 Saurabh Chaudhary1†
Saurabh Chaudhary1† Waqas Khokhar1†
Waqas Khokhar1† Ibtissam Jabre1†
Ibtissam Jabre1† Anireddy S. N. Reddy2Lee J. Byrne1
Anireddy S. N. Reddy2Lee J. Byrne1 Cornelia M. Wilson1
Cornelia M. Wilson1 Naeem H. Syed1*
Naeem H. Syed1*- 1School of Human and Life Sciences, Canterbury Christ Church University, Canterbury, United Kingdom
- 2Department of Biology and Program in Cell and Molecular Biology, Colorado State University, Fort Collins, CO, United States
Plants, unlike animals, exhibit a very high degree of plasticity in their growth and development and employ diverse strategies to cope with the variations during diurnal cycles and stressful conditions. Plants and animals, despite their remarkable morphological and physiological differences, share many basic cellular processes and regulatory mechanisms. Alternative splicing (AS) is one such gene regulatory mechanism that modulates gene expression in multiple ways. It is now well established that AS is prevalent in all multicellular eukaryotes including plants and humans. Emerging evidence indicates that in plants, as in animals, transcription and splicing are coupled. Here, we reviewed recent evidence in support of co-transcriptional splicing in plants and highlighted similarities and differences between plants and humans. An unsettled question in the field of AS is the extent to which splice isoforms contribute to protein diversity. To take a critical look at this question, we presented a comprehensive summary of the current status of research in this area in both plants and humans, discussed limitations with the currently used approaches and suggested improvements to current methods and alternative approaches. We end with a discussion on the potential role of epigenetic modifications and chromatin state in splicing memory in plants primed with stresses.
Introduction
Plants have evolved various developmental and physiological strategies to control daily activities that respond to variable and extreme environmental conditions (Gratani, 2014; Becklin et al., 2016). To maximize efficiency under diverse conditions, the crosstalk between multiple layers of gene regulation including co-transcriptional, post-transcriptional, and post-translational regulation is crucial for plants (Reddy et al., 2013; Guerra et al., 2015; Skelly et al., 2016). Alternative splicing (AS) is one such mechanism, which is widespread in plants and humans, generates two or more mRNAs from the same precursor-mRNA (pre-mRNA) and is thought to significantly contribute toward protein diversity (Nilsen and Graveley, 2010; Syed et al., 2012; Reddy et al., 2013). The basic mechanism of AS in higher eukaryotes is similar, however, some differences in gene architecture, splicing and transcription machinery between plants and animals suggest plant-specific regulation of AS (Kornblihtt et al., 2013; Irimia and Roy, 2014; Wang et al., 2014).
The advances in next-generation sequencing (NGS) technology and omics approaches in plants have revealed that up to 70% of multi-exon genes undergo AS (Filichkin et al., 2010; Lu et al., 2010; Marquez et al., 2012; Shen et al., 2014; Thatcher et al., 2014; Chamala et al., 2015; Zhang et al., 2017). Among all AS events, intron retention (IR) is the predominant mode of AS in plants (Filichkin et al., 2010; Kalyna et al., 2012; Drechsel et al., 2013), whereas exon-skipping (ES) is the major type in humans (Figure 1) (Sammeth et al., 2008; Wang et al., 2008). Interestingly, IR generates mostly non-sense mRNAs harboring premature terminal codons (PTC+) and are either degraded by the non-sense-mediated mRNA decay (NMD) pathway, or escape NMD to produce truncated proteins, thereby regulating the function and abundance of their full-length counterparts (Filichkin and Mockler, 2012; Kalyna et al., 2012; Drechsel et al., 2013; Filichkin S.A. et al., 2015). The NMD pathway is a post-transcriptional mRNA quality control mechanism which acts to degrade PTC+ mRNAs. Some studies suggest alternative roles for transcripts with IR, which are either sequestered in the nucleus and released on demand (Filichkin S.A. et al., 2015; Filichkin S. et al., 2015) or function as protein-coding introns known as exitrons (Figure 1), a new class of retained introns with some features of exons (Marquez et al., 2015; Staiger and Simpson, 2015).
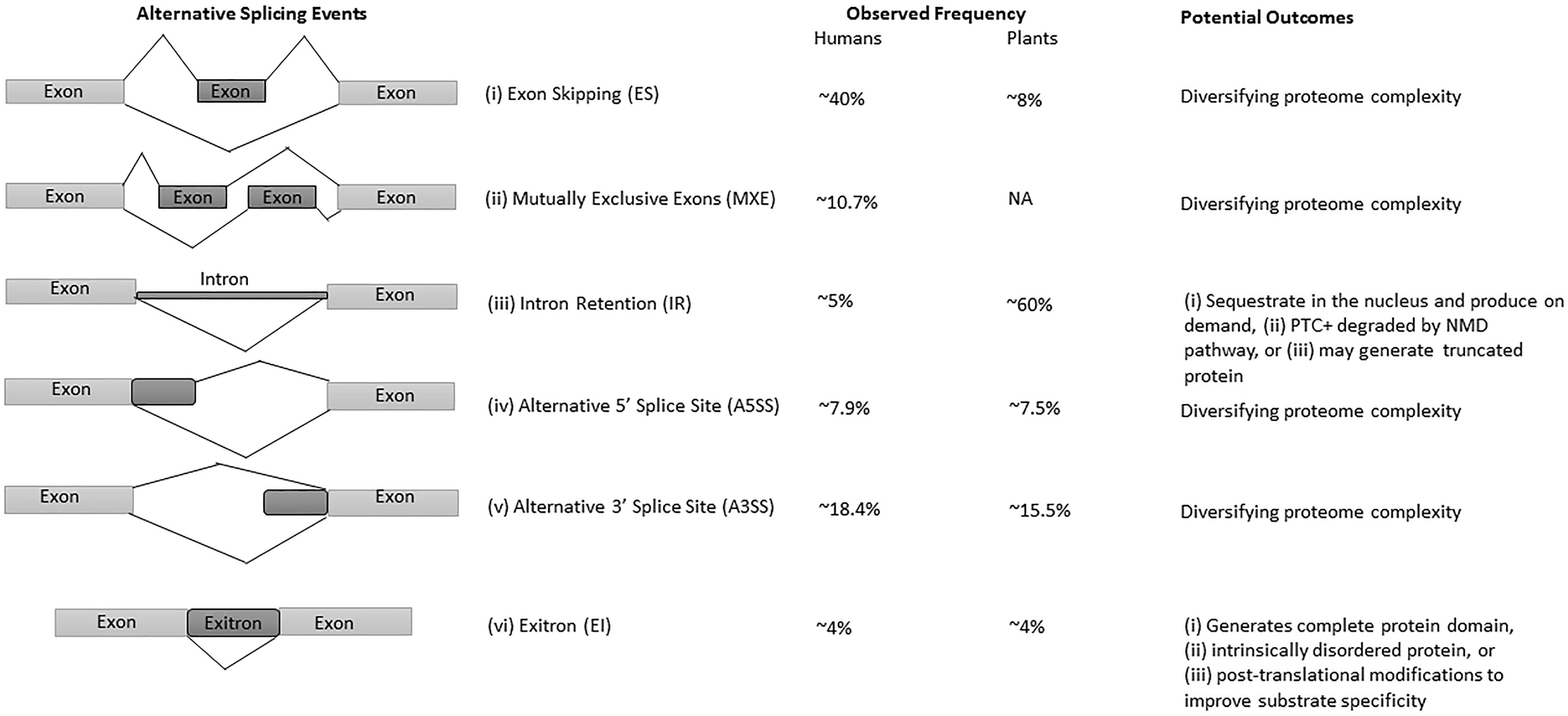
Figure 1. Major types of AS events, their frequency, and potential outcomes in humans and plants. (i) exon skipping (ES) or cassette exon, in which single or multiple exons are spliced out or retained; (ii) mutually exclusive exons (MXE), in which only one of the two exons is retained; (iii) intron retention (IR), where an intron remains in the mature transcript; (iv, v) alternative donor/acceptor site or 5′/3′ splice junction is used to alter the boundary of exons, and (vi) exitrons are a variety of IR with some feature of exons. Constitutive and alternatively spliced exons are represented as light and dark gray blocks, respectively. The observed frequencies represented here are approximate values, and may differ in different species, tissues and conditions. The presented data on AS events frequency are from Reddy et al. (2013), Marquez et al. (2015).
Plants modulate their gene expression patterns via AS coupled to NMD during different developmental stages, abiotic and/or biotic stresses and the circadian clock function (James et al., 2012; Kalyna et al., 2012; Drechsel et al., 2013; Kwon et al., 2014; Filichkin S.A. et al., 2015; Sureshkumar et al., 2016). Stressful conditions control not only the ratios but the timing of both sense and non-sense AS transcripts (Filichkin S.A. et al., 2015; Filichkin et al., 2018). However, it is unclear how environmental signals modulate splicing ratios and timing to help plants acclimate to such stresses in the short and long term. Furthermore, it is largely unknown to what extent AS transcripts are recruited for translation to be functionally significant at the proteomic level in plants.
Alternative splicing regulates essential functions in humans such as autophagy, apoptosis, protein localization, enzymatic activities and interaction with ligands, transcription factors activity and mRNA abundance, etc. (Kelemen et al., 2013; Paronetto et al., 2016; Gallego-Paez et al., 2017). Hence, it is not surprising that any aberrant or dysregulation in AS can cause several human diseases including cancer, neurological disorders, heart, and skeletal muscle abnormalities, and multiple genetic disorders (Matlin et al., 2005; Poulos et al., 2011; Kelemen et al., 2013; Sveen et al., 2016). Recent transcriptome (RNA-Seq), translatome (ribosomal foot-printing), and proteome data have shown a significant contribution of AS toward protein diversity in humans (Weatheritt et al., 2016; Liu et al., 2017). On the other hand, some proteomic studies suggest that AS may not significantly contribute to protein diversity and only single dominant isoforms are represented at the protein level for most of the protein-coding genes (Ezkurdia et al., 2015; Tress et al., 2017a). Apparently, these contradictions stem from the lower depth and limitations of mass spectrometry (MS) techniques to detect changes in protein domains as a result of AS (Wang et al., 2018; Chaudhary et al., 2019). In this review, basic differences in the mechanism of AS and its contribution toward protein diversity in plants and humans are discussed. We also discuss some emerging aspects of IR, NMD pathway, chromatin structure, and splicing memory in plants.
Coupling of Transcription and Splicing in Plants and Humans
Plant spliceosome machinery is not well characterized due to the unavailability of in vitro systems. However, in a recent study, an attempt has been made to develop an in vitro pre-mRNA splicing assay using plant nuclear extracts, and it may help to delineate and characterize components of the plant spliceosome machinery (Albaqami and Reddy, 2018). Sequence similarity based analyses suggest conserved regulation of AS in higher eukaryotes. Briefly, splicing is carried out by the spliceosome, which consists of five small nuclear ribonucleoprotein particles (snRNPs) designated as U1, U2, U4, U5, and U6 and additional spliceosome-associated non-snRNP proteins (Will and Lührmann, 2011; Matera and Wang, 2014; Wang et al., 2014). The cis-acting elements present on pre-mRNA include 5′ splice sites (5′SS), 3′ splice sites (3′SS), polypyrimidine tracts (PPT) and branch point sequences, which are recognized by the trans-acting factors such as splicing factors (SFs) mainly SR proteins and hnRNPs. The trans-acting SFs and cis-regulatory elements guide and modulate the spliceosome to recognize differential splice sites present on pre-mRNA (Koncz et al., 2012; Reddy et al., 2013; Chen and Moore, 2015). The details on the assembly of the spliceosome and regulation of AS has been reviewed extensively and readers are referred to excellent articles on this topic (Will and Lührmann, 2011; Reddy et al., 2013; Chen and Moore, 2015).
