskaBEbleJATomlinsonMGet al. G6b-B inhibits constitutive and agonist-induced signaling by glycoprotein VI and CLEC-2. J Biol Chem. (2008) 283:35419–27. 10.1074/jbc.M806895200
38.
EngelholmLHListKNetzel-ArnettSCukiermanEMitolaDJAaronsonHet al. uPARAP/Endo180 is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J Cell Biol. (2003) 160:1009–15. 10.1083/jcb.200211091
39.
JürgensenHJMadsenDHIngvarsenSMelanderMCGårdsvollHPatthyLet al. A novel functional role of collagen glycosylation: interaction with the endocytic collagen receptor uparap/ENDO180. J Biol Chem. (2011) 286:32736–48. 10.1074/jbc.M111.266692
40.
JürgensenHJJohanssonKMadsenDHPorseAMelanderMCSørensenKRet al. Complex determinants in specific members of the mannose receptor family govern collagen endocytosis. J Biol Chem. (2014) 289:7935–47. 10.1074/jbc.M113.512780
41.
MadsenDHEngelholmLHIngvarsenSHilligTWagenaar-MillerRAKjøllerLet al. Extracellular collagenases and the endocytic receptor, urokinase plasminogen activator receptor-associated protein/Endo180, cooperate in fibroblast-mediated collagen degradation. J. Biol Chem. (2007) 282:27037–45. 10.1074/jbc.M701088200
42.
EngelholmLHMelanderMCHaldAPerssonMMadsenDHJürgensenHJet al. Targeting a novel bone degradation pathway in primary bone cancer by inactivation of the collagen receptor uPARAP/Endo180. J Pathol. (2016) 238:120–33. 10.1002/path.4661
43.
BartonSPMarksR. Measurement of collagen-fibre diameter in human skin. J Cutan Pathol. (1984) 11:18–26. 10.1111/j.1600-0560.1984.tb00345.x
44.
RauchFGlorieuxFH. Osteogenesis imperfecta. Lancet (2004) 63:1377–85. 10.1016/S0140-6736(04)16051-0
45.
ForlinoACabralWABarnesAMMariniJC. New perspectives on osteogenesis imperfecta. Nat Rev Endocrinol. (2011) 7:540–57. 10.1038/nrendo.2011.81
46.
Fratzl-ZelmanNMisofBMRoschgerPKlaushoferK. Classification of osteogenesis imperfecta. Wien Med Wochenschr. (2015) 165:264–70. 10.1007/s10354-015-0368-3
47.
MyllyharjuJ. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann Med. (2008) 40:402–17. 10.1080/07853890801986594
48.
GorresKLRainesRT. Prolyl 4-hydroxylase. Crit Rev Biochem Mol Biol. (2010) 45:106–24. 10.3109/10409231003627991
49.
MariniJCCabralWABarnesAMChangW. Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle (2007) 6:1675–81. 10.4161/cc.6.14.4474
50.
Fratzl-ZelmanNMorelloRLeeBRauchFGlorieuxFHMisofBMet al. CRTAP deficiency leads to abnormally high bone matrix mineralization in a murine model and in children with osteogenesis imperfecta type VII. Bone (2010) 46:820–6. 10.1016/j.bone.2009.10.037
51.
BarnesAMChangWMorelloRCabralWAWeisMEyreDRet al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med. (2006) 355:2757–64. 10.1056/NEJMoa063804
52.
CabralWAPerdivaraIWeisMTerajimaMBlissettARChangWet al. Abnormal type I collagen post-translational modification and crosslinking in a cyclophilin B KO mouse model of recessive osteogenesis imperfecta. PLoS Genet. (2014) 10:e1004465. 10.1371/journal.pgen.1004465
53.
van DijkFSNesbittIMZwikstraEHNikkelsPGPiersmaSRFratantoniSAet al. PPIB mutations cause severe osteogenesis imperfecta. Am J Hum Genet. (2009) 85:521–7. 10.1016/j.ajhg.2009.09.001
54.
