 Tim Würger1,2
Tim Würger1,2 Christian Feiler1
Christian Feiler1 Félix Musil3
Félix Musil3 Gregor B. V. Feldbauer4Daniel Höche1,5
Gregor B. V. Feldbauer4Daniel Höche1,5 Sviatlana V. Lamaka1
Sviatlana V. Lamaka1 Mikhail L. Zheludkevich1,6
Mikhail L. Zheludkevich1,6 Robert H. Meißner1,2*
Robert H. Meißner1,2*- 1MagIC - Magnesium Innovation Centre, Institute of Materials Research, Helmholtz Centre for Materials and Coastal Research, Geesthacht, Germany
- 2Institute of Polymers and Composites, Hamburg University of Technology, Hamburg, Germany
- 3Laboratory of Computational Science and Modelling, Institute of Materials, École Polytechnique Fédérale de Lausanne, Lausanne, Switzerland
- 4Institute of Advanced Ceramics, Hamburg University of Technology, Hamburg, Germany
- 5Computational Material Design, Faculty of Mechanical Engineering, Helmut-Schmidt-University, Hamburg, Germany
- 6Faculty of Engineering, Institute for Materials Science, University of Kiel, Kiel, Germany
Magnesium exhibits a high potential for a variety of applications in areas such as transport, energy and medicine. However, untreated magnesium alloys are prone to corrosion, restricting their practical application. Therefore, it is necessary to develop new approaches that can prevent or control corrosion and degradation processes in order to adapt to the specific needs of the application. One potential solution is using corrosion inhibitors which are capable of drastically reducing the degradation rate as a result of interactions with the metal surface or components of the corrosive medium. As the sheer number of potential dissolution modulators makes it impossible to obtain a detailed atomistic understanding of the inhibition mechanisms for each additive, other measures for inhibition prediction are required. For this purpose, a concept is presented that combines corrosion experiments, machine learning, data mining, density functional theory calculations and molecular dynamics to estimate corrosion inhibition properties of still untested molecules. Concomitantly, this approach will provide a deeper understanding of the fundamental mechanisms behind the prevention of corrosion events in magnesium-based materials and enables more accurate continuum corrosion simulations. The presented concept facilitates the search for molecules with a positive or negative effect on the inhibition efficiency and could thus significantly contribute to the better control of magnesium / electrolyte interface properties.
1. Introduction
Light-weight materials such as magnesium and its alloys are of high interest for the industrial sector. Potential applications can be found in the automobile industry as structural component (Kulekci, 2008), in batteries as anode material (Aurbach et al., 2000; Höche et al., 2018) and in medical engineering as biocompatible, resolvable implant (Brar et al., 2009). However, dealing with corrosion is a challenging task in various engineering disciplines. Durability and versatility strongly depend on the corrosion properties of the applied material and for most applications as structural component, corrosion activity has to be minimized. Yet, for other approaches the corrosion properties have to be adapted to fit the desired application. As for example, introducing magnesium as battery or implant material requires the corrosion or degradation to proceed with a certain rate. Consequently, the development of reliable, predictive models and methods for general dissolution control is crucial.
There are several concepts to protect magnesium from corrosion, ranging from alloying to surface coatings (Gray and Luan, 2002; Blawert et al., 2006; Jia et al., 2016). Recent studies strongly suggest that the re-deposition of released noble impurities (e.g., iron) results in higher corrosion rates as the size of cathodically active sites at the magnesium surface increases over time up to a state of equilibrium (Höche et al., 2016; Li et al., 2016; Mercier et al., 2018; Michailidou et al., 2018). Concerning the iron re-deposition mechanism, a promising strategy to prevent or control corrosion in magnesium-based materials is the introduction of chemical substances that either form stable complexes with the released iron species or block their access to the surface (Lamaka et al., 2016; Yang et al., 2018).
Novel methods for inhibition prediction of not yet tested compounds based on modern data science techniques are in high demand to predict whether a molecule is a potential inhibitor or even further promotes dissolution of the used material. Hence, the high experimental effort and costs of testing multiple compounds for their corrosion inhibition potential can be circumvented. The molecular structure of potential corrosion inhibiting additives is easily obtained nowadays and thus, represents a promising starting point to identify property-structure relationships as well as to predict the inhibition efficiencies of uninvestigated additives. Following this strategy, Ceriotti et al. developed sophisticated methods to vividly illustrate property-structure landscapes by employing SOAP (Smooth Overlap of Atomic Positions) kernels (Bartók et al., 2013) to create a high-dimensional similarity measure and reducing it to a two-dimensional visualization with the dimensionality reduction algorithm “sketch-map” (Ceriotti et al., 2011, 2013). Moreover, this approach is particularly suited for high-dimensionality data from atomistic simulations as it was already successfully applied to molecular crystals (Musil et al., 2018) and high-throughput structural databases (De et al., 2016, 2017).
In this study, the capabilities of the SOAP kernel and sketch-map are focused on a corrosion inhibition database for multiple molecular compounds to improve the understanding of the inhibition-structure relationship. Furthermore, obtained results can be directly used to qualitatively predict the inhibition properties of not yet tested compounds, thus allowing for a data-driven design of anti-corrosion additives for magnesium-based materials.
