 Hua Zhong
Hua Zhong Weilong Kong
Weilong Kong Ziyun Gong
Ziyun Gong Xinyi Fang
Xinyi Fang Xiaoxiao Deng
Xiaoxiao Deng- State Key Laboratory of Hybrid Rice, Key Laboratory for Research and Utilization of Heterosis in Indica Rice, Ministry of Agriculture, College of Life Sciences, Wuhan University, Wuhan, China
The SPL (SQUAMOSA promoter binding protein-like) gene family is one of the plant-specific transcription factor families and controls a considerable number of biological functions, including floral development, phytohormone signaling, and toxin resistance. However, the evolutionary patterns and driving forces of SPL genes in the Oryza genus are still not well-characterized. In this study, we investigated a total of 105 SPL genes from six AA genome Oryza representative species (O. barthii, O. glumipatula, O. nivara, O. rufipogon, O. glaberrima, and O. sativa). Phylogenetic and motif analyses indicated that SPL proteins could be divided into two distinct lineages (I and II), and further studies showed lineage II consisted of three clades (IIA, IIB, and IIC). We found that clade I had comparable structural features with clade IIA, whereas genes in clade IIC displayed intrinsic differences, such as lower exon numbers and the presence of miR156 regulation elements. Nineteen orthologous groups of OsSPLs in Oryza were also identified, and most exons within those genes maintained constant length, whereas length of intron changed relatively. All groups were constrained by stronger purifying selection and diversified continually including alterative gene number, intron length, and miR156 regulation. Subsequently, cis-acting element analyses revealed the potential role of SPLs in wild rice, which might participate in light-responsive, phytohormone response, and plant growth and development. Our results shed light on that different evolutionary rates and duplication events might result in divergent evolutionary patterns in each lineage of SPL genes, providing a guide in exploring diverse function in the rice gene family among six closely related Oryza species.
Introduction
Rice (Oryza sativa L.), one of the most important staple foods, provides more than one-fifth of the calories consumed worldwide (Khush, 2003). As an important model organism in monocot plants, Oryza sativa belongs to the Oryza genus. The genus Oryza probably originated within a relatively short time scale about 15 million years ago (MYA), which includes 23 species categorized into 10 genomic types (AA, BB, BBCC, CC, CCDD, EE, FF, GG, HHJJ, and HHKK) (Aggarwal et al., 1997; Ge et al., 1999; Khush, 2003; Vaughan et al., 2003; Jacquemin et al., 2014). As the most important genetic resources for rice breeding, the Oryza AA genome contains two cultivated rice (Oryza sativa and Oryza glaberrima) and six wild species (Oryza rufipogon, Oryza nivara, Oryza barthii, Oryza longistaminata, Oryza meridionalis, and Oryza glumaepatula) (Vaughan et al., 2003). With an abundant infrastructure of high-quality plant genomes, further studies of gene families at the genome scale are now available in the Oryza genus especially among those in the AA genome.
Plant DNA binding transcription factors are capable of regulating mRNA transcriptional initiation, which significantly impact a broad range of plant developmental processes and response to environmental changes (Schwechheimer et al., 1998; Henríquez-valencia et al., 2018). Transcription factors have previously been classified into families, such as MYB (Baisakh et al., 2016), bHLH (Feller et al., 2011), MADS-box (Smaczniak et al., 2012), and SBP-box families (Klein et al., 1996). It is common knowledge that the SPL (SQUAMOSA promoter binding protein-like) gene encodes a highly conserved DNA binding domain, which is known as the SBP domain (Klein et al., 1996; Cardon et al., 1999). This domain contains two non-interleaved zinc-binding sites consisting of Cys3HisCys2HisCys or Cys6HisCys sequence motif (Yamasaki et al., 2004). As a CCHC-type zinc finger, the first zinc-binding site (Zn1) is essential in the folding of the overall tertiary structure. The second zinc-binding site (Zn2) at the C-terminal site is responsible for the DNA binding (Yamasaki et al., 2006). In addition, the bipartite nuclear localization signal (NLS) motif is highly conserved in the SPLs and other families (Dingwall and Laskey, 1991). The first two SBPs were discovered in Antirrhinum majus (AmSBP1 and AmSBP2) based on their function to interact with the floral meristem identity gene SQUAMOSA (Klein et al., 1996).
So far, numerous SBP-box genes have been identified and characterized in plants, including Arabidopsis thaliana (Cardon et al., 1999), maize (Wei et al., 2018), apple (Li et al., 2013), caster bean (Zhang and Ling, 2014), and pepper (Zhang et al., 2016). AtSPL14 has been demonstrated to regulate floral transition negatively and display resistance to Fumonisin B1 (Stone et al., 2005). Likewise, overexpression of AtSPL3/4/5 results in the promotion of the reproductive transition in response to photoperiod and GA signals (Porri et al., 2012; Yu et al., 2012; Preston and Hileman, 2013). Among the 19 identified OsSPLs in rice (Yang et al., 2008), multiple essential and divergent developmental processes have been influenced. For example, OsSPL3 can increase cold stress tolerance (Zhou and Tang, 2018). Chen used near isogenic lines for mapping a minor QTL for heading date, qHd1, which contained OsSPL2 (Chen et al., 2014). Higher expression of OsSPL16/GW8 promotes cell division and grain filling, which determines grain shape, rice quality, and yield (Wang S. et al., 2012; Wang Y. et al., 2012; Wang et al., 2015). Additionally, OsSPL14, which is synonymous with IPA1, reveals a complex network to regulate plant architecture (Jiao et al., 2010; Lu et al., 2013) and promotes both grain yield and disease resistance (Wang et al., 2018). Those findings stressed that SPL gene family, of great significance in rice breeding, represents an important strategy to enhance rice yield performance simultaneously.
MircoRNAs are generally a kind of 20–24 nt non-coding small RNAs. They can bind to their complementary mRNAs and reduce protein expression level (Rogers and Chen, 2013). The regulational mechanism of plant miRNA-guided silencing is ancient and significant (Rhoades et al., 2002). Xie identified 11 miR156 targets from rice SPL genes and revealed tissue-specific interactions between miR156 and OsSPLs (Xie et al., 2006). The overexpression of miR156 delayed moderately in flowering and decreased, obviously, apical dominance through modulating SPL genes (Schwab et al., 2005). Nevertheless, it is still unclear whether miR156 regulation retains conserved in the Oryza genus and if the SPL gene with a miR156 target site expands in rice domestication. Moreover, despite the great progress made in this field, the origin and evolutionary process of the SBP-box gene family in Oryza have not been largely undefined.
In this study, SPL genes from six Oryza species, representing the main AA lineage, were identified. In addition, phylogenetic analysis and classification were performed to explore the evolution of the SBP-box gene family. Detailed gene information, including the exon–intron structure, the pattern of the conserved motifs, the role of the miR156 target, as well as the divergence of functions were also discussed systematically.
Materials and Methods
Identification and Phylogenetic Tree Construction of SPL Genes
The protein and cDNA sequences of six representative Oryza species, namely, O. barthii, O. glaberrima, O. glumipatula, O. nivara, O. rufipogon, and O. sativa, were downloaded from Ensembl Plants release 41 (Bolser et al., 2017) and Phytozome v12 (Goodstein et al., 2012). The hidden Markov model (HMM) profile of the SBP domain (Accession No. PF03110) was downloaded from the PFAM database (Finn et al., 2014). All these candidate proteins were separately obtained by HMMER 3.2.1 (Finn et al., 2011) and BLASTP (Camacho et al., 2009). Then, the Pfam tool was used to confirm the highly conserved SBP domains (Kong et al., 2018). All the DNA sequences and core motifs of the SPL genes were mapped to genome data using the GeneDoc software (Nicholas et al., 1997). ProtParam tools in the ExPASy server was used to calculate physicochemical parameters (Gasteiger et al., 2005).
