Abstract
We present a comprehensive analysis of the electronic structure of the PF anion, a prototypical octahedral molecular system with high symmetry. Using symmetry-adapted linear combinations of atomic orbitals and group theoretical techniques, we construct molecular orbitals and provide a systematic classification according to irreducible representations of the point group. The role of the P-centered , , and orbitals, together with the symmetry-adapted 2 and 2 orbitals of the six surrounding fluorine atoms, is explicitly analyzed. The electronic structure is described both within the theory of molecular orbitals and with the picture based on hybridization. Maximally localized Wannier functions derived from first-principles density functional theory calculations using the siesta and wannier90 codes are computed. The constructed Wannier functions accurately reflect the expected molecular symmetries and provide a natural minimal basis for tight-binding and second-principles modeling. A detailed comparison is made between bonding, nonbonding, and antibonding orbitals, as well as their energetic ordering. Our results demonstrate the interplay between symmetry, bonding, and electronic structure in molecular systems with high cubic symmetry and set the stage for the development of accurate minimal models for such systems.
1 Introduction
Understanding the electronic structure of molecules with high symmetry is crucial for rationalizing their stability, reactivity, and spectroscopic properties. The hexafluorophosphate anion, , serves as a paradigmatic system in this regard. Its octahedral geometry, with six fluorine atoms located at the vertices of a regular octahedron around a central phosphorus atom, leads to an ideal point group symmetry. This high symmetry allows for a rigorous analysis of its electronic structure using group-theoretical techniques, providing valuable insights into orbital interactions and chemical bonding.
The anion has attracted significant attention due to its widespread use in electrochemical and energy storage applications. Its high thermal stability, low nucleophilicity, and chemically inert character make it a common component in electrolytes for lithium-ion and lithium-air batteries, as well as in ionic liquids and superacid chemistry. At electrode interfaces, modulates local electronic environments, influencing O–O bond activity and improving kinetics in Li/O2 battery systems [1].
The choice of anion plays a critical role in determining battery performance, with offering an optimal balance of ionic conductivity and electrochemical stability compared to and other salts [2]. Luan et al. [3] reported that decomposition at the graphite/electrolyte interface affects solid electrolyte interphase stability, a key factor in long-term cell performance. In lithium-ion electrolytes, is valued for its combined conductivity, stability, and compatibility with electrodes. Beyond ionic transport, influences solvation-sheath structure and interfacial chemistry [4].
Recent investigations indicate that actively participates in interfacial reactions rather than acting as a passive anion. Spotte-Smith et al. [5] revealed decomposition through P–F bond cleavage, while Ruan et al. [6] and Qi et al. [7] demonstrated that governs solvation-sheath composition and charge-transfer kinetics. Wang et al. [8] showed that optimized co-solvents can stabilize under high-voltage operation.
At the cathode interface, Peiris et al. [9] and Xing et al. [10] highlighted that anion identity dictates oxygen redox activity and stability. Moreover, adsorption or substitutional interaction with graphene surfaces has been reported to reduce accumulation by altering charge distribution and promoting toroidal growth, thereby mitigating cathode passivation and enhancing cycle reversibility [7, 9].
However, most theoretical treatments still idealize as a uniform anion, neglecting its localized electronic structure and symmetry-dependent orbital interactions. A deeper understanding of these features is essential for accurate modeling of electrochemical behavior.
Simulating an entire electrochemical cell at the first-principles level remains computationally prohibitive due to its size and complexity. Hence, reduced-order frameworks such as second-principles methods [11] based on tight-binding Hamiltonians constructed from maximally localized Wannier functions (MLWFs) enable efficient yet chemically accurate modeling of such systems. First steps in this direction have been taken in our group, where (a key discharge product in non-aqueous lithium-air systems) has been extensively studied [12]. In particular, the coupling between lattice distortions and electronic states was characterized.
In this work, we present a comprehensive study of the isolated anion, focusing on the construction of symmetry-adapted MOs and a compact, chemically intuitive MLWF basis. The MOs are analyzed using both symmetry-adapted linear combination (SALC) methods and first-principles density functional theory calculations, implemented with siesta and wannier90. We emphasize the hybridization between phosphorus 3, 3, 3 and fluorine 2, 2 orbitals under symmetry. The resulting MLWFs bridge molecular orbital and real-space bonding pictures, providing a transparent foundation for building low-energy Hamiltonians. All pseudopotentials, basis sets, and parameters are made openly available for future second-principles modeling.
The rest of this paper is organized as follows. Section 2 outlines the computational methodology. Section 3 describes the relaxed atomic structure. In Section 4, we analyze the electronic states and their symmetry classification. Section 5 discusses the construction and properties of the MLWFs, along with the resulting tight-binding Hamiltonian. The paper concludes with a summary of the main findings and perspectives for future work.