2. Materials and Methods
2.1. Corrosion Experiments
The balance between magnesium dissolution and hydrogen evolution dominates the aqueous magnesium corrosion process. Due to the processing of magnesium with various methods (Pekguleryuz et al., 2013), noble impurities, as for example iron, are impossible to avoid. Thus, local galvanic cells are induced into the material that locally promote the corrosion, resulting in increased magnesium dissolution, hydrogen evolution and the release of impurities, such as iron. Finding molecules that form stable soluble or insoluble complexes with the released impurities is a promising way to screen for dissolution modulators and provides the basis for our workflow.
In a systematic screening for magnesium corrosion inhibitors (Lamaka et al., 2017), the influence of various organic molecules on the hydrogen evolution rate in magnesium corrosion was investigated. Here, the compounds were either previously reported as magnesium corrosion inhibitors or chosen based on their ability to form stable soluble complexes with Fe2+/3+ in order to prevent iron re-deposition (Höche et al., 2016; Lamaka et al., 2016). Hydrogen evolution tests were performed for six different alloys as well as three grades of pure magnesium. Based on the resulting hydrogen evolution rate, the inhibitors were ranked by their inhibition efficiencies, where positive values, up to 99% correspond to suppressed Mg corrosion (referred to as corrosion inhibitors) and negative values to promoted dissolution of Mg (referred to as corrosion promoters) with respect to a reference experiment in 0.5% NaCl electrolyte without any additives. The potential inhibitors were dissolved in 0.5% NaCl to obtain concentrations of 0.05 M and the initial pH was adjusted to the values in the range of 5.5−7.2. Further experimental details can be found in the original publication (Lamaka et al., 2017). In this study, only inhibition results for commercial purity magnesium (CP-Mg) with 220 ppm iron content are considered.
2.2. Molecular Similarity
SMILES (simplified molecular-input line-entry system) strings of the experimentally investigated compounds are used to create molecular structures using the small molecule topology generator STaGE (Lundborg and Lindahl, 2015). As implemented in the high-throughput workflow of STaGE, the structures are geometry optimized with GAMESS/US (Schmidt et al., 1993; Gordon and Schmidt, 2005) using the B3LYP functional (Becke, 1993; Stephens et al., 1994) with 6-311++G(d,p) basis set and a SCF convergence criterion of 106. As the inhibitor molecules are experimentally tested in solution, the optimizations are performed using a polarizable water model (c-PCM) (Barone and Cossi, 1998; Cossi et al., 2002, 2003; Wang and Li, 2009). Further information on the computational details is given in the Supplementary Material.
We quantify the structural and chemical similarity between inhibitor structures using the SOAP-REMatch kernel (Bartók et al., 2013; De et al., 2016) to investigate the relation between their structure and associated properties. The SOAP kernel compares local atomic environments and the REMatch (Regularized Entropy Match) kernel condenses the local similarities between two structures into a global similarity measure. A local environment is defined within a spherical region of radius rc centered on an atom and is built by a superposition of Gaussian functions with width ξ. The larger rc is chosen, the more structural information surrounding the atom is included. The SOAP kernel measures the rotationally and translationally invariant overlap between two such local environments and can be raised to a power ζ to discriminate more between large (~0.9) and medium (< 0.6) similarities. The combination of the local similarities can be tuned by the hyper parameter γ of the REMatch kernel. For large values (γ ~ 10) more equal weights are assigned to the local similarities while for small values (γ ~ 0.01) only the best matching pairs of local environments are selected to compute the global similarity (see De et al., 2016 for more details).
To help the visualization of potential structure-property relationship, we consider each structure to lie in a high-dimensional space defined by the SOAP-REMatch kernel, which is transformed into a distance (Berg et al., 1984), and we project this information on a two-dimensional map using sketch-map (Ceriotti et al., 2011). This dimensionality reduction technique allows to focus the distortions of the space so that close/distant, i.e., similar/dissimilar, structures in the high-dimensional space keep this relationship in the low dimensional space. This behavior is achieved by a sigmoid function that is applied to the distances and is mainly influenced by the switching distance σ, as well as a and b as tuning parameters (see Supplementary Material).
Thus, it is possible to create a two-dimensional similarity landscape that allows assessing the molecular similarity by analyzing relative positions and cluster formations. However, due to the form of the sigmoid function, far apart points can be arbitrarily far apart in the lower dimensional projection–making a physical interpretation of distances between basins in the low dimensional projection impossible.
2.3. The Inhibition Prediction Workflow
When combining the presented methods, it is possible to visualize the relationship between the molecular structures and the corresponding inhibition efficiencies in a property-structure landscape where all experimentally tested structures act as landmark points. Subsequently, the inhibition efficiencies of uninvestigated compounds can be predicted following the proposed workflow (Figure 1).