Multiple sequence alignment of full-length SPL protein sequences was performed by ClustalW (Thomopson et al., 1994), and an unrooted phylogenetic relationship was constructed by MEGA7 (Kumar et al., 2016) using the neighbor-joining (NJ) method with the Jones–Taylor–Thornton (JTT) model based on 1,000 bootstrap replicates.
Gene Structure, Motif, and Homology Analysis
Exon/intron site and length data were extracted based on six respective genome annotation GFF files from Ensembl Plants (Bolser et al., 2017). The software MEME Suite 5.0.2 (Bailey et al., 2009) was employed to identify conserved motifs with the maximum number 20. To predict putative functions of identified motifs, the consensus sequences were subjected to search against the Interpro database (Mitchell et al., 2018). The phylogenetic tree combined with motif arrangement was drawn by EvolView v2 (He et al., 2016), and exon/intron structures were shown by TBtools v0.665 proportionally (Chen et al., 2018).
The orthologous groups between diverse SPL genes were deduced by OrthMCL (Li et al., 2003). The collinearity relationships were obtained using BLAST search with the default parameters and generated using a procedure in ColinearScan by the MCScanX toolkit (Wang S. et al., 2012). All Oryza SPL genes were then classified into various types of duplications with a “duplicate_gene_classifier” procedure.
Prediction of miR156-Targeted Genes and Substitution Rates Estimation
All mature sequences of miR156 genes were downloaded from miRBase release 22 (Kozomara and Griffiths-Jones, 2014). Binding sites on SPL gene transcripts were identified using the online psRNATarget server (Dai and Zhao, 2011) with default settings. The codon sequence alignments of each species SPLs were generated using ClustalW (Thomopson et al., 1994). DnaSP v5 software (version 5.101) was used to calculate non-synonymous (Ka) and synonymous (Ks) substitutions and ratios of Ka/Ks (Librado and Rozas, 2009).
cis-Acting Element Analysis and Expression Analyses of SPL Genes in Three Oryza Plants
The 2-kbp upstream of the transcription start site from each SPL coding sequence was examined, which was regarded as putative promoter sequence. Then, the cis-elements were analyzed using the PlantCARE program2 (Lescot, 2002).
Three transcriptome data (O. barthii, O. glaberrima, and O. rufipogon) were downloaded from NCBI SRA databases (SRP151515) to investigate the expression profiles of SPL genes in six different tissues, roots, tiller base, leaf blades, panicles (<1; 1–5; >5 cm), leaf sheaths, and leaf pulvini. TopHat2 (Kim et al., 2013) was used to map paired reads to their own reference genomes3. Then, the gene expression level was calculated by Cufflinks (Trapnell et al., 2012; Kong et al., 2017). The heatmaps were illustrated by TBtools v0.665 (Chen et al., 2018), and the rice anatomogram picture was downloaded from online website Expression Atlas (Petryszak et al., 2016).
Results
Identification of SPL Genes in Six Oryza Genus Species
SBP transcription factor coding genes in Oryza barthii (Ob), Oryza glumipatula (Oglu), Oryza nivara (On), Oryza rufipogon (Or), Oryza sativa subsp. japonica (Os), and Oryza glaberrima (Ogla) are convinced by Pfam through searching the SBP-box domain. Finally, 16, 16, 18, 19, 17, and 19 SPL genes in Ob, Ogla, Oglu, On, Or, and Os were identified, respectively. Names of putative Oryza SPLs were assigned based on chromosomal order in each genome in accordance with previous rice SPLs study. These SPLs in each species were unevenly distributed on the chromosomes. However, only the ORGLA08G0230100 gene could not be mapped to any chromosome conclusively and was named as OglaSPL14 (Supplementary Figure S2 and Supplementary Table S1). The copy number of SPLs and the percentage to total genes in each genome were also displayed (Figure 1). We found that SBP-box gene family members in Asian cultivated rice (Os) showed an obvious gene number expansion compared to wild varieties. Although Asian rice (Os, Or, and On) had more SPL gene copy number than African rice (Ob and Ogla), the SPL number proportion of the latter was larger because of their relatively small genome sizes.
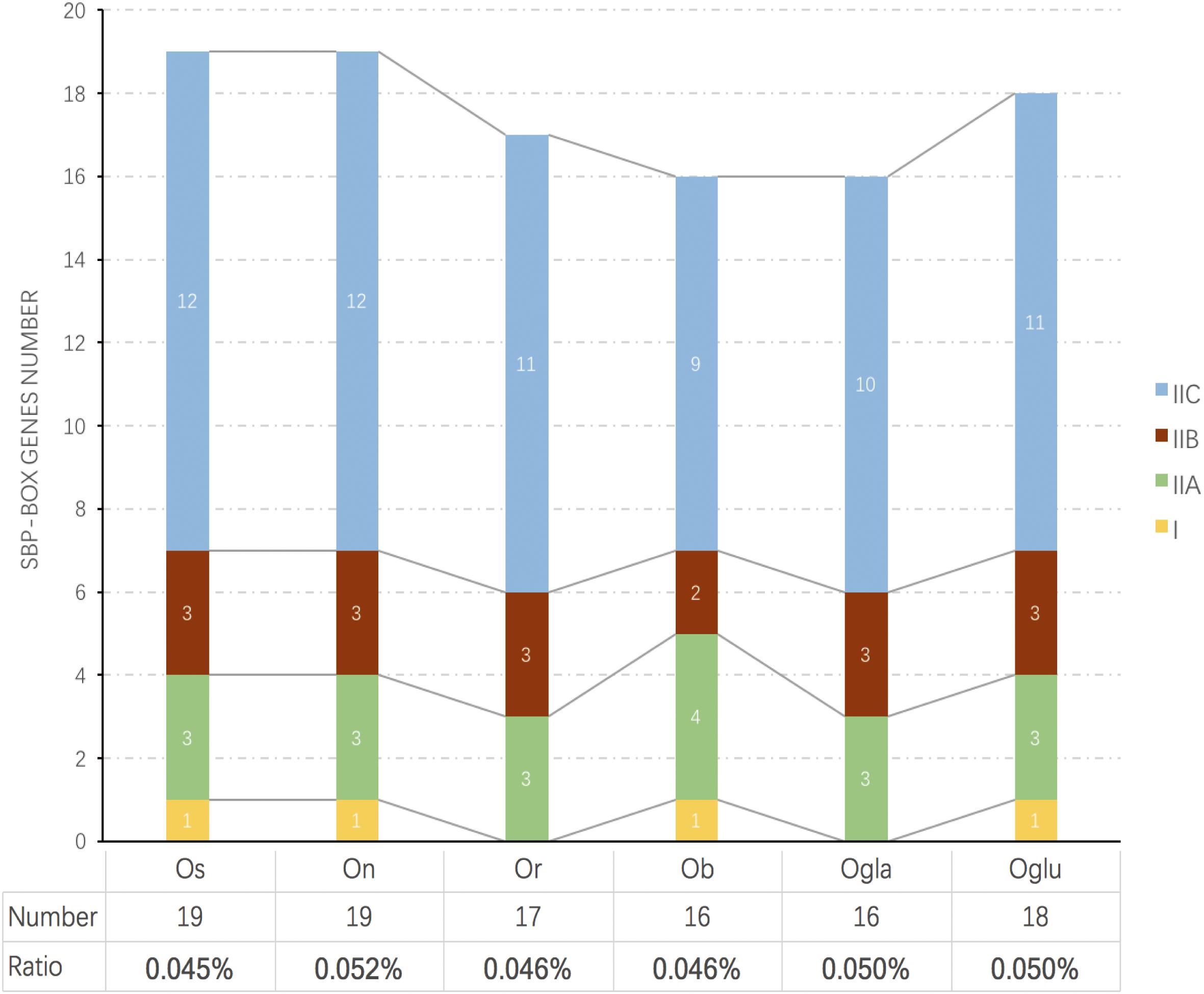
Figure 1. Comparison of the number and ratio of SPL genes in six Oryza species. The species names with the prefixes “Ob,” “Ogla,” “Oglu,” “On,” “Or,” and “Os” indicate Oryza barthii, Oryza glaberrima, Oryza glumipatula, Oryza nivara, Oryza rufipogon, and Oryza sativa subsp. japonica, respectively. The groups I and II are displayed in different colored boxes. The below chart represents SBP number and their ratios to total protein numbers in different species.