Recent evidence from metazoans indicates that the process of splicing is largely co-transcriptional (Shukla and Oberdoerffer, 2012; Brugiolo et al., 2013; Merkhofer et al., 2014). Extensive studies in animals and emerging data in plants show that the splicing process for the majority of genes is predominantly co-transcriptional in nature (Figure 2) (Nojima et al., 2015, 2018; Wang et al., 2016; Wong et al., 2017; Zhu et al., 2018; Jabre et al., 2019). The co-transcriptional behavior of splicing means that the chromatin environment such as methylation status, histone modifications, nucleosome occupancy and RNA Polymerase II (RNAPII) processivity has a strong influence on splicing outcomes (Listerman et al., 2006; Khodor et al., 2011; Pajoro et al., 2017; Jabre et al., 2019).
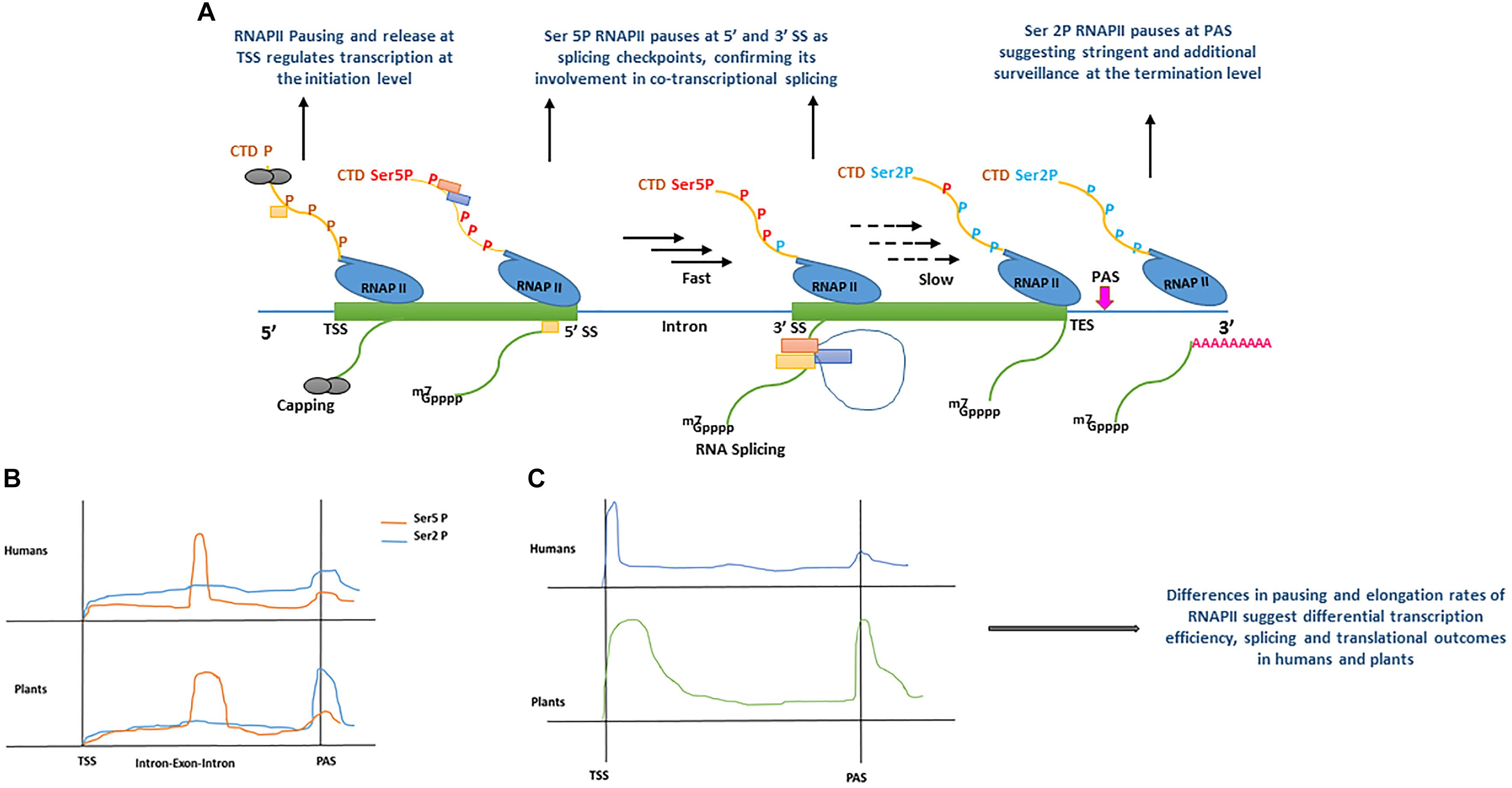
Figure 2. (A) Model displaying the role of RNA polymerase II (RNAPII) C-terminal domain (CTD) phosphorylation during co-transcriptional splicing regulation in human (Nojima et al., 2015), and plants (Zhu et al., 2018). During transcription initiation, the serine residues of RNAPII heptad repeat (yellow line) remain un-phosphorylated (brown ‘P’) around transcription start site (TSS) allowing core spliceosome recruitment (yellow rectangle) and capping (gray circles). During the elongation stage, serine 5 residues of RNAPII heptad repeat (red Ser5P) are phosphorylated around the 5′ splice sites (5′ SS) allowing the recruitment of additional components of the spliceosome machinery (orange and blue rectangles) and enhance RNAPII speed (black arrows). RNAPII elongation slows down (black dotted arrows) promotionally with the decrease of Ser5 phosphorylation toward the 3′ splice site (3′SS). Toward the transcription end site (TES), phosphorylation of serine 2 residues increase significantly resulting in RNAPII pausing before mRNA release (green line). m7GPPP and pink ‘repeated A’ represent 5′ cap and poly A tail, respectively. (B) Comparison of RNAPII CTD serine 2 and 5 residues phosphorylation levels accumulation between human (Nojima et al., 2015) and plants (Zhu et al., 2018). In human and plants, both serine 5 and serine 2 phosphorylation show significant increase after the transcription start site (TSS), only Ser 5P displays a sharp peak at exon–intron boundaries. For instance, a sharp peak of Ser2 P is only shown at polyadenylation site (PAS) in plants, whereas it remains less prominent in humans. (C) Comparison of RNAPII accumulation between humans and plants based on GRO-Seq experiments (Hetzel et al., 2016). In humans and plants, RNAPII occupancy is lower during the elongation stage and marginally increases around PAS. In contrast, plants show a broad peak after TSS, as compared with humans, and a more pronounced increase at PAS, suggesting a surveillance mechanism before a transcript is released. All Graphs are modified from published data to depict peaks.
Interestingly, long non-coding RNAs (lncRNAs) can also influence the splicing dynamics of their target genes either directly and/or after processing into short interfering or micro RNAs (Romero-Barrios et al., 2018). Non-coding RNAs can affect AS via modulating chromatin structure (Luco et al., 2011; Romero-Barrios et al., 2018), splicing factor recruitment and altering the phosphorylation status of spliceosomal proteins (Misteli et al., 1998; Romero-Barrios et al., 2018). Circular RNAs which are generated by the so-called non-canonical “backsplicing” of pre-mRNAs are known to regulate AS in animals and examples from plants are beginning to emerge as well. CircRNAs could make DNA:RNA hybrids with the genomic DNA to generate the so-called R-loop. Indeed, a circRNA derived from exon 6 of the SEPALLATA3 (SEP3) gene forms an R-loop via direct interaction with the SEP3 locus (Conn et al., 2017). The R-loop formation around exon 6 of the SEP3 gene results in skipping of this exon and affects petal and stamen number in Arabidopsis (Conn et al., 2017).
Plant promoters are largely devoid of nucleosomes, as a result of lower GC content (high AT enrichment) as compared with humans (Narang et al., 2005; Yang et al., 2007; Hetzel et al., 2016). Therefore, the dynamics of transcription initiation are fundamentally different between humans and plants (Hetzel et al., 2016). Depending upon the chromatin context in animals and plants, RNAPII is recruited at a promoter to form the pre-initiation complex (PIC), however, its processivity is inherently dependent on the chromatin structure along gene bodies and influences RNA-processing during transcription (Guo and Price, 2013; Grasser and Grasser, 2018; Jabre et al., 2019). Techniques such as native elongation transcript sequencing (NET-Seq) (Churchman and Weissman, 2011) in mammals (mNET-Seq) (Nojima et al., 2015) and plants (pNET-Seq) (Zhu et al., 2018) and global run-on sequencing (GRO-seq) (Hetzel et al., 2016), have revealed some important aspects of RNAPII elongation and structural features during transcription and RNA-processing, in humans and plants, respectively. The carboxyl-terminal domain (CTD) of the largest subunit of RNAPII contains a heptad repeat “Tyr1-Ser2-Pro3-Thr4-Ser5-Pro6-Ser7.” The Ser2 and Ser5 of this heptad repeat undergoes phosphorylation and plays a key role in the coordination of transcription and other RNA processing activities (Harlen and Churchman, 2017). In mNET-Seq, phosphorylation-specific antibodies were used to study immunoprecipitated RNAPII transcripts in humans (Nojima et al., 2015, 2018). The comparative analysis of un-phosphorylated (unph) or low-phosphorylated and phosphorylated CTD of RNAPII revealed the accumulation of different forms at differential positions on protein-coding genes. For instance, the RNAPII unph-CTD shows a peak at the transcription start site (TSS), whereas RNAPII Ser5P CTD accumulates at the 5′SS of exon–intron boundaries and its density reduces as the RNAPII elongation proceeds downstream toward the 3′ end of the intron (Figure 2A) (Nojima et al., 2015, 2018). Similarly, RNAPII Ser2P CTD spreads over gene bodies (GB) and shows accumulation at the transcription end site (TES) (Figure 2A) (Nojima et al., 2015, 2018). Moreover, genes that undergo co-transcriptional splicing, such as TARS in humans, show a major peak of RNAPII Ser5P CTD at 5′SS, suggesting pausing at the exon to allow time for the spliceosome to catalyze the first splicing reaction (Nojima et al., 2015). Similar to humans, the dynamics of RNAPII in plants is also established during transcription (Erhard et al., 2015; Hetzel et al., 2016; Zhu et al., 2018). As shown in the proposed model of co-transcriptional splicing in Figure 2A, plants RNAPII CTD is phosphorylated as transcription proceeds. However, in both humans and plants, unph RNAPII is recruited at the promoter region to form the PIC. After initiation, phosphorylation of RNAPII Ser5 CTD and Ser2 CTD begins as transcription proceeds toward the 3′ end. The RNAPII Ser5P CTD pauses at 5′SS, whereas RNAPII Ser2P CTD shows accumulation immediately after polyadenylation site (PAS), suggesting their role in splicing and transcription termination, respectively (Nojima et al., 2015; Zhu et al., 2018).
Despite similarities in the dynamics of RNAPII during transcription and co-transcriptional splicing among plants and humans, significant differences have also been reported, suggesting species-specific regulation of transcription and splicing (Hetzel et al., 2016). For instance, the engaged RNAPII profiles suggest, promoter-proximal pausing and divergent transcripts in Arabidopsis and maize are absent, whereas, these are prominent features of the human transcription (Core et al., 2008; Preker et al., 2008; Erhard et al., 2015; Zhu et al., 2015; Hetzel et al., 2016). In plants, the lack of promoter-proximal-pausing and a high correlation between transcription and steady-state RNA suggests initiation level regulation of transcription as compared to humans (Hetzel et al., 2016). In contrast to GRO-Seq analysis, the combination of GRO-Seq and pNET-Seq data in Arabidopsis show that RNAPII pauses or slows down in some genes after initiation of transcription (Zhu et al., 2018). However, unlike humans, which show RNAPII pausing in narrow regions (20–25 nt), plant RNAPII pausing in the promoter-proximal-regions is much broader (Figure 2C) (Zhu et al., 2018). Additionally, a strong positive correlation has been observed between RNAPII pausing at PAS, CpG methylation and longer genes in plants than in humans, which further suggests plant-specific regulation of transcription and splicing regulation (Hetzel et al., 2016).