2 Methodology
Our calculations were carried out within the framework of density functional theory (DFT) [13], employing the generalized gradient approximation (GGA) for the treatment of exchange and correlation effects. The simulations relied on a numerical atomic orbital (NAO) basis, as implemented in the siesta code [14, 15]. We adopted the Perdew-Burke-Ernzerhof (PBE) GGA functional [16] to describe exchange-correlation interactions, utilizing the implementation provided by the libxc library [17, 18]. Given that semilocal GGA functionals suffer from self-interaction errors and deviations from piecewise linearity (thereby violating the Koopmans condition) we additionally performed calculations using the range-separated HSE06 hybrid functional [19], which mitigates these shortcomings and provides a more reliable estimate of the HOMO–LUMO gap by exploiting the newly implemented hybrid-functional capabilities of siesta.
Core electrons were modeled using norm-conserving pseudopotentials in the fully separable Kleinman-Bylander form [20]. In particular, we employed the optimized norm-conserving Vanderbilt pseudopotentials developed by Hamann [21], provided in the psml format [22], as distributed by the Pseudo-Dojo project [23, 24]. For fluorine and phosphorus atoms, the valence electrons included the and orbitals (F) and the and orbitals (P), respectively.
The electronic states were expanded using a basis set of strictly confined numerical atomic orbitals [25, 26], obtained as eigenfunctions of the isolated atoms subjected to a soft confinement potential [27]. The basis quality was varied by choosing between double- and triple- sets, corresponding to two or three radial components per valence orbital (i.e., , for F and , for P). To improve the angular flexibility of the basis, we added polarization functions with character on both species, including either one or two radial functions per angular shell, depending on the desired level of polarization (single or double). All basis set parameters were variationally optimized for the relaxed molecular geometry, using the procedure described in Ref. [27] and reference data obtained from fully converged plane-wave calculations.
All real-space integrals–such as the electronic density, the Hartree potential, and the exchange-correlation contributions–were evaluated on a uniform grid. The grid spacing was chosen to be equivalent to a plane-wave cutoff energy of 600 Ry, ensuring sufficient numerical accuracy [14].
To simulate the anion in isolation, we placed the molecule in a cubic supercell with 20 Å of side, sufficiently large to suppress interactions between periodic images. The system was given a net charge of to account for the additional electron. For charged cells under periodic boundary conditions, electrostatic artifacts arise from the interaction between the charge and its periodic images. These were corrected by subtracting the leading-order Madelung energy, following the approach of Ref. [28].
Geometrical optimizations were performed using the conjugate gradient method, and the atomic coordinates were relaxed until all residual forces were below 10 meV/Å.
To validate the completeness of the NAO basis sets, we benchmarked our results against those from the abinit package [29–31], which uses a plane-wave basis. The comparison was designed to be as consistent as possible. Both siesta and abinit utilized the same pseudopotentials in psml format, incorporating an identical separation between the local potential and the nonlocal Kleinman-Bylander projectors. The same exchange-correlation functional was used in both codes, sourced from a common version of libxc. The primary difference lies in the basis representation: while siesta employed numerical atomic orbitals, abinit used a plane-wave basis with a cutoff energy of 60 Ha, sufficient to ensure convergence of total energies and forces.
3 Structural properties
The isolated is octahedral in shape. Each fluorine atom sits on one of the six vertices of a regular octahedron with the phosphorus atom in the centre. Therefore, it exhibits symmetry in its ideal isolated form. The only internal degree of freedom is the distance between the P and the F atoms. Numerous experimental and theoretical studies have reported values for the P–F bond distance. X-ray diffraction measurements on crystalline salts such as [N()4 [32] indicate a slightly distorted octahedral environment, with axial P–F distances ranging from 1.585 to 1.592Å, and equatorial bonds of 1.568 Å. The associated F–P–F bond angles were found to be 90 and 180 within experimental uncertainty. In other compounds, such as P()4, the measured P–F distances are slightly shorter, in the range of 1.555–1.556 Å. It is important to note that direct experimental observation of the anion in the gas phase or in solution is not feasible with current techniques. As such, experimental bond lengths obtained from crystals should be viewed as reference values for comparison. Computational investigations of the isolated molecule have also been reported. Xuan et al. [33] conducted a detailed analysis using basis sets of Gaussian-type orbitals and varying levels of theory. The computed P–F bond length spans a range from 1.5897 Å at the Hartree-Fock level to 1.6292 Å using the hybrid B3LYP functional, and 1.6348 Å within the MP2 formalism. In siesta, using a TZDP basis set within the HSE06 range-separated hybrid functional, the equilibrium distance amounts to 1.625 Å.
Table 1 presents the convergence behavior of numerical atomic orbital (NAO) basis sets for the anion in the gas phase. Results obtained using progressively larger, variationally optimized basis sets are compared against a reference calculation performed with a plane-wave (PW) basis set at a cutoff energy of 60 Ha, which serves as the converged-basis limit. All other computational parameters, including the pseudopotentials and exchange-correlation functional, were kept identical across all simulations to ensure consistency. It is important to emphasize that this reference PW result differs from lower-cutoff plane-wave calculations frequently employed in the literature, which may not fully eliminate basis-set incompleteness errors. A clear and systematic convergence trend is observed as the NAO basis size increases. The fully converged plane-wave result, where remaining errors are primarily associated with the choice of exchange-correlation functional and the pseudopotentials, yields a total energy marginally lower (by approximately 0.4%) than the most accurate NAO calculation tried here. As previously discussed, direct comparison with experimental bond lengths is not straightforward due to the lack of measurements for the isolated anion in the gas phase. Nevertheless, the P–F distance obtained with the siesta calculations overestimates the experimental values by roughly 4%, a deviation that falls within the expected range for simulations based on the PBE functional.