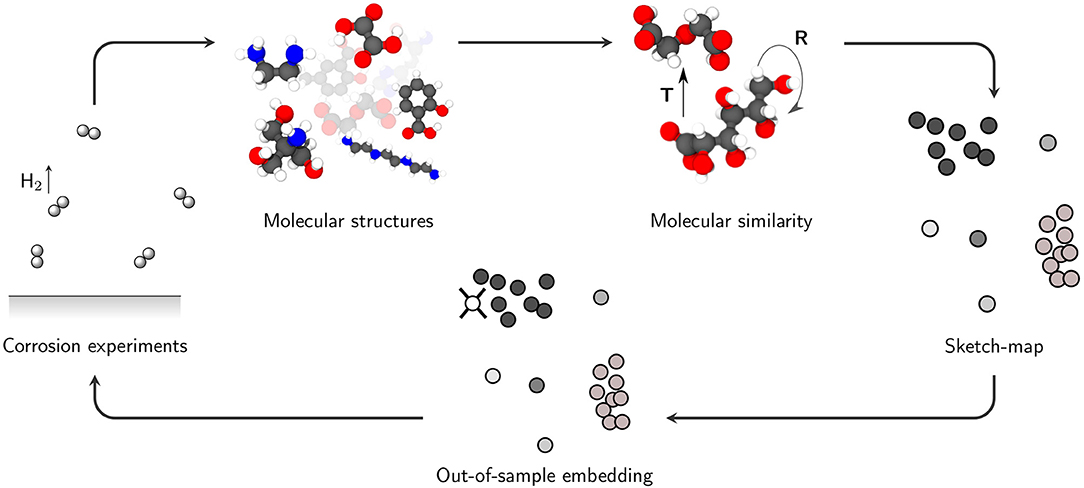
Figure 1. The workflow diagram. Molecular structures from an inhibitor database (Lamaka et al., 2017) are used to generate a two-dimensional sketch-map. Inhibition efficiencies determined in the hydrogen evolution experiments, as well as data generated in DFT computations are used to investigate the property-structure relationship. Unknown structures can be projected into the sketch map to get a first indication on their potential inhibition efficiency. Potentially interesting structures can be then tested again in corrosion experiments to extend the inhibitor database.
Experimental inhibition efficiencies obtained from a corrosion inhibitor databank (Lamaka et al., 2017), as well as molecular similarity measures determined with the SOAP-REMatch kernel can be combined to create a two-dimensional property-structure landscape for the tested inhibitor molecules. Here, clusters can indicate correlations between the inhibition efficiency and inhibitor structure, allowing to relate certain molecular structures to potentially promoting or inhibiting corrosion properties. For now, the small sample size favors an unsupervised machine learning technique where the decision boundaries are drawn by human, instead of a supervised learning algorithm (Kotsiantis et al., 2007).
Consequently, to predict the inhibition properties of an untested compound its structural relationship to the landmark points has to be determined. This can be accomplished by out-of-sample embedding, where the new structure is projected into the generated sketch-map by reproducing the distances to the previously defined landmark points (Ceriotti et al., 2011). Once the structure is projected into the property-structure landscape, its relative position to previously identified clusters can help to assess the impact on corrosion events. Concurrently, this approach indicates whether it is reasonable to examine untested additives in further corrosion experiments, hence saving a tremendous amount of time and resources compared to an experimental high-throughput approach.
3. Results
To create a sketch-map displaying the relationship between corrosion inhibition efficiency and inhibitor structure, a total of 80 compounds was chosen out of the 151 experimentally tested structures provided in a corrosion inhibitor database (Lamaka et al., 2017). All structures were chosen based on a mutual inhibitor concentration of 0.05 M during the hydrogen evolution experiment to avoid concentration dependencies. Before conducting any analysis, the dataset has been further subdivided into 74 plus 6 randomly selected training and test structures – 74 structures for creating the sketch-map and six structures for validating the inhibition workflow.
After geometry optimization, we measure the structural and chemical similarity between these structures using the SOAP-REMatch kernel (De et al., 2016). In order to improve the understanding of the property-structure relationship, the influence of the respective parameters is examined. Indeed, to achieve a wide range of applicability, the SOAP-REMatch kernel and sketch-map technique rely on a few hyper parameters that need to be tuned accordingly (see De et al., 2017; Musil et al., 2018 for a more comprehensive discussion). Depending on the choice of hyper parameters, structural data points are either divided into clusters based on an observable similarity or appear completely scattered. Hence, the parameters have a strong impact on the identifiability of correlations between structure and investigated property. After thorough investigation of the parameter behavior (see Supplementary Material), a set of parameters is chosen that allows the division of structural data points with similar corrosion properties into clusters.
To put a higher focus on the local atomic structure for the similarity determination using SOAP, a cutoff radius rc = 3.0 Åis chosen which includes all moieties of interest but neglects the overall molecular structure for most of the investigated dissolution modulators. For a good balance between strict similarity requirements and a sufficient number of pairs of local environments, the Gaussian width is set to ξ = 0.3. By choosing ζ = 2.0 the discrimination between large and medium similarities is increased, thus amplifying clustering effects. Based on the parameter γ = 2.0, a broad selection of well matching pairs of local environments—as determined by the SOAP kernel—is taken into account to compute the global similarity using the SOAP-REMatch kernel.
As the dimensionality reduction with sketch-map is based on a sigmoid function, the corresponding parameters have to be optimized for the given data. Again, optimizing the parameters with respect to cluster formations of the structural data points, choosing σ = 2 as well as the tuning parameters a = b = 3 results in the sketch-maps shown in Figures 2, 3. The data points originating from the input structures are divided into two elongated “islands,” a small island in the lower left and a larger island in the upper right part of the sketch-map. It is noteworthy that aromatic compounds are solely found in the lower region whereas aliphatic compounds are distributed in the upper island of the sketch-map. This leads us to the conclusion, that the chosen parameters are well suited to generate a sketch-map of the investigated molecule database.