Comparative Phylogenetic and Motif Composition Analyses
We constructed a NJ phylogenetic tree for the 105 SPL genes from six Oryza species. In this tree, the SPL genes of Oryza plants were clustered into two main lineages (I and II), and we observed an apparent difference in the SPL gene copy number among these two groups. Only four genes were classified into clade I. Meanwhile, clade II contained the remaining genes, and it was further grouped into three main subclades (IIA, IIB, and IIC), which had several members in each species (Figure 1). Of 101 SPL genes in clade II, over 64% gene copies were found in clade IIC, and the number of genes was relatively conserved among the six species: Os had 12 genes, and On, Oglu, Or, Ogla, and Ob had 12, 11, 11, 10, and 9 genes, respectively (Figure 1). In contrast, there were comparatively less than four gene copies in clade IIA. These results indicated that the SPL family expansion was mainly the result of duplication of group II genes, especially in clade IIC.
Twenty putative motifs (Figure 2 and Supplementary Table S2) were found through the MEME suite, and these results indicated that the SBP domain was composed of three motifs: Zn1 (motifs 2 and 3), Zn2 (motif 4), and NLS (motif 1). The domain structures of Oryza SPLs were further analyzed by multiple sequence alignment with full-length protein sequences. As shown in Supplementary Figure S1, the C-terminal zinc finger of both clades I and II had the same C2HC motif, while their N-terminal zinc finger showed different signatures, C4 in clade I and C3H in clade II. Moreover, the Oryza SPL proteins in the same clade exhibited similar motif composition. Motifs-5, -7, and -16 were group-specific elements in clade IIA and I, as motif-9 only existed in clade IIA, which made a transmembrane helix. Meanwhile, clade IIC and IIB SPL genes contained other unique motifs accept highly conserved SBP domains. The Ankyrin repeats (motif-8) were also observed nearby the SBP domain in clade IIA, indicating that those domains might mediate protein–protein interactions (Michaely and Bennett, 1992; Li et al., 2006). Most SPL genes’ exon sequence in clade IIC possessed a unique motif (motif-10), which could be recognized and regulated by a conserved peptide element ALSLLS of miR156, except for OsSPL4, OsSPL13, and their orthologous genes (Supplementary Table S2), with binding sites located in the 3′ untranslated regions (UTR) (Xie et al., 2006). We also used the online prediction tool (psRNATarget) to screen miR156-targeting sequences in SPL transcripts, and 58 SPL genes were indicated as the putative targets. It was comparable with the result of motif-10 prediction that all in clade IIC had conserved functional sites either in the last exon or the 3′-UTR (Supplementary Table S3).
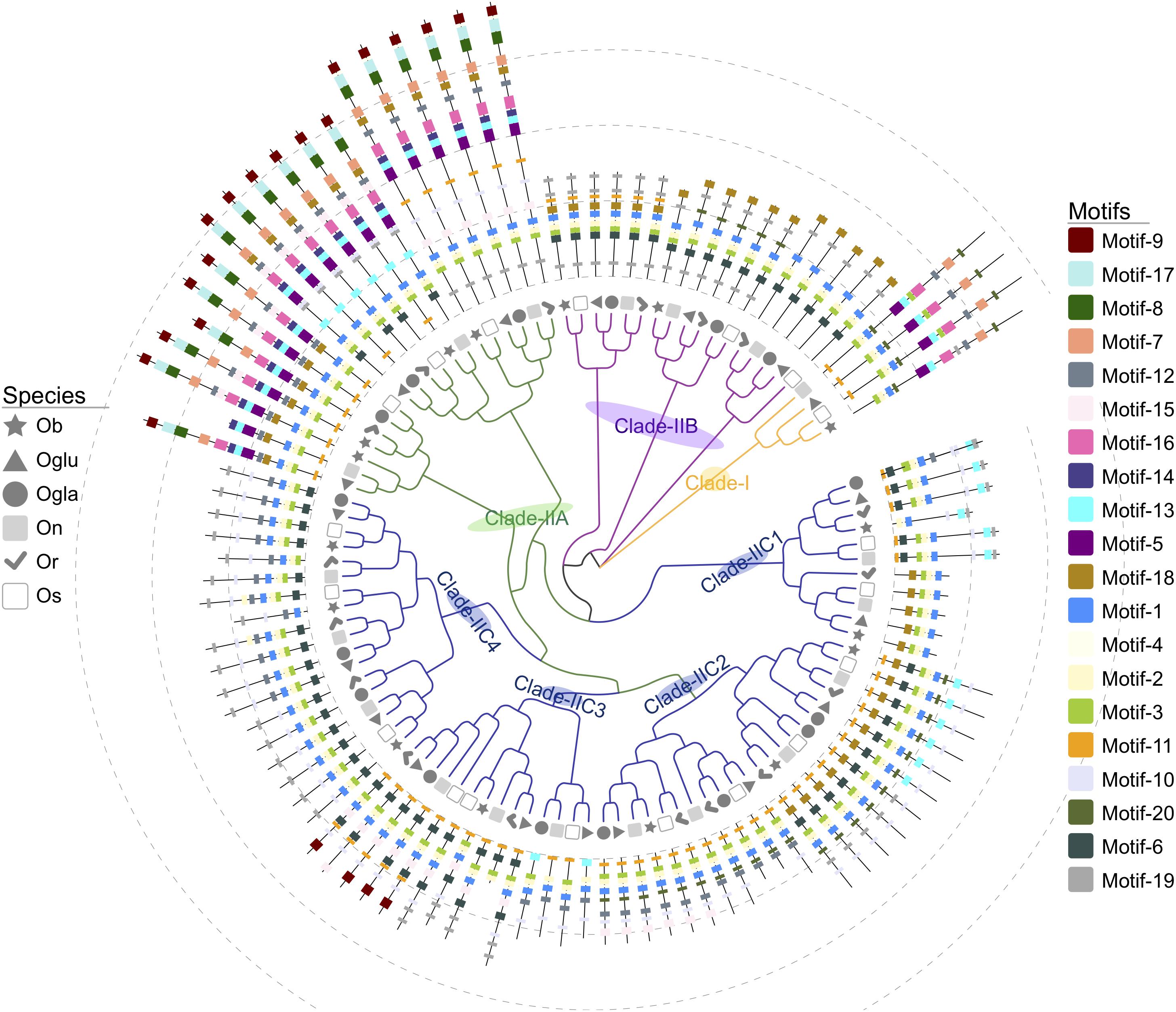
Figure 2. Unrooted phylogenetic tree and motif architectures of SPL genes in six Oryza plants. The different-colored cluster lines indicate different groups of SPL genes. The different-shaped arcs indicate different species. The motifs, numbers 1–20, are displayed in different colored boxes.