MariniJCCabralWABarnesA. M. Null mutations in LEPRE1 and CRTAP cause severe recessive osteogenesis imperfecta. Cell Tissue Res. (2010) 339:59–70. 10.1007/s00441-009-0872-0
55.
PerdivaraIYamauchiMTomerKB. Molecular characterization of collagen hydroxylysine O-glycosylation by mass spectrometry: current status. Aust J Chem. (2013) 66:760–9. 10.1071/CH13174
56.
CantyEGKadlerKE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. (2005) 118:1341–53. 10.1242/jcs.01731
57.
DuranINevarezLSarukhanovAWuSLeeKKrejciPet al. HSP47 and FKBP65 cooperate in the synthesis of type I procollagen. Hum Mol Genet. (2015) 24:1918–28. 10.1093/hmg/ddu608
58.
AlanayYAvayganHCamachoNUtineGEBodurogluKAktasDet al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. (2010) 86:551–9. 10.1016/j.ajhg.2010.02.022
59.
KelleyBPMalfaitFBonafeLBaldridgeDHomanESymoensSet al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J. Bone Miner Res. (2011) 26:666–72. 10.1002/jbmr.250
60.
BarnesAMCabralWAWeisMMakareevaEMertzELLeikinSet al. Absence of FKBP10 in recessive type XI osteogenesis imperfecta leads to diminished collagen cross-linking and reduced collagen deposition in extracellular matrix. Hum. Mutat. (2012) 33:1589–98. 10.1002/humu.22139
61.
KesslerETakaharaKBiniaminovLBruselMGreenspanDS. Bone morphogenetic protein-1: the type I procollagen C-proteinase. Science (1996) 271:360. 10.1126/science.271.5247.360
62.
ProckopDJSieronALLiSW. Procollagen N-proteinase and procollagen C-proteinase. Two unusual metalloproteinases that are essential for procollagen processing probably have important roles in development and cell signaling. Matrix Biol. (1998) 16:399–408. 10.1016/S0945-053X(98)90013-0
63.
ColigeAVandenbergheIThiryMLambertCAVanBeeumen JLiSWet al. Cloning and characterization of ADAMTS-14, a novel ADAMTS displaying high homology with ADAMTS-2 and ADAMTS-3. J Biol Chem. (2002) 277:5756–66. 10.1074/jbc.M105601200
64.
LindahlKBarnesAMFratzl-ZelmanNWhyteMPHefferanTEMakareevaEet al. COL1 C-propeptide cleavage site mutations cause high bone mass osteogenesis imperfecta. Hum. Mutat. (2011) 32:598–609. 10.1002/humu.21475
65.
Hoyer-KuhnHSemlerOSchoenauERoschgerPKlaushoferKRauchF. Hyperosteoidosis and hypermineralization in the same bone: bone tissue analyses in a boy with a homozygous BMP1 mutation. Calcif Tissue Int. (2013) 93:565–70. 10.1007/s00223-013-9799-2
66.
BeckerJSemlerOGilissenCLiYBolzHJGiuntaCet al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet. (2011) 88:362–71. 10.1016/j.ajhg.2011.01.015
67.
HomanEPRauchFGrafeILietmanCDollJADawsonBet al. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J Bone Miner Res. (2011) 26:2798–803. 10.1002/jbmr.487
68.
RauchFHusseiniARoughleyPGlorieuxFHMoffattP. Lack of circulating pigment epithelium-derived factor is a marker of osteogenesis imperfecta type VI. J Clin Endocrinol Metab. (2012) 97:E1550–6. 10.1210/jc.2012-1827
69.
Al-JalladHPalomoTRoughleyPGlorieuxFHMcKeeMDMoffattPet al. The effect of SERPINF1 in-frame mutations in osteogenesis imperfecta type VI. Bone (2015) 76:115–20. 10.1016/j.bone.2015.04.008
70.
EriksenEFCharlesPMelsenFMosekildeLRisteliLRisteliJ. Serum markers of type I collagen formation and degradation in metabolic bone disease: correlation with bone histomorphometry. J Bone Miner Res. (1993) 8:127–32. 10.1002/jbmr.5650080202
71.