Many features of transcription are conserved between humans and plants, however, some important differences exist between them. For example, there is a higher RNAPII elongation rate and AS in the presence of light than dark, demonstrating coupling between AS transcription and growth conditions, which is an important mechanism for plants to respond to different environmental conditions (Petrillo et al., 2014; Godoy Herz et al., 2019). Thus, the role of RNAP II processivity and its impact on AS needs to be analyzed in a tissue- and condition-dependent manner in plants. In the last decade, significant progress has been made to understand the co-transcriptional behavior of splicing/AS in animals, and yeast systems (Shukla and Oberdoerffer, 2012; Merkhofer et al., 2014; Saldi et al., 2016). However, this area is relatively new in plants and more studies are required to illuminate the co-transcriptional dynamics and its impact on RNA processing in tissue- and condition-specific manner.
Aspects of IR and NMD in Plants and Humans
Intron retention is the most prevalent AS event in plants with observed frequencies between 28% to as high as 64% (Figure 1) depending upon growth condition, tissue type and the coverage of transcriptome data (Filichkin et al., 2010; Kalyna et al., 2012; Marquez et al., 2012; Mandadi and Scholthof, 2015). In comparison with plants, only 5% of IR events were observed in humans (Figure 1) (Keren et al., 2010; Reddy et al., 2012), owing to the large size of animal introns, sequencing depth and bioinformatics challenges to detect them. As a consequence, IR had received limited interest in humans until recently (Wong et al., 2013; Braunschweig et al., 2014; Boutz et al., 2015), whereas in plants IR has been found to be an important regulator in growth, development, physiology, and stress responses (Kalyna et al., 2012; Syed et al., 2012; Drechsel et al., 2013; Filichkin S.A. et al., 2015). However, recent research is unveiling various menace regulatory functions of IR in humans. For example, in addition to physiologically regulated events, any mutation in the splice site or splicing regulatory sequences cause aberrant IR, which further results in perturbed splicing patterns and potentially cause diseases (Jung et al., 2015; Wong et al., 2015; Jacob and Smith, 2017).
In humans, possible causes of IR and its abundance in response to cell differentiation and stresses have been studied recently (Wong et al., 2013; Braunschweig et al., 2014; Boutz et al., 2015). For instance, to predict the prevalence of IR, and their regulation and biological significance, a deep quantitative survey using Poly(A+) RNA-Seq data from 40 human and mouse tissue samples was conducted (Braunschweig et al., 2014). This study involved the quantitative measurement and comparison of reads across unspliced (exon–intron) and spliced (exon–exon) junctions, as well as, reads within introns in terms of “percent intron retention” (PIR) (Braunschweig et al., 2014). These findings suggest a large number of multiexonic genes are affected by the variable frequency of IR events processed in different tissues, which is much higher in comparison with previously estimated values (Pan et al., 2008; Wang et al., 2008). Comparative analysis across various species revealed tissue-specific IR events in neurons and immune cells. Furthermore, IR in neurons is highly conserved as compared with other AS events (Barbosa-Morais et al., 2012; Merkin et al., 2012; Braunschweig et al., 2014). In contrast with previous studies, IR was prevalent and mainly enriched in untranslated regions (UTRs), non-coding RNAs, depleted protein coding regions, and/or at the 3′ end of RNAs among different tissues in humans (Bicknell et al., 2012; Jacob and Smith, 2017). Moreover, the frequency of IR in the nucleus was observed to be higher than the cytoplasm, suggesting nuclear sequestration or coupling with the NMD pathway (Wong et al., 2013; Braunschweig et al., 2014; Boutz et al., 2015; Edwards et al., 2016).
In comparison with humans, the prevalence and significance of IR in plants and its role in development, stress and tissue-specific physiology are well documented. The observed frequency of IR in plants is as high as 64%, and potentially fine-tunes the transcriptome functionality (Filichkin et al., 2010, 2018; Kalyna et al., 2012; Drechsel et al., 2013; Filichkin S.A. et al., 2015). However, the mechanisms behind the high occurrence of IR in plants are still not very clear, yet many studies emphasize its significance in plants under normal, stress and various development and growth conditions. For example, the expression of INTERMINATE DOMAIN 14 (IDD 14) isoforms controlled via IR mediate starch accumulation and utilization under cold stress in Arabidopsis (Seo et al., 2011). Similarly, cold-dependent IR in clock genes such as CIRCADIAN CLOCK ASSOCIATED 1 (CCA1), LATE ELONGATED HYPOCOTYL (LHY) and PSEUDO-RESPONSE REGULATOR7 (PRR7), modulate their transcript and protein abundance for CCA1 (Seo et al., 2011; James et al., 2012). In wheat, the PECTIN METHYL ESTERASE INHIBITOR (PMEI), which secretes pectin for the cell wall, is also regulated by IR. Although, PMEI IR isoforms are found in almost all tissues but only anthers contained mature transcripts without IR, suggesting possible tissue-specific functionality of these transcripts (Rocchi et al., 2012). Similarly, studies in a Marsilea vestita (Boothby et al., 2013) and Arabidopsis (Filichkin and Mockler, 2012; Filichkin S.A. et al., 2015) provide a useful model to explain unproductive AS via IR. It has been demonstrated in M. vestita that some NMD insensitive IR transcripts remain in the nucleus as un-spliced mRNAs. Subsequently, these IR transcripts could be spliced and their translation results in a specific function, such as gamete development (Boothby et al., 2013).
Interestingly, many of IR PTC+ transcripts are not subjected to NMD in plants (Kalyna et al., 2012), suggesting regulatory functions. Components of the NMD machinery are highly conserved between plants and humans and its efficiency is strongly influenced by the pioneer round of translation (activity of ribosomes) (Shaul, 2015). However, it is intriguing that NMD responses are much less pronounced under stressful conditions in humans and plants, affecting the expression and translation of stress-responsive genes and splice variants (Trcek et al., 2013; Shaul, 2015). For example, inhibition of NMD mediates plant defense response during pathogen attack in Arabidopsis NMD mutants as they constitutively make more salicylic acid (SA) and show a heightened response after infection with Pseudomonas syringae (Rayson et al., 2012). However, mechanistic details of AS and its role via protein diversity in subverting a pathogen attack is not clear. Since the NMD pathway is translation dependent, slow engagement of different non-canonical transcripts with the ribosomal machinery may be the cause of their degradation. Intriguingly, in several model species including Arabidopsis, PTCs in the first and last intron appear earlier in their sequence than expected by chance alone, to keep the metabolic cost of producing truncated proteins and their subsequent degradation (Behringer and Hall, 2016). This data supports the notion that the appearance of earlier PTCs in introns seems to be favored by selection. Presence of PTCs in the first and last introns also points toward multiple features favoring degradation of non-sense transcripts (Behringer and Hall, 2016).
Interestingly, introns in plants UTRs also play a crucial role by affecting translation efficiency via a process called intron-mediated enhancement (IME). IME was proposed as a conserved phenomenon enhancing the translation efficiency of IR transcripts (Parra et al., 2011; Gallegos and Rose, 2015). For example, analysis of 5′ UTR introns identified an intron element in transcripts of the Mg2+/H+ ion exchange (MHX) gene in Arabidopsis, which further show an increase in translation efficiency (Akua and Shaul, 2013). In summary, differences in the frequencies of IR events suggest a varied mode of downstream processing and fates of IR transcripts in plants and humans. However, further work is needed to illuminate the mechanistic details of the IME mechanism.
As and Protein Diversity in Humans: Supporting Evidence
Higher eukaryotes are diverse with varying degrees of biological complexity, nonetheless, the number of protein-coding genes is comparable between different species (Chen et al., 2014). Comparative sequencing and evolutionary studies between different eukaryotic species (including complex avian and mammals to species with fewer cell types) suggest a strong correlation between AS and organism complexity (Chen et al., 2014). AS plays a crucial role to enrich the expression of many genes and mediates various biological functions, pathways, and processes (Merkin et al., 2012; Weatheritt et al., 2012; Irimia et al., 2014). In humans, despite significant advancements in the field of transcriptome and proteome analysis techniques, the extent to which AS transcripts contribute to protein diversity remains unclear. However, renewed interest in humans has led to concerted efforts to illuminate this phenomenon in the recent past (Table 1). For example, isolation and sequencing of ribosome-bound transcripts have enabled researchers to delineate how the variety and abundance of mRNAs correlate with ribosomal recruitment (potentially translating mRNA). In a recent study, the ribosomal-engaged landscape of AS transcripts was surveyed using ribosomal-profiling in humans (Weatheritt et al., 2016). The ribosomal profiling data suggest transcripts with exon skipping events are present in medium to high abundance and thus likely to be translated. On the contrary, transcripts present in low abundance at the transcriptome level were not engaged with the ribosomes. This might be due to either the presence of introns in the low abundance transcripts, which remain in the nucleus (Braunschweig et al., 2014; Boutz et al., 2015) or incomplete RNA processing, preventing ribosomal engagement (Weatheritt et al., 2016). Similarly, other studies using Frac-Seq (subcellular fractionation and RNA-sequencing) (Sterne-Weiler et al., 2013) and TrIP-Seq (transcript isoforms in polysomes sequencing) (Floor and Doudna, 2016), also detected a large proportion of splice variants in the polyribosome fractions suggesting spliced isoforms play a significant role in controlling protein output in human cells. However, the degree to which ribosomal bound AS transcripts are translated and represented at the protein level is unclear. For example, pre-mRNA processing in the nucleus influences an isoform’s association with polyribosomes (Sterne-Weiler et al., 2013). Approximately 30% of mRNA processing events are differentially partitioned between cytoplasmic and polyribosome fractions (Sterne-Weiler et al., 2013). Moreover, differences in the polyribosome association are the result of a change in the cis-regulatory landscapes such as inclusion or exclusion of uORFs and Alu-elements in the 5′UTR, and microRNA target sites in the 3′UTR by AS (Sterne-Weiler et al., 2013). Similarly, TrIP-Seq analysis revealed that each transcript isoform harbors special regulatory features controlling ribosome occupancy and translation (Floor and Doudna, 2016). Floor and Doudna (2016) found robust translational control by 5′ UTRs between cell lines, whereas 3′ UTRs impact cell type-specific expression. This work also suggested that transcript isoform diversity must be considered when associating RNA and protein levels.