Homologous Relationship, Gene Structure, and Selective Forces
Homologous genes were mainly classified as orthologous and paralogous types. For one gene family, tandem and segmental duplication events are the main reasons for gene expansion (Coghlan et al., 2005). MCScanX toolkit was used to scan each Oryza genome for identifying putative paralogous chromosomal regions. In the OsSPL gene family, five homologous gene pairs of Oryza sativa (OsSPL3/12, OsSPL4/11, OsSPL5/10, OsSPL14/17, and OsSPL16/18) were reported to locate within segmental duplication region (Yang et al., 2008). Besides that, five, four, three, four, and four paralogous pairs of SPL genes were identified in Or, On, Ob, Oglu, and Ogla, respectively. Interestingly, we found that paralogs of the SBP-box family in Asian rice (Os, Or, and On) were larger than those of the African rice (Ob and Ogla) (Supplementary Table S4). Except for one tandem duplicate gene pair (ObSPL1/ObSPL2), we called the other paralogous genes as ohnologs, which derived specifically from WGD (whole genome duplication).
In order to understand the evolution pattern of the SPL genes in whole Oryza AA genus, we used the OrthoMCL software to investigate orthologous target gene pairs with a basic codon substitution model. In total, 19 orthologous groups were identified termed corresponding with OsSPL names (Figure 3B and Supplementary Figure S3). Intron/exon numbers might represent splicing variants and were used to classify genes (Roy and Gilbert, 2006). Thus, intron/exon structures of each orthologous group were generated based on genome sequences and corresponding coding sequences (Figure 3A). In clade I and IIA, SPLs contained more than 10 exons, while genes in IIB only harbored three exons; according to this, proteins of clade I and IIA had the long C terminus with more than 700 aa residues (Supplementary Figure S3 and Table S1). Despite that exon copy number was constant in clade IIB or clade IIA, the first two introns elongated or shortened in distinct groups. In clade IIC, we found all groups harbored four or fewer exons and divided them into four subclades IIC-1 to -4 based on phylogenetic and ohnologous relationships (Figure 3B). Most genes in one group containing a similar structure (exon/intron number and length) even belonged to different species. There was one exception in group 4, where the last exon of cultivated rice SPLs (OsSPL4 and OglaSPL4) possessed a shorter length than other wild rice SPLs. In general, introns exhibited significant length change, whereas most exon maintained a relatively constant length during the course of the Oryza evolution. Some closely related groups might suffer exon/intron gain or loss event in one clade, such as group 13 lost one exon compared to group 2 (Figure 3C). It should be noted that group 19 had extreme long introns, which included OsSPL19. Since OsSPL19 was suspected of being likely a pseudogene (Xie et al., 2006; Yang et al., 2008), the function of the orthologous group SPL19 in Oryza needs more experimental validation.
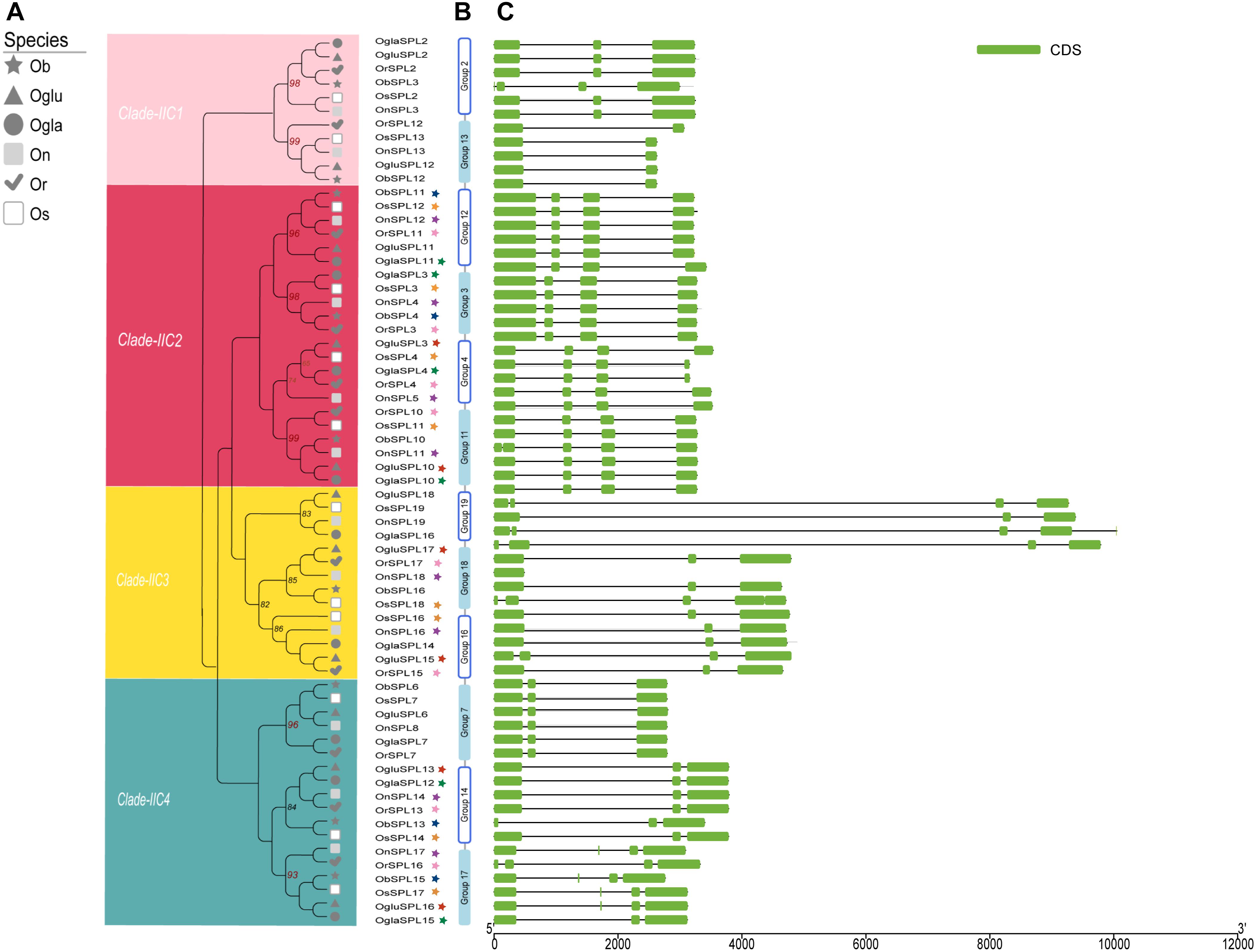
Figure 3. Gene structure of four subgroups and 12 orthologs in clade IIC. (A) A phylogenetic tree of IIC was constructed. Details of four clusters are shown in different colors, and the number above the branch indicates the bootstrap values over 50%. Colorful stars display paralogous pairs in each species. (B) Orthologous groups in clade IIC. (C) Exons–introns and untranslated regions (UTRs) of SPL genes. Green boxes indicate exons; black lines indicate introns. The length of protein can be estimated using the scale at the bottom.
To investigate the selective pressure of SPLs, we calculated the number of non-synonymous substitutions per non-synonymous site (Ka), synonymous (Ks), and Ka/Ks ratio in each ortholog group. A statistically significant Ka/Ks ratio, equal to 1.0, meant neutral or absence of evolution. Whereas lower than or greater than that represent purifying selection and positive selection, separately. The infinite value of group 18 (Ks = 0) was excluded for computing values; thereby, our findings indicated that mean Ka/Ks values of all groups were lower than 1.0, which meant under purifying selection. Genes in groups 3, 13, and 15 exhibited low Ka/Ks ratio, suggesting significantly strong signs of positive selection. In contrast, a relative weak positive selection sign was found in group 2 (Supplementary Table S5). At the same time, the classification was also performed with evolutionary parameters (Ka, Ks, and Ka/Ks) between two datasets: miR156-targeted genes (clade IIC) and miR156-non-target genes (clades I, IIA, and IIB; might undergo distinct evolutionary rates and selection pressures (Supplementary Figure S4).