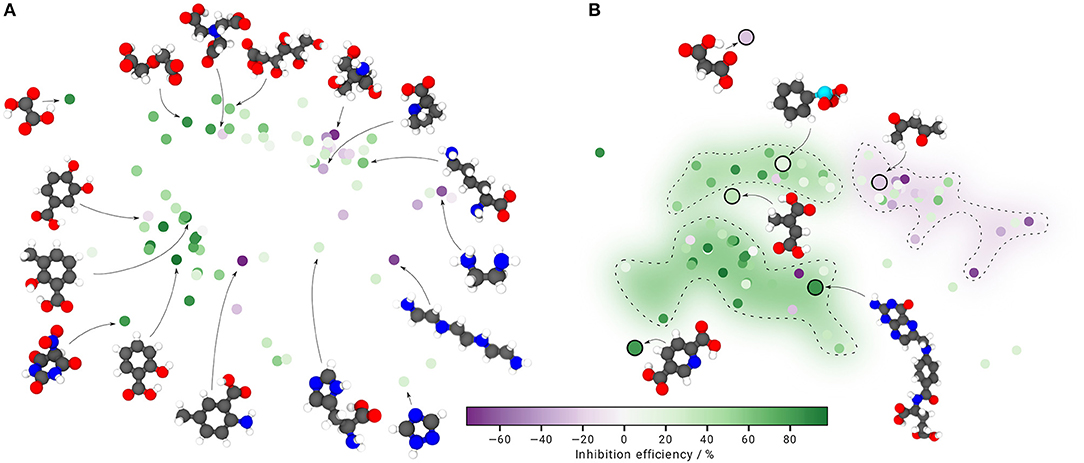
Figure 2. Property-structure landscape based on the SOAP-REMatch kernel and sketch-map. (A) The structural data points are colored according to their corresponding inhibition efficiency in the experiment by Lamaka et al. (2017) (green corrosion inhibition, purple corrosion promotion). Selected molecular structures are shown to illustrate cluster origins. (B) Out-of-sample embedding to predict inhibition properties. New structures are projected into the generated sketch-map from (A) and related to previously identified inhibitor clusters, marked by dashed lines. The clusters are colored according to their median inhibition efficiency in the respective region. Landmarks are depicted as smaller circles; the new test structures as larger, black-rimmed circles along with illustrations of their according molecular structure. Atom color code: redoxygen, graycarbon, bluenitrogen, whitishhydrogen, cyan phosphorus.
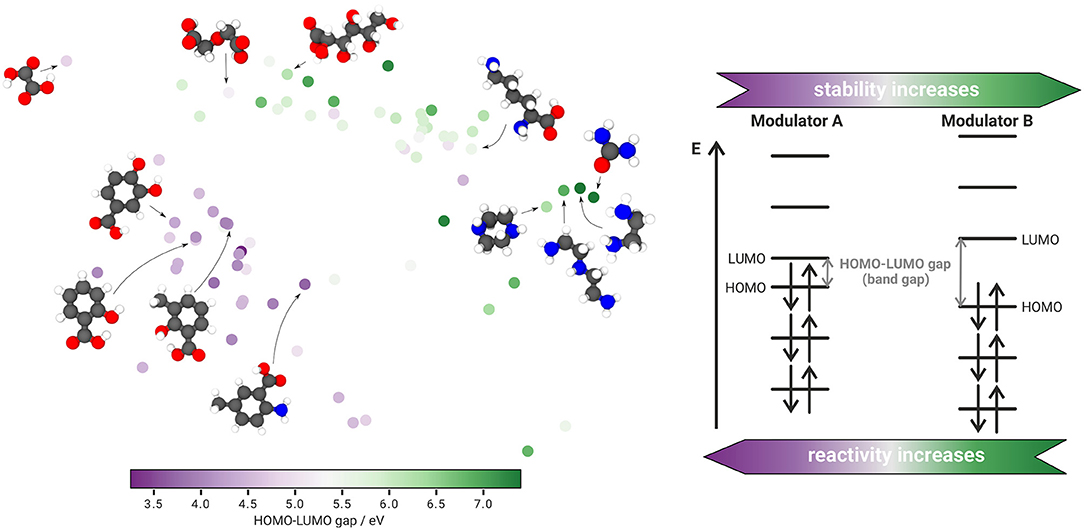
Figure 3. Property-structure landscape based on the SOAP-REMatch kernel and sketch-map. The structural data points are colored according to their corresponding HOMO-LUMO gap, as presented schematically. The color code clearly highlights the separation of aromatic compounds with low and aliphatic compounds with high HOMO-LUMO gaps.
When coloring the structural data points according to their corresponding inhibition efficiency (Figure 2A), the upper right island is further divided into two clusters, where the left cluster is populated by corrosion inhibitors (green) and the right cluster mostly by corrosion promoters (purple) or moderately inhibiting (light green) additives. The lower left island is dominantly populated by corrosion inhibitors, except for three structures on its outer edge. Cluster formations clearly indicate a property-structure relationship, allowing to cautiously correlate inhibition efficiency and molecular structure.
For the inhibition prediction it is desired to project not yet tested compounds into the generated sketch-map and relate their position to the three identified clusters. When a new structure is projected into an area with dominantly corrosion inhibitors or promoters, it is assumed to share similar inhibition properties and can be further investigated experimentally if desired. For purposes of validation, six structures of the experimental database are randomly chosen and projected into the sketch-map by determining their global similarity including all structures. Subsequently, the distance of the new structures is related to the 74 defined landmarks and used to compute the required projections. As a guide for the eye, the three previously identified clusters are outlined with dashed lines and colored according to the median inhibition efficiency in the respective region (Figure 2B).