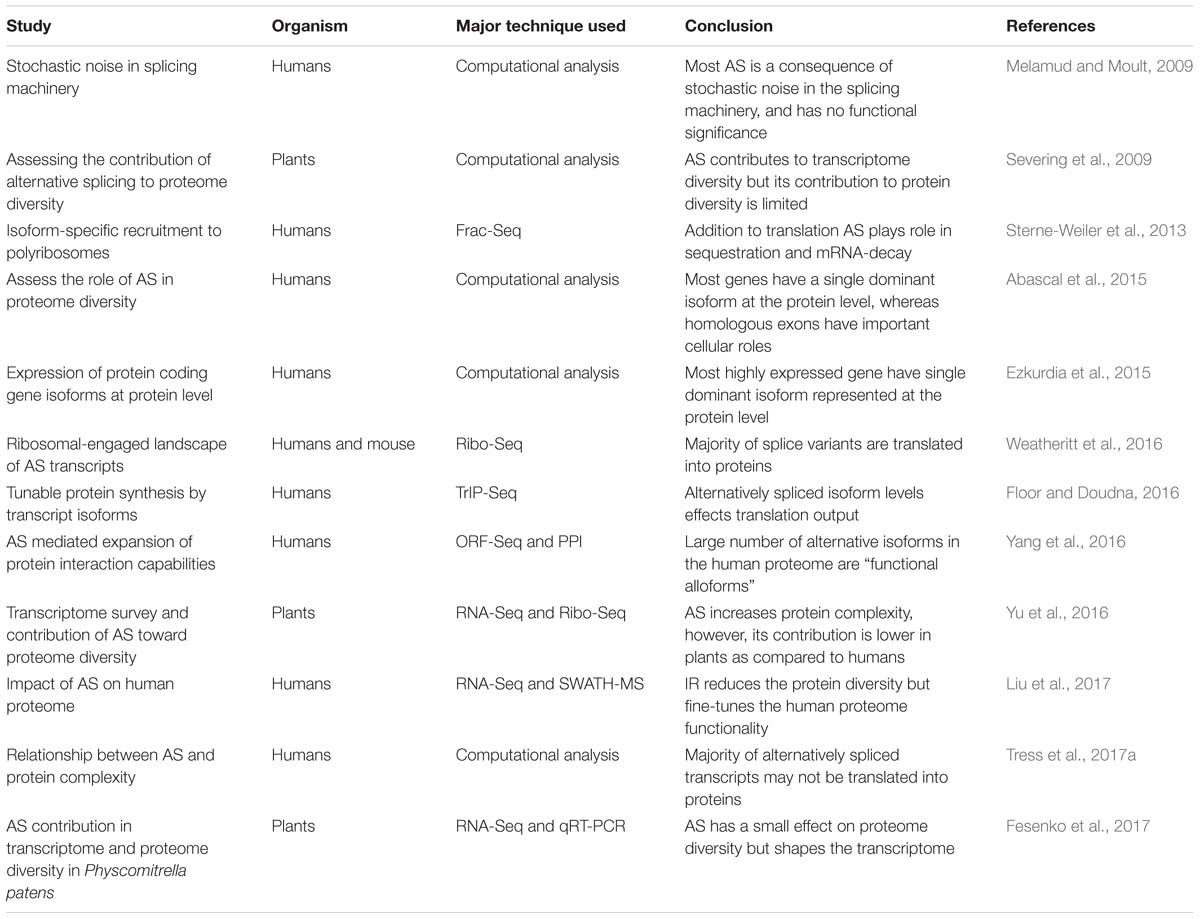
Table 1. Major studies deciphering the role of AS in protein diversity in humans and plants using different technique.
Some proteomic studies contradict ribosome profiling data and argue that only a small fraction of splice variants are represented at the protein level (Abascal et al., 2015; Ezkurdia et al., 2015; Tress et al., 2017a). Furthermore, the shotgun MS techniques used in many proteomic studies have their own limitations of coverage and sensitivity to detect low abundance splice variants at the protein level (Bensimon et al., 2012; Rost et al., 2015). To improve isoforms detection efficiency, alternative approaches need to be developed to overcome the limitations of the techniques used at present. Toward this goal, full-length ORFs of AS isoforms from a large number of human genes were cloned and protein–protein interaction (PPI) profiling was performed to demonstrate the functionality of hundreds of protein isoforms (Yang et al., 2016). This study demonstrated vastly different interaction profiles among isoforms as a result of AS. Strikingly, the isoforms encoded by the same genes exhibit widespread functional differences in the PPI network analysis. Since differences between protein isoforms are as high as observed between different genes, isoforms-specific partners could have different expression and functional characteristics. Yang et al. (2016) proposed that a vast diversity of “functional alloforms” are generated that contribute to different physiological and developmental processes (Yang et al., 2016).
In humans, a number of studies have been conducted to identify protein isoforms that result from AS by comparing transcriptome and proteome data (Brosch et al., 2011; Ezkurdia et al., 2012; Lopez-Casado et al., 2012; Sheynkman et al., 2013). However, most of these studies were carried out in a steady state manner and do not explain the consequences of perturbation in splicing to protein diversity. To overcome these limitations, an integrated approach was developed to illuminate how variation in mRNA splicing patterns could subsequently change the proteome composition in a systematic manner (Liu et al., 2017). Selectively depleted spliceosome U5 component PRPF8 (Wickramasinghe et al., 2015) orchestrated changes at the transcriptome and proteome level that were determined using RNA-Seq and Sequential Window Acquisition of all Theoretical Spectra-Mass Spectrometry (SWATH-MS), respectively. After PRPF8 depletion, quantification of splice variants and a large fraction of proteome identified 1,542 proteins that displayed at least one peptide with altered expression. Functional annotation revealed that transcripts with altered splicing patterns possess similar cellular functions and processes (such as RNA splicing, the mitotic cell cycle and ubiquitination) as those found in proteins with altered levels. Thus, splicing variants at the transcriptomic level were found to be functionally represented at the protein level (Liu et al., 2017). Furthermore, to identify the differentially spliced event at the transcriptome level, the authors used a transcript-centric approach, in which a transcript is considered as a whole unit (Liu et al., 2017). Firstly, transcript expression is estimated, followed by identification of differentially used transcripts and expressed genes. The correlation analysis between fold changes in the expression level after PRPF8 depletion suggests protein expression levels are exclusively associated with the alternatively spliced transcripts involving differential transcripts usage (DTU). Interestingly, IR events, which are considered as one of the major regulatory events for gene expression, had reduced representation at the protein level (Liu et al., 2017). Although, around 75% of multi-exon genes are affected by IR and help in regulating transcript levels (Braunschweig et al., 2014), its impact on protein expression is inverse because an increase in the level of IR transcripts, throughout the genome, is associated with PRPF8 depletion (Wickramasinghe et al., 2015). The peptide expression of 270 genes with retained introns showed downregulation of protein expression coded by genes with IR. Moreover, the relative abundance of transcripts also plays a significant role in protein expression as the low abundance transcripts with IR do not affect the protein expression until they are present in high abundance. These observations suggest IR reduces the protein diversity but fine-tunes the human proteome functionality. However, this finding may not be strictly applicable to plants as IR is the predominant mode of AS and may fine-tune the proteome function via modulating its abundance, especially in stressful conditions.
Collectively, various studies in the recent past such as ribosomal profiling (Weatheritt et al., 2016), PPI interaction analysis of spliced isoforms (Yang et al., 2016), and integrative analysis using perturbed systems (Liu et al., 2017) suggest a strong correlation between AS and protein diversity in humans. Moreover, these studies provide an alternative to MS techniques, which have limitations of coverage and sensitivity to detect low level splice isoforms at the protein level and could be useful to study plant systems in the future.
As and Protein Diversity in Humans: Opposing Evidence
The contribution of AS toward protein diversity in humans is well documented (Weatheritt et al., 2016; Yang et al., 2016; Blencowe, 2017; Liu et al., 2017). However, recent data from some proteomic studies in humans supports the opposing view and suggest that AS may not be the key contributor to protein diversity (Tress et al., 2017a,b). Substantial amount of AS data has been generated in various RNA-Seq experiments in humans, however, most of the alternative isoforms in proteomic experiments are undetectable even in large-scale MS-based analyses (Ezkurdia et al., 2015; Tress et al., 2017a,b). Moreover, some studies suggest that AS is the result of noise in the splicing machinery and does not contribute to protein diversity as expected. For example, Melamud and Moult (2009) proposed a stochastic noise model of splicing machinery, which explained that AS events arise as a result of noise in the splicing machinery (Melamud and Moult, 2009). The idea of noise in the splicing machinery has also been supported by other studies as well, suggesting a large proportion of alternative isoforms are non-functional (Modrek et al., 2001; Kan et al., 2002; Neverov et al., 2005). Further, it was recently demonstrated that the majority of expressed genes have a single major isoform represented at the protein level (Abascal et al., 2015; Ezkurdia et al., 2015). This was supported by monitoring peptide evidence from eight large-scale MS experiments and observing that only one main protein isoform was dominant at the protein level from almost all coding genes (Tress et al., 2017a). On the other hand, several reports have supported the presence of a small number of alternative protein isoforms in humans (Tanner et al., 2007), drosophila (Tress et al., 2008), and mouse (Brosch et al., 2011) in large-scale proteomic studies. However, AS events such as ES detected in RNA-Seq studies have revealed subtle effects on the structure and function of proteins. Tress et al. argue that it is the gene expression that is conserved across species, have strong tissue dependence, and are translated to detectable proteins but not the alternatively spliced isoforms (Tress et al., 2017a,b). Clearly, more work and evidence is needed to illuminate the relationship between AS and protein diversity in tissue- and condition-dependent manner.
The efficiency of the MS also needs to be enhanced because current MS techniques cannot reliably detect changes in protein domains as a result of AS (Wang et al., 2018; Chaudhary et al., 2019). For example, lysine and arginine coding triplets are the most abundant amino acids at the end of exons or exon–exon junctions (Wang et al., 2018), and are the preferential sites for trypsin, which is the most common enzyme used in MS analyses (Olsen et al., 2004). Since trypsin digests exon–exon junctions, it hinders with the detection of novel AS derived peptides in MS-based proteome analysis (Ning and Nesvizhskii, 2010; Sheynkman et al., 2013; Wang et al., 2013). To improve efficiency, enzymes such as chymotrypsin can be used as an alternative to improve the detection of AS-derived peptides in proteome studies (Wang et al., 2018; Chaudhary et al., 2019).
The Contribution of as Toward Protein Diversity in Plants
The role of AS in the expansion of functional protein diversity is less clear in plants as compared to humans (Kim et al., 2007). However, in the absence of in-depth proteomic studies to elucidate the role of AS toward protein diversity is tenuous. Recently, some studies have evaluated the influence of AS on protein diversity in plants. For example, hypoxia in Arabidopsis mediates an increase in the number of IR events in many mRNA isoforms, and show ribosomal engagement and potentially influence protein variety and abundance (Juntawong et al., 2014). Interestingly, transcriptome and translatome profiling among shoot apical meristem (SAM) and leaf domains, suggest 751 genes isoforms show domain-specific enrichment in the translatome data (Tian et al., 2019). Another study in Arabidopsis has shown that 35% of AS events are represented among the polysome-bound mRNAs and expected to undergo translation (Yu et al., 2016). Among all transcripts, IR is the least representative among translated transcripts, compared with untranslated transcripts, suggesting a variable role of IR in regulating transcript level via NMD machinery or sequestration in the nucleus and further processing on demand (Filichkin S.A. et al., 2015; Filichkin S. et al., 2015). In contrast, other splicing events such as ES, 5′AD, and 3′AA have higher proportions among transcripts that may be translated (Yu et al., 2016). Sequence analysis of translated transcripts suggests that any alteration in the CDS by AS could lead to a change in protein sequences (Yu et al., 2016). Interestingly, a large proportion of a new class of exon-like introns called exitrons (Marquez et al., 2015) (Figure 1) was found at the transcriptome as well as translatome level, suggesting these unique events of AS may contribute to protein diversity (Yu et al., 2016). A recent report in Physcomitrella patens suggests that AS shapes the transcriptome rather than the proteome (Fesenko et al., 2017), because only 85 isoform-specific peptides, representing only 25 differentially AS genes, were found in moss cells. Among all, only five genes unambiguously showed two or more protein isoforms from the same locus. The number of AS genes identified in this study was substantially large (approximately 66 times) as compared to proteomic datasets, nonetheless, only support a small contribution of AS on protein diversity. Collectively, these data support the view that AS increases protein complexity, however, its contribution is found to be lower as compared with humans (Yu et al., 2016). Further, supporting as well as the opposing evidence presented above for the notion, “AS contributes toward protein diversity,” suggests that the exact number of splice isoforms represented at the proteome level in humans as well as in plants is still elusive. On the other hand, IR events are the predominant AS type in plants and may not be translated due to nuclear sequestration or degradation by the NMD pathway and thus remain poorly represented in MS experiments (Gohring et al., 2014; Hartmann et al., 2018). Since limited information is available at the proteome level, we envisage that strategies like cloning of spliced isoforms and PPI profiling (like in humans Yang et al., 2016), could be beneficial and may uncover different aspects of AS contribution toward protein diversity in plants.