Analyses of cis-Elements and Expression Divergence
Regulation of gene expression via specific cis-elements in the promoter region elements has become a major adaptive mechanism to respond to different environmental conditions (Walther et al., 2007). We searched the PlantCARE database to identify potential cis-acting elements in the 2,000 bp upstream promoter regions of SPL genes. A large number of cis-elements in the promoter regions of Oryza SPLs were detected and then were classified into four subdivisions: light responsiveness, plant growth, phytohormone, and abiotic stress response (Figure 4B). More than half of predicted cis-elements were classified in the phytohormone response category, including the P-box, TATC-box, GARE motif (gibberellin-responsive elements), and TGA (auxin-responsive element). Among them, ABRE (involved in abscisic acid response) was covered the largest portion (55%), followed by the TCA element (related to salicylic acid; 20%). As for the light responsiveness category, cis-acting elements were distributed widely throughout the promoter regions, including a series of elements that participate in part of light responsive (GA motif, GATA motif, LS7, TCT motif, and I-box) MRE (MYB binding site involved in light responsiveness). GT1 motif was the most common (31%), whose proportion was a little higher than Sp1 (30%). In the abiotic stress response category, three main stress-related cis-acting elements were identified, known as the GC motif (anoxic-specific inducibility), TC-rich repeats (defense and stress responsiveness), and LTR (response to low temperature). Furthermore, plant growth-related elements HD-Zip 1 (differentiation of the palisade mesophyll cells) and GCN4 motif accounted for 5% (responsible for endosperm expression) in all cis-elements, which mainly placed in groups 18, 3, and 12 (Figure 4A).
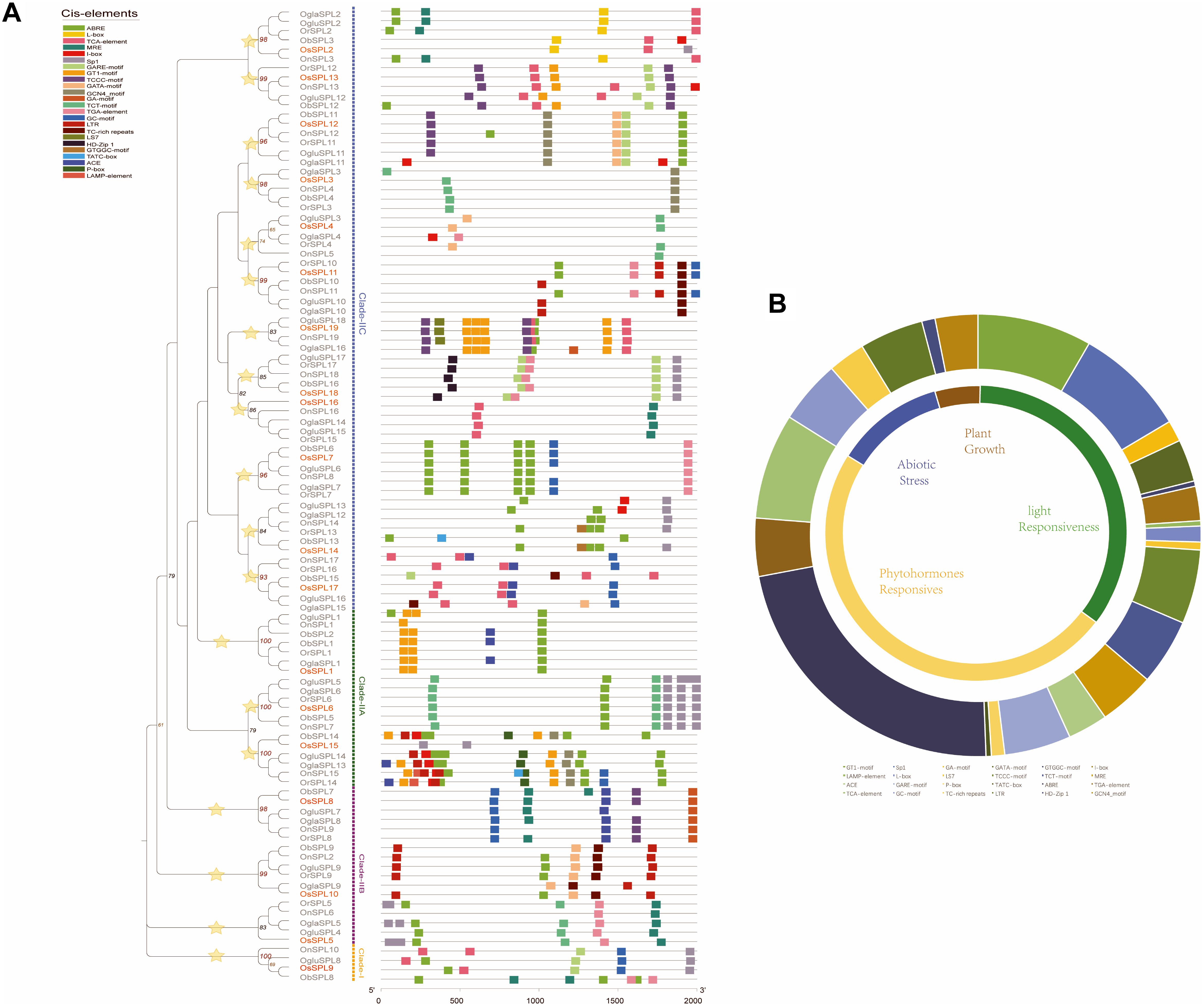
Figure 4. Identification of cis-acting elements in all Oryza SPL genes. (A) Cis-acting elements of all SPLs based on a phylogenetic tree. The different colored boxes indicate different promoter elements in each SPL gene. Yellow stars indicate orthologous groups. (B) Pie charts of different sizes represent the ratio of each promoter element in each category.
In our results, SPL genes in different orthologous groups were heterogeneous, which might have same functions due to differential types of cis-acting elements in their promoter regions (Figure 4A). For example, group 16 only contained two types of cis-acting elements involved in the phytohormone responsiveness. Intriguingly, there was a considerable difference between Asian rice (Os, Or, and On) and the other rice (Ob, Ogla, and Oglu) in group 11. The former had more three elements (ABRE, GC motif, and TGA element) than the latter (LTR and TC-rich repeats), suggesting more functional regulation of phytohormone and abiotic stress response in the Asian rice (Figure 4A). To explore the expression pattern of wild rice SPLs, we also obtained the expression profiles of SPL genes in O. barthii, O. glaberrima, and O. rufipogon from the NCBI SRA databases. In these organisms, a total of six major tissues were examined: roots, tiller base, leaf blades, panicles, leaf sheaths, and leaf pulvini, respectively. We observed that the SPLs with miR156-targeted sites represented organ-specific expression patterns, comparing with the SPL genes not bound by miRNA (Supplementary Figure S5). Although these transcriptomic data provided us clues to the functional conservation and significance of rice SPLs, it remains to be experimentally verified in future studies.
Discussion
Evolution of SPL Genes in the Oryza AA Genus
Previous results reflected that SPL genes were specific in the plant and might predate the divergence of the green algae (Guo et al., 2008; Zhang et al., 2015). The ancestor SPL gene originally formed two distinct lineages, I and II, by altering the Zn1 from C3H to C4. Clade I contained longer protein length and more exons than clade II. Then, lineage II might be duplicated into three copies, namely, clades IIB, IIA, and IIC, caused by losing exons and introns. Likewise, genes in clade IIA had similar structure properties to clade I with some continuous zero phase introns (Figure 3C), which might be lost more easily. Especially in IIB, the copy number of SPLs was relatively small, hinting fewer duplication events occurred. Strikingly, all members in clade IIC had the conserved miRNA target site and even conserved function.