As the new structures differ strongly in topology, it is natural that the computed projections lead to differing positions in the sketch-map. In relation to the landmarks, structures containing unusually coordinated atoms, additional atom species or an unusual number of functional groups are projected in regions far away from the observed islands, indicating discrepancies in similarity. However, except for one structure at the top of the sketch-map, structural similarities to the defined landmarks result in projections within or close to the generated sketch-map, where the corresponding inhibition efficiencies match the relative positioning to the inhibitor and promoter clusters fairly well.
The generated sketch-map can also be used to correlate the dissolution modulator structure to other properties, as for instance the HOMO-LUMO gap (HL gap). The energetic difference between the highest occupied and lowest unoccupied molecular orbital (HOMO and LUMO) is indicative for the affinity of the investigated corrosion modulators to transition metals (Griffith and Orgel, 1957), where formation of these complexes is more likely with lower HL gaps as this allows for a energetically more favorable overlap of the involved orbitals. Moreover, the HL gap is a sound indicator for chemical reactivity as the stability of a molecule increases with larger HL gaps. Concomitantly, the reactivity of the dissolution modulator decreases (Aihara, 2000). Consequently, aromatic ligands (e.g., pyridine derivatives) are more likely to form complexes with transition metals (e.g., Fe, Ni) than aliphatic ligands that, in general, exhibit larger HL gaps. Hence, the HL gap might be an important parameter that has to be taken into account in future studies to adequately predict the capability of an untested compound to prevent the re-deposition of noble impurities like iron (Höche et al., 2016; Lamaka et al., 2016). The HL gaps were calculated on the TPSSh/def2SVP level of density functional theory using Turbomole 7.2 (TURBOMOLE, 2017) for each of the 80 compounds (Figure 3). As computing the HL gaps using the B3LYP/6-311++G** level of theory that was employed for the STaGE calculations is computationally rather demanding, TPSSh/def2SVP is chosen here as a fast and accurate alternative. Comparing the optimized geometries for each functional, no structural discrepancies could be observed.
Coloring the sketch-map according to the calculated HL gaps, puts further emphasis on the expected separation between aromatic and aliphatic compounds in the investigated dataset. Aromatic structures in the lower left island are assigned with rather low values of 3.2–5.3eV whereas aliphatic compounds in the top right island correspond to rather high energy gaps of 5.5–7.4eV. Albeit this outcome corroborates our current working hypothesis, further work is required to quantitatively correlate the HL gap to the inhibition efficiency of potential inhibitor molecules based on the employed sketch-map approach.
4. Discussion
The acquired property-structure landscape in Figure 2A uncovers a clear relationship between inhibitor structure and inhibition efficiency, whereas only a few outliers in the defined corrosion inhibitor and promoter clusters are observed. Furthermore, almost all new molecules that are projected into the sketch-map, matching the landmarks in similarity, are correctly positioned within or close to the defined clusters according to their corresponding inhibition efficiency. Hence we are confident, that the presented concept is suitable to predict the potential of uninvestigated corrosion inhibitors or promoters based on their resemblance to a defined landmark structure.
However, similarity values obtained using the SOAP-REMatch kernel depend strongly on the chemistry of the input structures. The direct effect can be observed in Figure 2B. Molecules that differ strongly in similarity—due to unusually coordinated atoms, varying atom species or an unusual number of functional groups—are positioned far away from the observed islands. The origin for this behavior lies within the SOAP-REMatch kernel where similarity measures are computed based on the overlap of local atomic environments. Hence, comparing a relatively large molecule to a high number of relatively small molecules leads to low similarity values, and thus a large distance in high-dimensional space, given that a large cutoff radius rc is provided. Also, variations for the number and type of functional groups are affected by this behavior. For the given case, a relatively small cutoff radius rc = 3.0 Åis chosen, leading to a higher focus on local atomic bonds than on the overall molecular structure. Thus, for a significant similarity between local atomic bond networks of landmarks and projections, also large structures can be assigned to clusters of smaller molecules within the sketch-map. For similarity measures between structures containing different elements, a separate density is built for each atomic species and an overlap of differing local environments corresponds to zero (De et al., 2016). Therefore, molecules containing atomic species varying from the ones included in the landmark structures are also more likely to be projected further away. However, here the only investigated structure containing a different atom species (phosphorus in phenylphosphonic acid) is projected directly into the cluster of aliphatic corrosion inhibitors, even though it contains an aromatic ring. A possible reason for this behavior is the low cutoff radius rc that gives greater weight to the similarity of the oxygen arrangement within the phosphonic acid functional group than to the phenyl ring. Since no other structure containing phosphorus is provided, the structure thus appears most similar to the aliphatic compounds. Following the same reasoning, the projected structure at the top of the sketch-map, as well as the landmark structure at the far left, are spaced further away from the aliphatic cluster as their local structure (arrangement of carbon and oxygen atoms) differs significantly. With respect to the proposed inhibition prediction workflow, the presented results already suggest important factors for future hydrogen evolution experiments. Accordingly, using out-of-sample embedding to find structures that match the already defined clusters, potential corrosion inhibitors or promoters can be identified. However, as the proposed inhibition prediction is to be understood more as the formulation of a first clue with respect to the inhibition properties, the predicted inhibition efficiency still has to be validated experimentally. To improve the prediction potential of the proposed concept, more data point from hydrogen evolution experiments are required. With an increasing number of tested compounds, the presented sketch-map can be extended by newly tested structures, thus facilitating the search for new inhibitor molecules with new properties even further. Moreover, structures projected into unexplored regions may indicate promising starting points for the discovery of novel additives with interesting inhibition properties that would not have been considered for testing otherwise.