TABLE 1
| Basis set | Energy | |
|---|---|---|
| DZP | 1.652 | −4220.540302 |
| TZP | 1.647 | −4220.691276 |
| TZDP | 1.647 | −4220.760023 |
| PW | 1.641 | −4222.390066 |
| Expt | 1.568–1.592 |
Structural properties of molecule. refers to the internal coordinate (P-F distance), in Å. Energy is the total energy of the relaxed structure (in eV). DZP, TZP, and TZDP stands for double- polarized, triple- polarized, and triple- double polarized, respectively. PW stands for a plane wave calculation carried out with the abinit code, with a cutoff of 60 Ha. The experimental geometry is taken from Ref. [32].
4 Electronic structure
The anion adopts a highly symmetric octahedral geometry, described by the point group as illustrated in Figure 1. Its electronic structure can be efficiently analyzed by decomposing the valence atomic orbitals of phosphorus and fluorine into SALC that transform as irreducible representations of . This group-theoretical framework allows a systematic construction of MOs classified by symmetry, and highlights the role of bonding, nonbonding, and antibonding interactions in the stability of the anion.
FIGURE 1
4.1 Symmetry-adapted linear combinations of the phosphorus-centered valence orbitals
The valence shell of the central phosphorus atom includes the 3 and 3 orbitals, both centered at the molecular origin. Under symmetry, these atomic orbitals transform according to (i) 3: transforms as the totally symmetric representation ; and (ii) 3: transforms as the threefold degenerate vector representation .
Additionally, although not occupied in the neutral atom, the 3 orbitals of phosphorus are included in the simulations. The 3, 3, and 3 are of symmetry, while the 3 and 3 transform as .
These orbitals serve as the central symmetry anchors for the MOs and can interact with compatible SALCs formed from the valence orbitals of fluorine.
4.2 Symmetry-adapted linear combinations of the fluorine valence orbitals
The six fluorine atoms in the anion are arranged at the vertices of a regular octahedron centered at the phosphorus atom. In the following we shall discuss the SALCs for the valence 2 and 2 orbitals.
4.2.1 Symmetry-adapted linear combinations of F 2 orbitals
Each fluorine contributes a 2 orbital, denoted as with , where the atoms are labeled according to their position along the Cartesian directions, as shown in Figure 1.
A basis of six SALCs can be formed from these 2 orbitals by decomposing their reducible representation into irreducible components. Under the point group, the decomposition yields towhich corresponds to (i) one totally symmetric combination ; (ii) a triply degenerate vector-like set ; and (iii) a doubly degenerate set of quadrupolar character , as shown in Equation 1.
The totally symmetric combination , invariant under all symmetry operations of , is given byEquation 2 indicates the overlaps with the 3 orbital of the central phosphorus atom.
The SALCs transform like the , , and components of a vector and are antisymmetric with respect to inversion and are represented in Equations 3–5.These interacts with 3 orbitals of P with the same symmetry.
Finally, the two SALCs of symmetry have quadrupolar character and can be expressed as in Equations 6, 7.These combinations can interact with the and orbitals of phosphorus, which also belong to the representation.
A compact summary of the SALCs derived from the 2 orbitals is given in Table 2, including their symmetry and analytical expressions.
TABLE 2
| Symmetry | SALC expression |
|---|---|
Symmetry-adapted linear combinations of the six F 2 orbitals in , classified according to the irreducible representations of the point group.
4.2.2 Symmetry-adapted linear combinations of F 2 orbitals
Each fluorine atom in contributes three 2 valence orbitals, aligned along the local Cartesian axes: one orbital oriented radially toward the central phosphorus atom and two perpendicular orbitals , tangent to the molecular surface. With six fluorine atoms, this results in a total of 18 -type atomic orbitals.
To exploit the full symmetry of the molecule, these 18 functions can be recombined into SALCs that transform as irreducible representations of the point group. The symmetry decomposition of this reducible representation reads as in Equation 8:consisting of seven distinct irreducible representations: one nondegenerate , one doubly degenerate , and five triply degenerate (, , , , and ). The superscript or refers to the orientation of the orbitals that enter in a given combination during the generation of the symmetry adapted one.
The six orbitals (those directed along the P–F bonds) contribute to the , , and representations. These SALCs are analogous in symmetry and form to those constructed from the F 2 orbitals, as discussed in Section 4 2, and are responsible for the formation of -bonding MOs with the P 3, 3, and 3/3 orbitals.
The remaining twelve orbitals (two per fluorine, lying in planes orthogonal to the P–F bonds) contribute SALCs transforming as , , , and .
The combinations transform like the , , and orbitals, and form -type bonding interactions with the phosphorus 3, 3, 3 shell. The and combinations are symmetry-allowed but often correspond to nonbonding or weakly interacting MOs. In particular, they do not have a counterpart among the valence orbitals of the central atom and typically form nonbonding combinations. The remaining are distinct from the components already listed, but they transform under the same irrep and are orthogonal combinations within the same symmetry type.
