 Yuxin Pan
Yuxin Pan Fanbo Meng1
Fanbo Meng1 Xiyin Wang
Xiyin Wang- 1Center for Genomics and Computational Biology, North China University of Science and Technology, Tangshan, China
- 2National Key Laboratory for North China Crop Improvement and Regulation, Agriculture University of Hebei, Baoding, China
Cotton is a major fiber plant, which provides raw materials for clothing, protecting humans from the harsh environment of cold or hot weathers, enriching the culture and custom of human societies. Due to its importance, the diploid and tetraploid genomes of different cotton plants have been repeatedly sequenced to obtain their complete and fine genome sequences. These valuable genome data sets revealed the evolutionary past of the cotton plants, which were recursively affected by polyploidization, with a decaploidization contributing to the formation of the genus Gossypium, and a neo-tetraploidization contributing to the formation of nowadays widely cultivated cotton plants. Post-polyploidization genome instability resulted in numerous structural changes of the genomes, such as gene loss, DNA inversion and translocation, illegitimate recombination, and accumulation of repetitive sequences, and functional innovation accompanied by elevated evolutionary rates of genes. Many these changes have been asymmetric between subgnomes of the tetraploid cottons, rendering their divergent profiles of biological regulation and function. The availability of whole-genome sequences has now paved the way to identify and clone functional genes, e.g., those relating to fiber development, and to enhance breeding efforts to cultivate cottons to produce high-yield and high-quality fibers, and to resist environmental and biological stress.
Introduction
Cotton is the world economic crop for its natural textile fiber, averaging about 25% of total world fiber use. Cotton seeds are rich in oil and proteins, and therefore used for oil production and as feed supplement for cattle and sheep, or as raw materials to manufacture industrial products, such as soaps and cosmetics. More than 80 countries produce cotton, distributed in arid and semi-arid regions of the tropics and sub-tropics. The top three cotton producers are India, China, and the United States. World consumption of cotton fiber and seed oil was approximately 119.4 million bales and 43.67 million metric tons in 2018, respectively (U.S. Department of Agriculture, https://usda.library.cornell.edu/).
Classification of Cotton Species
Cotton (Gossypium spp.) is an outstanding model species system for the plant cell elongation, cell wall, cellulose biosynthesis, and polyploidy research (Kim and Triplett, 2001). It is from Malvaceae family, including four subgenera, G. subg. Gossypium, G. subg. Houzingenia, G. subg. Karpas, and G. subg. Sturtia, and having approximately 45 diploid (2n = 2X = 26) and seven tetraploid (2n = 4X = 52) species (Hawkins et al., 2006; Salmon et al., 2010; Grover et al., 2015; Ditta et al., 2018). About 12.5 million years ago (Mya), Gossypium was diverged from its closer relatives Kokia and Gossypioides during the Miocene (Cronn et al., 2002).
Eight diploid cytogenetic genomes, designated as A to G and K, have been found in the world (Wang et al., 2012). The A, B, E, and F genomes occur naturally in Africa and Asia (Cronn et al., 2003). G. herbaceum (A1) is originated from Africa, and the primitive G. arboreum (A2) from India. D genome occurs in Americas, with G. raimondii (D5) initially found in Peru. The C, G, and K genomes are found in Australia (Chen et al., 2007). The haploid genome sizes vary from 2,500 Mb in the K genome, to about 800 Mb in the D genome (Hawkins et al., 2006).
The A genome, African species much like modern G. herbaceum (A1) and G. arboreum (A2), and D genome like American diploid species, G. raimondii (D5), were reunited by trans-oceanic dispersal and chromosome doubling to give rise to allotetraploid cotton species (Paterson et al., 2012). Five widely-recognized tetraploid species include G. hirsutum (AD1), or “Upland cotton”, G. barbadense (AD2), “Pima” or “Egyptian” cotton, and three other exclusively wild polyploid species, G. tomentosum (AD3), G. mustelinum (AD4), and G. darwinii (AD5), are endemic to coastal and island habitats (Salmon et al., 2010). G. hirsutum (AD1) and G. barbadense (AD2), widely cultivated tetraploid cotton species, arose in the American continent, with the former in Mexico and the latter in Peru. These facts suggest that parallel and convergent domestication occurred (Figure 1).
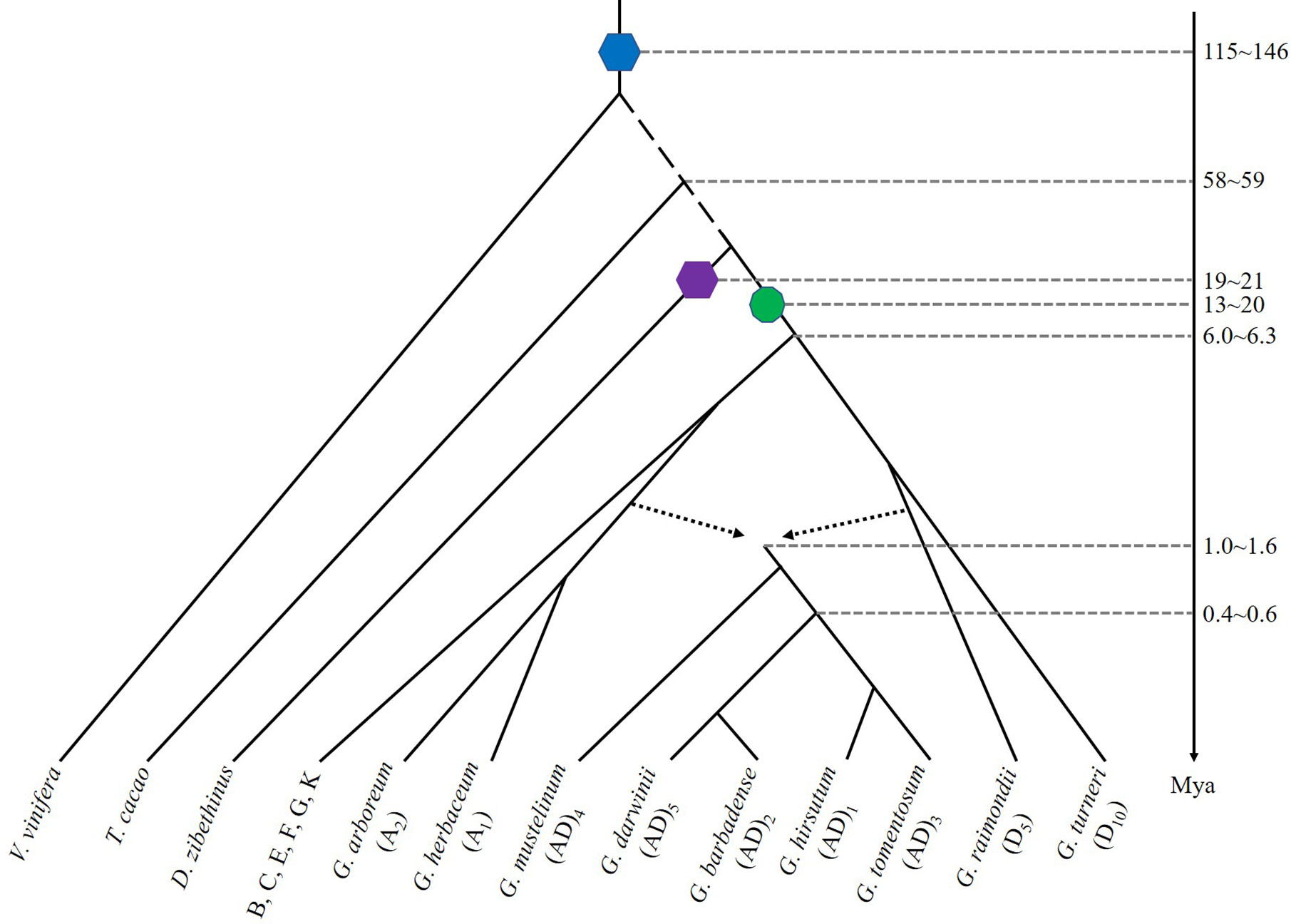
Figure 1 Evolutionary relationships of diploid and tetraploid species of Gossypium and related plants. The blue hexagon denotes a hexaploidization event shared by major eudicots, purple hexagon denotes a hexaploidization in Durian, and green decagon denotes a decaploidization shared by Gossypium species. Broken arrowed lines show a hybridization to produce the ancestral allotetraploid cotton. A time line is shown with approximate dates of key evolutionary events. Mya: millions years ago.