Splicing Memory and Plant Stress Tolerance
Successful attempts have been made in plant systems to understand the impact of stress, its tolerance and the development of genetically engineered stress tolerant crops (Vinocur and Altman, 2005; Pereira, 2016). However, the majority of studies are restricted to acute and single stress only (Zhu, 2016). Since stresses are usually multiple, recurring and chronic, plants have evolved sophisticated defense mechanisms to deal with a variety of stresses. Plants have the ability to acquire tolerance to chronic stress through establishing “molecular stress memory” to confer tolerance through a phenomenon referred to as priming or acclimation, in response to previous exposure to a mild stress (Sani et al., 2013; Conrath et al., 2015; Hilker et al., 2016). Priming establishes a new cellular state in plants, which is different from the naïve or unexposed plants (Sani et al., 2013; Conrath et al., 2015; Hilker et al., 2016). In recent years, it has become increasingly apparent that various epigenetic features, such as chromatin modifications, nucleosome positioning, and DNA methylation, are important components of adaptation and play a role in stress memory (Boyko et al., 2010; Ding et al., 2012; Lämke and Bäurle, 2017; Friedrich et al., 2018). Since the splicing process is largely co-transcriptional in nature, the chromatin structure has a strong influence on the transcriptional as well as the splicing processes (Listerman et al., 2006; Khodor et al., 2011; Jabre et al., 2019). Recent DNase I-Seq data suggest enrichment of IR in DNase I hypersensitivity sites (DHSs) in both Arabidopsis and rice (Ullah et al., 2018). Since RNAPII elongation speed is high in regions with open chromatin, the spliceosome machinery has less time to recognize introns, resulting in more IR during co-transcriptional splicing (Braunschweig et al., 2014; Naftelberg et al., 2015). Furthermore, condition-dependent variation in the chromatin environment under different stresses and environmental cues plays an additional regulatory and fine-tuning role (Struhl and Segal, 2013; Zentner and Henikoff, 2013). Moreover, along with the positioning and spacing of the nucleosome, posttranslational modifications and DNA methylation also affect the transcriptional and splicing dynamics (Naftelberg et al., 2015; Friedrich et al., 2018; Zhang et al., 2018). Hence, various epigenetic modifications may provide a basic regulatory mechanism to orchestrate stress and splicing memory (Figure 3) in the same or future generations to respond to recurring stress more efficiently.
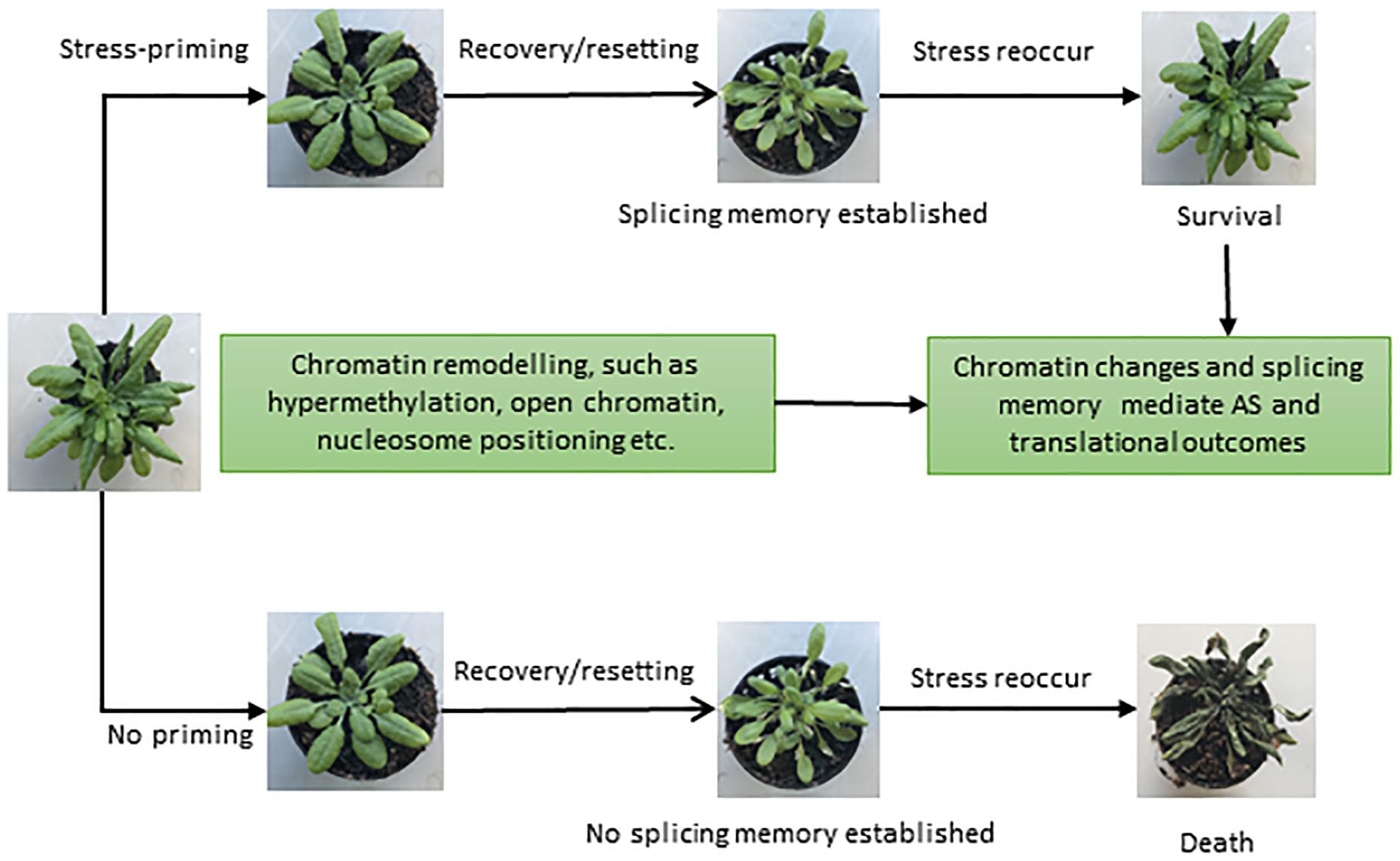
Figure 3. Different phenotypes representing the importance of splicing memory in plants. Once exposed to stressful conditions, plants develop an adaptive componenet of induced resistance defined as stress-priming. Stress-induced chromatin modifications plays a crucial role in stress-priming and likely help in establishing a splicing memory, which in turn facilitates plant survival upon exposure to recurring stresses (upper panel). In the absence of priming (lower panel) and splicing memory, plants may die once the stress reoccur. Different phenotypes shown are based on Ling et al. (2018) and Sanyal et al. (2018).
Not surprisingly, a recent study uncovered splicing memory response to heat stress priming in Arabidopsis as revealed by genome-wide differentially expressed genes (DEGs) and AS patterns (Ling et al., 2018; Sanyal et al., 2018). DEGs in response to heat stress were identified for different stages of priming, and genes responsible for potentially controlling heat stress memory were selected. With the help of gene networking analysis, heat and abiotic responsive genes were found to be involved in stress memory (Ling et al., 2018). Importantly, IR was found to be the most prevalent event under heat stress and contributed significantly toward establishing the splicing memory in response to heat. The primed plants produced comparable splicing patterns and efficiency compared with control plants, which were not exposed to heat stress before. In contrast, non-primed plants showed a significant increase in IR and produced splicing variants in heat conditions. Therefore, the primed plants, after relief from the second exposure to heat stress, maintain the splicing memory and perform in a similar manner to the control plants under non-stressful conditions (Ling et al., 2018). Ling et al. (2018) suggested that heat stress priming might be established at the post-transcriptional level and maintains splicing memory, which is crucial for plant survival and adaptation under stress. It is tempting to speculate that exposure to multiple stresses and coordination of gene expression and splicing patterns mediated by the chromatin environment may influence predictable responses and adaptive solutions in the long term. However, further research is needed to explore splicing memory and the underlying molecular mechanisms in response to different stresses in plants. We envisage that in addition to its contribution to protein diversity, AS may also play regulatory roles, and after repeated episodes of stress, splicing memory may also fine-tune stress-specific protein diversity to enhance plants networking capability to cope with given stress.
Conclusion
Emerging evidence indicates that the splicing process is also predominantly co-transcriptional in plants as in humans (Zhu et al., 2018). In plants, environmental fluctuations modulate chromatin structure, which in turn, could influence the co-transcriptional splicing process. Intriguingly, recent work indicates that plants can establish splicing memory in response to higher temperature conditions and thus may “remember” a particular stress, likely through specific epigenetic signatures. This strategy may allow plants to engender an appropriate and reproducible response to a given stress. Further, IR transcripts are prevalent in plants and a majority of these are “trapped” in the nucleus. In addition, IR and many other AS transcripts are NMD sensitive and potentially degraded by the NMD pathway. It is clear that AS modulates transcriptome composition and splicing ratios, however, its role in diversifying proteome complexity is far from being understood.
It was a surprising discovery to find that the human genome codes for only ∼20,000 to 21,000 protein-coding genes (Willyard, 2018), which is comparable with a weed (Arabidopsis, which has over 27,000 protein-coding genes) with a much smaller genome (Swarbreck et al., 2008). Since 95% of human genes and over 70% of genes in some plants are alternatively spliced, they can potentially make multiple proteins from each gene and considerably increase their proteome complexity (Kim et al., 2007; Pan et al., 2008). Although it is clear that AS does increase proteome complexity, the extent to which it enhances proteome diversity is far from clear. Multiple proteomic studies do not support a linear relationship between splicing and proteome complexity in humans (Tress et al., 2017a,b). Therefore, in-depth proteome analyses in multiple tissues and conditions, in conjunction with the variable expression of corresponding genes, need to be performed to illuminate the relationship between AS and proteome complexity in plants.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the funding agencies for research support. Leverhulme Trust (RPG-2016-014); DOE Office of Science, Office of Biological and Environmental Research (BER) (DE-SC0010733); National Science Foundation; and the U.S. Department of Agriculture (ASNR).