The full set of SALCs, including all 18 linear combinations, is shown in Table 3, where each function is labeled by its symmetry and expressed analytically in terms of the original atomic orbitals , with labeling the fluorine site and indicating orientation.
TABLE 3
| Symmetry | SALC expression |
|---|---|
Complete set of SALCs of the 18 fluorine 2 orbitals in , classified according to the irreducible representations of the point group. Each denotes a 2 orbital on fluorine atom pointing along direction . The horizontal line separates the SALCs for the and orbitals.
4.3 Molecular orbital structure
The MO diagram of , shown in Figure 2, results from the interaction between the phosphorus-centered orbitals (3, 3, 3) and the SALCs of the six surrounding fluorine atoms.
FIGURE 2
The SALCs of fluorine 2 orbitals transform as , , and irreducible representations under the group. These match the symmetry of the phosphorus 3, 3, and 3- valence orbitals, respectively, enabling the formation of bonding MOs. Specifically, the SALC forms a deep bonding MO () with P 3, the SALC couples to the P 3 orbitals forming the triply degenerate set, and the symmetry SALC gives rise to the pair via weak overlap with P 3 and 3 orbitals. These three bonding combinations accommodate twelve valence electrons and constitute the low-energy part of the MO diagram in Figure 2.
Regarding the high-energy part of the MO diagram, the key features are: (i) the formation of strong bonding MOs that emerge from combinations of P 3, 3, and 3- with , , and SALCs, forming filled bonding orbitals , , and ; (ii) interactions between P 3- orbitals and F SALCs give rise to filled nonbonding levels; (iii) additional nonbonding levels such as , , and primarily retain fluorine character essentially of the same character as the , , and , and lie deeper in energy; and (iv) antibonding levels (, ) remain unoccupied.
With 48 valence electrons, attains a closed-shell configuration in which all bonding and nonbonding orbitals are filled, and antibonding states are empty.’ molecule’s high thermodynamic stability, weak coordinating behavior, and classification as a prototypical superhalogen anion.
It is important to note that symmetry provides a powerful framework for classifying MOs according to their irreducible representations and gaining insight into their character and bonding interactions. However, determining the precise energetic ordering of these orbitals requires accurate electronic-structure calculations.
4.4 Valence bond hybridization picture and electron counting
Although the electronic structure of the molecule is best described using symmetry-adapted MOs, it is instructive to consider an approximate valence-bond picture based on localized hybrid orbitals to rationalize the bonding and geometry of the anion. In this framework, the phosphorus atom undergoes hybridization by combining its valence 3, 3, and two 3 orbitals (of symmetry in ). The resulting six hybrid orbitals are oriented toward the corners of an octahedron and are singly occupied due to the presence of five valence electrons in the neutral P atom and one additional electron from the anionic charge, yielding six electrons available for bonding. Each fluorine atom contributes one 2 valence electron to form a bond with the corresponding hybrid orbital of phosphorus. This leads to the formation of six equivalent P–F bonds, each containing a pair of bonding electrons. In this picture, 12 electrons are involved in P–F bonding. The remaining valence electrons of fluorine (six F atoms six electrons = 36 electrons) reside in nonbonding orbitals. Rather than invoking hybridization at each fluorine site, these electrons are more accurately described as occupying atomic-like 2 and 2 orbitals, which are reorganized into SALCs under the point group. These SALCs form nonbonding or weakly antibonding MOs, classified according to irreducible representations such as , , , and , among others (see Section 4.2.2; Figure 2). This hybridization-based picture accounts for all 48 valence electrons in : 12 in bonding P–F orbitals and 36 in nonbonding combinations primarily localized on fluorine atoms. Although the phosphorus atom formally exceeds the octet rule by accommodating 12 electrons in its valence shell, this is permitted for third-row elements and beyond, which can utilize low-lying orbitals. The resulting electronic configuration is closed-shell and consistent with the exceptional chemical stability and weakly coordinating character of the anion. Nonetheless, this localized valence-bond picture offers only a qualitative approximation and neglects the full delocalization, symmetry, and orbital mixing present in the true MO description required to accurately capture the electronic structure of a high-symmetry anion such as .
5 Maximally localized wannier functions
Localized MOs, such as those described in Section 4, have a long-standing tradition in the chemistry literature as a powerful and intuitive framework for analyzing chemical bonding in molecular systems. These orbitals offer not only an insightful picture of bonding characteristics, coordination, and hybridization but also serve as a compact and efficient basis for high-accuracy electronic structure calculations. Maximally localized Wannier functions (MLWFs) [34, 35] represent a natural generalization of this concept to extended systems, providing a localized and orthonormal representation of the electronic structure in crystals and molecules alike. MLWFs retain much of the chemical interpretability of localized MOs while offering additional advantages: they preserve crystal symmetry, reflect the spatial distribution and coordination of electronic states, and enable the identification of transferable chemical building blocks. In this work, starting from the Kohn–Sham eigenstates computed with the siesta code, we construct MLWFs using the wannier 90 package [36].