Based on the structures of already investigated dissolution modulators within the inhibitor clusters, yet unexplored molecules can be identified that might yield promising corrosion inhibition or promotion properties. In this manner, a small number of unknown structures has been selected that shall be tested in future hydrogen experiments–comprising the sodium salt of 6-hydroxypyridine-3-carboxylic acid and quercetin (based on the aromatic cluster) as well as the sodium salt of hexanoic acid (based on the aliphatic cluster). Using out-of-sample embedding to get a first indication of the inhibition performance (see Figure S4), the sodium salt of 6-hydroxypyridine-3-carboxylic acid and quercetin can be identified as potential corrosion inhibitors, whereas the sodium salt of hexanoic acid is expected to promote the corrosion rate.
Even though the proposed workflow works well for the considered data, there are still certain factors to be aware of. On the one hand, the used input structures are all geometrically optimized with an implicit solvent model which might not represent the actual molecular geometry on the surface or in coordination complexes at all. On the other hand, the large number of tunable parameters when using the SOAP-REMatch kernel and sketch-map makes it difficult to fully understand its outcome, as the fine-tuning process contains a lot of trial and error as well as visual inspection (see Supplementary Material). For the given case, this strategy is still reasonable as the aim of finding a property-structure relationship with respect to the inhibition efficiency, as well as predicting the inhibition performance of new compounds is accomplished. However, a comprehensive understanding of the underlying physical concepts behind the occurring inhibition mechanisms still requires further work.
Due to the data-hungry nature of most machine learning applications, like sketch-map, more input structures are desired to improve its validity and prediction abilities since the proposed inhibition prediction workflow is highly dependent on the provided experimental input data. Thus, possible outliers within the inhibition efficiencies are to be expected without a sufficient amount of data points. However, the generation of new experimental data points is limited by costly and time-consuming hydrogen evolution investigations. Experimental conditions have to be accurately defined as small discrepancies in the experimental environment of the chosen structures may already have a severe impact on the predictive performance of the generated property-structure landscape. Consequently, we aim to employ high-throughput MD or DFT computations to identify properties that correlate well with the experimentally determined inhibition efficiencies. A promising starting point for this in silico approach are the presented HL gaps (Figure 3). The separation between lower and higher energy gaps for aromatic and aliphatic compounds, respectively, matches the spatial separation due to the SOAP-REMatch kernel and sketch-map. Looking at the property-structure landscape more carefully, small point clusters within the islands can be identified that indicate some property-structure relationship. An example are the four data points in the far right of the sketch-map, provided with their corresponding molecular structures. The further right the structure lies within the sketch-map, the higher the HL gap of the respective compound becomes. Of course, the property-structure landscape does not allow investigations of this behavior in more detail. Nevertheless, it represents a potential relation between the molecular structure and the energy gap of the frontier orbitals, that can be further examined using other measures. Hence, we are currently investigating if the calculated HL gaps will help to detect a relationship between the HL gap and the inhibition efficiency as well.
Since the molecular compounds are tested in solution, another interesting parameter is the free energy of solvation. However, no obvious relationship to the inhibition properties can be observed so far (Figure S3A). Consequently, future works will focus on the determination of the free energy of solvation for corrosive species (e.g., Mg or Fe ions) in a solution containing the dissolution modulators to yield more accurate—and correlatable—results with respect to the occurring inhibition mechanisms. Here, STaGE is a mighty tool to screen free energies of solvation for high numbers of molecular compounds requiring very few input parameters (Lundborg and Lindahl, 2015). Moreover, even simpler properties as the number of certain functional moieties within an inhibitor molecule can provide a deeper insight on a potential correlation to the experimentally determined inhibition efficiency. For instance, the property-structure landscape in Figure S3B indicates that nitrogen plays an immediate role in the corrosion inhibition mechanism of aliphatic compounds.
In subsequent steps, by providing material and system parameters like the free energy of solvation or adsorption for inhibitor molecules, by predicting HOMO-LUMO gaps or by computing energy levels related to coordination complexes, physico-chemical entities at nano- and microscale, relevant for mathematically based system modeling, can be derived. For example, the shift in the electrochemical potential due to changes of the free energy of adsorption (Groß, 2018) or efficient (ion-) transport parameters like diffusion coefficients can be calculated. Furthermore, based on the molecule data, the cluster formation and its interaction with the surface can be analyzed more accurately by molecular dynamic studies. As a consequence, more precise calculations of elemental surface coverage, concentration distributions of chemical species or averaged, system relevant surface kinetic parameters are possible and more profound input data applicable in upscaled continuum corrosion models (Höche, 2015). Typically, such kind of information is experimentally difficult to access but of main interest for setting up advanced non-empirical corrosion models which are required to enhance computational corrosion and system engineering capabilities. The developed data science based concept can be applied for analyzing or even learning from corrosion simulation results by correlating simulation predictions and molecular structures.
In conclusion, it was possible to create a property-structure landscape based on the results of hydrogen evolution measurements, that vividly demonstrates the relationship between corrosion inhibition efficiency and corresponding molecular structure of magnesium corrosion inhibitors. After creating a high-dimensional similarity measure with the SOAP-REMatch kernel between 74 tested compounds, the similarity matrix is reduced to a two-dimensional visualization with sketch-map, providing a reference to qualitatively predict the inhibition behavior of yet to be tested molecules. Aside from the inhibition efficiency, also other properties as the HL gap were correlated with the inhibitor structure, matching impressively well the spatial separation into aliphatic and aromatic compounds. The predictive performance of the proposed workflow is still limited by the relatively low amount of available experimental input data. However, the discovered corrosion inhibitor and promoter clusters provide a valuable reference for inhibition prediction and identification of yet unexplored structures – thus facilitating the search for potential corrosion inhibitors and increasing the efficiency of corrosion inhibition experiments and corrosion models.