References
Abascal, F., Ezkurdia, I., Rodriguez-Rivas, J., Rodriguez, J. M., del Pozo, A., Vázquez, J., et al. (2015). Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comput. Biol. 11:e1004325. doi: 10.1371/journal.pcbi.1004325
Akua, T., and Shaul, O. (2013). The Arabidopsis thaliana MHX gene includes an intronic element that boosts translation when localized in a 5′ UTR intron. J. Exp. Bot. 64, 4255–4270. doi: 10.1093/jxb/ert235
Albaqami, M., and Reddy, A. S. N. (2018). Development of an in vitro pre-mRNA splicing assay using plant nuclear extract. Plant Methods 14:1. doi: 10.1186/s13007-017-0271-6
Barbosa-Morais, N. L., Irimia, M., Pan, Q., Xiong, H. Y., Gueroussov, S., Lee, L. J., et al. (2012). The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593. doi: 10.1126/science.1230612
Becklin, K. M., Anderson, J. T., Gerhart, L. M., Wadgymar, S. M., Wessinger, C. A., and Ward, J. K. (2016). Examining plant physiological responses to climate change through an evolutionary lens. Plant Physiol. 172, 635–649. doi: 10.1104/pp.16.00793
Behringer, M. G., and Hall, D. W. (2016). Selection on position of nonsense codons in Introns. Genetics 204, 1239–1248. doi: 10.1534/genetics.116.189894
Bensimon, A., Heck, A. J. R., and Aebersold, R. (2012). Mass spectrometry-based proteomics and network biology. Annu. Rev. Biochem. 81, 379–405. doi: 10.1146/annurev-biochem-072909-100424
Bicknell, A. A., Cenik, C., Chua, H. N., Roth, F. P., and Moore, M. J. (2012). Introns in UTRs: why we should stop ignoring them. BioEssays 34, 1025–1034. doi: 10.1002/bies.201200073
Blencowe, B. J. (2017). The relationship between alternative splicing and proteomic complexity. Trends Biochem. Sci. 42, 407–408. doi: 10.1016/j.tibs.2017.04.001
Boothby, T. C., Zipper, R. S., Van der Weele, C. M., and Wolniak, S. M. (2013). Removal of retained introns regulates translation in the rapidly developing gametophyte of Marsilea vestita. Dev. Cell 24, 517–529. doi: 10.1016/j.devcel.2013.01.015
Boutz, P. L., Bhutkar, A., and Sharp, P. A. (2015). Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 29, 63–80. doi: 10.1101/gad.247361.114
Boyko, A., Blevins, T., Yao, Y., Golubov, A., Bilichak, A., Ilnytskyy, Y., et al. (2010). Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of dicer-like proteins. PLoS One 5:e0009514. doi: 10.1371/journal.pone.0009514
Braunschweig, U., Barbosa-Morais, N. L., Pan, Q., Nachman, E. N., Alipanahi, B., Gonatopoulos-Pournatzis, T., et al. (2014). Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 24, 1774–1786. doi: 10.1101/gr.177790.114
Brosch, M., Saunders, G. I., Frankish, A., Collins, M. O., Yu, L., Wright, J., et al. (2011). Shotgun proteomics aids discovery of novel protein-coding genes, alternative splicing, and “resurrected” pseudogenes in the mouse genome. Genome Res. 21, 756–767. doi: 10.1101/gr.114272.110
Brugiolo, M., Herzel, L., and Neugebauer, K. M. (2013). Counting on co-transcriptional splicing. F1000Prime Rep. 5:9. doi: 10.12703/P5-9
Chamala, S., Feng, G., Chavarro, C., and Barbazuk, W. B. (2015). Genome-wide identification of evolutionarily conserved alternative splicing events in flowering plants. Front. Bioeng. Biotechnol. 3:33. doi: 10.3389/fbioe.2015.00033
Chaudhary, S., Jabre, I., Reddy, A. S. N., Staiger, D., and Syed, N. H. (2019). Perspective on alternative splicing and proteome complexity in plants. Trends Plant Sci. 24, 496–506. doi: 10.1016/j.tplants.2019.02.006
Chen, L., Bush, S. J., Tovar-Corona, J. M., Castillo-Morales, A., and Urrutia, A. O. (2014). Correcting for differential transcript coverage reveals a strong relationship between alternative splicing and organism complexity. Mol. Biol. Evol. 31, 1402–1413. doi: 10.1093/molbev/msu083
Chen, W., and Moore, M. J. (2015). Spliceosomes. Curr. Biol. 25, R181–R183. doi: 10.1016/j.cub.2014.11.059
Churchman, L. S., and Weissman, J. S. (2011). Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 469, 368–373. doi: 10.1038/nature09652
Conn, V. M., Hugouvieux, V., Nayak, A., Conos, S. A., Capovilla, G., Cildir, G., et al. (2017). A circRNA from SEPALLATA3 regulates splicing of its cognate mRNA through R-loop formation. Nat. Plants 3:17053. doi: 10.1038/nplants.2017.53
Conrath, U., Beckers, G. J. M., Langenbach, C. J. G., and Jaskiewicz, M. R. (2015). Priming for enhanced defense. Annu. Rev. Phytopathol. 53, 97–119. doi: 10.1146/annurev-phyto-080614-120132
Core, L. J., Waterfall, J. J., and Lis, J. T. (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. doi: 10.1126/science.1162228
Ding, Y., Fromm, M., and Avramova, Z. (2012). Multiple exposures to drought “train” transcriptional responses in Arabidopsis. Nat. Commun. 3:740. doi: 10.1038/ncomms1732
Drechsel, G., Kahles, A., Kesarwani, A. K., Stauffer, E., Behr, J., Drewe, P., et al. (2013). Nonsense-mediated decay of alternative precursor mrna splicing variants is a major determinant of the Arabidopsis steady state transcriptome. Plant Cell 25, 3726–3742. doi: 10.1105/tpc.113.115485
Edwards, C. R., Ritchie, W., Wong, J. J.-L., Schmitz, U., Middleton, R., An, X., et al. (2016). A dynamic intron retention program in the mammalian megakaryocyte and erythrocyte lineages. Blood 127, e24–e34. doi: 10.1182/blood-2016-01-692764
Erhard, K. F., Talbot, J. E. R. B., Deans, N. C., McClish, A. E., and Hollick, J. B. (2015). Nascent transcription affected by RNA polymerase IV in Zea mays. Genetics 199, 1107–1125. doi: 10.1534/genetics.115.174714
Ezkurdia, I., Del Pozo, A., Frankish, A., Rodriguez, J. M., Harrow, J., Ashman, K., et al. (2012). Comparative proteomics reveals a significant bias toward alternative protein isoforms with conserved structure and function. Mol. Biol. Evol. 29, 2265–2283. doi: 10.1093/molbev/mss100
Ezkurdia, I., Rodriguez, J. M., Carrillo-De Santa Pau, E., Vázquez, J., Valencia, A., and Tress, M. L. (2015). Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res. 14, 1880–1887. doi: 10.1021/pr501286b
Fesenko, I., Khazigaleeva, R., Kirov, I., Kniazev, A., Glushenko, O., Babalyan, K., et al. (2017). Alternative splicing shapes transcriptome but not proteome diversity in Physcomitrella patens. Sci. Rep. 7, 2698. doi: 10.1038/s41598-017-02970-z
Filichkin, S. A., Priest, H. D., Givan, S. A., Shen, R., Bryant, D. W., Fox, S. E., et al. (2010). Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 20, 45–58. doi: 10.1101/gr.093302.109
Filichkin, S. A., and Mockler, T. C. (2012). Unproductive alternative splicing and nonsense mRNAs: a widespread phenomenon among plant circadian clock genes. Biol. Direct 7:20. doi: 10.1186/1745-6150-7-20
Filichkin, S. A., Cumbie, J. S., Dharmawardhana, P., Jaiswal, P., Chang, J. H., Palusa, S. G., et al. (2015). Environmental stresses modulate abundance and timing of alternatively spliced circadian transcripts in Arabidopsis. Mol. Plant 8, 207–227. doi: 10.1016/j.molp.2014.10.011
Filichkin, S., Priest, H. D., Megraw, M., and Mockler, T. C. (2015). Alternative splicing in plants: directing traffic at the crossroads of adaptation and environmental stress. Curr. Opin. Plant Biol. 24, 125–135. doi: 10.1016/j.pbi.2015.02.008
Filichkin, S. A., Hamilton, M., Dharmawardhana, P. D., Singh, S. K., Sullivan, C., Ben-Hur, A., et al. (2018). Abiotic stresses modulate landscape of poplar transcriptome via alternative splicing, differential intron retention, and isoform ratio switching. Front. Plant Sci. 9:5. doi: 10.3389/fpls.2018.00005
Floor, S. N., and Doudna, J. A. (2016). Tunable protein synthesis by transcript isoforms in human cells. eLife 5, e10921. doi: 10.7554/eLife.10921.001
Friedrich, T., Faivre, L., Bäurle, I., and Schubert, D. (2018). Chromatin-based mechanisms of temperature memory in plants. Plant. Cell Environ. 42, 762–770. doi: 10.1111/pce.13373
Gallego-Paez, L. M., Bordone, M. C., Leote, A. C., Saraiva-Agostinho, N., Ascensão-Ferreira, M., and Barbosa-Morais, N. L. (2017). Alternative splicing: the pledge, the turn, and the prestige: the key role of alternative splicing in human biological systems. Hum. Genet. 136, 1015–1042. doi: 10.1007/s00439-017-1790-y
Gallegos, J. E., and Rose, A. B. (2015). The enduring mystery of intron-mediated enhancement. Plant Sci. 237, 8–15. doi: 10.1016/j.plantsci.2015.04.017
Godoy Herz, M. A., Kubaczka, M. G., BrzyŻek, G., Servi, L., Krzyszton, M., Simpson, C., et al. (2019). Light regulates plant alternative splicing through the control of transcriptional elongation. Mol. Cell 73:1066-1074.e3. doi: 10.1016/j.molcel.2018.12.005
Gohring, J., Jacak, J., and Barta, A. (2014). Imaging of endogenous messenger RNA splice variants in living cells reveals nuclear retention of transcripts inaccessible to nonsense-mediated decay in Arabidopsis. Plant Cell 26, 754–764. doi: 10.1105/tpc.113.118075
Grasser, M., and Grasser, K. D. (2018). The plant RNA polymerase II elongation complex: a hub coordinating transcript elongation and mRNA processing. Transcription 9, 117–122. doi: 10.1080/21541264.2017.1356902
Gratani, L. (2014). Plant phenotypic plasticity in response to environmental factors. Adv. Bot. 2014, 1–17. doi: 10.1155/2014/208747
Guerra, D., Crosatti, C., Khoshro, H. H., Mastrangelo, A. M., Mica, E., and Mazzucotelli, E. (2015). Post-transcriptional and post-translational regulations of drought and heat response in plants: a spiderâ€TMs web of mechanisms. Front. Plant Sci. 6:57. doi: 10.3389/fpls.2015.00057
Guo, J., and Price, D. H. (2013). RNA polymerase II transcription elongation control. Chem. Rev. 113, 8583–8603. doi: 10.1021/cr400105n
Harlen, K. M., and Churchman, L. S. (2017). The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 18, 263–273. doi: 10.1038/nrm.2017.10
Hartmann, L., Wießner, T., and Wachter, A. (2018). Subcellular compartmentation of alternatively-spliced transcripts defines SERINE/ARGININE-RICH PROTEIN 30 Expression. Plant Physiol. 176, 2886–2903. doi: 10.1104/pp.17.01260
Hetzel, J., Duttke, S. H., Benner, C., and Chory, J. (2016). Nascent RNA sequencing reveals distinct features in plant transcription. Proc. Natl. Acad. Sci. U.S.A. 113, 12316–12321. doi: 10.1073/pnas.1603217113
Hilker, M., Schwachtje, J., Baier, M., Balazadeh, S., Bäurle, I., Geiselhardt, S., et al. (2016). Priming and memory of stress responses in organisms lacking a nervous system. Biol. Rev. 91, 1118–1133. doi: 10.1111/brv.12215
Irimia, M., and Roy, S. W. (2014). Origin of spliceosomal introns and alternative splicing. Cold Spring Harb. Perspect. Biol. 6, a016071. doi: 10.1101/cshperspect.a016071