5.1 Wannierization of each group of molecular orbitals
First of all, we have performed separate Wannierizations for each group of degenerate molecular orbitals, as listed in Table 4. Specifically, we considered: (i) One manifold for each irreducible representation: a singlet (e.g., ), a doublet , or a triplet (, , , ); (ii) For each manifold, the Wannierization was constrained to a fixed subspace formed by the corresponding molecular orbitals, ensuring no mixing with orbitals of other symmetry; (iii) Under these conditions, the resulting MLWFs are, by construction, equivalent to the original symmetry-adapted molecular orbitals, and the corresponding Hamiltonian matrices are strictly diagonal within each manifold, and the elements of the diagonal equal the eigenvalues of the Kohn–Sham Hamiltonian, given in Table 4. The resulting MLWFs are plotted in Table 4, where the clearly resemblances with the MOs described in Section 4.3 are obvious. Figure 2 shows the resulting Wannier functions, where their expected transformation properties under the point group can be clearly identified. Due to the symmetry of the molecule, all the MLWfs are centered at the origin, and the spreadings range between 2.34 and 3.17 Å2. While this approach is pedagogically useful (highlighting the symmetry and spatial character of each molecular orbital) it does not exploit the full flexibility of the Wannier construction in terms of compactness and spatial localization.
TABLE 4
| Molecular orbital | Eigenvalue (eV) | Molecular orbital/MLWF shape |
|---|---|---|
| 4 | −28.53 |  |
| 3 | −26.59 |  |
| 2 | −25.79 | 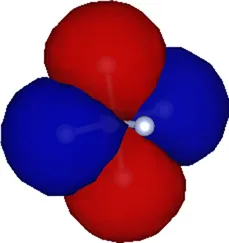 |
| 5 | −13.51 | 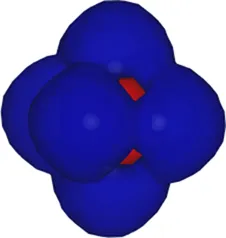 |
| 4 | −10.47 |  |
| 1 | −8.33 | 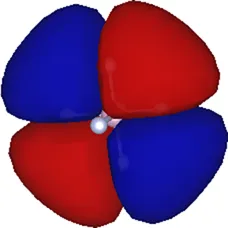 |
| 3 | −8.05 | 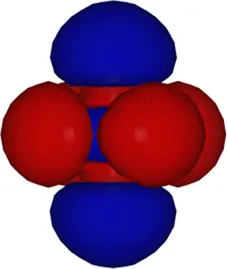 |
| 5 | −6.57 |  |
| 1 | −6.37 | 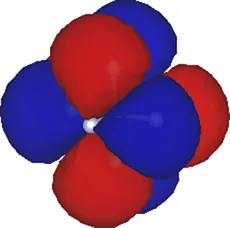 |
| 1 | −5.49 |  |
Kohn–Sham eigenvalues for the molecular orbitals of the molecule obtained using a TZDP basis set at the optimized geometry reported in Table 1. The right column displays the corresponding maximally localized Wannier functions, whose spatial shapes coincide with those of the MOs. All energies are given in eV.
5.2 Global wannierization of all occupied orbitals
To complement the symmetry-resolved construction discussed above, we have also performed a global Wannierization of all 24 occupied molecular orbitals of the anion. In this case, we used the full set of fluorine and orbitals (four per F atom, for a total of 24) as initial projections. Importantly, we allowed full mixing between molecular orbitals of different symmetry, as no constraint was imposed on the subspace. This approach leads to a set of 24 compact MLWFs, each strongly localized around a specific fluorine atom. The resulting Wannier functions naturally group into six equivalent subsets, one per fluorine atom. A careful inspection of their spatial structure reveals additional chemically intuitive features: the Wannier function located farthest from the phosphorus atom exhibits a clear -bonding character, while the Wannier function closest to the phosphorus shows a -antibonding character, as evidenced by the position of the nodal plane that cuts through the P–F bond. Furthermore, the two remaining Wannier functions, which appear as a degenerate pair, display character, identifiable from the orientation of their lobes, which lie in directions perpendicular to the corresponding P–F bond. The spreads of these MLWFs, listed in Table 5, range from 0.37 to 0.53 Å2, much smaller than those obtained from the symmetry-constrained scheme, reflecting the increased compactness enabled by full mixing among orbitals. In this fully localized Wannier basis, the tight-binding Hamiltonian is no longer diagonal, but includes inter-orbital hopping terms within and between fluorine centers. These couplings can be used as input for second-principles simulations of more complex -containing materials [11]. When the Hamiltonian expressed in this fully localized Wannier basis is mapped onto a tight-binding model, the resulting eigenvalues reproduce exactly those reported in Table 4, confirming the internal consistency of the Wannier construction and its suitability as a minimal basis for model Hamiltonians. These localized Wannier functions provide a compact representation of the occupied states and reproduce the Kohn–Sham eigenvalues accurately, validating the fidelity of the projection. This result highlights the versatility of the Wannier construction: symmetry-adapted orbitals offer insight into chemical bonding and orbital interactions, while globally localized Wannier functions provide a minimal, transferable, and efficient basis for modeling. Although this study focuses on the isolated anion, the resulting Wannier Hamiltonian offers a chemically intuitive framework for embedding into extended systems. In particular, MLWFs from global localization are well-suited for second-principles or tight-binding models of -containing environments, such as crystalline salts, solvated clusters, and electrochemical interfaces. This facilitates future studies of bonding, polarization, and charge transfer under realistic conditions.