Cotton Genome Sequencing Efforts
In view of cotton’s importance in human life, intensive efforts have been focused on uncovering the genome mysteries of cotton species. Three diploid cottons and two tetraploid cottons were sequenced, including G. arboreum (Li et al., 2014; Du et al., 2018; Huang et al., 2020), G. herbaceum (Huang et al., 2020), G. raimondii (Wang et al., 2012; Paterson et al., 2012; Udall et al., 2019), G. hirsutum (Li et al., 2015; Zhang et al., 2015; Hu et al., 2019; Wang M. et al., 2019; Chen et al., 2020; Huang et al., 2020) and G. barbadense (Yuan et al., 2015; Liu et al., 2015; Hu et al., 2019; Wang M. et al., 2019; Chen et al., 2020). Detailed genome information is shown in Tables 1 and 2.
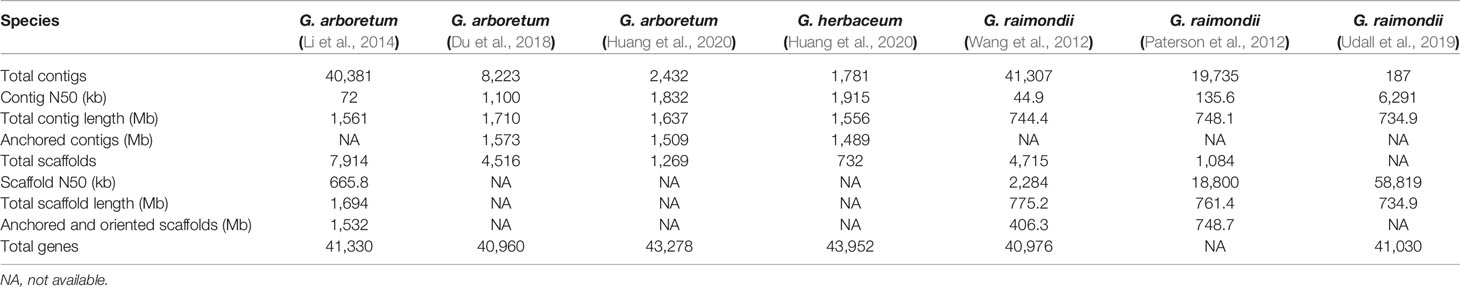
Table 1 Characteristics of G. arboreum, G. herbaceum, and G. raimondii genome sequences.

Table 2 Characteristics of G. barbadense and G. hirsutum genome sequences.
Having relatively smaller sizes, the diploid genomes were firstly sequenced to provide a reference to explore the tetraploid genomes. Using Illumina HiSeq 2000 platform, the genome of G. raimondii was deciphered and assembled into 775.2 Mb length (Wang et al., 2012). At the meantime, by using Sanger, Roche 454 XLR and Illumina Genome Analyzer IIx platform, another G. raimondii genome was sequenced, and based on the cotton genetic and physical maps, 761.4 Mb data was assembled and oriented to 98.3% of the expected genome size (Paterson et al., 2012). Seven years later, deciphered using PacBio long-read technology, HiC, and Bionano optical mapping, the third genome sequence of G. raimondii was published (Udall et al., 2019). In this assembled genome, the number of contigs decreased and the length of contig N50 was much elongated (6.3 Mb). The A genome from G. arboreum has a genome size almost twice of that of the D genome from G. raimondii. Using Illumina HiSeq 2000 platform, G. arboreum genome was sequenced and a total of 1,694 Mb was assembled with 90% oriented onto 13 pseudochromosomes (Li et al., 2014). In 2018, using PacBio and Hi-C technology, G. arboreum genome was re-sequenced again, and a total of 1,710 Mb contigs was assembled and 92% of contigs oriented (Du et al., 2018). Very recently, an improved genome sequence of G. arboreum with 1,637 Mb assembled DNA was published (with 92% oriented) (Huang et al., 2020). G.herbaceum var. africanum, another A-genome diploid cotton, was sequenced using Illumina, PacBio and Hi-C sequence technology. The assembled size was 1,556 Mb with 95.69% (1,489 Mb) of all sequenced oriented and organized into 13 chromosomes (Huang et al., 2020).
G. hirsutum, known for its high lint production than any other cultivated cotton species, accounts for more than 90% of commercial cotton production worldwide (Cronn et al., 2003). Acc. Texas Marker-1 (TM-1) is a genetic standard for G. hirsutum genome. Using Ilumina and BAC sequencing, G. hirsutum genome was sequenced to produce a total of 2,173 Mb assembled sequence (V1.0), with 88.5% anchored and oriented to 26 pseudochromosomes. The anchored At-subgenome was 1,170 Mb with 35,056 genes, whereas the Dt-subgenome was 753 Mb with 37,086 genes (Li et al., 2015). By integrating Illumina, Sanger-sequenced BAC-end sequences, the assembled TM-1 genome sequence (V1.0), and an ultra-dense genetic map, a 2,432.7 Mb TM-1 genome sequence (V1.1) was produced. In the assembly V1.1, 218 misassembled scaffolds (442.2 Mb) in the assembly V1.0 was corrected (Zhang et al., 2015). Very recently, by using PacBio, Illumina HiSeq and Hi-C, a much improved G. hirsutum genome sequence was published (Huang et al., 2020).