Aimed at dissecting the evolutionary dynamic and function divergence of the SBP-box gene family in the Oryza genus, we identified SPL genes in six rice species and determined the phylogenetic relationships of this family. In accordance with the classification of previous research (Guo et al., 2008), we discovered three main clades, IIA, IIB, and IIC, of all 105 Oryza SPLs, as outgroup was clade I. Furthermore, 19 orthologous groups of OsSPLs were marked in this phylogenetic tree under purifying selection mostly. Group I contained a gene expansion of seven homologs at a smaller scale including six orthologs and one paralogous pair ObSPL1/ObSPL2, while the other nine groups had exactly one ortholog in each species. Compared to groups 9 and 19 with barely four species, five groups (groups 3, 4, 5, 13, and 16) had SPLs in the absence of one species. For example, ObSPLs did not pair with orthologs in other rice sequences in groups 4, 5, and 16. In many high-copy number gene families, such as NB-ARC, F-Box, and B3 gene families, tandem duplication (TD) and gene transposition duplication (GTD) have been reported to frequently occur (Freeling, 2009). In the present study, we identified 25 ohnologous genes, but there was only one paralogous pair (ObSPL1 and ObSPL2) caused by TD considering that the SBP gene family has middle scale of gene copy number (less than 20). As OsSPL19 has been cognized as a putative pseudogene (Xie et al., 2006), we deduced its orthologs in group 19 (OgluSPL18, OnSPL19, and OglaSPL16) were pseudogenes. These findings implied that pseudogenization was preserved by loss-of-function mutations in some wild rice, and the acts of Oryza SPL genes were redundant and complex. Combined with a detailed comparison of gene structure and motif composition, our results suggested that intra-group genes showed approximate gene structures and conserved motifs in coding regions. While those structural differences among species were less than among the orthologous groups. Thus, we proposed that the SPL family is relatively conservative in Oryza.
Functional Diversity in the Oryza SPL Gene Family
The SPL family has been characterized to have diverse biological processes, governing many fundamental aspects of plant growth and development (Preston and Hileman, 2013). Recent studies have revealed that some group members of the SPL gene family regulate various yield-related traits in rice (Wang and Zhang, 2017). For example, OsSPL8, known as OsLG1, was reported for controlling leaf angle in wild rice and regulating a closed panicle trait in domesticated rice (Ishii et al., 2013). In addition, OsSPL14/IPA1 promotes panicle branching and grain productivity, which define ideal plant architecture in rice. OsSPL16/GW8 has been identified as an important QTL improving rice yield and grain quality (Wang S. et al., 2012; Wang et al., 2015). Afterward, OsSPL13/GLW7 enhances rice grain length and yield by positively regulating cell size in the grain hull (Si et al., 2016). Those researches provided us some hints that SPL genes may have great promise for improving many significant agricultural traits in crop species.
In past cases of other species, the BnaSPLs (Brassica napus) and ZmSPLs (maize) in the same phylogenetic clade showed conserved gene structures, which have similar expression profiles with AtSPLs (Cheng et al., 2016; Wei et al., 2018). Herein, wild rice SPLs had same cis-acting elements in the same orthologous group, suggesting that they might perform similar biological tasks to OsSPLs. The preliminary in silico analysis of gene expression of three wild rice SPLs might help us to acquire their tissue-specific expression patterns substantially. Though the wild rice germplasm has been a crucial resource to clone agronomically useful genes for domesticated rice (Doebley et al., 2006), it would be worthy to explore the exact role and possible utilization of wild rice SPLs by functional characterization given the lack of related experimental verification on SPL genes in wild rice. However, we have to note that redundant orthologous genes do not always predict function considering the negative effect of subfunctionalization (partition original functions) or beneficial effect of neo-functionalization (create new roles) (Force et al., 1999; Teichmann and Babu, 2004). It is widely accepted that the gain of novel function by neo-functionalization is rare, while many redundant genes will become pseudogenes finally at a relatively high probability (Jacquemin et al., 2014). On the other hand, the complexity of SPL-related regulatory network also must be taken into account (Teichmann and Babu, 2004; Wang et al., 2015).
Conclusion
Diversification of SPL genes in six Oryza genomes was observed from many aspects, including phylogenesis, genomic structure, and the location of the miR156 target site. The SPL gene family within the Oryza genus displayed size variation between six rice species. All 105 SPL genes were divided into lineages I and II, and clade II were further grouped into three clades (IIA, IIB, and IIC) through several rounds of duplication. Genes in one clade experienced similar conserved evolutionary features and cis-acting elements, implying similarity of plant biological function potentially. Nineteen orthologous groups of OsSPLs in Oryza were identified, and all groups suffered relaxed purifying selection and diversified continually including alterative gene number, intron length, and miR156 regulation. Taken together, our results will interpret a comprehensive understanding of the molecular characteristics and evolutionary pattern of the SPL gene family in Oryza AA genome plants.
Author Contributions
HZ and WK conceptualized and designed the research. HZ performed most of the analyses and wrote the manuscript. WK carried out the gene expression analysis. ZG, CL, XD, and XF helped in the statistical analyses and revision. YL guided the study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was financially supported by the National Special Key Project for Transgenic Breeding (Grant No. 2016ZX08001001), the National Key Research and Development Program of China (Grant No. 2016YFD0100400), the 863 Program (Grant No. 2014AA10A604-9), and the Key Grant Project of the Chinese Ministry of Education (Grant No. 313039).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00565/full#supplementary-material
FIGURE S1 | Alignment of multiple SBP domain amino acid sequences in Oryza SPLs. (A) The overall height of each stack represents the degree of conservation at each position, while the height of the letters within each stack indicates the relative frequency of the corresponding amino acid. (B) The two conserved zinc-finger structures (C3H/C4, C2HC) and nuclear localization signal (NLS) are indicated on the top. Multiple sequence alignment of full-length protein sequences in the SBP domain was performed.
FIGURE S2 | Chromosomal localization of Oryza SPLs. (A–F) Chromosome mapping of SPL genes in the six Oryza genomes. Chromosome numbers are indicated at the bottom of each bar. Scale is represented in mega bases (Mb), and lines in the background chromosome indicate all genes within the genomes.
FIGURE S3 | Gene structure and othologous groups in clades I, IIA, and IIB. (A) Exons–introns and UTRs and orthologous groups in clade IIA based on a phylogenetic relationship. (B) Exons–introns and orthologous groups in clade IIB based on a phylogenetic relationship. (C) Exons–introns and orthologous groups in clade I based on a phylogenetic tree. Colorful stars display paralogous pairs in each species; yellow boxes indicate exons; and black lines indicate introns. The length of protein can be estimated using the scale at the bottom, and intron phases (0, 1, and 2) are shown.
FIGURE S4 | Comparison of the mean Ka/Ks value in miR156-non-target and miR156-targeted genes.
FIGURE S5 | Tissue-specific expression analysis of SPL genes in (A) Oryza barthii, (B) Oryza glaberrima, and (C) Oryza rufipogon. (D) Analyzed tissues included roots, tiller base, leaf blades, panicles, leaf sheaths, and leaf pulvini.
TABLE S1 | Summary of information on SPL genes in this study.
TABLE S2 | Annotation of conserved motifs in Oryza SPL proteins.
TABLE S3 | The detail information of predicted targets for miRNA.
TABLE S4 | Segmentally and tandemly duplicated Oryza SPL gene pairs.
TABLE S5 | Comparison of the mean Ka/Ks value in each othologous group.