SeibelMJ. Biochemical markers of bone turnover part I: biochemistry and variability. Clin Biochem Rev. (2005) 26:97–122.
72.
EngelholmLHNielsenBSNetzel-ArnettSSolbergHChenXDLopezGarcia JMet al. The urokinase plasminogen activator receptor-associated protein/endo180 is coexpressed with its interaction partners urokinase plasminogen activator receptor and matrix metalloprotease-13 during osteogenesis. Lab Invest. (2001) 81:1403–14. 10.1038/labinvest.3780354
73.
AbdelgawadMESøeKAndersenTLMerrildDMHChristiansenPKjærsgaard-AndersenPet al. Does collagen trigger the recruitment of osteoblasts into vacated bone resorption lacunae during bone remodeling?Bone (2014) 67:181–8. 10.1016/j.bone.2014.07.012
74.
CsiszarK. Lysyl oxidases: a novel multifunctional amine oxidase family. Prog Nucleic Acid Res Mol Biol. (2001) 70:1–32. 10.1016/S0079-6603(01)70012-8
75.
RobinsSP. Fibrillogenesis and maturation of collagens, Chapter 2. In: SeibelMJRobinsSPBilezikianJP editors. Dynamics of Bone and Cartilage Metabolism, 2nd Edn.Burlington, ON: Academic Press (2006). p. 41–53.
76.
PerumalSAntipovaOOrgelJPRO. Collagen fibril architecture, domain organization, and triple-helical conformation govern its proteolysis. Proc Natl Acad Sci USA. (2008) 105:2824–9. 10.1073/pnas.0710588105
77.
NurminskayaMVBelkinAM. Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol. (2012) 294:1–97. 10.1016/B978-0-12-394305-7.00001-X
78.
FortunatiDChauDWangZCollighanRGriffinM. Cross-linking of collagen I by tissue transglutaminase provides a promising biomaterial for promoting bone healing. Amino Acids (2014) 46:1751–61. 10.1007/s00726-014-1732-0
79.
BoskeyAL. Mineralization, structure and function of bone, Chapter 12. In: SeibelMJRobinsSPBilezikianJP editors. Dynamics of Bone and Cartilage Metabolism, 2nd Edn.Burlington, ON: Academic Press (2006). p. 201–212.
80.
GeorgeAVeisA. Phosphorylated proteins and control over apatite nucleation, crystal growth, and inhibition. Chem Rev. (2008) 108:4670–93. 10.1021/cr0782729
81.
HelfrichMHHortonMA. Integrins and other adhesion molecules, Chapter 8. In: SeibelMJRobinsSPBilezikianJP editors. Dynamics of Bone and Cartilage Metabolism, 2nd Edn.Burlington, ON: Academic Press (2006). p. 129–151.
82.
DomonTOsanaiMYasudaMSekiETakahashiSYamamotoTet al. Mononuclear odontoclast participation in tooth resorption: the distribution of nuclei in human odontoclasts. Anat Rec. (1997) 249:449–57.
83.
BoyleWJSimonetWSLaceyDL. Osteoclast differentiation and activation. Nature (2003) 423:337–42. 10.1038/nature01658
84.
NakamuraHSatoGHirataAYamamotoT. Immunolocalization of matrix metalloproteinase-13 on bone surface under osteoclasts in rat tibia. Bone (2004) 34:48–56. 10.1016/j.bone.2003.09.001
85.
AtleyLMMortJSLalumiereMEyreDR. Proteolysis of human bone collagen by cathepsin K: characterization of the cleavage sites generating the cross-linked N-telopeptide neoepitope. Bone (2000) 26:241–7. 10.1016/S8756-3282(99)00270-7
86.
DelaisséJMAndersenTLEngsigMTHenriksenKTroenTBlavierL. Matrix metalloproteinases (MMP) and cathepsin K contribute differently to osteoclastic activities. Microsc Res Tech. (2003) 61:504–13. 10.1002/jemt.10374
87.