Author's Note
The datasets for this manuscript are not publicly available because they are published in a closed access journal (https://doi.org/10.1016/j.corsci.2017.07.011). Requests to access the datasets should be directed to c3ZpYXRsYW5hLmxhbWFrYUBoemcuZGU=. The sketch-maps used in this article are published on https://interactive.sketchmap.org/.
Author Contributions
TW, SL, CF, MZ, and RM: contributed the conception and design of the study. CF and GBVF: ran DFT simulations. FM: performed the statistical analysis. SL: provided experimental data. TW: wrote the first draft of the manuscript. RM, CF, GBVF, FM, and DH: wrote sections of the manuscript. All authors contributed to the manuscript revision, read and approved the submitted version.
Funding
Funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Projektnummer 192346071-SFB 986. GBVF acknowledges financial support by the Austrian Research Promotion Agency (FFG) within the project PL2N A, project number 865011. FM is supported by NCCR MARVEL, funded by the Swiss National Science Foundation. MMDi IDEA project funded by HZG is gratefully acknowledged.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Prof. D. Winkler from La Trobe University, Australia as well as Prof. M. Ceriotti from École Polytechnique Fédérale de Lausanne, Switzerland are acknowledged for discussions about setting up machine learning for magnesium dissolution modulators.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmats.2019.00053/full#supplementary-material
References
Aihara, J.-i. (2000). Correlation found between the homo–lumo energy separation and the chemical reactivity at the most reactive site for isolated-pentagon isomers of fullerenes. Phys. Chem. Chem. Phys. 2, 3121–3125. doi: 10.1039/b002601h
Aurbach, D., Lu, Z., Schechter, A., Gofer, Y., Gizbar, H., Turgeman, R., et al. (2000). Prototype systems for rechargeable magnesium batteries. Nature 407, 724. doi: 10.1038/35037553
Barone, V., and Cossi, M. (1998). Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001. doi: 10.1021/jp9716997
Bartók, A. P., Kondor, R., and Csányi, G. (2013). On representing chemical environments. Phys. Rev. B 87, 184115. doi: 10.1103/PhysRevB.87.184115
Becke, A. D. (1993). Density-functional thermochemistry. iii. the role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Berg, C., Christensen, J. P. R., and Ressel, P. (1984). Harmonic Analysis on Semigroups. New York, NY: Springer.
Blawert, C., Dietzel, W., Ghali, E., and Song, G. (2006). Anodizing treatments for magnesium alloys and their effect on corrosion resistance in various environments. Adv. Eng. Mater. 8, 511–533. doi: 10.1002/adem.200500257
Brar, H. S., Platt, M. O., Sarntinoranont, M., Martin, P. I., and Manuel, M. V. (2009). Magnesium as a biodegradable and bioabsorbable material for medical implants. Jom 61, 31–34. doi: 10.1007/s11837-009-0129-0
Ceriotti, M., Tribello, G. A., and Parrinello, M. (2011). From the Cover: Simplifying the representation of complex free-energy landscapes using sketch-map. Proc. Natl. Acad. Sci. U.S.A. 108, 13023–13028. doi: 10.1073/pnas.1108486108
Ceriotti, M., Tribello, G. A., and Parrinello, M. (2013). Demonstrating the Transferability and the Descriptive Power of Sketch-Map. J. Chem. Theory Comput. 9, 1521–1532. doi: 10.1021/ct3010563c
Cossi, M., Rega, N., Scalmani, G., and Barone, V. (2003). Energies, structures, and electronic properties of molecules in solution with the c-pcm solvation model. J. Comput. Chem. 24, 669–681. doi: 10.1002/jcc.10189
Cossi, M., Scalmani, G., Rega, N., and Barone, V. (2002). New developments in the polarizable continuum model for quantum mechanical and classical calculations on molecules in solution. J. Chem. Phys. 117, 43–54. doi: 10.1063/1.1480445
De, S., Bartók, A. P., Csányi, G., and Ceriotti, M. (2016). Comparing molecules and solids across structural and alchemical space. Phys. Chem. Chem. Phys. 18, 13754–13769. doi: 10.1039/C6CP00415F
De, S., Musil, F., Ingram, T., Baldauf, C., and Ceriotti, M. (2017). Mapping and classifying molecules from a high-throughput structural database. J. Cheminformatics 9:6. doi: 10.1186/s13321-017-0192-4
Gordon, M. S., and Schmidt, M. W. (2005). “Advances in electronic structure theory: gamess a decade later,” in Theory and Applications of Computational Chemistry, eds C. E. Dykstra, G. Frenking, K. S. Kim, and G. E. Scuseria (Amsterdam: Elsevier), 1167–1189.