TABLE 5
| Wannier center (Å) | Spreading (Å2) | MLWF shape |
|---|---|---|
| (1.911, 0.000, 0.000) | 0.374 | 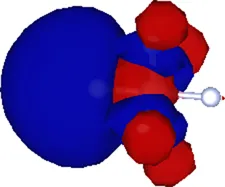 |
| (1.200, 0.000, 0.000) | 0.394 | 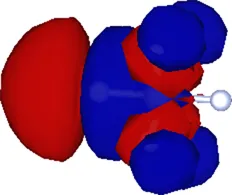 |
| (1.608, 0.000, 0.000) | 0.537 | 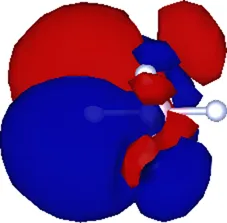 |
| (1.608, 0.000, 0.000) | 0.537 | 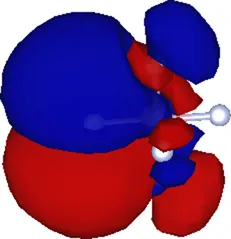 |
Cartesian coordinates of the center and spatial spreads of the maximally localized Wannier functions (MLWFs) obtained when all occupied molecular orbitals are simultaneously included in the Wannierization procedure. Only the four Wannier functions centered near the atom (as labeled in Figure 1) are shown. For reference, the equilibrium position of the atom in the relaxed geometry using the TZDP basis is (1.641, 0.000, 0.000) Å.
5.3 Differences in energy between HOMO and LUMO
The remarkable stability and low reactivity of the PF anion, often emphasized in the literature, can be quantitatively rationalized through the computed HOMO–LUMO energy separation (1–6). Within the PBE functional, this gap amounts to 8.35 eV for , compared with 7.47 eV for the anion commonly used in battery electrolytes. Within the HSE06 functional, the HOMO-LUMO gap increases up to 11.96 eV for the TZDP basis set quality. This sizable separation supports the experimentally observed chemical inertness of and provides a simple electronic-structure indicator of its exceptional stability relative to other anions of similar functionality.
6 Comparison with previous MO and population-based treatments
The electronic structure of the anion has been extensively discussed in the literature using traditional molecular orbital (MO) theory and population-based methods, including Mulliken charges, NBO analysis, and energy-level diagrams. While these approaches offer chemically intuitive insights into bonding, they lack both real-space localization and symmetry-enforced orbital orthogonality. As such, they are not readily transferable to multiscale models or tight-binding Hamiltonians.
In contrast, the present work employs both symmetry-constrained and globally localized maximally localized Wannier functions (MLWFs), derived from first-principles DFT calculations. The symmetry-constrained MLWFs reproduce the expected irreducible representations of the Oh point group, providing a rigorous foundation for classifying molecular orbitals. More significantly, the global Wannierization procedure allows for full mixing across symmetry manifolds, producing a minimal and chemically insightful basis localized around each fluorine atom.
Each ligand-centered MLWF naturally decomposes into bonding, antibonding, or -character, as revealed by their spatial shape and nodal structure. These MLWFs exhibit significantly reduced spatial spreads (down to 0.37 Å2), far below those of symmetry-constrained orbitals (2.3–3.2 Å2). Moreover, the resulting tight-binding Hamiltonian–constructed from the MLWF overlaps–reproduces the full Kohn–Sham eigenvalue spectrum with high fidelity, enabling physically grounded coarse-grained modeling.
To the best of our knowledge, no previous study has implemented a global Wannierization of to derive a transferable minimal basis suitable for model Hamiltonians. This approach thus bridges the gap between chemical intuition and first-principles electronic structure theory, and positions as a prototypical system for symmetry-aware multiscale simulation frameworks.
7 Conclusion
In this work, we have analyzed in detail the electronic structure of the anion, combining group theory, chemical intuition, and first-principles calculations. The high symmetry of the molecule permits the construction of SALCs of the fluorine orbitals, which form a natural basis for building MOs with well-defined symmetry properties. We have examined the contributions of the phosphorus 3, 3, and 3 orbitals and their interaction with the F 2 and 2 SALCs, identifying the resulting bonding, nonbonding, and antibonding combinations. The comparison between the traditional hybridization model and the MO picture derived from symmetry provides complementary insights into the bonding nature of the system. By projecting the Kohn–Sham states onto MLWFs, we obtained a minimal orthogonal basis that reproduces the symmetry and bonding character of the system and is suitable for tight-binding and model Hamiltonian construction. The obtained MLWFs confirm the expected irreducible representations and spatial localizations. Overall, the methodology employed here–combining symmetry analysis, localized orbital construction, and first-principles simulations–offers a robust and general approach to studying the electronic structure of molecular and extended systems with high symmetry, and lays the groundwork for efficient multiscale modeling strategies. The previous results on the structural and electronic properties of validates the combined use of the optimized norm-conserving Vanderbilt pseudopotentials proposed in Ref. [21] in combination with a basis of numerical atomic orbitals [37].