RawlingsNDBarrettAJBatemanA. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. (2011) 40:D343–50. 10.1093/nar/gkr987
88.
KafienahWBrömmeDButtleDJCroucherLJHollanderAP. Human cathepsin K cleaves native type I and II collagens at the N-terminal end of the triple helix. Biochem J. (1998) 331:727–32. 10.1042/bj3310727
89.
NovinecMLenarcicB. Cathepsin K: a unique collagenolytic cysteine peptidase. Biol Chem. (2013) 394:1163–79. 10.1515/hsz-2013-0134
90.
AgudaAHPanwarPDuXNguyenNTBrayerGDBrömmeD. Structural basis of collagen fiber degradation by cathepsin K. Proc Natl Acad Sci USA. (2014) 111:17474–9. 10.1073/pnas.1414126111
91.
BossardMJTomaszekTAThompsonSKAmegadzieBYHanningCRJonesCet al. Proteolytic activity of human osteoclast cathepsin K. Expression, purification, activation, and substrate identification. J Biol Chem. (1996) 271:12517–24. 10.1074/jbc.271.21.12517
92.
LiZKienetzMCherneyMMJamesMNGBrömmeD. The crystal and molecular structures of a cathepsin K: chondroitin sulfate complex. J Mol Biol. (2008) 383:78–91. 10.1016/j.jmb.2008.07.038
93.
ChapurlatRD. Odanacatib for the treatment of postmenopausal osteoporosis. Expert Opin Pharmacother. (2014) 15:97–102. 10.1517/14656566.2014.853038
94.
NallasethFSLecailleFLiZBrömmeD. The role of basic amino acid surface clusters on the collagenase activity of cathepsin K. Biochemistry (2013) 52:7742–52. 10.1021/bi401051j
95.
SharmaVPanwarPO'DonoghueAJCuiHGuidoRVCraikCSet al. Structural requirements for the collagenase and elastase activity of cathepsin K and its selective inhibition by an exosite inhibitor. Biochem J. (2015) 465:163–73. 10.1042/BJ20140809
96.
PanwarPDuXSharmaVLamourGCastroMLiHet al. Effects of cysteine proteases on the structural and mechanical properties of collagen fibers. J Biol Chem. (2013) 288:5940–50. 10.1074/jbc.M112.419689
97.
JohnsonMRPolymeropoulosMHVosHLOrtizde Luna RIFrancomanoCA. A nonsense mutation in the cathepsin K gene observed in a family with pycnodysostosis. Genome Res. (1996) 6:1050–5. 10.1101/gr.6.11.1050
98.
HouWSBrömmeDZhaoYMehlerEDusheyCWeinsteinHet al. Characterization of novel cathepsin K mutations in the pro and mature polypeptide regions causing pycnodysostosis. J Clin Invest. (1999) 103:731–8. 10.1172/JCI653
99.
SaftigPHunzikerEWehmeyerOJonesSBoydeARommerskirchWet al. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci USA. (1998) 95:13453–8. 10.1073/pnas.95.23.13453
100.
SaftigPHunzikerEEvertsVJonesSBoydeAWehmeyerOet al. Functions of cathepsin K in bone resorption: Lessons from cathepsin K deficient mice. In: LangnerJAnsorgeS, editors. Cellular Peptidases in Immune Functions and Diseases 2, New York, Boston, Dordrecht, London, Moscow: Kluwer Academic/Plenum Publishers (2000).
101.
KivirantaRMorkoJAlataloSLNicAmhlaoibhRRisteliJLaitala-LeinonenTet al. Impaired bone resorption in cathepsin K-deficient mice is partially compensated for by enhanced osteoclastogenesis and increased expression of other proteases via an increased RANKL/OPG ratio. Bone (2004) 36:159–72. 10.1016/j.bone.2004.09.020
102.
BühlingFRöckenCBraschFHartigRYasudaYSaftigPet al. Pivotal role of cathepsin K in lung fibrosis. Am J Pathol. (2004) 164:2203–16. 10.1016/S0002-9440(10)63777-7
103.