G. barbadense (Pima cotton) is famous for its extra-long, strong and fine fiber, and its accessions 3–79 were sequenced, being annotated 80,876 protein-coding genes in total. The subgenome At (1.50 Gb) was found to have a double size as to the Dt (853 Mb) (Yuan et al., 2015). And also, researchers sequenced G. barbadense cv. Xinhai21, through using three next-generation sequencing platforms, Roche 454, Illumina Hiseq2000, and PacBio SMRT (Liu et al., 2015). This genome sequence covered 1.395 Gb of the subgenome At and 0.776 Gb of the Dt.
The draft genome sequences of allotetraploid cotton species had been highly fragmented and incomplete. Especially, telomere, centromere, and repeat-rich regions were often poorly assembled. Therefore, allotetraploid cottons, G. hirsutum accession TM-1, and G. barbadense accession 3–79, were re-sequenced and annotated by integrating single-molecule real-time sequencing, BioNano optical mapping, and high-throughput chromosome conformation capture techniques (Hi-C). These re-sequencing efforts revealed 70,199 genes in G. hirsutum and 71,297 genes in G. barbadense (Wang M. et al., 2019). At the meantime, G. hirsutum accession TM-1 and G. barbadense accession Hai7124 were also re-sequenced and assembled by integrating non-PCR-based short-read sequencing, long-read-based gap closure, scaffolding, and orientation based on 3D proximity information derived from Hi-C data and from optical and genetic maps. 72,761 and 75,071 protein-coding genes were identified in TM-1 and Hai7124 (V1.1), respectively. Contiguity and completeness for regions with high content of repeats were improved, especially with centromeric regions for each chromosome (Hu et al., 2019). Very recently, based on single-molecule real-time, Illumina and Hi-C, another five allotetraploid cotton genomes were sequenced. The assembled genomes range in size from 2.2 to 2.3 Gb (Chen et al., 2020).
Each assembly genome has its advantages and significance. The newly (re)sequenced and assembled genomes, G. arboreum, G. herbaceum, G. hirsutum (Huang et al., 2020), G. raimondii (Udall et al., 2019), and G. barbadense (Wang M. et al., 2019) provided chromosome-scale references. Two prominent databases were set up to store these genome data sets, CottonGen (https://www.cottongen.org/) (Yu et al., 2014) and CottonFGD (https://cottonfgd.org/) (Zhu et al., 2017).
Based on the assembling information, we compared the gene pairs between the same species of the relatively newly sequenced genomes using Blastn (sequence identity > 99% and aligned length >80% of each gene) (Table 3). As to the two compared D genome sequences, the number of common genes is only 11,541, accounting up to about 1/3 of the total annotated genes in each genome. A comparison of two A genome sequences revealed ~3/5 of commonly annotated genes. Comparisons of tetraploid cottons also found prominent fractions of non-common or specific genes. These comparisons alert a careful use of the gene annotation, in case the same gene was not well decoded, with its full length, or exon/intron composition. Besides, a pan-genus re-annotation of cotton genes integrating de novo prediction software and homology searching, supported by complete transcriptome information, is indispensable to assist further biological and agricultural exploration. We also inferred orthologous genes (23,906–28,334 between diploid cottons and 44,672–62,708 between tetraploid cottons), which also implies much divergence in gene composition among different cottons (Table 4).

Table 3 Comparison of annotated genes in the updated or newly sequenced cotton genomes.

Table 4 Orthologous genes between diploid or tetraploid cotton genomes.
Polyploidization and Cotton Origination
Polyploidy is a significant evolutionary drive force in plants (Bowers et al., 2003; Tang et al., 2008a). Cotton is ideal for investigating polyploidy for having been affected by recursive polyploidization.
Gossypium shared an ancient hexaploidization event with the other core eudicots (Tang et al., 2008b). Shortly after divergence from cacao, the Gossypium lineage experienced multiple-fold ploidy increase (Paterson et al., 2012). Through detecting gene collinearity, five-times of duplicated regions in cotton to those in cacao and grape suggested a paleo-decaploidy, or penta-plication of the ancestral genome, implying a rather complex nature of the cotton genome, as compared to many other eudicot plants (Wang et al., 2016). The obscurity whether cotton and the other Malvaceae plants share the event were discussed by comparing genomic information from durian and Bombax (Teh et al., 2017; Conover et al., 2019). Exploration of the collinear genes, including inter-genomic ratio of retained homologs, homologous gene tree topology, and gene retention levels suggested that the above-mentioned decaploidy was not shared with durian, which was affected by an independent hexaploidization (Wang J. et al., 2019). Actually, the decaploidy might have directly contributed to the origination and divergence of the Gossypium plants. Besides, it might continue to drive their evolution and functional innovation in that much increased mutations were observed in the duplicated genes, resulting in aberrant topology of gene trees (Wang J. et al., 2019). At least 83.2% of phylogenetic trees constructed with collinear homologs, including grape, cotton, and cacao, did not conform to the expected topology, clearly due to elevated evolutionary rates of certain duplicated gene copies (Meng et al., 2020).
However, it has been controversial about the time of cotton origination and speciation. Molecular phylogenetic analyses suggested that the common ancestor of G. arboreum and G. raimondii was diverged from T. cacao around 18–58 Mya (Li et al., 2014). By examining 745 single-copy gene families from nine sequenced plant genomes, G. raimondii and T. cacao were inferred to have probably been diverged approximately 33.7 Mya from a common ancestor (Wang et al., 2012). A latest estimation suggested that the divergence between Gossypium and T. cacao occurred about 58−59 Mya (Chen et al., 2020). Using synonymous substitution rates (Ks) obtained from 3,195 paralogous gene pairs in the G. raimondii and T. cacao genomes, two Ks value peaks were observed at 0.40–0.60 and 1.5–1.90. Corresponding to the peak with smaller Ks, the decaploidization event was proposed to occurred approximately 16.6 (13.3–20.0) Mya in the Gossypium lineages. The second peak corresponds to the more ancient hexaploidization event shared by core eudicots, having occurred approximately 130.8 (115.4–146.1) Mya (Tang et al., 2008b; Van de Peer et al., 2009). In G. arboreum, two coincident polyploidization events shared by G. raimondii were estimated using Ks of 1,917 paralogous gene pairs of similar age, with one peak around 0.17 synonymous transversions per site and a second peak at about 0.54 was observed. This information showed the two polyploidization events to have occurred at 13–20 and 115–146 Mya (Li et al., 2014). In a comparative analysis of cotton to cacao and durian, the cotton decaploidization was inferred to have occurred ~13–14 Mya and split from cacao ~21–24 Mya (Wang J. et al., 2019). Homeologous exchanges had occurred throughout the polyploid divergence and speciation in cotton (Salmon et al., 2010), possibly resulting in gene conversion, which made it even more complex to perform a reasonable evolutionary dating with gene or protein sequences (Guo et al., 2014).