Footnotes
- ^ http://www.ub.edu/dnasp/
- ^ http://www.dna.affrc.go.jp/PLACE/signalup.html
- ^ http://plants.ensembl.org/info/website/ftp/index.html
References
Aggarwal, R. K., Brar, D. S., and Khush, G. S. (1997). Two new genomes in the Oryza complex identified on the basis of molecular divergence analysis using total genomic DNA hybridization. Mol. Gen. Genet. 254, 1–12. doi: 10.1007/s004380050384
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). MEME suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Baisakh, N., Prasad, M., Grima-Pettenati, J., Cao, Y., Han, Y., Li, D., et al. (2016). MYB transcription factors in Chinese pear (Pyrus bretschneideri Rehd).: genome-wide identification, classification, and expression profiling during fruit. Development 7:577. doi: 10.3389/fpls.2016.00577
Bolser, D. M., Staines, D. M., Perry, E., and Kersey, P. J. (2017). Ensembl plants: integrating tools for visualizing, mining, and analyzing plant genomic data. Methods Mol. Biol. 1374, 115–140. doi: 10.1007/978-1-4939-6658-5_1
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinform. 10:421. doi: 10.1186/1471-2105-10-421
Cardon, G., Höhmann, S., Klein, J., Nettesheim, K., Saedler, H., and Huijser, P. (1999). Molecular characterisation of the Arabidopsis SBP-box genes. Gene 237, 91–104. doi: 10.1016/S0378-1119(99)00308-X
Chen, C., Xia, R., Chen, H., and He, Y. (2018). TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface. bioRxiv [Preprint]. doi: 10.1101/289660
Chen, J. Y., Guo, L., Ma, H., Chen, Y. Y., Zhang, H. W., Ying, J. Z., et al. (2014). Fine mapping of qHd1, a minor heading date QTL with pleiotropism for yield traits in rice (Oryza sativa L). Theor. Appl. Genet. 127, 2515–2524. doi: 10.1007/s00122-014-2395-7
Cheng, H., Hao, M., Wang, W., Mei, D., Tong, C., Wang, H., et al. (2016). Genomic identification, characterization and differential expression analysis of SBP-box gene family in Brassica napus. BMC Plant Biol. 16:196. doi: 10.1186/s12870-016-0852-y
Coghlan, A., Eichler, E. E., Oliver, S. G., Paterson, A. H., and Stein, L. (2005). Chromosome evolution in eukaryotes: a multi-kingdom perspective. Trends Genet. 21, 673–682. doi: 10.1016/j.tig.2005.09.009
Dai, X., and Zhao, P. X. (2011). PsRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159. doi: 10.1093/nar/gkr319
Dingwall, C., and Laskey, R. A. (1991). Nuclear targeting sequences—a consensus? Trends Biochem. Sci. 16, 478–481. doi: 10.1016/0968-0004(91)90184-W
Doebley, J. F., Gaut, B. S., and Smith, B. D. (2006). The molecular genetics of crop domestication. Cell 127, 1309–1321. doi: 10.1016/j.cell.2006.12.006
Feller, A., MacHemer, K., Braun, E. L., and Grotewold, E. (2011). Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 66, 94–116. doi: 10.1111/j.1365-313X.2010.04459.x
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39(Suppl._2), W29–W37. doi: 10.1093/nar/gkr367
Force, A., Lynch, M., Pickett, F. B., Amores, A., Yan, Y. L., and Postlethwait, J. (1999). Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545.
Freeling, M. (2009). Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 60, 433–453. doi: 10.1146/annurev.arplant.043008.092122
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., et al. (2005). “Protein identification and analysis tools on the ExPASy server,” in The Proteomics Protocols Handbook, ed. J. M. Walker (New York, NY: Humana Press).
Ge, S., Sang, T., Lu, B.-R., and Hong, D.-Y. (1999). Phylogeny of rice genomes with emphasis on origins of allotetraploid species. Proc. Natl. Acad. Sci. U.S.A. 96, 14400–14405. doi: 10.1073/pnas.96.25.14400
Goodstein, D. M., Shu, S., Howson, R., Neupane, R., Hayes, R. D., Fazo, J., et al. (2012). Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186. doi: 10.1093/nar/gkr944
Guo, A. Y., Zhu, Q. H., Gu, X., Ge, S., Yang, J., and Luo, J. (2008). Genome-wide identification and evolutionary analysis of the plant specific SBP-box transcription factor family. Gene 418, 1–8. doi: 10.1016/j.gene.2008.03.016
He, Z., Zhang, H., Gao, S., Lercher, M. J., Chen, W. H., and Hu, S. (2016). Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res. 44, W236–W241. doi: 10.1093/nar/gkw370
Henríquez-valencia, C., Arenas-m, A., Medina, J., and Canales, J. (2018). Integrative transcriptomic analysis uncovers novel gene modules that underlie the sulfate response in Arabidopsis thaliana. Front. Plant Sci. 9:470. doi: 10.3389/fpls.2018.00470
Ishii, T., Numaguchi, K., Miura, K., Yoshida, K., Thanh, P. T., Htun, T. M., et al. (2013). OsLG1 regulates a closed panicle trait in domesticated rice. Nat. Genet. 45, 462–465. doi: 10.1038/ng.2567
Jacquemin, J., Ammiraju, J. S. S., Haberer, G., Billheimer, D. D., Yu, Y., Liu, L. C., et al. (2014). Fifteen million years of evolution in the Oryza genus shows extensive gene family expansion. Mol. Plant. 7, 642–656. doi: 10.1093/mp/sst149
Jiao, Y., Wang, Y., Xue, D., Wang, J., Yan, M., Liu, G., et al. (2010). Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 42, 541–544. doi: 10.1038/ng.591
Khush, G. S. (2003). Productivity improvements in rice. Nutr. Rev. 61(6 Pt 2), S114–S116. doi: 10.131/nr.2003.jun.S114
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Klein, J., Saedler, H., and Huijser, P. (1996). A new family of DNA binding proteins includes putative transcriptional regulators of the Antirrhinum majus floral meristem identity gene SQUAMOSA. Mol. Gen. Genet. 47, 152–154. doi: 10.1016/j.rama.2015.03.002
Kong, W., Bendahmane, M., and Fu, X. (2018). Genome-wide identification and characterization of aquaporins and their role in the flower opening processes in carnation (Dianthus caryophyllus). Molecules 23:1895. doi: 10.3390/molecules23081895
Kong, W., Yang, S., Wang, Y., Bendahmane, M., and Fu, X. (2017). Genome-wide identification and characterization of aquaporin gene family in Beta vulgaris. PeerJ 5:e3747. doi: 10.7717/peerj.3747
Kozomara, A., and Griffiths-Jones, S. (2014). MiRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. doi: 10.1093/nar/gkt1181
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lescot, M. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327. doi: 10.1093/nar/30.1.325
Li, J., Hou, H., Li, X., Xiang, J., Yin, X., Gao, H., et al. (2013). Genome-wide identification and analysis of the SBP-box family genes in apple (Malus × domestica Borkh). Plant Physiol. Biochem. 70, 100–114. doi: 10.1016/j.plaphy.2013.05.021
Li, J., Mahajan, A., and Tsai, M. D. (2006). Ankyrin repeat: a unique motif mediating protein–protein interactions. Biochemistry 45, 15168–15178. doi: 10.1021/bi062188q
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lu, Z., Yu, H., Xiong, G., Wang, J., Jiao, Y., Liu, G., et al. (2013). Genome-wide binding analysis of the transcription activator IDEAL PLANT ARCHITECTURE1 reveals a complex network regulating rice plant architecture. Plant Cell 25, 3743–3759. doi: 10.1105/tpc.113.113639
Michaely, P., and Bennett, V. (1992). The ANK repeat: a ubiquitous motif involved in macromolecular recognition. Trends Cell Biol. 2, 127–129. doi: 10.1016/0962-8924(92)90084-Z
Mitchell, A. L., Attwood, T. K., Babbitt, P. C., Blum, M., Bork, P., Bridge, A., et al. (2018). InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47, D351–D360. doi: 10.1093/nar/gky1100
Nicholas, K. B., Nicholas, H. B., Deerfield, D. W., America, B., Street, M., Francisco, S., et al. (1997). GeneDoc: analysis and visualization of genetic variation. EMBnet News 4:14.