ZhangDLeungNWeberESaftigPBrömmeD. The effect of cathepsin K deficiency on airway development and TGF-b1 degradation. Respir Res. (2011) 12:72. 10.1186/1465-9921-12-72
104.
DauthSSîrbulescuRFJordansSRehdersMAvenaLOswaldJet al. Cathepsin K deficiency in mice induces structural and metabolic changes in the central nervous system that are associated with learning and memory deficits. BMC Neurosci. (2011) 12:74. 10.1186/1471-2202-12-74
105.
ChiusaroliRMaierAKnightMCByrneMCalviLMBaronRet al. Collagenase cleavage of type I collagen is essential for both basal and parathyroid hormone (PTH)/PTH-related peptide receptor-induced osteoclast activation and has differential effects on discrete bone compartments. Endocrinology (2003) 144:4106–16. 10.1210/en.2003-0254
106.
Fanjul-FernándezMFolguerasARCabreraSLópez-OtínC. Matrix metalloproteinases: evolution, gene regulation and functional analysis in mouse models. Biochim Biophys Acta (2010) 1803:3–19. 10.1016/j.bbamcr.2009.07.004
107.
KraneSMInadaM. Matrix metalloproteinases and bone. Bone (2008) 43:7–18. 10.1016/j.bone.2008.03.020
108.
HiroseTPattersonCPourmotabbedTMainardiCLHastyKA. Structure-function relationship of human neutrophil collagenase: identification of regions responsible for substrate specificity and general proteinase activity. Proc Natl Acad Sci USA. (1993) 90:2569–73. 10.1073/pnas.90.7.2569
109.
ChungLShimokawaKIDinakarpandianDGramsFFieldsGBNagaseH. Identification of the 183RWTNNFREY191Region as a critical segment of matrix metalloproteinase 1 for the expression of collagenolytic activity. J Biol Chem. (2000) 275:29610–7. 10.1074/jbc.M004039200
110.
ChungLDinakarpandianDYoshidaNLauer-FieldsJLFieldsGBVisseRet al. Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. (2004) 23:3020–30. 10.1038/sj.emboj.7600318
111.
SarkarSKMarmerBGoldbergGNeumanKC. Single-molecule tracking of collagenase on native type I collagen fibrils reveals degradation mechanism. Curr Biol. (2012) 22:1047–56. 10.1016/j.cub.2012.04.012
112.
VincentiMPBrinckerhoffCE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. (2002) 4:157–64. 10.1186/ar401
113.
LiuHKatoYErzingerSAKiriakovaGMQianYPalmieriDet al. The role of MMP-1 in breast cancer growth and metastasis to the brain in a xenograft model. BMC Cancer (2012) 12:583. 10.1186/1471-2407-12-583
114.
HayamiTKapilaYLKapilaS. MMP-1 (collagenase-1) and MMP-13 (collagenase-3) differentially regulate markers of osteoblastic differentiation in osteogenic cells. Matrix Biol. (2008) 27:682–92. 10.1016/j.matbio.2008.07.005
115.
SasakiKTakagiMKonttinenYTSasakiATamakiYOginoTet al. Upregulation of matrix metalloproteinase (MMP)-1 and its activator MMP-3 of human osteoblast by uniaxial cyclic stimulation. J Biomed Mater Res B Appl Biomater. (2007) 80B:491–8. 10.1002/jbm.b.30622
116.
NeidhartMBaraliakosXSeemayerCZelderCGayREMichelBAet al. Expression of cathepsin K and matrix metalloproteinase 1 indicate persistent osteodestructive activity in long-standing ankylosing spondylitis. Ann Rheum Dis. (2009) 68:1334–9. 10.1136/ard.2008.092494
117.
KaspirisAKhaldiLGrivasTBVasiliadisEKouvarasIDagkasSet al. Subchondral cyst development and MMP-1 expression during progression of osteoarthritis: an immunohistochemical study. Orthop Traumatol Surg Res. (2013) 99:523–9. 10.1016/j.otsr.2013.03.019
118.