All diploid cotton species retain 13 common chromosomes and largely collinear gene order. The A-genome and D-genome diploids were inferred to share a common ancestor about 5–10 Mya (Wendel and Albert, 1992; Argout et al., 2011; Wang et al., 2012). A most recent estimation of divergence time between the A and D was inferred to be 6.0–6.3 Mya (Zhang et al., 2015), or 6.2–7.1 Mya (Hu et al., 2019). A more recent estimation of 4.7–5.2 Mya was recently obtained (Chen et al., 2020). The common ancestor of the A1 and A2 clade was phylogenetically a sister to the At- subgenomes of (AD)1 and of (AD)2, and the divergence time for A1 and A2 was estimated to be ~0.7 Mya, suggesting that allotetraploid formation (~1.0−1.6 Mya) preceded the speciation of A1 and A2 (Huang et al., 2020) (Figure 1).
Gene collinearity analysis between G. hirsutum and G. barbadense genome suggested that both were originated from a common allotetraploid ancestor (Figure 1). The hybridization between A- and D- genomes was initially inferred to occur 1 to 2 Mya (Fawcett et al., 2009). By comparing the sequenced G. hirsutum, G. arboreum, and G. raimondii orthologous genes, the ancestral allotetraploid was estimated to form about 1–1.5 Mya (Li et al., 2015). The divergence of the two allotetraploid cottons was inferred to occur ~1 Mya (Liu et al., 2015). An updated analysis with allotetraploid genomes inferred the tetraploidization to occur ~1.7–1.9 Mya and the divergence of G. barbadense and G. hirsutum was 0.4–0.6 Mya (corresponding to Ks peaks at 0.002 and 0.003, respectively) (Hu et al., 2019). Recently, the allotetraploid formation was estimated to ~1.0–1.6 Mya (Huang et al., 2020) and five allotetraploid genome sequences further illustrated the divergence between tetraploid and diploid clades occurred about the same time (Chen et al., 2020).
Chromosomal Structural Conservative and Variations
Polyploidization may have impact on the architecture of the genome and structural changes may lead to phenotypic variation. Actually, 780 collinear blocks were detected between G. raimondii [JGI, (Paterson et al., 2012)] and G. arboreum. Based on inferred gene collinearity between orthologous chromosomes, large-scale rearrangements were inferred on chromosomes 2 and 3 of G. raimondii, while deletions and insertions were found on chromosomes 7 and 8 of G. arboreum (Li et al., 2014). With a comparative analysis of A1 (G. herbaceum) and A2 (G. arboreum) genomes, a reciprocal translocation was revealed on each of chromosomes 1 and 2, and two large-scale inversions were detected in chromosomes 10 (~18.4 to ~61.3 Mb in A1 and ~18.79 to ~58.96 Mb in A2) and 12 (~15.96 Mb to ~77.61 Mb in A1 and 15.66 to 84.69Mb in A2) of A1- and A2-genomes (Huang et al., 2020).
Owing to high gene collinearity between G. hirsutum and G. barbadense and that of each with their diploid progenitors or within themselves, DNA translocation and inversion were inferred in these genomes (Zhang et al., 2015; Huang et al., 2020). The At and Dt subgenomes had similar number of rearrangements (19 versus 18) (Zhang et al., 2015). Whereas, the Hi-C data indicates that chromosome rearrangements occurred in all 13 chromosomes between G. babadense and G. hirsutum genome sequences. A total of 170.2 Mb inverted DNA regions were identified and the majority were shared by both At- subgenomes of G. hirsutum and G. barbadense. Four chromosomes exhibited paracentric inversions and eleven chromosomes showed pericentric inversions in heterochromatin. There were four large inversions, including three paracentric inversions and one pericentric inversion in the A06 chromosome between G. hirsutum and G. barbadense (Wang M. et al., 2019). Compared to G. raimondii (JGI, (Paterson et al., 2012), unique and shared structural variants were detected in G. hirsutum and G, barbadense, such as one large inversion in G. barbadense chromosome D05 and G. hirsutum D12, and a shared large inversion in the D09 chromosome was shared by both tetraploids. A large pericentric inversion was revealed in G. barbadense D05 (41.48–51.18 Mb) and G. hirsutum D12 (14.15–31.54 Mb), and a large inversion in the D09 was shared by both tetraploids (Wang M. et al., 2019). The A1- and A2-genomes were found to have two and three translocations, respectively, as compared to the At1 subgenome (G. hirsutum), which is between chromosomes 2 and 3, and 4 and 5 in the tetraploid At1 subgenome. An inversion in chromosome 10 occurred between A1 and At1 (~18.4 Mb to ~61.3 Mb in A1 and ~23.09 Mb to ~97.42 Mb in At1) (Huang et al., 2020). The Dt-subgenomes had fewer and smaller inversions than the At-subgenomes among five tetraploid cotton genomes, except for a few small inversions in D10 of Gt–Gm and Gm–Gb and D12 of Gd–Gt–Gm (Chen et al., 2020). Using the D5 genome [JGI, (Paterson et al., 2012)] as a reference, 77 putative translocation sites were observed in the 13 chromosomes of Dt and chromosomal translocations occurred more frequently in sub-telomeric regions in G. barbadense (Yuan et al., 2015). In the G. hirsutum, at least nine translocations and 28 inversions were identified. Two large reciprocal translocations were found between A02 and A03 and between A04 and A05, and three inversions found on the A12 and D12 homoeologous chromosomes (Zhang et al., 2015). Between G. babadense and G. hirsutum genome sequence, there were 3,820 translocations revealed (Wang M. et al., 2019). Also, there were translocations along chromosomes 1–3 and chromosomes 4 and 5 in G. hirsutum and G. barbadense (Hu et al., 2019).
Expansion of Transposable Elements
Transposable elements (TEs) have important roles in driving genome evolution (Galindo-Gonzalez et al., 2017). They account up to 57% (441Mb) of the G. raimondii genome, mainly the gypsy and copia-like long terminal repeat (LTR), a type of retrotransposons, which explains much of the expansion of the G. raimondii genome. Data analysis indicated that the G. arboreum genome tended to harbor more LTRs inserted than G. raimondii genome during the last 0.5 million years (Cheng et al., 2019). G. arboreum genome had the greatest amount of repeat-containing sequences among sequenced cottons, and LTRs accounted for 95.12% of all repeat sequences (Cheng et al., 2019). In comparison to the G. raimondii genome, the G. arboreum genome had noticeable proliferation of Gorge elements. LTR retrotransposons in G. arboreum appeared to cluster near centromeres. Thus, LTR expansion seemed to contribute after a two-fold increase in G. arboreum genome (Li et al., 2014).