Irimia, M., Weatheritt, R. J., Ellis, J. D., Parikshak, N. N., Gonatopoulos-Pournatzis, T., Babor, M., et al. (2014). A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523. doi: 10.1016/j.cell.2014.11.035
Jabre, I., Reddy, A. S. N., Kalyna, M., Chaudhary, S., Khokhar, W., Byrne, L. J., et al. (2019). Does co-transcriptional regulation of alternative splicing mediate plant stress responses? Nucleic Acids Res. 47, 2716–2726. doi: 10.1093/nar/gkz121
Jacob, A. G., and Smith, C. W. J. (2017). Intron retention as a component of regulated gene expression programs. Hum. Genet. 136, 1043–1057. doi: 10.1007/s00439-017-1791-x
James, A. B., Syed, N. H., Bordage, S., Marshall, J., Nimmo, G. A., Jenkins, G. I., et al. (2012). Alternative splicing mediates responses of the Arabidopsis circadian clock to temperature changes. Plant Cell 24, 961–981. doi: 10.1105/tpc.111.093948
Jung, H., Lee, D., Lee, J., Park, D., Kim, Y. J., Park, W. Y., et al. (2015). Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 47, 1242–1248. doi: 10.1038/ng.3414
Juntawong, P., Girke, T., Bazin, J., and Bailey-Serres, J. (2014). Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 111, E203–E212. doi: 10.1073/pnas.1317811111
Kalyna, M., Simpson, C. G., Syed, N. H., Lewandowska, D., Marquez, Y., Kusenda, B., et al. (2012). Alternative splicing and nonsense-mediated decay modulate expression of important regulatory genes in Arabidopsis. Nucleic Acids Res. 40, 2454–2469. doi: 10.1093/nar/gkr932
Kan, Z., States, D., and Gish, W. (2002). Selecting for functional alternative splices in ESTs. Genome Res. 12, 1837–1845. doi: 10.1101/gr.764102
Kelemen, O., Convertini, P., Zhang, Z., Wen, Y., Shen, M., Falaleeva, M., et al. (2013). Function of alternative splicing. Gene 514, 1–30. doi: 10.1016/j.gene.2012.07.083
Keren, H., Lev-Maor, G., and Ast, G. (2010). Alternative splicing and evolution: diversification, exon definition and function. Nat. Rev. Genet. 11, 345–355. doi: 10.1038/nrg2776
Khodor, Y. L., Rodriguez, J., Abruzzi, K. C., Tang, C. H. A., Marr, M. T., and Rosbash, M. (2011). Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 25, 2502–2512. doi: 10.1101/gad.178962.111
Kim, E., Magen, A., and Ast, G. (2007). Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 35, 125–131. doi: 10.1093/nar/gkl924
Koncz, C., deJong, F., Villacorta, N., Szakonyi, D., and Koncz, Z. (2012). The spliceosome-activating complex: molecular mechanisms underlying the function of a pleiotropic regulator. Front. Plant Sci. 3:9. doi: 10.3389/fpls.2012.00009
Kornblihtt, A. R., Schor, I. E., Alló, M., Dujardin, G., Petrillo, E., and Muñoz, M. J. (2013). Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 14, 153–165. doi: 10.1038/nrm3525
Kwon, Y.-J., Park, M.-J., Kim, S.-G., Baldwin, I. T., and Park, C.-M. (2014). Alternative splicing and nonsense-mediated decay of circadian clock genes under environmental stress conditions in Arabidopsis. BMC Plant Biol. 14:136. doi: 10.1186/1471-2229-14-136
Lämke, J., and Bäurle, I. (2017). Epigenetic and chromatin-based mechanisms in environmental stress adaptation and stress memory in plants. Genome Biol. 18:124. doi: 10.1186/s13059-017-1263-6
Ling, Y., Serrano, N., Gao, G., Atia, M., Mokhtar, M., Woo, Y. H., et al. (2018). Thermopriming triggers splicing memory in Arabidopsis. J. Exp. Bot. 69, 2659–2675. doi: 10.1093/jxb/ery062
Listerman, I., Sapra, A. K., and Neugebauer, K. M. (2006). Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat. Struct. Mol. Biol. 13, 815–822. doi: 10.1038/nsmb1135
Liu, Y., Gonzàlez-Porta, M., Santos, S., Brazma, A., Marioni, J. C., Aebersold, R., et al. (2017). Impact of alternative splicing on the human proteome. Cell Rep. 20, 1229–1241. doi: 10.1016/j.celrep.2017.07.025
Lopez-Casado, G., Covey, P. A., Bedinger, P. A., Mueller, L. A., Thannhauser, T. W., Zhang, S., et al. (2012). Enabling proteomic studies with RNA-Seq: the proteome of tomato pollen as a test case. Proteomics 12, 761–774. doi: 10.1002/pmic.201100164
Lu, T., Lu, G., Fan, D., Zhu, C., Li, W., Zhao, Q., et al. (2010). Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 20, 1238–1249. doi: 10.1101/gr.106120.110
Luco, R. F., Allo, M., Schor, I. E., Kornblihtt, A. R., and Misteli, T. (2011). Epigenetics in alternative pre-mRNA splicing. Cell 144, 16–26. doi: 10.1016/j.cell.2010.11.056
Mandadi, K. K., and Scholthof, K.-B. G. (2015). Genome-wide analysis of alternative splicing landscapes modulated during plant-virus interactions in Brachypodium distachyon. Plant Cell 27, 71–85. doi: 10.1105/tpc.114.133991
Marquez, Y., Brown, J. W. S., Simpson, C., Barta, A., and Kalyna, M. (2012). Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res. 22, 1184–1195. doi: 10.1101/gr.134106.111
Marquez, Y., Höpfler, M., Ayatollahi, Z., Barta, A., and Kalyna, M. (2015). Unmasking alternative splicing inside protein-coding exons defines exitrons and their role in proteome plasticity. Genome Res. 25, 995–1007. doi: 10.1101/gr.186585.114
Matera, A. G., and Wang, Z. (2014). A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 15, 108–121. doi: 10.1038/nrm3742
Matlin, A. J., Clark, F., and Smith, C. W. J. (2005). Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol. 6, 386–398. doi: 10.1038/nrm1645
Melamud, E., and Moult, J. (2009). Stochastic noise in splicing machinery. Nucleic Acids Res. 37, 4873–4886. doi: 10.1093/nar/gkp471
Merkhofer, E. C., Hu, P., and Johnson, T. L. (2014). Introduction to cotranscriptional RNA splicing. Methods Mol. Biol. 1126, 83–96. doi: 10.1007/978-1-62703-980-2_6
Merkin, J., Russell, C., Chen, P., and Burge, C. B. (2012). Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338, 1593–1599. doi: 10.1126/science.1228186
Misteli, T., Cáceres, J. F., Clement, J. Q., Krainer, A. R., Wilkinson, M. F., and Spector, D. L. (1998). Serine phosphorylation of SR proteins is required for their recruitment to sites of transcription in vivo. J. Cell Biol. 143, 297–307. doi: 10.1083/jcb.143.2.297
Modrek, B., Resch, A., Grasso, C., and Lee, C. (2001). Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 29, 2850–2859. doi: 10.1093/nar/29.13.2850
Naftelberg, S., Schor, I. E., Ast, G., and Kornblihtt, A. R. (2015). Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu. Rev. Biochem. 84, 165–198. doi: 10.1146/annurev-biochem-060614-034242
Narang, V., Sung, W. K., and Mittal, A. (2005). Computational modeling of oligonucleotide positional densities for human promoter prediction. Artif. Intell. Med. 35, 107–119. doi: 10.1016/j.artmed.2005.02.005
Neverov, A. D., Artamonova, I. I., Nurtdinov, R. N., Frishman, D., Gelfand, M. S., and Mironov, A. (2005). Alternative splicing and protein function. BMC Bioinformatics 6:266. doi: 10.1186/1471-2105-6-266
Nilsen, T. W., and Graveley, B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463. doi: 10.1038/nature08909
Ning, K., and Nesvizhskii, A. I. (2010). The utility of mass spectrometry-based proteomic data for validation of novel alternative splice forms reconstructed from RNA-Seq data: a preliminary assessment. BMC Bioinformatics 11(Suppl. 11):S14. doi: 10.1186/1471-2105-11-S11-S14
Nojima, T., Gomes, T., Grosso, A. R. F., Kimura, H., Dye, M. J., Dhir, S., et al. (2015). Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161, 526–540. doi: 10.1016/j.cell.2015.03.027
Nojima, T., Rebelo, K., Gomes, T., Grosso, A. R., Proudfoot, N. J., and Carmo-Fonseca, M. (2018). RNA polymerase II phosphorylated on CTD Serine 5 interacts with the spliceosome during co-transcriptional splicing. Mol. Cell 72:369-379.e4. doi: 10.1016/j.molcel.2018.09.004
Olsen, J. V., Ong, S.-E., and Mann, M. (2004). Trypsin cleaves exclusively C-terminal to arginine and lysine residues. Mol. Cell. Proteomics 3, 608–614. doi: 10.1074/mcp.T400003-MCP200
Pajoro, A., Severing, E., Angenent, G. C., and Immink, R. G. H. (2017). Histone H3 lysine 36 methylation affects temperature-induced alternative splicing and flowering in plants. Genome Biol. 18:102. doi: 10.1186/s13059-017-1235-x
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., and Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415. doi: 10.1038/ng.259
Paronetto, M. P., Passacantilli, I., and Sette, C. (2016). Alternative splicing and cell survival: from tissue homeostasis to disease. Cell Death Differ. 23, 1919–1929. doi: 10.1038/cdd.2016.91
Parra, G., Bradnam, K., Rose, A. B., and Korf, I. (2011). Comparative and functional analysis of intron-mediated enhancement signals reveals conserved features among plants. Nucleic Acids Res. 39, 5328–5337. doi: 10.1093/nar/gkr043
Pereira, A. (2016). Plant abiotic stress challenges from the changing environment. Front. Plant Sci. 7:1123. doi: 10.3389/fpls.2016.01123
Petrillo, E., Godoy Herz, M. A., Fuchs, A., Reifer, D., Fuller, J., Yanovsky, M. J., et al. (2014). A chloroplast retrograde signal regulates nuclear alternative splicing. Science 344, 427–430. doi: 10.1126/science.1250322
Poulos, M. G., Batra, R., Charizanis, K., and Swanson, M. S. (2011). Developments in RNA splicing and disease. Cold Spring Harb. Perspect. Biol. 3, a000778. doi: 10.1101/cshperspect.a000778
Preker, P., Nielsen, J., Kammler, S., Lykke-Andersen, S., Christensen, M. S., Mapendano, C. K., et al. (2008). RNA exosome depletion reveals transcription upstream of active human promoters. Science 322, 1851–1854. doi: 10.1126/science.1164096
Rayson, S., Arciga-Reyes, L., Wootton, L., de Torres Zabala, M., Truman, W., Graham, N., et al. (2012). A role for nonsense-mediated mrna decay in plants: pathogen responses are induced in Arabidopsis thaliana nmd mutants. PLoS One 7:e0031917. doi: 10.1371/journal.pone.0031917
Reddy, A. S. N., Marquez, Y., Kalyna, M., and Barta, A. (2013). Complexity of the alternative splicing landscape in plants. Plant Cell 25, 3657–3683. doi: 10.1105/tpc.113.117523
Reddy, A. S. N., Rogers, M. F., Richardson, D. N., Hamilton, M., and Ben-Hur, A. (2012). Deciphering the plant splicing code: experimental and computational approaches for predicting alternative splicing and splicing regulatory elements. Front. Plant Sci. 3:18. doi: 10.3389/fpls.2012.00018
Rocchi, V., Janni, M., Bellincampi, D., Giardina, T., and D’Ovidio, R. (2012). Intron retention regulates the expression of pectin methyl esterase inhibitor (Pmei) genes during wheat growth and development. Plant Biol. 14, 365–373. doi: 10.1111/j.1438-8677.2011.00508.x
Romero-Barrios, N., Legascue, M. F., Benhamed, M., Ariel, F., and Crespi, M. (2018). Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 46, 2169–2184. doi: 10.1093/nar/gky095