Petryszak, R., Keays, M., Tang, Y. A., Fonseca, N. A., Barrera, E., Burdett, T., et al. (2016). Expression atlas update—an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 44, D746–D752. doi: 10.1093/nar/gkv1045
Porri, A., Torti, S., Romera-Branchat, M., and Coupland, G. (2012). Spatially distinct regulatory roles for gibberellins in the promotion of flowering of Arabidopsis under long photoperiods. Development 139, 2198–2209. doi: 10.1242/dev.077164
Preston, J. C., and Hileman, L. C. (2013). Functional evolution in the plant SQUAMOSA-PROMOTER BINDING PROTEIN-LIKE (SPL) gene family. Front. Plant Sci. 4:80. doi: 10.3389/fpls.2013.00080
Rhoades, M. W., Reinhart, B. J., Lim, L. P., Burge, C. B., Bartel, B., and Bartel, D. P. (2002). Prediction of plant microRNA targets. Cell 110, 513–520. doi: 10.1016/S0092-8674(02)00863-2
Rogers, K., and Chen, X. (2013). Biogenesis, turnover, and mode of action of plant MicroRNAs. Plant Cell 25, 2383–2399. doi: 10.1105/tpc.113.113159
Roy, S. W., and Gilbert, W. (2006). The evolution of spliceosomal introns: patterns, puzzles and progress. Nat. Rev. Genet. 7, 211–221. doi: 10.1038/nrg1807
Schwab, R., Palatnik, J. F., Riester, M., Schommer, C., Schmid, M., and Weigel, D. (2005). Specific effects of microRNAs on the plant transcriptome. Dev. Cell. 8, 517–527. doi: 10.1016/j.devcel.2005.01.018
Schwechheimer, C., Zourelidou, M., and Bevan, M. W. (1998). Plant transcription factor studies. Annu. Rev. Plant Physiol. Plant Mol. Biol. 49, 127–150. doi: 10.1146/annurev.arplant.49.1.127
Si, L., Chen, J., Huang, X., Gong, H., Luo, J., Hou, Q., et al. (2016). OsSPL13 controls grain size in cultivated rice. Nat. Genet. 48, 447–456. doi: 10.1038/ng.3518
Smaczniak, C., Immink, R. G. H., Angenent, G. C., and Kaufmann, K. (2012). Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139, 3081–3098. doi: 10.1242/dev.074674
Stone, J. M., Liang, X., Nekl, E. R., and Stiers, J. J. (2005). Arabidopsis AtSPL14, a plant-specific SBP-domain transcription factor, participates in plant development and sensitivity to Fumonisin B1. Plant J. 41, 744–754. doi: 10.1111/j.1365-313X.2005.02334.x
Teichmann, S. A., and Babu, M. M. (2004). Gene regulatory network growth by duplication. Nat. Genet. 36, 492–496. doi: 10.1038/ng1340
Thomopson, J. D., Higgins, D. G., and Gibson, T. J. (1994). ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1007/s10750-012-1385-5
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with topHat and cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Vaughan, D. A., Morishima, H., and Kadowaki, K. (2003). Diversity in the Oryza genus. Curr. Opin. Plant Biol. 6, 139–146. doi: 10.1016/S1369-5266(03)00009-8
Walther, D., Brunnemann, R., and Selbig, J. (2007). The regulatory code for transcriptional response diversity and its relation to genome structural properties in A. thaliana. PLoS Genet. 3:e11. doi: 10.1371/journal.pgen.0030011
Wang, J., Zhou, L., Shi, H., Chern, M., Yu, H., Yi, H., et al. (2018). A single transcription factor promotes both yield and immunity in rice. Science 361, 1026–1028. doi: 10.1126/science.aat7675
Wang, L., and Zhang, Q. (2017). Boosting rice yield by fine-tuning SPL gene expression. Trends Plant Sci. 22, 643–646. doi: 10.1016/j.tplants.2017.06.004
Wang, S., Li, S., Liu, Q., Wu, K., Zhang, J., Wang, S., et al. (2015). The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 47, 949–954. doi: 10.1038/ng.3352
Wang, S., Wu, K., Yuan, Q., Liu, X., Liu, Z., Lin, X., et al. (2012). Control of grain size, shape and quality by OsSPL16 in rice. Nat. Genet. 44, 950–954. doi: 10.1038/ng.2327
Wang, Y., Tang, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, 1–14. doi: 10.1093/nar/gkr1293
Wei, H., Zhao, Y., Xie, Y., and Wang, H. (2018). Exploiting SPL genes to improve maize plant architecture tailored for high-density planting. J. Exp. Bot. 69, 4675–4688. doi: 10.1093/jxb/ery258
Xie, K., Wu, C., and Xiong, L. (2006). Genomic organization, differential expression, and interaction of SQUAMOSA promoter-binding-like transcription factors and microRNA156 in rice. Plant Physiol. 142, 280–293. doi: 10.1104/pp.106.084475
Yamasaki, K., Kigawa, T., Inoue, M., Tateno, M., Yamasaki, T., Yabuki, T., et al. (2004). A novel zinc-binding motif revealed by solution structures of DNA-binding domains of Arabidopsis SBP-family transcription factors. J. Mol. Biol. 337, 49–63. doi: 10.1016/j.jmb.2004.01.015
Yamasaki, K., Kigawa, T., Inoue, M., Yamasaki, T., Yabuki, T., Aoki, M., et al. (2006). An Arabidopsis SBP-domain fragment with a disrupted C-terminal zinc-binding site retains its tertiary structure. FEBS Lett. 580, 2109–2116. doi: 10.1016/j.febslet.2006.03.014
Yang, Z., Wang, X., Gu, S., Hu, Z., Xu, H., and Xu, C. (2008). Comparative study of SBP-box gene family in Arabidopsis and rice. Gene. 407, 1–11. doi: 10.1016/j.gene.2007.02.034
Yu, S., Galvao, V. C., Zhang, Y.-C., Horrer, D., Zhang, T.-Q., Hao, Y.-H., et al. (2012). Gibberellin regulates the Arabidopsis floral transition through miR156-Targeted SQUAMOSA PROMOTER BINDING-LIKE transcription factors. Plant Cell 24, 3320–3332. doi: 10.1105/tpc.112.101014
Zhang, H.-X., Jin, J.-H., He, Y.-M., Lu, B.-Y., Li, D.-W., Chai, W.-G., et al. (2016). Genome-wide identification and analysis of the SBP-box family genes under Phytophthora capsici stress in pepper (Capsicum annuum L). Front. Plant Sci. 7:504. doi: 10.3389/fpls.2016.00504
Zhang, S. D., and Ling, L. Z. (2014). Genome-wide identification and evolutionary analysis of the SBP-box gene family in castor bean. PLoS One 9:e86688. doi: 10.1371/journal.pone.0086688
Zhang, S. D., Ling, L. Z., and Yi, T. S. (2015). Evolution and divergence of SBP-box genes in land plants. BMC Genomics 16:787. doi: 10.1186/s12864-015-1998-y
Keywords: evolution, SBP-box family genes, motif, miR156, Oryza genus
Citation: Zhong H, Kong W, Gong Z, Fang X, Deng X, Liu C and Li Y (2019) Evolutionary Analyses Reveal Diverged Patterns of SQUAMOSA Promoter Binding Protein-Like (SPL) Gene Family in Oryza Genus. Front. Plant Sci. 10:565. doi: 10.3389/fpls.2019.00565
Received: 25 January 2019; Accepted: 15 April 2019;
Published: 08 May 2019.
Edited by:
Yuannian Jiao, Chinese Academy of Sciences, ChinaReviewed by:
Liangsheng Zhang, Fujian Agriculture and Forestry University, ChinaAles Kovarik, Academy of Sciences of the Czech Republic (ASCR), Czechia
Copyright © 2019 Zhong, Kong, Gong, Fang, Deng, Liu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yangsheng Li, bHlzaDIwMDFAd2h1LmVkdS5jbg==