TEs of the Dt-subgenome tend to be more active than those of the At-subgenome after the tetraploidization. Both copia and gypsy were more actively transcribed in the Dt-subgenome. Copia elements were remarkably more active than gypsy in the recent 0~1 My time, with higher proportions of copia located near coding genes than gypsy (Li et al., 2015). Two different methods inferred similar TE proportion in its genome (1,339 Mb versus 1,445 Mb, 64.8 versus 66%) (Li et al., 2015; Zhang et al., 2015). There were more TEs in the At-subgenome (at least 843.5 Mb) than in the Dt-subgenome (at least 433 Mb). Among them, the number of gypsy retro elements was 3-fold higher in the At-subgenome than in the Dt-subgenome. The TE types and relative proportions of At- and Dt-subgenome were similar to their corresponding genomes of G. arboreum and G. raimondii, whereas the retrotransposon frequencies were different (52.29 versus 62.81%). The TE divergence time was inferred to be older than 1.5 My, suggesting that most TEs expanded before the formation of allotetraploid cotton (Zhang et al., 2015). Moreover, TE expansion occurred in the progenitor genomes and was retained after allotetraploid formation. Copia elements had been remarkably more active than gypsy, with copia more frequently located near coding genes than gypsy. Some copia- and gypsy-like elements were observed to be present in the Dt while absent in the D-genome (Chen et al., 2020). One of the two G. barbadense genomes were observed to have 83.5 and 82.2% LTRs in At and Dt (Liu et al., 2015), and the other detected 1,778.6 Mb of TEs (69.1% of the assembly), including 1,098.0 Mb of TE sequences in At (representing 73.5% of the subgenome) and 541.6 Mb of TEs in Dt (representing 63.5% of the subgenome) (Yuan et al., 2015). The latest data showed that At had 4.0–5.9% lower repetitive DNA content than the A-genome, whereas the Dt had 1.5–2.9% higher content than the D-genome. These changes may affect the downsizing and equilibration in allotetraploids (Chen et al., 2020).
A large number (9.15%) of LTR bursted at 5 Mya and decreased thereafter in At, whereas a substantially lower and flat peak appeared 3−5 Mya in Dt (Liu et al., 2015). Whereas, the timing of insertion for LTRs was peaked within 1.9 Mya in Dt and approximately 3.1 Mya ago in At (Yuan et al., 2015). The above research suggested that the most expansions of extant LTRs independently occurred after the lineage separation but before tetraploidization (Wu et al., 2017). The two-fold expansion of A1 and A2-genomes and the G. hirsutum At1-subgenome was found to be highly correlated to TE bursts. LTR families in Gossypium had been greatly expanded in comparison to durian [52.42% of the Dt1-subgenome (G. hirsutum), 53.2% of the D5-genome, 26.2% of durian]. More than 72% genome is LTRs in A-genome (72.57% of the A1-genome and 73.62% of the A2-genome). LTR retrotransposons in Gossypium experienced continuing and more recent amplification bursts occurred about 0–2 Mya. Using the representative LTR/Gypsy sequences, TE burst time was evaluated to be related to cotton genome divergence. The earliest burst occurred ~5.7 Mya, which coincided the expected speciation time for A- and D- genome. The next one occurred ~2.0 Mya, observed specifically in Dt1- and At1-genomes (G. hirsutum), coinciding the formation of allotetraploid cotton (Huang et al., 2020).
Asymmetric Evolution of the A and D Genome
The A genome species produce spinnable fiber, whereas the D genome species do not (Applequist et al., 2001), showing divergent contribution to phynotypes. Actually, with genome sequences, asymmetric evolution was found to have characterized the difference between genomes, including the above-mentioned difference in TE composition and expansion.
In G. hirsutum, the Dt-subgenome had higher mutation rates than the At-subgenome. The single-nucleotide variation rate in Dt versus D was greater than that in At versus A in intergenic collinear regions. Further analysis revealed that A- and At-genomes had been undergoing greater positive selection than the D- and Dt-genomes (Li et al., 2015). For example, the average Ks for collinearity-supported gene pairs in At-and Dt-subgenomes were found to be substantially lower than those of the A and D diploid genomes, respectively. In addition, Dt/D versus T. cacao had lower dN/dS ratios than At/A versus T. cacao. These data suggested that the genetic redundancy generated by allotetraploidy may have allowed relaxed purifying selection in both the At- and Dt-subgenomes (Li et al., 2015). There were similar levels of rearrangements between chromosomes and those between the At- and Dt-subgenomes (19 versus 18). However, the length of total rearrangements was revealed to be larger in the At-subgenome (372.6 Mb) than in the Dt-subgenome (82.6 Mb). The SNP frequency between G. hirsutum and G. barbadense was slightly larger in the At-subgenome with 8,131,276 (5.95 per kb), than that in the Dt-subgenome, with 4,685,422 (5.81 per kb) (Wang M. et al., 2019). All above information revealed that At-subgenome had experienced more relaxed selection pressure (Yuan et al., 2015; Liu et al., 2015). In the same way, allele-SNPs within subgenomes, between accessions of six cotton tetraploid species, were detected to be higher in the At-than Dt-genomes. In both AD1 and AD2, the number of At-subgenome allele-SNPs was observed about 1.5X that of Dt-genome allele-SNPs (Page et al., 2016).
A systematic characterization of presence/absence variations (PAVs) between the two tetraploid accessions was also performed. With the PAVs, a lot of genes unique to G. barbadense were highly expressed during fiber development. Besides, there were also inversions between G. hirsutum and G. barbadense, including 120.4 Mb of At-subgenome and 49.8 Mb of Dt. By using Hi-C, large inversions were shown to have paracentric and pericentric inversions in the chromosomes A06 and D12 (Wang M. et al., 2019). Besides, introgressions were detected in AD1 and AD2 genomes owing to the attempts of breeders for transfer of genes for disease resistance, fiber quality, and other traits between (AD)1 and (AD)2 (Page et al., 2016).
Asymmetric DNA duplication phenomenon in tetraploid subgenomes was discovered through comparing accessions of G. hirsutum and G. barbadense. At DNA duplications were more (2X) conserved than Dt DNA duplications in G. hirsutum cultivars, although not in G. barbadense. Besides, At DNA deletions were more conserved than Dt DNA deletions in G. barbadense but not in G. hirsutum. Both these findings implied the existence of independent domestication events for these two species (Page et al., 2016).
Asymmetric changes in At- and Dt-subgenomes were also reflected in biased At- or Dt-homeolog expression (Yoo and Wendel, 2014). For example, more transcription factor genes (such as MYB family members) were expressed in the At homeologs, suggesting their important roles in fiber development. This might have led to subfunctionalization of the At-Dt paralogs (Li et al., 2015).