Rost, H. L., Malmstrom, L., and Aebersold, R. (2015). Reproducible quantitative proteotype data matrices for systems biology. Mol. Biol. Cell 26, 3926–3931. doi: 10.1091/mbc.E15-07-0507
Saldi, T., Cortazar, M. A., Sheridan, R. M., and Bentley, D. L. (2016). Coupling of RNA polymerase II transcription elongation with Pre-mRNA splicing. J. Mol. Biol. 428, 2623–2635. doi: 10.1016/j.jmb.2016.04.017
Sammeth, M., Foissac, S., and Guigó, R. (2008). A general definition and nomenclature for alternative splicing events. PLoS Comput. Biol. 4:e1000147. doi: 10.1371/journal.pcbi.1000147
Sani, E., Herzyk, P., Perrella, G., Colot, V., and Amtmann, A. (2013). Hyperosmotic priming of Arabidopsis seedlings establishes a long-term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 14, R59. doi: 10.1186/gb-2013-14-6-r59
Sanyal, R. P., Misra, H. S., and Saini, A. (2018). Heat-stress priming and alternative splicing-linked memory. J. Exp. Bot. 69, 2431–2434. doi: 10.1093/jxb/ery111
Seo, P. J., Ryu, J., Kang, S. K., and Park, C. M. (2011). Modulation of sugar metabolism by an indeterminate domain transcription factor contributes to photoperiodic flowering in Arabidopsis. Plant J. 65, 418–429. doi: 10.1111/j.1365-313X.2010.04432.x
Severing, E. I., van Dijk, A. D. J., Stiekema, W. J., and van Ham, R. C. H. J. (2009). Comparative analysis indicates that alternative splicing in plants has a limited role in functional expansion of the proteome. BMC Genomics 10:154. doi: 10.1186/1471-2164-10-154
Shaul, O. (2015). Unique aspects of plant nonsense-mediated mrna decay. Trends Plant Sci. 20, 767–779. doi: 10.1016/j.tplants.2015.08.011
Shen, Y., Zhou, Z., Wang, Z., Li, W., Fang, C., Wu, M., et al. (2014). Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 26, 996–1008. doi: 10.1105/tpc.114.122739
Sheynkman, G. M., Shortreed, M. R., Frey, B. L., and Smith, L. M. (2013). Discovery and mass spectrometric analysis of novel splice-junction peptides using RNA-Seq. Mol. Cell. Proteomics 12, 2341–2353. doi: 10.1074/mcp.O113.028142
Shukla, S., and Oberdoerffer, S. (2012). Co-transcriptional regulation of alternative pre-mRNA splicing. Biochim. Biophys. Acta 1819, 673–683. doi: 10.1016/j.bbagrm.2012.01.014
Skelly, M. J., Frungillo, L., and Spoel, S. H. (2016). Transcriptional regulation by complex interplay between post-translational modifications. Curr. Opin. Plant Biol. 33, 126–132. doi: 10.1016/j.pbi.2016.07.004
Staiger, D., and Simpson, G. G. (2015). Enter exitrons. Genome Biol. 16:136. doi: 10.1186/s13059-015-0704-3
Sterne-Weiler, T., Martinez-Nunez, R. T., Howard, J. M., Cvitovik, I., Katzman, S., Tariq, M. A., et al. (2013). Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome Res. 23, 1615–1623. doi: 10.1101/gr.148585.112
Struhl, K., and Segal, E. (2013). Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 20, 267–273. doi: 10.1038/nsmb.2506
Sureshkumar, S., Dent, C., Seleznev, A., Tasset, C., and Balasubramanian, S. (2016). Nonsense-mediated mRNA decay modulates FLM-dependent thermosensory flowering response in Arabidopsis. Nat. Plants 2:16055. doi: 10.1038/NPLANTS.2016.55
Sveen, A., Kilpinen, S., Ruusulehto, A., Lothe, R. A., and Skotheim, R. I. (2016). Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 35, 2413–2427. doi: 10.1038/onc.2015.318
Swarbreck, D., Wilks, C., Lamesch, P., Berardini, T. Z., Garcia-Hernandez, M., Foerster, H., et al. (2008). The Arabidopsis information resource (TAIR): gene structure and function annotation. Nucleic Acids Res. 36, 1009–1014. doi: 10.1093/nar/gkm965
Syed, N. H., Kalyna, M., Marquez, Y., Barta, A., and Brown, J. W. S. (2012). Alternative splicing in plants - coming of age. Trends Plant Sci. 17, 616–623. doi: 10.1016/j.tplants.2012.06.001
Tanner, S., Shen, Z., Ng, J., Florea, L., Guigó, R., Briggs, S. P., et al. (2007). Improving gene annotation using peptide mass spectrometry. Genome Res. 17, 231–239. doi: 10.1101/gr.5646507
Thatcher, S. R., Zhou, W., Leonard, A., Wang, B.-B., Beatty, M., Zastrow-Hayes, G., et al. (2014). Genome-wide analysis of alternative splicing in Zea mays: landscape and genetic regulation. Plant Cell Online 26, 3472–3487. doi: 10.1105/tpc.114.130773
Tian, C., Wang, Y., Yu, H., He, J., Wang, J., Shi, B., et al. (2019). A gene expression map of shoot domains reveals regulatory mechanisms. Nat. Commun. 10:141. doi: 10.1038/s41467-018-08083-z
Trcek, T., Sato, H., Singer, R. H., and Maquat, L. E. (2013). Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev. 27, 541–551. doi: 10.1101/gad.209635.112
Tress, M. L., Bodenmiller, B., Aebersold, R., and Valencia, A. (2008). Proteomics studies confirm the presence of alternative protein isoforms on a large scale. Genome Biol. 9, R162. doi: 10.1186/gb-2008-9-11-r162
Tress, M. L., Abascal, F., and Valencia, A. (2017a). Alternative Splicing may not be the key to proteome complexity. Trends Biochem. Sci. 42, 98–110. doi: 10.1016/j.tibs.2016.08.008
Tress, M. L., Abascal, F., and Valencia, A. (2017b). Most alternative isoforms are not functionally important. Trends Biochem. Sci. 42, 408–410. doi: 10.1016/j.tibs.2017.04.002
Ullah, F., Hamilton, M., Reddy, A. S. N., and Ben-Hur, A. (2018). Exploring the relationship between intron retention and chromatin accessibility in plants. BMC Genomics 19:21. doi: 10.1186/s12864-017-4393-z
Vinocur, B., and Altman, A. (2005). Recent advances in engineering plant tolerance to abiotic stress: achievements and limitations. Curr. Opin. Biotechnol. 16, 123–132. doi: 10.1016/j.copbio.2005.02.001
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Wang, X., Codreanu, S. G., Wen, B., Li, K., Chambers, M. C., Liebler, D. C., et al. (2018). Detection of proteome diversity resulted from alternative splicing is limited by trypsin cleavage specificity. Mol. Cell. Proteomics 17, 422–430. doi: 10.1074/mcp.RA117.000155
Wang, X., Hu, L., Wang, X., Li, N., Xu, C., Gong, L., et al. (2016). DNA methylation affects gene alternative splicing in plants: an example from rice. Mol. Plant 9, 305–307. doi: 10.1016/j.molp.2015.09.016
Wang, X., Zhang, B., and Wren, J. (2013). customProDB: an R package to generate customized protein databases from RNA-Seq data for proteomics search. Bioinformatics 29, 3235–3237. doi: 10.1093/bioinformatics/btt543
Wang, Y., Liu, J., Huang, B., Xu, Y., Li, J., Huang, L., et al. (2014). Mechanism of alternative splicing and its regulation (Review). Biomed. Reports. 3, 152–158. doi: 10.3892/br.2014.407
Weatheritt, R. J., Davey, N. E., and Gibson, T. J. (2012). Linear motifs confer functional diversity onto splice variants. Nucleic Acids Res. 40, 7123–7131. doi: 10.1093/nar/gks442
Weatheritt, R. J., Sterne-Weiler, T., and Blencowe, B. J. (2016). The ribosome-engaged landscape of alternative splicing. Nat. Struct. Mol. Biol. 23, 1117–1123. doi: 10.1038/nsmb.3317
Wickramasinghe, V. O., Gonzàlez-Porta, M., Perera, D., Bartolozzi, A. R., Sibley, C. R., Hallegger, M., et al. (2015). Regulation of constitutive and alternative mRNA splicing across the human transcriptome by PRPF8 is determined by 5’ splice site strength. Genome Biol. 16:201. doi: 10.1186/s13059-015-0749-3
Will, C. L., and Lührmann, R. (2011). Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 3, 1–2. doi: 10.1101/cshperspect.a003707
Willyard, C. (2018). New human gene tally reignites debate. Nature 558, 354–355. doi: 10.1038/d41586-018-05462-w
Wong, J. J.-L. L., Au, A. Y. M., Ritchie, W., and Rasko, J. E. J. (2015). Intron retention in mRNA: no longer nonsense. BioEssays 38, 41–49. doi: 10.1002/bies.201500117
Wong, J. J. L., Ritchie, W., Ebner, O. A., Selbach, M., Wong, J. W. H., Huang, Y., et al. (2013). Orchestrated intron retention regulates normal granulocyte differentiation. Cell 154, 583–595. doi: 10.1016/j.cell.2013.06.052
Wong, M. M., Chong, G. L., and Verslues, P. E. (2017). Epigenetics and RNA processing: connections to drought, salt, and ABA? Methods Mol. Biol. 1631, 3–21. doi: 10.1007/978-1-4939-7136-7_1
Yang, C., Bolotin, E., Jiang, T., Sladek, F. M., and Martinez, E. (2007). Prevalence of the initiator over the TATA box in human and yeast genes and identification of DNA motifs enriched in human TATA-less core promoters. Gene 389, 52–65. doi: 10.1016/j.gene.2006.09.029
Yang, X., Coulombe-Huntington, J., Kang, S., Sheynkman, G. M., Hao, T., Richardson, A., et al. (2016). Widespread expansion of protein interaction capabilities by alternative splicing. Cell 164, 805–817. doi: 10.1016/j.cell.2016.01.029
Yu, H., Tian, C., Yu, Y., and Jiao, Y. (2016). Transcriptome survey of the contribution of alternative splicing to proteome diversity in Arabidopsis thaliana. Mol. Plant 9, 749–752. doi: 10.1016/j.molp.2015.12.018
Zentner, G. E., and Henikoff, S. (2013). Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259–266. doi: 10.1038/nsmb.2470
Zhang, H., Lang, Z., and Zhu, J. K. (2018). Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 19, 489–506. doi: 10.1038/s41580-018-0016-z
Zhang, R., Calixto, C. P. G., Marquez, Y., Venhuizen, P., Tzioutziou, N. A., Guo, W., et al. (2017). A high quality Arabidopsis transcriptome for accurate transcript-level analysis of alternative splicing. Nucleic Acids Res. 45, 5061–5073. doi: 10.1093/nar/gkx267
Zhu, B., Zhang, W., Zhang, T., Liu, B., and Jiang, J. (2015). Genome-wide prediction and validation of intergenic enhancers in Arabidopsis using open chromatin signatures. Plant Cell 27, 2415–2426. doi: 10.1105/tpc.15.00537
Zhu, J.-K. (2016). Abiotic stress signaling and responses in plants. Cell 167, 313–324. doi: 10.1016/j.cell.2016.08.029
Keywords: alternative splicing, co-transcriptional splicing, protein diversity, intron retention, NMD, splicing memory, epigenetic modifications
Citation: Chaudhary S, Khokhar W, Jabre I, Reddy ASN, Byrne LJ, Wilson CM and Syed NH (2019) Alternative Splicing and Protein Diversity: Plants Versus Animals. Front. Plant Sci. 10:708. doi: 10.3389/fpls.2019.00708
Received: 08 March 2019; Accepted: 13 May 2019;
Published: 12 June 2019.
Edited by:
Ezequiel Petrillo, CONICET Instituto de Fisiología, Biología Molecular y Neurociencias (IFIBYNE), ArgentinaReviewed by:
Federico Damian Ariel, Instituto de Agrobiotecnología del Litoral (IAL), ArgentinaJohn William Slessor Brown, University of Dundee, United Kingdom
Copyright © 2019 Chaudhary, Khokhar, Jabre, Reddy, Byrne, Wilson and Syed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naeem H. Syed, bmFlZW0uc3llZEBjYW50ZXJidXJ5LmFjLnVr
†These authors have contributed equally to this work