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
PM: Writing – original draft, Methodology, Investigation, Writing – review and editing, Conceptualization, Software. ET: Writing – review and editing, Writing – original draft, Investigation. PR: Writing – original draft, Investigation, Writing – review and editing, Supervision. VS: Investigation, Conceptualization, Supervision, Writing – review and editing, Writing – original draft. BM: Writing – review and editing, Conceptualization, Writing – original draft, Supervision. JJ: Writing – review and editing, Supervision, Investigation, Conceptualization, Software, Methodology, Writing – original draft.
Funding
The author(s) declared that financial support was received for this work and/or its publication. We acknowledge support from Erasmus+ KA-107 action and the Vice-Rector for Internationalisation and Global Engagement of the University of Cantabria. JJ acknowledges financial support from Grant No. PID2022-139776NB-C63 funded by MCIN/AEI/10.13039/501100011033 and by ERDF “A way of making Europe” by the European Union.
Conflict of interest
The author(s) declared that this work was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declared that generative AI was not used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
LaoireCOMukerjeeSAbrahamKMPlichtaEJHendricksonMA (2009). Elucidating the mechanism of oxygen reduction for lithium-air battery applications. J Phys Chem C. 113:20127–34. 10.1021/jp908090s
2.
GunasekaraIMukerjeeSPlichtaEJHendricksonMAAbrahamKM. A study of the influence of lithium salt anions on oxygen reduction reactions in Li-air batteries. J Electrochem Soc (2015) 162:A1055–A1066. 10.1149/2.0841506jes
3.
LuanYHuRFangYZhuKChengKYanJet alNitrogen and phosphorus dual-doped multilayer graphene as universal anode for full carbon-based lithium and potassium ion capacitors. Nanomicro Lett (2019) 11:30. 10.1007/s40820-019-0260-6
4.
DingJYangCHuWLiuXZhangAPengDet alStabilizing the electrode–electrolyte interface for high-voltage Li——LiCoO2 cells using dual electrolyte additives. Chem Sci (2025) 16:13723–30. 10.1039/d5sc03120f
5.
Spotte-SmithEWCPetrocelliTBPatelHDBlauSMPerssonKA. Elementary decomposition mechanisms of lithium hexafluorophosphate in battery electrolytes and interphases. ACS Energy Lett. (2023) 8:347–55. 10.1021/acsenergylett.2c02351
6.
RuanDTanLChenSFanJNianQChenLet alSolvent versus anion chemistry: unveiling the structure-dependent reactivity in tailoring electrochemical interphases for lithium-metal batteries. JACS Au (2023) 3:953–63. 10.1021/jacsau.3c00035
7.
QiSTangXHeJLiuJMaJ. Construction of localized high-concentration region for suppressing NCM622 cathode failure at high voltage. Small Methods (2023) 7:2201693. 10.1002/smtd.202201693
8.
WuWBaiYWangXWuC. Sulfone-based high-voltage electrolytes for high energy density rechargeable lithium batteries: progress and perspective. Chin Chem Lett (2021) 32:1309–15. 10.1016/j.cclet.2020.10.009
9.
PeirisMHCLiepinyaDLiuHSmeuM. Electrolyte reactivity, oxygen states, and degradation mechanisms of nickel-rich cathodes. Cell Rep. Phys. Sci. (2024) 5:102039. 10.1016/j.xcrp.2024.102039
10.
XingPSanglierPZhangXLiJLiYSuB-L. Advances in cathode materials for Li-O2 batteries. J Energy Chem. (2024) 95:126–67. 10.1016/j.jechem.2024.03.016
11.
García-FernándezPWojdełJCÍñiguezJJunqueraJ. Second-principles method for materials simulations including electron and lattice degrees of freedom. Phys Rev B (2016) 93:195137. 10.1103/PhysRevB.93.195137
12.
MasanjaPMFernández-RuizTTarimoEJCarral-SainzNKanaka RaoPVSinghVet alStructural and electronic properties of bulk Li2O2: first-principles simulations based on numerical atomic orbitals. J Phys Condens Matter (2025) 37:165502. 10.1088/1361-648X/adbaa6
13.
HohenbergPKohnW. Inhomogeneous electron gas. Phys Rev (1964) 136:B864–B871. 10.1103/physrev.136.b864
14.
SolerJMArtachoEGaleJDGarcíaAJunqueraJOrdejónPet alThe siesta method for ab initio order-N materials simulation. J Phys Condens Matter (2002) 14:2745–79. 10.1088/0953-8984/14/11/302
15.
GarcíaAPapiorNAkhtarAArtachoEBlumVBosoniEet alSiesta: recent developments and applications. J Chem Phys (2020) 152. 10.1063/5.0005077
16.
PerdewJPBurkeKErnzerhofM. Generalized gradient approximation made simple. Phys Rev Lett (1996) 77:3865–8. 10.1103/PhysRevLett.77.3865
17.