Unidirectional DNA exchanges were the predominant mechanism responsible for allelic differences between the Gossypium tetraploids and their diploid progenitors. At-to-Dt conversion was enriched in heterochromatin, which was highly correlated with GC content and transposon distribution, and may silence abundant A-genome-derived retrotransposons (Guo et al., 2014).
Genomic Insight for Cotton Fiber Gene Expression and Development
Cotton fibers, which are highly elongated epidermal cells, undergoing four over-lapping development, initiation, elongation (Primary cell wall, PCW), secondary cell wall (SCW) synthesis and maturation (Basra and Malik, 1984). The elongation and SCW synthesis stages are key stages for fiber length and strength. At SCW stage, cross-linking of cellulose microfibrils and non-cellulosic matrices presumably “fix” the structure of the PCW, resulting in the first significant increase in fiber strength (Wilkins and Arpat, 2005).
Different development features in Gossypium genus may lead to typical fiber traits. G. barbadense, Hai7124, had an extended time longer than G. hirsutum, TM-1, in fiber elongation period. The stage lasted about 15 days from 5 to 25 days post-anthesis (DPA) in TM-1, whereas this lasted from 5 to 30 DPA in Hai7124. During the PCW-SCW transition period, expression of most genes is earlier in TM-1 than in Hai7124 (Zhu et al., 2011). Also, there are amount of expression data revealing the difference in the regulatory pathway. For example, translation and ribosome biosynthesis pathway genes are enriched during fiber elongation in Upland cotton and during cellulose biosynthesis in Pima cotton (Chen et al., 2020).
Based on genome sequences, more and more genes with structure and qualitative transcript differences in fiber development were identified. Actually, fiber elongates more than 2000-fold after initiation, which is regulated by cell turgor. The plasmodesmata on/off switch, together with sucrose and potassium ions (K+) transporter is curial for fiber cells (Ruan et al., 2004). Several genes associate with membrane transport, transcription, and glycan biosynthesis and carbon metabolism are significantly expressed in Hai7124, as compared to TM-1 fibers. Furthermore, genes regulating Hai7124 fiber biosynthesis, such as those encoding the sucrose transporter (GbTST1), Na+/H+ antiporter (GbNHX1), aluminum-activated malate transporter (GbALMT16), the vacuole-localized vacuolar (GbVIN1) and plasmodesmata (PD) opening have a longer period of expression than the corresponding genes in TM-1 (Hu et al., 2019).
The fiber SCW is consisted of >94% cellulose(Wilkins and Arpat, 2005). Sucrose synthase gene (sus) expressed in the later stage of fiber development plays a role in cell wall cellulose (Suppression of sucrose synthase gene expression represses cotton fiber cell initiation, elongation, and seed development). Between G. raimondii and G. hirsutum cotton ovules at 3 DPA, three Sus genes (SusB, Sus1 and SusD) are expressed at substantially higher levels in G. hirsutum than in G. raimondii (Wang et al., 2012). Cellulose synthase A (CesA), synthesis cellulose, acts a key role in the regulating of the secondary cell wall thickening. Through genome-wide analysis, 32 CesA and 64 cellulose synthase-like (CSL) genes were detected in G. hirsutum, and 37 CesA genes identified in G. barbadense. The CesA genes can be classified into two major groups with six branches. One group is expressed during primary cell wall development, whereas the other group is expressed during secondary cell wall biosynthesis. Genes encoding CesA, UGD, UGP and UER, important for cotton fiber growth, locate in the At-subgenome, are expressed highly during either the primary or the secondary cell wall biosynthesis stages (Yuan et al., 2015; Li et al., 2015).
Transcription factors are important for cotton fiber development. MYB is one of the most abundant transcription factors in cotton, and play diverse roles during cotton growth and evolution (Salih et al., 2016). A total of 219 and 524 MYB genes were identified in the G. raimondii and G. hirsutum genome (Wang et al., 2012; Salih et al., 2016). In G. hirsutum, two groups of MYB genes are expressed. One exhibits low or undetectable expression levels and the other express higher during -1 to 10DPA. A large number of MYB genes are expressed more predominantly in G. hirsutum ovules than G. raimondii. Some of MYB MIXTA-like (GhMML) homeologs are also highly expressed during fiber initiation in G. hirsutum, but down-regulated in fiberless mutants. All the above information indicates that some MYB genes might be required for early fiber development. WRKY, another transcription factor, participates in fiber development. In total, 112 and 109 WRKY genes were identified in G. raimondii and G. arboreum. Many SNPs are distributed unequally in exon and intron regions in these genes (Ding et al., 2015).
Ethylene is a key signaling modulator of cotton fiber cell growth (Shi et al., 2006; Pang et al., 2010). Among these, 1-aminocyclopropane-1-carboxylic acid oxidase (ACO) is the last enzyme in the ethylene synthesis. High amounts of transcripts relating to ACO activities were discovered from G. raimondii at the 3-DPA stage. ACO3 and ACO1 are 500- and 1000-fold higher than G. arboreum, respectively. CaACO loss of MYB-binding sites may lead to lower transcripts in G. arboreum. Compared to G. raimondii, ACO expression and ethylene are both lower in G. hirsutum. The ACO expression peak and ethylene burst occurred in the later stage of G. hirsutum. Very high levels of ACO transcripts will give rise to ethylene burst and may force an early fiber senescence phenotype, whereas the inactivation of ACO in G. arboreum ovules may be responsible of the short-fiber phenotype in this species. A compromise ACO expression may be the power for fiber traits in G. hirsutum. Thus, ACO is suggestive of a major role for the plant hormone ethylene during early fiber cell development (Wang et al., 2012; Li et al., 2014).
A total of 591 PSGs (72.8%) genes with the codons subjected to positive selection, express during the fiber development. Many of the A-homeologous PSGs are enriched in the synthesis of ethylene and very-long-chain fatty acids, sucrose metabolism and beta-d-glucan biosynthetic pathway to produce UDP-glucose, whereas the D-homeologous PSGs are enriched in carbohydrate transport, response to superoxide and other abiotic stresses. These results suggest that allotetraploid cotton domestication is associated with intensive human selection for fiber yield and quality on the A homeologs from fiber-producing species and for wider adaptation on the D homeologs from non-poor species. These findings suggested that At-subgenome have contributed more to fiber improvement (Zhang et al., 2015).
Conclusion and Future Prospects
The availability of multiple cotton genomes shed precious light on their genome structures and evolution, especially genome instability after recursive rounds of paleo-polyploidization. They contributed to understanding genes regulating the formation and elongation of fibers.
In this era of information technology and big data, biology-related technology advances every day and data is increasing at a fast pace. The big data produced requires to be quickly and deeply analyzed to yield useful knowledge. In the future, we will have a lot of opportunity to elucidate the cotton genome and other omics data to clarify the biology of cotton, especially the regulation of fiber biosynthesis. We look forward to seeing the big genome data can be fully analyzed and then applied to agricultural practices to improve the cotton yield quality, contributing to humans’ harmonious life on the earth.