Gray, J., and Luan, B. (2002). Protective coatings on magnesium and its alloysa critical review. J. Alloys Compounds 336, 88–113. doi: 10.1016/S0925-8388(01)01899-0
Griffith, J. S., and Orgel, L. E. (1957). Ligand-field theory. Q. Rev. Chem. Soc. 11, 381. doi: 10.1039/qr9571100381
Groß, A. (2018). Fundamental challenges for modeling electrochemical energy storage systems at the atomic scale. Top. Curr. Chem. 376, 1–22. doi: 10.1007/978-3-030-00593-1_1
Höche, D. (2015). Simulation of corrosion product deposit layer growth on bare magnesium galvanically coupled to aluminum. J. Electrochem. Soc. 162, C1–C11. doi: 10.1149/2.0071501jes
Höche, D., Blawert, C., Lamaka, S. V., Scharnagl, N., Mendis, C., and Zheludkevich, M. L. (2016). The effect of iron re-deposition on the corrosion of impurity-containing magnesium. Phys. Chem. Chem. Phys. 18, 1279–1291. doi: 10.1039/C5CP05577F
Höche, D., Lamaka, S., Vaghefinazari, B., Braun, T., Petrauskas, R., Fichtner, M., et al. (2018). Performance boost for primary magnesium cells using iron complexing agents as electrolyte additives. Sci. Rep. 8, 7578–7578. doi: 10.1038/s41598-018-25789-8
Jia, Z., Xiong, P., Shi, Y., Zhou, W., Cheng, Y., Zheng, Y., et al. (2016). Inhibitor encapsulated, self-healable and cytocompatible chitosan multilayer coating on biodegradable mg alloy: a ph-responsive design. J. Mater. Chem. B 4, 2498–2511. doi: 10.1039/C6TB00117C
Kotsiantis, S. B., Zaharakis, I., and Pintelas, P. (2007). Supervised machine learning: A review of classification techniques. Emerg. Artif. Intel. Appl. Comput. Eng. 160, 3–24.
Kulekci, M. K. (2008). Magnesium and its alloys applications in automotive industry. Int. J. Adv. Manuf. Technol. 39, 851–865. doi: 10.1007/s00170-007-1279-2
Lamaka, S. V., Höche, D., Petrauskas, R. P., Blawert, C., and Zheludkevich, M. L. (2016). A new concept for corrosion inhibition of magnesium: suppression of iron re-deposition. Electrochem. Commun. 62, 5–8. doi: 10.1016/j.elecom.2015.10.023
Lamaka, S. V., Vaghefinazari, B., Mei, D., Petrauskas, R. P., Höche, D., and Zheludkevich, M. L. (2017). Comprehensive screening of Mg corrosion inhibitors. Corrosion Sci. 128, 224–240. doi: 10.1016/j.corsci.2017.07.011
Li, J., Sun, W., Hurley, B., Luo, A. A., and Buchheit, R. G. (2016). Cu redistribution study during the corrosion of az91 using a rotating ring-disk collection experiment. Corrosion Sci. 112, 760–764. doi: 10.1016/j.corsci.2016.08.011
Lundborg, M., and Lindahl, E. (2015). Automatic GROMACS topology generation and comparisons of force fields for solvation free energy calculations. J. Phys. Chem. B 119, 810–823. doi: 10.1021/jp505332p
Mercier, D., Światowska, J., Zanna, S., Seyeux, A., and Marcus, P. (2018). Role of segregated iron at grain boundaries on mg corrosion. J. Electrochem. Soc. 165, C42–C49. doi: 10.1149/2.0621802jes
Michailidou, E., McMurray, H., and Williams, G. (2018). Quantifying the role of transition metal electrodeposition in the cathodic activation of corroding magnesium. J. Electrochem. Soc. 165, C195–C205. doi: 10.1149/2.0251805jes
Musil, F., De, S., Yang, J., Campbell, J. E., Day, G. M., and Ceriotti, M. (2018). Machine learning for the structure-energy-property landscapes of molecular crystals. Chem. Sci. 9, 1289–1300. doi: 10.1039/C7SC04665K
Pekguleryuz, M. O., Kainer, K., and Kaya, A. A. (2013). Fundamentals of Magnesium Alloy Metallurgy. Cambridge: Elsevier.
Schmidt, M. W., Baldridge, K. K., Boatz, J. A., Elbert, S. T., Gordon, M. S., Jensen, J. H., et al. (1993). General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363. doi: 10.1002/jcc.540141112
Stephens, P., Devlin, F., Chabalowski, C., and Frisch, M. J. (1994). Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627. doi: 10.1021/j100096a001
TURBOMOLE, V. (2017). A Development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2018, TURBOMOLE GmbH, since 2007. Available online at: http://www.turbomole.com (accessed December, 2017).
Wang, Y., and Li, H. (2009). Smooth potential energy surface for cavitation, dispersion, and repulsion free energies in polarizable continuum model. J. Chem. Phys. 131, 206101. doi: 10.1063/1.3268921
Keywords: machine learning, property-structure relationship, high-throughput screening, corrosion inhibition, density functional theory, magnesium, dimensionality reduction
Citation: Würger T, Feiler C, Musil F, Feldbauer GBV, Höche D, Lamaka SV, Zheludkevich ML and Meißner RH (2019) Data Science Based Mg Corrosion Engineering. Front. Mater. 6:53. doi: 10.3389/fmats.2019.00053
Received: 04 January 2019; Accepted: 14 March 2019;
Published: 05 April 2019.
Edited by:
Surya R. Kalidindi, Georgia Institute of Technology, United StatesReviewed by:
Tim Kowalczyk, Western Washington University, United StatesRuifeng Lu, Nanjing University of Science and Technology, China
Copyright © 2019 Würger, Feiler, Musil, Feldbauer, Höche, Lamaka, Zheludkevich and Meißner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert H. Meißner, cm9iZXJ0Lm1laXNzbmVyQHR1aGguZGU=