MarquesMAOliveiraMJBurnusT. Libxc: a library of exchange and correlation functionals for density functional theory. Comput Phys Commun (2012) 183:2272–81. 10.1016/j.cpc.2012.05.007
18.
LehtolaSSteigemannCOliveiraMJMarquesMA. Recent developments in Libxc–a comprehensive library of functionals for density functional theory. SoftwareX (2018) 7:1–5. 10.1016/j.softx.2017.11.002
19.
KrukauAVVydrovOAIzmaylovAFScuseriaGE. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J Chem Phys (2006) 125:224106. 10.1063/1.2404663
20.
KleinmanLBylanderDM. Efficacious form for model pseudopotentials. Phys Rev Lett (1982) 48:1425–8. 10.1103/physrevlett.48.1425
21.
HamannD. Optimized norm-conserving vanderbilt pseudopotentials. Phys Rev B (2013) 88:085117. 10.1103/physrevb.88.085117
22.
GarcíaAVerstraeteMJPouillonYJunqueraJ. The PSML format and library for norm-conserving pseudopotential data curation and interoperability. Comput Phys Commun (2018) 227:51. 10.1016/j.cpc.2018.02.011
23.
Van SettenMGiantomassiMBousquetEVerstraeteMJHamannDRGonzeXet alThe PseudoDojo: training and grading a 85 element optimized norm-conserving pseudopotential table. Comput Phys Commun (2018) 226:39–54. 10.1016/j.cpc.2018.01.012
24.
ONCVPs (2018). The scalar relativistic oncvpsp v0.4.1 pseudopotentials with stringent accuracy were used.
25.
SankeyOFNiklewskiDJ. Ab initio multicenter tight-binding model for molecular-dynamics simulations and other applications in covalent systems. Phys Rev B (1989) 40:3979–95. 10.1103/physrevb.40.3979
26.
ArtachoESánchez-PortalDOrdejónPGarcíaASolerJM. Linear-scaling ab-initio calculations for large and complex systems. Phys Stat Sol.(b) (1999) 215:809.
27.
JunqueraJPazOSánchez-PortalDArtachoE. Numerical atomic orbitals for linear-scaling calculations. Phys Rev B (2001) 64:235111. 10.1103/physrevb.64.235111
28.
MakovGPayneMC. Periodic boundary conditions in ab initio calculations. Phys Rev B (1995) 51:4014–22. 10.1103/physrevb.51.4014
29.
GonzeXAmadonBAngladeP-MBeukenJ-MBottinFBoulangerPet alABINIT: first-Principles approach to material and nanosystem properties. Comput Phys Commun (2009) 180:2582–615. 10.1016/j.cpc.2009.07.007
30.
GonzeXJolletFAbreu AraujoFAdamsDAmadonBApplencourtTet alRecent developments in the abinit software package. Comput Phys Commun (2016) 205:106–31. 10.1016/j.cpc.2016.04.003
31.
GonzeXAmadonBAntoniusGArnardiFBaguetLBeukenJ-Met alThe abinit project: impact, environment and recent developments. Comput Phys Commun (2020) 248:107042. 10.1016/j.cpc.2019.107042
32.
WangYCalvertLDBrownsteinSK. The structure of tetramethylammonium hexafluorophosphate. Acta Crystallogr B (1980) 36:1523. 10.1107/s0567740880006498
33.
XuanXWangJWangH. Theoretical insights into and its alkali metal ion pairs: geometries and vibrational frequencies. Electrochim Acta (2005) 50:4196–201. 10.1016/j.electacta.2005.01.045
34.
MarzariNVanderbiltD. Maximally localized generalized wannier functions for composite energy bands. Phys Rev B (1997) 56:12847–65. 10.1103/physrevb.56.12847
35.
MarzariNMostofiAAYatesJRSouzaIVanderbiltD. Maximally localized wannier functions: theory and applications. Rev Mod Phys (2012) 84:1419–75. 10.1103/revmodphys.84.1419
36.
PizziGVitaleVAritaRBlügelSFreimuthFGérantonGet alWannier90 as a community code: new features and applications. J Phys Condens Matter (2020) 32:165902. 10.1088/1361-648X/ab51ff
37.
Data Avalibity Statement (2020). The basis set used in this study is available upon reasonable request.
Summary
Keywords
density functional (DFT), electronic structure, group theory, PF6-, wanniers functions
Citation
M. Masanja P, Tarimo EJ, Rao PVK, Singh V, Mwankemwa B and Junquera J (2026) Symmetry, bonding, and wannier function construction in the molecule: a first-principles case study. Front. Phys. 14:1764641. doi: 10.3389/fphy.2026.1764641
Received
10 December 2025
Revised
04 February 2026
Accepted
10 February 2026
Published
26 February 2026
Volume
14 - 2026
Edited by
Adrian Esteban Feiguin, Northeastern University, United States
Reviewed by
Jun Yang, Dartmouth College, United States
Saikiran Kotaru, Emory University, United States
Updates
Copyright
© 2026 M. Masanja, Tarimo, Rao, Singh, Mwankemwa and Junquera.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul M. Masanja, masanjapaul2012@gmail.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.