Author Contributions
XW conceived and led the research. YP and FM performed the analysis or joined discussion. XW and YP wrote the paper.
Funding
We appreciate financial support from Hebei Province Science and Technology Support Program (14962905D to YP), and Department of Education of Hebei Province (Y2012025 to YP), the Ministry of Science and Technology of the People’s Republic of China (2016YFD0101001 to XW), National Natural Science Foundation of China (3117022 to XW), and Tangshan Key Laboratory Project to XW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the helpful discussion with researchers at the iGeno co Ltd, China.
References
Applequist, W. L., Cronn, R., Wendel, J. F. (2001). Comparative development of fiber in wild and cultivated cotton. Evol. Dev. 3, 3–17. doi: 10.1046/j.1525-142x.2001.00079.x
Argout, X., Salse, J., Aury, J. M., Guiltinan, M. J., Droc, G., Gouzy, J., et al. (2011). The genome of Theobroma cacao. Nat. Genet. 43, 101–108. doi: 10.1038/ng.736
Basra, A. S., Malik, C. P. (1984). Development of the cotton fiber. Int. Rev. Cvtol 89, 65–113. doi: 10.1016/S0074-7696(08)61300-5
Bowers, J. E., Chapman, B. A., Rong, J., Paterson, A. H. (2003). Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438. doi: 10.1038/nature01521
Chen, Z. J., Scheffler, B. E., Dennis, E., Triplett, B. A., Zhang, T., Guo, W., et al. (2007). Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 145, 1303–1310. doi: 10.1104/pp.107.107672
Chen, Z. J., Sreedasyam, A., Ando, A., Song, Q., De Santiago, L. M., Hulse-Kemp, A. M., et al. (2020). Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 52, 525–533. doi: 10.1038/s41588-020-0614-5
Cheng, H., Sun, G., He, S., Gong, W., Peng, Z., Wang, R., et al. (2019). Comparative effect of allopolyploidy on transposable element composition and gene expression between Gossypium hirsutum and its two diploid progenitors. J. Integr. Plant Biol. 61, 45–59. doi: 10.1111/jipb.12763
Conover, J. L., Karimi, N., Stenz, N., Ane, C., Grover, C. E., Skema, C., et al. (2019). A Malvaceae mystery: A mallow maelstrom of genome multiplications and maybe misleading methods? J. Integr. Plant Biol. 61, 12–31. doi: 10.1111/jipb.12746
Cronn, R. C., Small, R. L., Haselkorn, T., Wendel, J. F. (2002). Rapid diversification of the cotton genus (Gossypium: Malvaceae) revealed by analysis of sixteen nuclear and chloroplast genes. Am. J. Bot. 89, 707–725. doi: 10.3732/ajb.89.4.707
Cronn, R., Small, R. L., Haselkorn, T., Wendel, J. F. (2003). Cryptic repeated genomic recombination during speciation in Gossypium gossypioides. Evolution 57, 2475–2489. doi: 10.1111/j.0014-3820.2003.tb01493.x
Ding, M., Chen, J., Jiang, Y., Lin, L., Cao, Y., Wang, M., et al. (2015). Genome-wide investigation and transcriptome analysis of the WRKY gene family in Gossypium. Mol. Genet. Genomics 290, 151–171. doi: 10.1007/s00438-014-0904-7
Ditta, A., Zhou, Z., Cai, X., Wang, X., Okubazghi, K. W., Shehzad, M., et al. (2018). Assessment of genetic diversity, population structure, and evolutionary relationship of uncharacterized genes in a novel germplasm collection of diploid and allotetraploid Gossypium accessions using EST and genomic SSR markers. Int. J. Mol. Sci. 19, 2401. doi: 10.3390/ijms19082401
Du, X., Huang, G., He, S., Yang, Z., Sun, G., Ma, X., et al. (2018). Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 50, 796–802. doi: 10.1038/s41588-018-0116-x
Fawcett, J. A., Maere, S., Van De Peer, Y. (2009). Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc. Natl. Acad. Sci. U. S. A. 106, 5737–5742. doi: 10.1073/pnas.0900906106
Galindo-Gonzalez, L., Mhiri, C., Deyholos, M. K., Grandbastien, M. A. (2017). LTR-retrotransposons in plants: Engines of evolution. Gene 626, 14–25. doi: 10.1016/j.gene.2017.04.051
Grover, C. E., Zhu, X., Grupp, K. K., Jareczek, J. J., Gallagher, J. P., Szadkowski, E., et al. (2015). Molecular confirmation of species status for the allopolyploid cotton species, Gossypium ekmanianum Wittmack. Genet. Resour. Crop Evol. 62, 103–114. doi: 10.1007/s10722-014-0138-x
Guo, H., Wang, X., Gundlach, H., Mayer, K. F., Peterson, D. G., Scheffler, B. E., et al. (2014). Extensive and biased intergenomic nonreciprocal DNA exchanges shaped a nascent polyploid genome, Gossypium (cotton). Genetics 197, 1153–1163. doi: 10.1534/genetics.114.166124
Hawkins, J. S., Kim, H., Nason, J. D., Wing, R. A., Wendel, J. F. (2006). Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium. Genome Res. 16, 1252–1261. doi: 10.1101/gr.5282906
Hu, Y., Chen, J., Fang, L., Zhang, Z., Ma, W., Niu, Y., et al. (2019). Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51, 739–748. doi: 10.1038/s41588-019-0371-5
Huang, G., Wu, Z., Percy, R. G., Bai, M., Li, Y., Frelichowski, J. E., et al. (2020). Genome sequence of Gossypium herbaceum and genome updates of Gossypium arboreum and Gossypium hirsutum provide insights into cotton A-genome evolution. Nat. Genet. 52, 516–524. doi: 10.1038/s41588-020-0607-4
Kim, H. J., Triplett, B. A. (2001). Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol. 127, 1361–1366. doi: 10.1104/pp.010724
Li, F., Fan, G., Wang, K., Sun, F., Yuan, Y., Song, G., et al. (2014). Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 46, 567–572. doi: 10.1038/ng.2987
Li, F., Fan, G., Lu, C., Xiao, G., Zou, C., Kohel, R. J., et al. (2015). Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530. doi: 10.1038/nbt.3208
Liu, X., Zhao, B., Zheng, H. J., Hu, Y., Lu, G., Yang, C. Q., et al. (2015). Gossypium barbadense genome sequence provides insight into the evolution of extra-long staple fiber and specialized metabolites. Sci. Rep. 5, 14139. doi: 10.1038/srep14139