KennedyAMInadaMKraneSMChristiePTHardingBLópez-OtínCet al. MMP13 mutation causes spondyloepimetaphyseal dysplasia, Missouri type (SEMDMO). J Clin Invest. (2005) 115:2832–42. 10.1172/JCI22900
119.
LauschEKepplerRHilbertKCormier-DaireVNikkelSNishimuraGet al. Mutations in MMP9 and MMP13 determine the mode of inheritance and the clinical spectrum of metaphyseal anadysplasia. Am J Hum Genet. (2009) 85:168–78. 10.1016/j.ajhg.2009.06.014
120.
TakaishiHKimuraTDalalSOkadaYD'ArmientoJ. Joint diseases and matrix metalloproteinases: a role for MMP-13. Curr Pharm Biotechnol. (2008) 9:47–54. 10.2174/138920108783497659
121.
LittleCBBaraiABurkhardtDSmithSMFosangAJWerbZet al. Matrix metalloproteinase 13–deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheumat. (2009) 60:3723–33. 10.1002/art.25002
122.
NeuholdLAKillarLZhaoWSungMLAWarnerLKulikJet al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J Clin Invest. (2001) 107:35–44. 10.1172/JCI10564
123.
StickensDBehonickDJOrtegaNHeyerBHartensteinBYuYet al. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development (2004) 131:5883–95. 10.1242/dev.01461
124.
Vanden Steen PEDuboisBNelissenIRuddPMDwekRAOpdenakkerG. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit Rev Biochem Mol Biol. (2002) 37:375–536. 10.1080/10409230290771546
125.
OkadaYNakaKKawamuraKMatsumotoTNakanishiIFujimotoNet al. Localization of matrix metalloproteinase 9 (92-kilodalton gelatinase/type IV collagenase = gelatinase B) in osteoclasts: implications for bone resorption. Lab Invest. (1995) 72:311–322.
126.
VuTHShipleyJMBergersGBergerJEHelmsJAHanahanDet al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell (1998) 93:411–22. 10.1016/S0092-8674(00)81169-1
127.
EngsigMTChenQJVuTHPedersenACTherkidsenBLundLRet al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J. Cell Biol. (2000) 151:879–90. 10.1083/jcb.151.4.879
128.
LiZHouWSEscalante-TorresCRGelbBDBrommeD. Collagenase activity of cathepsin K depends on complex formation with chondroitin sulfate. J. Biol Chem. (2002) 277:28669–76. 10.1074/jbc.M204004200
129.
EngelholmLHIngvarsenSJurgensenHJHilligTMadsenDHNielsenBSet al. The collagen receptor uPARAP/Endo180. Front Biosci. (2009) 14:2103–14. 10.2741/3365
130.
EyreD. New biomarkers of bone resorption. J Clin Endocrinol Metab. (1992) 74:470A–C. 10.1210/jcem.74.3.1740477
Summary
Keywords
bone, cathepsin K, collagen type I, discoidin domain receptors, integrins, leukocyte immunoglobulin-like receptor complex, matrix metalloproteinases, uPARAP/Endo180
Citation
Boraschi-Diaz I, Wang J, Mort JS and Komarova SV (2017) Collagen Type I as a Ligand for Receptor-Mediated Signaling. Front. Phys. 5:12. doi: 10.3389/fphy.2017.00012
Received
19 December 2016
Accepted
25 April 2017
Published
16 May 2017
Volume
5 - 2017
Edited by
Ewald Moser, Medical University of Vienna, Austria
Reviewed by
Fei Geng, McMaster University, Canada; Vincenza Ferraro, Institut National de la Recherche Agronomique (INRA), France
Updates
Copyright
© 2017 Boraschi-Diaz, Wang, Mort and Komarova.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svetlana V. Komarova svetlana.komarova@mcgill.ca
This article was submitted to Biomedical Physics, a section of the journal Frontiers in Physics
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.