Meng, F., Pan, Y., Wang, J., Yu, J., Liu, C., Zhang, Z., et al. (2020). Cotton duplicated genes produced by polyploidy show significantly elevated and unbalanced evolutionary rates, overwhelmingly perturbing gene tree topology. Front. Genet. 11, 239. doi: 10.3389/fgene.2020.00239
Page, J. T., Liechty, Z. S., Alexander, R. H., Clemons, K., Hulse-Kemp, A. M., Ashrafi, H., et al. (2016). DNA sequence evolution and rare homoeologous conversion in tetraploid cotton. PLoS Genet. 12, e1006012. doi: 10.1371/journal.pgen.1006012
Pang, C. Y., Wang, H., Pang, Y., Xu, C., Jiao, Y., Qin, Y. M., et al. (2010). Comparative proteomics indicates that biosynthesis of pectic precursors is important for cotton fibre and Arabidopsis root hair elongation. Mol. Cell Proteomics 9, 2019–2033. doi: 10.1074/mcp.M110.000349
Paterson, A. H., Wendel, J. F., Gundlach, H., Guo, H., Jenkins, J., Jin, D., et al. (2012). Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 492, 423–427. doi: 10.1038/nature11798
Ruan, Y. L., Xu, S. M., White, R., Furbank, R. T. (2004). Genotypic and developmental evidence for the role of plasmodesmatal regulation in cotton fiber elongation mediated by callose turnover. Plant Physiol. 136, 4104–4113. doi: 10.1104/pp.104.051540
Salih, H., Gong, W., He, S., Sun, G., Sun, J., Du, X. (2016). Genome-wide characterization and expression analysis of MYB transcription factors in Gossypium hirsutum. BMC Genet. 17, 129. doi: 10.1186/s12863-016-0436-8
Salmon, A., Flagel, L., Ying, B., Udall, J. A., Wendel, J. F. (2010). Homoeologous nonreciprocal recombination in polyploid cotton. New Phytol. 186, 123–134. doi: 10.1111/j.1469-8137.2009.03093.x
Shi, Y. H., Zhu, S. W., Mao, X. Z., Feng, J. X., Qin, Y. M., Zhang, L., et al. (2006). Transcriptome profiling, molecular biological, and physiological studies reveal a major role for ethylene in cotton fiber cell elongation. Plant Cell 18, 651–664. doi: 10.1105/tpc.105.040303
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., Paterson, A. H. (2008a). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Tang, H., Wang, X., Bowers, J. E., Ming, R., Alam, M., Paterson, A. H. (2008b). Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 18, 1944–1954. doi: 10.1101/gr.080978.108
Teh, B. T., Lim, K., Yong, C. H., Ng, C. C. Y., Rao, S. R., Rajasegaran, V., et al. (2017). The draft genome of tropical fruit durian (Durio zibethinus). Nat. Genet. 49, 1633–1641. doi: 10.1038/ng.3972
Udall, J. A., Long, E., Hanson, C., Yuan, D., Ramaraj, T., Conover, J. L., et al. (2019). De novo genome sequence assemblies of Gossypium raimondii and Gossypium turneri. G3 (Bethesda) 9, 3079–3085. doi: 10.1534/g3.119.400392
Van de Peer, Y., Fawcett, J. A., Proost, S., Sterck, L., Vandepoele, K. (2009). The flowering world: a tale of duplications. Trends Plant Sci. 14, 680–688. doi: 10.1016/j.tplants.2009.09.001
Wang, K., Wang, Z., Li, F., Ye, W., Wang, J., Song, G., et al. (2012). The draft genome of a diploid cotton Gossypium raimondii. Nat. Genet. 44, 1098–1103. doi: 10.1038/ng.2371
Wang, X., Guo, H., Wang, J., Lei, T., Liu, T., Wang, Z., et al. (2016). Comparative genomic de-convolution of the cotton genome revealed a decaploid ancestor and widespread chromosomal fractionation. New Phytol. 209, 1252–1263. doi: 10.1111/nph.13689
Wang, J., Yuan, J., Yu, J., Meng, F., Sun, P., Li, Y., et al. (2019). Recursive paleohexaploidization shaped the durian genome. Plant Physiol. 179, 209–219. doi: 10.1104/pp.18.00921
Wang, M., Tu, L., Yuan, D., Zhu, D., Shen, C., Li, J., et al. (2019). Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 51, 224–229. doi: 10.1038/s41588-018-0282-x
Wendel, J. F., Albert, V. A. (1992). Phylogenetics of the Cotton genus (Gossypium): Character-state weighted parsimony analysis of chloroplast-DNA restriction site data and its systematic and biogeographic implications. Syst. Bot. 17, 115–143. doi: 10.2307/2419069
Wilkins, T. A., Arpat, A. B. (2005). The cotton fiber transcriptome. Physiol. Plant. 295, 295–300. doi: 10.1111/j.1399-3054.2005.00514.x
Wu, Z., Yang, Y., Huang, G., Lin, J., Xia, Y., Zhu, Y. (2017). Cotton functional genomics reveals global insight into genome evolution and fiber development. J. Genet. Genomics 44, 511–518. doi: 10.1016/j.jgg.2017.09.009
Yoo, M. J., Wendel, J. F. (2014). Comparative evolutionary and developmental dynamics of the cotton (Gossypium hirsutum) fiber transcriptome. PLoS Genet. 10, e1004073. doi: 10.1371/journal.pgen.1004073
Yu, J., Jung, S., Cheng, C. H., Ficklin, S. P., Lee, T., Zheng, P., et al. (2014). CottonGen: a genomics, genetics and breeding database for cotton research. Nucleic Acids Res. 42, D1229–D1236. doi: 10.1093/nar/gkt1064
Yuan, D., Tang, Z., Wang, M., Gao, W., Tu, L., Jin, X., et al. (2015). The genome sequence of Sea-Island cotton (Gossypium barbadense) provides insights into the allopolyploidization and development of superior spinnable fibres. Sci. Rep. 5, 17662. doi: 10.1038/srep17662
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. doi: 10.1038/nbt.3207
Zhu, H., Han, X., Lv, J., Zhao, L., Xu, X., Zhang, T., et al. (2011). Structure, expression differentiation and evolution of duplicated fiber developmental genes in Gossypium barbadense and G. hirsutum. BMC Plant Biol. 11, 40. doi: 10.1186/1471-2229-11-40
Keywords: cotton, genome, phylogeny, transposable elements, fiber
Citation: Pan Y, Meng F and Wang X (2020) Sequencing Multiple Cotton Genomes Reveals Complex Structures and Lays Foundation for Breeding. Front. Plant Sci. 11:560096. doi: 10.3389/fpls.2020.560096
Received: 08 May 2020; Accepted: 19 August 2020;
Published: 16 September 2020.
Edited by:
Ruslan Kalendar, University of Helsinki, FinlandCopyright © 2020 Pan, Meng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiyin Wang, d2FuZ3hpeWluQHZpcC5zaW5hLmNvbQ==